

Pdi1 knockdown decreases apoB100 synthesis, reduces MTP activity and apoB100 lipidation, and impairs the oxidative folding of apoB100, which causes defective VLDL secretion. PDI1 promotes formation of disulfide bonds in apoB100 and serves as its disulfide isomerase.
Abstract
Apolipoprotein (apo) B is an obligatory component of very low density lipoprotein (VLDL), and its cotranslational and posttranslational modifications are important in VLDL synthesis, secretion, and hepatic lipid homeostasis. ApoB100 contains 25 cysteine residues and eight disulfide bonds. Although these disulfide bonds were suggested to be important in maintaining apoB100 function, neither the specific oxidoreductase involved nor the direct role of these disulfide bonds in apoB100-lipidation is known. Here we used RNA knockdown to evaluate both MTP-dependent and -independent roles of PDI1 in apoB100 synthesis and lipidation in McA-RH7777 cells. Pdi1 knockdown did not elicit any discernible detrimental effect under normal, unstressed conditions. However, it decreased apoB100 synthesis with attenuated MTP activity, delayed apoB100 oxidative folding, and reduced apoB100 lipidation, leading to defective VLDL secretion. The oxidative folding–impaired apoB100 was secreted mainly associated with LDL instead of VLDL particles from PDI1-deficient cells, a phenotype that was fully rescued by overexpression of wild-type but not a catalytically inactive PDI1 that fully restored MTP activity. Further, we demonstrate that PDI1 directly interacts with apoB100 via its redox-active CXXC motifs and assists in the oxidative folding of apoB100. Taken together, these findings reveal an unsuspected, yet key role for PDI1 in oxidative folding of apoB100 and VLDL assembly.
INTRODUCTION
Secretion of very low density lipoprotein (VLDL) from the liver is vital for maintaining circulating lipid homeostasis (Hamilton, 1972). Overproduction of hepatic VLDL particles is a common cause of systemic hyperlipidemia in humans, a high risk factor of cardiovascular disease in obesity and type 2 diabetes (Adiels et al., 2008). The fundamental cellular mechanism(s) that drive lipid-rich VLDL assembly are not fully understood (Sundaram and Yao, 2010).
Hepatic VLDL is secreted as a spherical particle containing a requisite structural protein, apolipoprotein B (apoB), which binds with different lipid species, mainly bulk neutral lipids, especially triglycerides (TGs; Yang et al., 1995). ApoB is synthesized by cotranslational lipidation in the rough endoplasmic reticulum (rER; Rutledge et al., 2010) and undergoes further posttranslational lipidation within the smooth ER (sER) lumen. Both cotranslational and posttranslational lipidations of apoB are mediated by the resident ER heterodimeric microsomal triglyceride transfer protein (MTP) complex, which comprises a large subunit (MTP) and a smaller subunit, protein disulfide isomerase 1 (PDI1; Wetterau et al., 1991). Previous genetic studies demonstrated that the large subunit of MTP is absolutely required for apoB lipidation, VLDL maturation, and secretion (Raabe et al., 1999). However, the role of PDI1 as a subunit of MTP complex in apoB synthesis and lipidation remains to be elucidated.
Each VLDL particle contains a single apoB molecule, in one of two forms, either apoB100 (the full-length protein) or apoB48 (the N-terminal 48% of apoB100). The two forms are encoded by the same ApoB gene, but RNA editing modifies ApoB mRNA at codon 2153, which converts a glutamine codon to a stop codon, giving rise to apoB48 (Blanc and Davidson, 2011). ApoB100 is the only form in human liver, although both apoB100 and 48 are present in rodent livers (Blanc and Davidson, 2003). ApoB100 is a large hydrophobic protein, with molecular weight >500 kDa (Gibbons et al., 2004). Folding of apoB is a complex process, involving an array of protein chaperones and posttranslational modifications including lipidation, glycosylation, and disulfide bond formation (Burch and Herscovitz, 2000). ApoB100 that fails to fold into its mature form is directed to proteasomal and/or autophagy-mediated degradation (Liao et al., 2003).
PDI1 is a resident foldase containing two redox-active thioredoxin domains that catalyze the formation and isomerization of disulfide bonds during protein folding in the ER. PDI1 was identified >35 years ago (Carmichael et al., 1977) and has been extensively studied in vitro (Hatahet and Ruddock, 2009). Biochemical studies with model substrates demonstrated that PDI1 increases the rate of disulfide bond formation without changing the protein-folding pathway (Bulleid and Freedman, 1988; Hatahet and Ruddock, 2009). In addition to being a molecular chaperone of MTP, PDI1 exhibits antiaggregation as well as antichaperone activities (Puig and Gilbert, 1994; Sideraki and Gilbert, 2000) in a concentration- and calcium-dependent manner that does not require its thiol-redox activity (Primm et al., 1996). Recent studies suggest that PDI retards disulfide bond formation and secretion of proinsulin (Rajpal et al., 2012). In addition, a requirement for PDI1 was demonstrated for reduction in infectivity of cholera toxin (Moore et al., 2010). However, it remains unknown whether PDI1 is required to promote oxidation of any substrate in vivo.
Previous studies demonstrated that disulfide bonds are required for folding and secretion of apoB100. ApoB100 contains 25 cysteines that form eight disulfide bonds that are largely clustered within the amino terminus (Shelness and Thornburg, 1996). Because apoB100 is a large protein and most of its disulfide bonds are formed between adjacent cysteine residues, it was believed that the shift in the migration of the disulfide-bonded form compared with its reduced form is practically undetectable (Burch and Herscovitz, 2000); therefore it remains unknown whether any or multiple PDI family members catalyze apoB100 disulfide bond formation in vivo. In this study, we successfully distinguished the disulfide-bonded form of apoB100 from its completely reduced form by electrophoresis on 5% polyacrylamide gels. We found that the smaller subunit of the MTP complex, PDI1, plays a key role in promoting disulfide bond formation within apoB100. Our data suggest that physiological lipidation of apoB100 requires correct disulfide bond formation in apoB100, which is mediated by PDI1. Taken together, our findings demonstrate an unsuspected role of PDI1 as a requisite disulfide isomerase for apoB100 processing.
RESULTS
Knockdown of PDI1 sensitizes cells to ER stress without altering ER homeostasis
To study the functional role of PDI1 in hepatic VLDL assembly, we first generated adenoviruses expressing Pdi1 short hairpin RNAs (shRNAs) to knock down PDI1 in rodent hepatoma cells. Adenovirus infects hepatoma cells with high efficiency, ensuring effective knockdown of target genes. Accordingly, Pdi1 mRNA levels were decreased by >95%, and PDI1 protein levels were almost undetectable in Hepa1-6 cells 3 d after adenoviral infection (confirmed with two sets of shRNAs; Supplemental Figure S1, A and B). For the purposes of this article, all studies were conducted with shRNA 1. Adenovirus-mediated Pdi1 knockdown was then tested in McA-RH7777 (McA) rat hepatoma cells, a widely used model for studying hepatic lipoprotein secretion (Boren et al., 1994; Liang et al., 2004; Yamaguchi et al., 2003). McA cells secrete predominantly apoB100 in VLDL particles with size and density distribution similar to that of human hepatocytes (Boren et al., 1994). Pdi1 mRNA and protein levels were decreased ∼80% after shRNA-mediated knockdown in McA cells (Figure 1, A and B). Moreover, knockdown of Pdi1 did not lead to compensatory up-regulation or off-target knockdown of other PDI family members, including ERp57 and ERp72 (Figure 1A and Supplemental Figure S1A) in both Hepa1-6 and McA cells. In addition, the Pdi1 knockdown mediated by adenovirus persisted at least 8 d after infection, allowing adequate time to perform the functional studies in Pdi1-knockdown cells (Supplemental Figure S2). These findings gave us confidence that our approach allows us to examine the phenotypes associated with Pdi1 knockdown in McA cells.
FIGURE 1:

Knockdown of Pdi1 sensitizes cells to ER stress. (A, B) Adenoviral expression of Pdi1 shRNA knocks down PDI1 levels in McA cells. (A) Relative abundance of mRNA expressed in control (NSi) and Pdi1-knockdown (PDI1i) cells. mRNA levels were normalized to actin levels. Values are relative to mRNA levels of control cells. *p < 0.01 vs. NSi. (B) PDI1 protein levels in control (NSi) and Pdi1-knockdown (PDI1i) cells. Cell lysates were subjected to immunoblotting analyses using PDI1 and vinculin antibodies. (C–E) Pdi1-knockdown cells are more sensitive to ER stress. (C, D) Cell survival was measured by cell counting after treatment with different concentration of Tm or Tg. *p < 0.01 vs. NSi in the same treatment group. (E) Protein levels of ER stress markers in control (NSi) and Pdi1-knockdown (PDI1i) cells. Cells were treated with 0.2 μM Tg for indicated time periods, and cell lysates were immunoblotted for cleaved caspase 3, PARP, eIF2α-P, CHOP, and BiP. (F) ER redox balance is not altered in Pdi1-knockdown cells. Cells expressing Grx-roGFP-iEER were treated with 10 mM DTT for 10 min and washed out. The oxidation status of Grx-roGFP-iEER was analyzed by nonreducing SDS–PAGE and immunoblotting with GFP, KDEL (GRP78 and GRP94), and PDI antibodies.
Because PDI1 is proposed to be an important ER protein chaperone and disulfide isomerase, we first tested whether Pdi1 knockdown disturbs ER homeostasis to activate the unfolded protein response (UPR) in either Hepa1-6 or McA cells. Our analyses revealed no significant changes in mRNA abundance for spliced Xbp1 or other ER chaperones, suggesting that the UPR is not activated upon Pdi1 knockdown in Hepa1-6 and McA cells (Figure 1A and Supplemental Figure S1A). However, after exposure to the ER stress inducer tunicamycin (Tm) or thapsigargin (Tg), we observed that Pdi1-knockdown cells (Hepa1-6 or McA cells) were more sensitive to ER stress, as indicated by decreased cell survival and increased levels of cleaved caspase 3 and PARP (Figure 1, C– E, and Supplemental Figure S1, C–E). Moreover, Pdi1 knockdown increased phosphorylation of eukaryotic initiation factor 2α (eIF2α) in response to Tg-induced ER stress and induced expression of C/EBP homologous protein (CHOP), a major UPR mediator of apoptosis, in both cell lines (Figure 1, C– E, and Supplemental Figure S1, C–E). Thus knockdown of Pdi1 increased sensitivity of cells to agents that cause ER stress.
To determine whether PDI deficiency alters the ER redox balance, which, in turn, affects ER homeostasis and oxidative protein folding, we generated McA cells that stably express in situ sensor molecules—green fluorescent protein (GFP) iE variant fused to ER-localized glutaredoxin-1 (Grx-roGFP-iEER; Birk et al., 2013). Grx-roGFP-iEER exists in reduced and oxidized forms, which reflect the redox status of the ER. We performed dithiothreitol (DTT) recovery experiments in PDI-deficiency cells to determine the oxidation potential as described in Materials and Methods. These experiments revealed that despite its demonstrated role as a major oxidoreductase in the ER (Appenzeller-Herzog et al., 2010), PDI1 knockdown did not change the redox status of Grx-roGFP-iEER (Figure 1F). We interpret these results to suggest that PDI1 knockdown does not cause systemic redox imbalance in the ER (Figure 1F). Taken together, the results indicate the knockdown of Pdi1 does not significantly alter ER homeostasis under normal conditions but sensitizes cells to ER stress–induced cell death.
Knockdown of PDI1 decreases apoB100 synthesis and secretion in McA cells
We next examined protein levels of apoB100 and MTP and found that Pdi1 knockdown drastically decreased intracellular apoB100 levels by ∼50% without altering its mRNA expression. Pdi1 knockdown had a relatively milder effect on MTP, with a ∼20% decrease in steady-state protein levels (Figure 2, A and B). Nonetheless, these observations indicate that PDI1 plays an essential role in maintaining intracellular apoB100 levels.
FIGURE 2:

Knockdown of Pdi1 decreases apoB synthesis and secretion in McA cells. (A, B) Pdi1 knockdown in McA cells reduces apoB100 protein but not mRNA levels. (A) Relative abundance of apoB and MTP mRNAs in control (NSi) and Pdi1-knockdown (PDI1i) cells. Relative abundance was normalized to actin mRNA levels. Values are relative to mRNA levels of control cells. *p < 0.01 vs. NSi (B) ApoB100 and MTP protein levels in control (NSi) and Pdi1-knockdown (PDI1i) cells. Cell lysates were immunoblotted for PDI1, apoB100, MTP, and actin. (C) Pdi1-knockdown (PDI1i) in McA cells reduces apoB100 synthesis and secretion. Pulse-chase analysis of intracellular and secreted apoB100 was performed in control (NSi) and Pdi1-knockdown (PDI1i) McA cells. Double asterisks represent a nonspecific band. (D) Proteasomal degradation reduces apoB100 levels in McA cells. Pulse-chase analysis of intracellular apoB100 and secreted apoB was performed in control (NSi) and Pdi1-knockdown (PDI1i) McA cells in the absence (control) or presence of 10 μM MG132. (E) The amount of apoB100 in intracellular and secreted fractions was quantified by ImageJ. Recovery rates of apoB100 are presented as percentage of the sum of apoB100 (intracellular plus secreted) over total apoB100 at zero time point of pulse-chase experiments. Pulse-chase experiments were repeated at least twice.
To determine how PDI1 expression affects the steady-state level of apoB100 protein, we first performed pulse-labeling experiments to monitor apoB synthesis using [35S]methionine/cysteine (Met/Cys) over a 25-min time course in control and PDI1-knockdown McA cells. ApoB synthesis was significantly reduced by Pdi1 knockdown at each time point tested, indicating that Pdi1 knockdown attenuates full-length apoB synthesis (Supplemental Figure S3A). We also performed pulse-chase analyses to compare rates of apoB secretion and synthesis in the presence or absence of PDI1. As shown in the pulse-labeling experiments, Pdi1- knockdown McA cells synthesized ∼50% less apoB100 than control cells, and the secretion of apoB100 was also proportionally decreased (Figure 2C and Supplemental Figure S3B). However, overall apoB100 recovery rates were not significantly different in these two cell lines (Supplemental Figure S3B), suggesting that posttranslational degradation of apoB100 is not accelerated in Pdi1-knockdown cells. In addition, the decrease in apoB100 synthesis was not due to a general decrease in protein synthesis (unpublished data). These observations suggest that PDI1 is important for the synthesis of full-length apoB100.
Because endogenous apoB100 is constitutively degraded cotranslationally (Pan et al., 2008), it was important to address the question of whether apoB cotranslational degradation reduces the cellular recovery of apoB100 in Pdi1-knockdown cells. Accordingly, we performed pulse-chase experiments in cells treated with MG132 to inhibit proteasome-mediated protein degradation. Indeed, MG132 stabilized intracellular accumulation of apoB100 in both control and Pdi1-knockdown cells based on the quantification results (Figure 2, D and E), indicating that a portion of apoB100 is subject to cotranslational degradation by the proteasome. However, compared with control cells, Pdi1-knockdown cells still contained significantly less labeled apoB100, suggesting that either apoB synthesis is compromised and/or nascent apoB undergoes nonproteasomal degradation in Pdi1-knockdown cells. These observations together suggest that PDI1 is important for apoB100 synthesis and stabilization.
Knockdown of PDI1 decreases apoB100 lipidation in McA cells
Poorly lipidated nascent apoB100 is readily degraded cotranslationally in a proteasome-dependent and/or -independent manner (Pan et al., 2008). To determine whether Pdi1 knockdown disrupts lipidation of apoB, we analyzed the density distribution of apoB100 particles secreted from control and Pdi1-knockdown cells. Cells were labeled continuously for 3 h with [35S]Met/Cys, and the labeled medium was subjected to density gradient ultracentrifugation (DGUC) to separate apoB100-containing particles based on their relative hydrated density. ApoB100-containing particles secreted from control cells migrated primarily in the VLDL fractions (fractions 1 and 2; Figure 3, A and B) either in the presence or absence of oleic acid in the media. In contrast, most of the apoB100-containing particles secreted from Pdi1 knockdown cells were distributed as low-density lipoproteins (fractions 3–6; Figure 3, A and B). We interpret this shift to suggest that the relative distribution of apoB100 in the lipid-rich VLDL fractions from Pdi1-knockdown cells was decreased, whereas the proportion of apoB100 migrating in lipid-poor particles correspondingly increased. In contrast to apoB100, apoB48 was mainly associated with the lipid-poor particles and was not altered by Pdi1 knockdown in McA cells (Figure 3, A and B). This result indicates that PDI1 is required for full lipidation of apoB100 and VLDL maturation.
FIGURE 3:

Knockdown of Pdi1 decreases apoB100 lipidation in McA cells. (A–D) Pdi1 knockdown reduces apoB100 lipidation. McA cells were continuously labeled with [35S]Met/Cys containing BSA (A) or 400 μM OA (C) for 4 h. The medium was collected for determination of buoyant density of secreted apoB-containing lipoproteins from control (NSi) and Pdi1 knockdown (PDI1i) McA cells by DGUC, followed by immunoprecipitation for apoB100. The amount of apoB100 in each fraction was quantified by ImageJ. Relative distributions were calculated according to the percentage of apoB100 in each fraction relative to its total intensity across the gel (B, D). (E, F) Efficient TG secretion requires catalytically active PDI1. TG secretion (E) and MTP activity (F) were decreased in Pdi1-knockdown McA cells. TG synthesis and secretion (E) and MTP activity (F) were measured in control and Pdi1-knockdown McA cells. *p < 0.01 vs. NSi. (G–I) Overexpression of human wild-type PDI1 but not catalytically inactive PDI1 rescues decreased TG secretion in Pdi1-knockdown McA cells. (G) Wild-type and mutant PDI1 are expressed at equal levels in Pdi1-knockdown McA cells. β-Galactosidase (β-gal), human wild-type PDI1 (PDI), and catalytically inactive PDI1 (PDImu) were expressed using adenoviruses, and their expression levels were measured by Western blotting analysis. TG synthesis and secretion (H) and MTP activity (I) were measured in control cells or Pdi1-knockdown cells with overexpression of β-gal, PDI, or PDImu McA cells. PBS denotes no adenovirus infection. *p < 0.01 vs. PBS, PDI1, or PDI1mu; **p < 0.01 vs. PBS or PDI1.
ApoB is the major carrier of TG secreted from hepatocytes, and therefore decreased apoB100 lipidation can directly decrease TG secretion in McA cells upon Pdi1 knockdown. To confirm that Pdi1 knockdown reduces TG secretion without altering TG synthesis, we measured TG synthesis and secretion by labeling cells with [3H]glycerol, followed by thin-layer chromatography (TLC) analyses. Analysis of intracellular TG levels indicated that TG synthesis was not altered, whereas TG secretion decreased >40% upon Pdi1 knockdown (Figure 3E). This observation is consistent with the finding that Pdi1-knockdown cells secrete apoB in lipid-poor particles (Figure 3, A and B).
Because PDI1 is a molecular chaperone for MTP, we next asked whether Pdi1 knockdown might decrease MTP activity to limit apoB lipidation. Indeed, MTP activity in Pdi1-knockdown McA cells decreased by >50% (Figure 3F). Thus our results indicate that PDI1 is required for McA cells to assemble and/or secrete lipid-rich apoB100 VLDL particles, at least in part, by maintaining MTP activity. Because the chaperone activity of PDI1 is independent of its isomerase activity (Wang et al., 1997), we next determined the extent to which the decreased apoB lipidation was due to reduced MTP activity in Pdi1-knockdown cells. For this purpose, we delivered adenoviruses expressing either wild-type human PDI1 or a catalytically inactive PDI1 in which all four redox-active cysteines are mutated to alanines (Uehara et al., 2006) into Pdi1-knockdown cells. These experiments revealed that expression of either wild-type or catalytically inactive PDI1 fully restored MTP activity, yet TG secretion was only partially restored by the catalytically inactive PDI1 in Pdi1-knockdown cells (Figure 3, G–I). In addition, we conducted pulse-chase experiments to measure apoB secretion in Pdi1-knockdown cells in which we rescued expression with either wild-type PDI1 or a catalytically inactive PDI1. These results showed that overexpression of the wild-type but not the catalytically inactive PDI1 restored apoB secretion in the PDI1-knockdown McA cells (Supplemental Figure S4), which is consistent with the findings regarding TG secretion. Therefore, from both apoB secretion and TG secretion experiments, we conclude that the secretion of apoB is at least in part dependent on catalytic oxidoreductase activity of PDI1 in McA cells. These findings suggest that PDI1 exerts an additional MTP-independent role in apoB synthesis, lipidation, and/or secretion (Figure 3, G–I).
Pdi1 knockdown decreases apoB100 oxidative folding in McA cells
Previous work suggested that the oxidoreductase activity of PDI1 is required for MTP-independent steps of apoB100 synthesis and secretion (Wang et al., 1997). To explore this possibility in more detail, we used pulse-chase experiments in which newly synthesized proteins were resolved by nonreducing gel electrophoresis to allow us to test whether Pdi1 knockdown modifies apoB disulfide bond formation. We found that intracellular and secreted apoB100 migrated more slowly at the later chase time points in Pdi1-knockdown cells, raising the possibility that disulfide bond formation is delayed for newly synthesized apoB100 (Figure 4A, asterisk). To our surprise, apoB100 with partially oxidized disulfide bonds were also secreted in Pdi1-knockdown cells, suggesting that these apoB100 molecules undergo sufficient lipidation to attenuate posttranslational degradation.
FIGURE 4:

Knockdown of Pdi1 decreases apoB100 oxidative folding in McA cells. (A) Pdi1 knockdown decreases oxidative folding of apoB100. Pulse-chase analysis of intracellular and secreted apoB100 in control (NSi) and Pdi1-knockdown (PDI1i) McA cells by nonreducing gel electrophoresis. The bracket indicates both oxidized and reduced form of apoB100 in nonreducing gel. (B) Pdi1-knockdown selectively reduces oxidative folding of apoB100. Oxidative folding of intracellular apoB100, albumin, and transferrin were measured by reducing (first two lanes) and nonreducing gel electrophoresis. Ox, oxidized form, and Re, reduced form, of the protein. (C) Oxidative folding of apoB100 requires catalytically active PDI1. Oxidative folding of intracellular apoB100, albumin, and transferrin was measured by nonreducing gel electrophoresis in control (NSi) cells, Pdi1-knockdown (PDI1i) and Pdi1-knockdown (PDI1i) cells with exogenous expression of HA-tagged substrate-trap mutant (huPDImu) or wild-type (huPDI) human PDI1. (D) Oxidative folding of apoB100 does not require MTP activity. Oxidative folding of intracellular apoB100 and albumin were analyzed by nonreducing gel electrophoresis in the absence or presence of an MTP inhibitor (CP-10447) at 0, 0.3, or 3 μM.
To compare further the kinetics of apoB100 disulfide bond formation between control and Pdi1 knockdown cells, we synchronized protein synthesis by puromycin pretreatment before the pulse-chase experiments. In addition, we used N-ethylmaleimide (NEM) to alkylate free thiols to prevent postlysis alterations in disulfide bonds. Furthermore, because apoB100 synthesis is decreased in Pdi1-knockdown cells, in order to have comparable levels of apoB100 to study apoB100 disulfide bond formation in both cells, we used more lysates from Pdi1- knockdown cells to conduct these experiments. First, we analyzed apoB100 on both reducing and nonreducing gels and demonstrated that the oxidized form of apoB100 (Ox in Figure 4B) migrated with greater mobility than the fully reduced form of apoB100 (Re in Figure 4B). Second, analysis of reducing gels demonstrated that the migration of apoB100 from control and Pdi1-knockdown cells was comparable, suggesting that the difference in migration is due to cysteine oxidation and not to another posttranslational modification (Figures 4, B [darker in Supplemental Figure S4] and D, and 5C). Furthermore, almost all of the intracellular apoB100 matured to the oxidized form upon translation in control cells, whereas a significant portion of apoB100 remained only partially oxidized until the end of the chase period in Pdi1-knockdown cells (Figure 4B).
In contrast to apoB100, the oxidative folding of other secretory proteins, including albumin and transferrin, was not altered in Pdi1-knockdown cells during the time course of our pulse-chase experiments (Figure 4B). We did not specifically investigate the disulfide bond formation of albumin and transferrin in a shorter time course of pulse-chase experiment in Pdi1- knockdown cells since it was previously demonstrated that PDI1 is not essential for oxidative folding of these two proteins in HepG2 cells (Rutkevich et al., 2010). Nevertheless, our preliminary observations in McA cells are consistent with this report (Rutkevich et al., 2010). Taken together, our results indicate that Pdi1 knockdown selectively delays apoB100 oxidative folding, indicating that PDI1 serves as a disulfide isomerase specifically for apoB100.
To confirm that the oxidative folding of apoB100 is dependent on PDI1, we stably expressed hemagglutinin (HA)-tagged wild-type human PDI1 (HA-PDI1) or a substrate-trap mutant of PDI1 (HA-PDI1CXXA) in McA cells (Rutkevich et al., 2010). We asked whether ectopic expression of human PDI1 can compensate for the delayed oxidation of apoB100 caused by Pdi1 knockdown in McA cells. Indeed, wild-type human PDI1, but not the substrate-trap mutant, promoted apoB100 oxidative folding in Pdi1-knockdown cells, indicating that disulfide bond formation of apoB100 is dependent on, at least in part, the oxidoreductase activity of PDI1 (Figure 4C). Although overexpression of wild-type human PDI1 appears to increase intracellular retention of albumin and transferrin, it does not affect their oxidative folding as it does for apoB100. Therefore the oxidoreductase activity of PDI1 is specifically required for apoB100 oxidative folding.
To test further whether PDI1 improves the oxidative folding of apoB100 independent of MTP activity, we treated McA cells with an MTP inhibitor, CP-10447 (Pan et al., 2002), at 0, 0.3, and 3 μM and analyzed the oxidative folding of apoB100 as before (Figure 4D). The MTP inhibitor decreased intracellular recovery of apoB100 in McA cells as previously described (Fukuda et al., 1983). However, it did not increase the amount of partially oxidized apoB100 compared with that observed upon Pdi1 knockdown (Figure 4D). Therefore PDI1 plays both MTP-dependent and MTP-independent roles in apoB100 synthesis and VLDL assembly.
PDI1 promotes apoB100 lipidation in McA cells
To confirm that apoB100 is a substrate for PDI1, we analyzed the apoB100 protein complex in Pdi1-knockdown McA cells that stably express either HA-PDI1 or HA-PDI1CXXA as described before. Cell lysates were immunoprecipitated using apoB antibody, and the immunocomplexes were analyzed by SDS–PAGE under nonreducing conditions, followed by immunoblotting with anti-HA antibody. If apoB100 is a substrate of PDI1, the substrate-trap mutant of PDI1 should be present in covalent association with apoB100 under nonreducing conditions. Indeed, we detected HA-PDI1CXXA in the apoB100 immunocomplex under nonreducing conditions (Figure 5A), indicating that the cysteines of the CXXA motif in substrate-trap mutant PDI1 can form disulfide bonds with cysteine residues of apoB100. This observation indicates that PDI1 promotes formation of disulfide bonds in apoB100 via intermolecular disulfide bonds. Thus PDI1 is important for stabilizing apoB100 structure by serving as a thiol-disulfide oxidoreductase.
FIGURE 5:
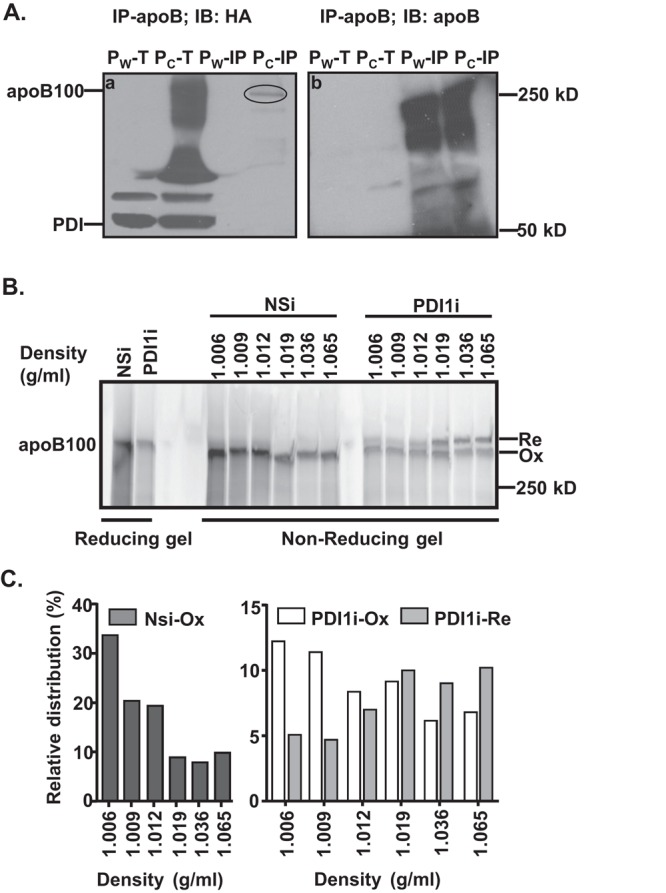
Reduced apoB100 oxidative folding decreases apoB lipidation. (A) PDI1 forms intermolecular disulfide bond(s) with apoB100. Lysates from cells that stably express HA-tagged wild-type (Pw: HA-PDI1) or substrate-trap mutant (Pc: HA-PDICXXA) of human PDI1 were immunoprecipitated with apoB100 antibody and analyzed by nonreducing SDS–PAGE and immunoblotting against HA antibody. The circle indicates PDI1 complexed with apoB100. IP, immunoprecipitated protein complex; T, total cell lysates. (B) Impaired apoB100 oxidative folding decreases apoB100 lipidation. Secreted apoB100-containing lipoproteins from McA cells were separated by DGUC as described in Figure 3 and analyzed by nonreducing gel electrophoresis. (C) The amount of apoB100 in each fraction was quantified by ImageJ. Relative distributions were calculated according to the percentage of apoB100 in each fraction relative to its total intensity across the gel.
To examine further whether reduced apoB100 oxidative folding could affect apoB100 lipidation in Pdi1-knockdown McA cells, we analyzed the DGUC-fractionated, apoB100-containing lipoprotein particles from the media using nonreducing gels. We found that oxidized apoB100 secreted by the control McA cells mainly migrated in fractions 1 and 2, which are associated with more abundant lipids, whereas reduced or partially oxidized apoB100 secreted by Pdi1-knockdown McA cells predominantly migrated in the lipid-poor fractions (fractions 4–6; Figure 5, C and D). On the basis of these findings, we conclude that PDI1-mediated apoB100 oxidative folding may facilitate apoB100 lipidation and in turn mitigate cotranslational and posttranslational degradation.
DISCUSSION
Disturbances of hepatic lipid metabolism from a variety of pathways may lead to excessive production of VLDL particles, which in turn contribute to atherogenic lipid and lipoprotein abnormalities (Dobiasova and Frohlich, 2001). ApoB100 is the key structural protein of VLDL, and a better understanding of the apoB100 folding and VLDL assembly process should provide new insights and avenues to treat hyperlipidemia and related diseases. Although the large subunit of MTP complex is known to be important for apoB100 lipidation (Kulinski et al., 2002), the role of the small subunit, PDI1, is not fully understood. PDI1 is generally believed to serve a chaperone function for MTP and maintain MTP activity to promote apoB100 lipidation (Bradbury et al., 1999). In this study, we provide evidence that PDI1 not only is important for MTP activity but also plays a key independent role in apoB100 oxidative folding. This conclusion is based on several sets of observations. First, Pdi1 knockdown reduced secretion of apoB100, which could be explained by either decreased apoB100 synthesis or accelerated apoB100 degradation. Second, Pdi1 knockdown reduced apoB100 lipidation, which was, at least in part, attributable to decreased MTP activity. Third, Pdi1 knockdown selectively delayed disulfide bond formation in newly synthesized apoB100, which in turn affected apoB100 lipidation. Together these findings provide new insight into the apoB100 oxidative folding process, which is tightly connected with apoB100 lipidation and lipid-rich VLDL assembly.
McA cells are widely used as a model for studying lipoprotein secretion because it is a cell line most similar to human hepatocytes in secretion of TG-rich VLDL (Boren et al., 1994; Ota et al., 2008). Here we knocked down Pdi1 in McA cells to explore the role for PDI1 in apoB100 oxidative folding and lipidation. We were able to distinguish apoB100 proteins with different oxidative folding states—complete to partially oxidized to reduced—based on the differential migration pattern on nonreducing gels, which was not previously described. However, because apoB100 contains eight disulfide bonds (Burch and Herscovitz, 2000), we were not able to identify how many disulfides are native or whether all of the apoB100 that migrated with the oxidized form contains the same number and/or correct pairing of disulfide bonds. Nevertheless, the migration pattern of apoB100 under nonreducing conditions was different between control and Pdi1-knockdown cells, indicating that PDI1 plays an indispensable role in the proper oxidation of at least some disulfide bonds. Previous studies showed that the disulfide bond involving cysteines 7 and 8 (residues 242 and 258, respectively) is most important for VLDL assembly and secretion (Tran et al., 1998). Because the reduced apoB100 appeared to be less lipidated in Pdi1-knockdown cells, our findings raise the interesting possibility that PDI1 could catalyze the disulfide bond formation between cysteines 7 and 8 (Tran et al., 1998).
Previous studies implicated both nonenzymatic and enzymatic roles of PDI1 in apoB100 lipidation and secretion (Wang et al., 1997). Here we provide direct evidence to pinpoint the dual role of PDI1 in apoB100 synthesis and secretion. PDI1 was characterized in the MTP complex to retain functional MTP in the ER to aid in apoB100 lipidation (Bradbury et al., 1999). We indeed observed decreased MTP activity in Pdi1-knockdown cells, which was restored by either wild-type PDI1 or a catalytically inactive form of PDI1, indicating a nonenzymatic requirement for PDI1 in regulation of MTP activity. On the other hand, we found that PDI1 also promotes oxidative folding of apoB100, indicating an enzymatic requirement for PDI1 in apoB100 synthesis and folding. The dual role of PDI1 couples apoB100 folding with lipidation so that efficient apoB100 lipidation either proceeds, is coincident with, or occurs immediately after proper disulfide bond formation in apoB100. Although it is not known whether PDI1 oxidizes apoB independently or in a complex with MTP, our findings suggest that PDI1 is an essential component for both apoB100 folding and lipidation.
The ER is the organelle where both apoB100 synthesis and lipidation occur (Wang and Kaufman, 2014). The ER lumen provides an optimal redox milieu for proper folding of apoB100 and an adequate lipid store for VLDL maturation (Rusinol et al., 1993). Recently, we reported that ER homeostasis maintained by the inositol-requiring transmembrane kinase/endoribonuclease 1α (IRE1α)-X-box–binding protein 1 (XBP1) arm of the UPR is important for lipid-rich VLDL assembly (Wang et al., 2012). The present study underscores our earlier conclusions that ER homeostasis plays a critical role in apoB100 folding to support the formation of disulfide bonds in apoB100. PDI1 appears to be the protein disulfide isomerase that catalyzes formation of disulfide bonds that are critical for apoB100 folding and lipidation. This conclusion is further supported by the observation that apoB100 forms an intermolecular disulfide with PDI1. However, the liver contains additional members of the PDI family, including ERp57 and ERp72 (Grubb et al., 2012). Their functions in catalyzing disulfide bond formation or rearrangement still need to be established.
In summary, our results provide new insight into the requirements and pathways for apoB100 folding and VLDL assembly and identify PDI1 as a critical isomerase for disulfide bond formation in apoB100. Given that PDI1 is an essential molecule for apoB100 synthesis and VLDL assembly, PDI1 inhibitors may provide an additional alternative to MTP inhibitors, antisense to apoB100, or inhibition of PCSK9 in treating hyperlipidemia (Koren et al., 2014), especially in individuals who do not respond well to statin therapy (Ahmad, 2014).
MATERIALS AND METHODS
Materials
Easytag radiolabel [35S]Cys/Met was from PerkinElmer (Waltham, MA), and [3H]glycerol was from American Radiolabeled Chemicals (St. Louis, MO). Antibody against human and rodent apoB was from Millipore (Billerica, MA, AB742); antibodies against albumin, transferrin, and tubulin were from Sigma-Aldrich (St. Louis, MO; A0433, T2027, A2228). Antibodies against GFP, MTP, and BiP were from BD Transduction Laboratories (Carlsbad, CA; 632375, 612022, 610979); antibodies against PDI1, caspase 3, and PARP were from Cell Signaling (Boston, MA; 3501, 9665, 9532); antibody against eIF2α-P was from Fisher Scientific (Carlsbad, CA; 44-728G); antibody against CHOP was from Santa Cruz Biotechnology (Santa Cruz, CA; SC-793). The MTP inhibitor, CP-10447, was purchased from Sigma-Aldrich.
Mammalian cell culture
Murine Hepa1-6 cells and rat McA-RH7777 hepatoma (McA) cells were obtained from the American Type Culture Collection (Manassas, VA) and maintained in DMEM (Corning, Corning, NY) with 10% fetal bovine serum (FBS) at 37°C in 5% CO2. The cells were passaged every other day. Both Hepa1-6 and McA cells were used to study the effect of PDI1 knockdown on the ER stress response. Cells were treated with different concentrations of Tm and Tg. Cell survival was measured by Cell Counting Kit-8 (Dojindo Molecular Technologies, Rockville, MD). McA cells that stably express HA-tagged wild-type (HA-PDI1) or the substrate-trap mutant of human PDI1 (HA-PDI1CXXA; Rutkevich et al., 2010) were generated by infection with retroviruses expressing each protein, followed by selection for G418 resistance (400 μg/ml). To generate McA cells that stably express Grx-roGFP-iEER (Birk et al., 2013), cells were transfected with plasmid DNA and selected for growth in G418 (400 μg/ml).
Adenovirus constructs
Two independent adenoviruses expressing shRNAs were constructed to target the sequences 5′-GGCTTCAAGGGCAAGATCCT-3′ and 5′-GGCGAACTTTGAAGAGGTCGC-3′ in PDI1. The nonspecific Ad-NS shRNA sequence was previously reported (Herzig et al., 2001).
RNA isolation and quantitative reverse transcription PCR
Total RNA was isolated from cells using Isol-RNA lysis reagent (Fisher Scientific, Carlsbad, CA). cDNA synthesis from total RNA was performed using iScript Reverse Transcription Supermix for quantitative reverse transcription PCR (Bio-Rad, Hercules, CA). All primers were synthesized by Integrated DNA Technologies (Coralville, IA). Primer sequences (5′ → 3′) were as follows: PDI1-a: forward, TCTGAGATTCGACTAGCAAAGGT, and reverse, GCCACGGACACCATACTGC; PDI1-b: forward, CCGTGGCTACCCCACAATC, and reverse, GCAGTGTCAGACAGGGTTGTA; ERp57: forward, CGCCTCCGATGTGTTGGAA, and reverse, CAGTGCAATCCACCTTTGCTAA; ERp72, forward, AGTCAAGGTGGTGGTGGGAAAG, and reverse, TGGGAGCAAAATAGATGGTAGGG; GRP94: forward, AATAGAAAGAATGCTTCGCC, and reverse, TCTTCAGGCTCTTCTTCTGG; apoB: forward, CAAGCACCTCCGAAAGTA, and reverse, CACGGTATCCAGGAACAA; MTP: forward, GCCCTAGTCAGGAAGCTGTG, and reverse, CCAGCAGGTACATTGTGGTG; XBP1 (spliced): forward, AAGAACACGCTTGGGAATGG, and reverse, CTGCACCTGCTGCGGAC; ERdj4: forward, TTAGAAATGGCTACTCCACAGTCA, and reverse, TTGTCCTGAACAATCAGTGTATGTAG; and P58IPK: forward, TCCTGGTGGACCTGCAGTACG, and reverse, CTGCGAGTAATTTCTTCCCC.
Protein isolation and Western blotting
Cells were lysed, and proteins were extracted using RIPA buffer (50 mM Tris-HCl, pH 7.4, buffer containing 1% Triton X-100, 0.5% sodium deoxycholate, and 0.5 mM EDTA). Proteins were separated by electrophoresis on criterion Tris-HCl gels (Bio-Rad, Hercules, CA) and electroblotted onto polyvinylidene fluoride membranes, which were blocked with nonfat dry milk. Membranes were then incubated with different primary antibodies (see Materials). The membranes were further incubated with appropriate horseradish peroxidase–conjugated secondary antibodies to develop the image using ECL reagents (Fisher Scientific, Carlsbad, CA).
Dithiothreitol recovery experiments
Cells were pretreated with 10 mM DTT in normal growth medium for 10 min at 37°C. Cells were then washed with phosphate-buffered saline (PBS) twice, followed by incubation with growth medium for 2, 5, and 10 min before collecting them in PBS containing 20 mM NEM for 20 min on ice. Cells were lysed using RIPA buffer containing 25 mM NEM, and lysates were analyzed by 12% SDS–PAGE under nonreducing conditions, followed by Western blotting analysis (Birk et al., 2013).
Pulse-chase analysis of apoB in McA cells
McA cells were incubated in media containing 0.4 mM bovine serum albumin (BSA)–conjugated oleic acid (OA) for 4 h before and during the entire duration of the pulse-chase experiment. Cells were incubated in Met- and Cys-free medium for 15 min, followed by 15-min labeling with 140 μCi/ml [35S]Met/Cys and 25- and 50-min chase in medium containing excess unlabeled Met and Cys (final concentration of 5 mM). After chase, both media and cell lysates were harvested for immunoprecipitation (IP) with apoB antibody. Labeled apoB immunocomplexes were analyzed by SDS–PAGE and exposed to a phosphoimager screen for autoradiography. Quantification was performed using ImageJ (Collins, 2007).
VLDL-TG synthesis and secretion in McA cells
TG synthesis and secretion were measured in McA cells by incubation for 3 h with OA (0.4 mM) and [3H]glycerol (1 μCi/ml). Labeled TG was extracted from both cells and media, separated by TLC, and analyzed by scintillation counting as described (Chen et al., 2010).
Density gradient analysis of secreted lipoproteins from McA cells
McA cells were exposed to 140 μCi/ml [35S]Met/Cys containing BSA (1.5%) or BSA-OA (400 μM) for 4 h. The density of labeled media was adjusted to 1.25 g/ml using KBr and layered below a KBr/NaCl solution with densities of 1.063, 1.019, and 1.006 g/ml (Chen et al., 2010). Lipoproteins were separated in the gradient by ultracentrifugation at 38,000 rpm overnight. Eleven fractions were harvested, immunoprecipitated with apoB antibody, and analyzed as described for pulse-chase analysis (Wang et al., 2012).
MTP activity assay
MTP activity was measured in McA cells using MTP Activity Assay Kit (Roar Biomedical, New York, NY) based on the published protocol (Athar et al., 2004; Wang et al., 2012).
Measurement of oxidative folding of apoB100 in McA cells
McA cells were treated with puromycin (100 μM) for 10 min before labeling with 140 μCi/ml [35S]Met/Cys to synchronize protein synthesis. Cells were incubated in PBS containing 25 mM NEM for 10 min and lysed in RIPA buffer containing 25 mM NEM. Cell lysates were immunoprecipitated with apoB antibody, and labeled apoB immunocomplexes were analyzed by 5% SDS–PAGE. The post-IP supernatants were used for immunoprecipitating albumin and transferrin and run on 7.5% SDS–PAGE under nonreducing conditions (Rutkevich et al., 2010).
Statistical analyses
Data in the figures are presented as mean ± SEM, applying the unpaired two-tailed t test. p < 0.01 is considered to be a significant difference.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants R37DK042394, R01DK088227, and R01HL052173 to R.J.K. and HL-38180, DK-56260, and DK-52574 to N.O.D. and an American Heart Association postdoctoral fellowship to S.W. We thank David Williams for human PDI constructs,Christian Appenzeller-Herzog for the Grx-roGFP-iEER construct, and Stewart Lipton for adenoviruses expressing human wild-type and a catalytically inactive PDI1.
Abbreviations used:
- apoB
apolipoprotein B
- McA
McA-RH7777
- MTP
microsomal triglyceride transfer protein
- PDI1
protein disulfide isomerase 1
- TG
triglycerides
- VLDL
very low density lipoprotein.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-08-1274) on December 17, 2014.
S.W. conceived and designed the experiments with R.J.K. and Z.C. S.W., S.P., and V.K.K. performed the experiments. S.W. wrote the article with N.O.D. and R.J.K. Z.C. and N.O.D. provided technical support.
REFERENCES
- Adiels M, Olofsson SO, Taskinen MR, Boren J. Overproduction of very low-density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008;28:1225–1236. doi: 10.1161/ATVBAHA.107.160192. [DOI] [PubMed] [Google Scholar]
- Ahmad Z. Statin intolerance. Am J Cardiol. 2014;113:1765–1771. doi: 10.1016/j.amjcard.2014.02.033. [DOI] [PubMed] [Google Scholar]
- Appenzeller-Herzog C, Riemer J, Zito E, Chin KT, Ron D, Spiess M, Ellgaard L. Disulphide production by Ero1alpha-PDI relay is rapid and effectively regulated. EMBO J. 2010;29:3318–3329. doi: 10.1038/emboj.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athar H, Iqbal J, Jiang XC, Hussain MM. A simple, rapid, and sensitive fluorescence assay for microsomal triglyceride transfer protein. J Lipid Res. 2004;45:764–772. doi: 10.1194/jlr.D300026-JLR200. [DOI] [PubMed] [Google Scholar]
- Birk J, Meyer M, Aller I, Hansen HG, Odermatt A, Dick TP, Meyer AJ, Appenzeller-Herzog C. Endoplasmic reticulum: reduced and oxidized glutathione revisited. J Cell Sci. 2013;126:1604–1617. doi: 10.1242/jcs.117218. [DOI] [PubMed] [Google Scholar]
- Blanc V, Davidson NO. C-to-U RNA editing: mechanisms leading to genetic diversity. J Biol Chem. 2003;278:1395–1398. doi: 10.1074/jbc.R200024200. [DOI] [PubMed] [Google Scholar]
- Blanc V, Davidson NO. Mouse and other rodent models of C to U RNA editing. Methods Mol Biol. 2011;718:121–135. doi: 10.1007/978-1-61779-018-8_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boren J, Rustaeus S, Olofsson SO. Studies on the assembly of apolipoprotein B-100- and B-48-containing very low density lipoproteins in McA-RH7777 cells. J Biol Chem. 1994;269:25879–25888. [PubMed] [Google Scholar]
- Bradbury P, Mann CJ, Kochl S, Anderson TA, Chester SA, Hancock JM, Ritchie PJ, Amey J, Harrison GB, Levitt DG, et al. A common binding site on the microsomal triglyceride transfer protein for apolipoprotein B and protein disulfide isomerase. J Biol Chem. 1999;274:3159–3164. doi: 10.1074/jbc.274.5.3159. [DOI] [PubMed] [Google Scholar]
- Bulleid NJ, Freedman RB. Defective co-translational formation of disulphide bonds in protein disulphide-isomerase-deficient microsomes. Nature. 1988;335:649–651. doi: 10.1038/335649a0. [DOI] [PubMed] [Google Scholar]
- Burch WL, Herscovitz H. Disulfide bonds are required for folding and secretion of apolipoprotein B regardless of its lipidation state. J Biol Chem. 2000;275:16267–16274. doi: 10.1074/jbc.M000446200. [DOI] [PubMed] [Google Scholar]
- Carmichael DF, Morin JE, Dixon JE. Purification and characterization of a thiol:protein disulfide oxidoreductase from bovine liver. J Biol Chem. 1977;252:7163–7167. [PubMed] [Google Scholar]
- Chen Z, Norris JY, Finck BN. Peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) stimulates VLDL assembly through activation of cell death-inducing DFFA-like effector B (CideB) J Biol Chem. 2010;285:25996–26004. doi: 10.1074/jbc.M110.141598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins TJ. ImageJ for microscopy. BioTechniques. 2007;43:25–30. doi: 10.2144/000112517. [DOI] [PubMed] [Google Scholar]
- Dobiasova M, Frohlich J. The plasma parameter log (TG/HDL-C) as an atherogenic index: correlation with lipoprotein particle size and esterification rate in apoB-lipoprotein-depleted plasma (FER(HDL)) Clin Biochem. 2001;34:583–588. doi: 10.1016/s0009-9120(01)00263-6. [DOI] [PubMed] [Google Scholar]
- Fukuda H, Iritani N, Tanaka T. Effects of high-fructose diet on lipogenic enzymes and their substrate and effector levels in diabetic rats. J Nutr Sci Vitaminol (Tokyo) 1983;29:691–699. doi: 10.3177/jnsv.29.691. [DOI] [PubMed] [Google Scholar]
- Gibbons GF, Wiggins D, Brown AM, Hebbachi AM. Synthesis and function of hepatic very-low-density lipoprotein. Biochem Soc Trans. 2004;32:59–64. doi: 10.1042/bst0320059. [DOI] [PubMed] [Google Scholar]
- Grubb S, Guo L, Fisher EA, Brodsky JL. Protein disulfide isomerases contribute differentially to the endoplasmic reticulum-associated degradation of apolipoprotein B and other substrates. Mol Biol Cell. 2012;23:520–532. doi: 10.1091/mbc.E11-08-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton RL. Synthesis and secretion of plasma lipoproteins. Adv Exp Med Biol. 1972;26:7–24. doi: 10.1007/978-1-4684-7547-0_2. [DOI] [PubMed] [Google Scholar]
- Hatahet F, Ruddock LW. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal. 2009;11:2807–2850. doi: 10.1089/ars.2009.2466. [DOI] [PubMed] [Google Scholar]
- Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–183. doi: 10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- Koren MJ, Giugliano RP, Raal FJ, Sullivan D, Bolognese M, Langslet G, Civeira F, Somaratne R, Nelson P, Liu T, et al. Efficacy and safety of longer-term administration of evolocumab (AMG 145) in patients with hypercholesterolemia: 52-week results from the Open-Label Study of Long-Term Evaluation Against LDL-C (OSLER) randomized trial. Circulation. 2014;129:234–243. doi: 10.1161/CIRCULATIONAHA.113.007012. [DOI] [PubMed] [Google Scholar]
- Kulinski A, Rustaeus S, Vance JE. Microsomal triacylglycerol transfer protein is required for lumenal accretion of triacylglycerol not associated with ApoB, as well as for ApoB lipidation. J Biol Chem. 2002;277:31516–31525. doi: 10.1074/jbc.M202015200. [DOI] [PubMed] [Google Scholar]
- Liang JJ, Oelkers P, Guo C, Chu PC, Dixon JL, Ginsberg HN, Sturley SL. Overexpression of human diacylglycerol acyltransferase 1, acyl-coa:cholesterol acyltransferase 1, or acyl-CoA:cholesterol acyltransferase 2 stimulates secretion of apolipoprotein B-containing lipoproteins in McA-RH7777 cells. J Biol Chem. 2004;279:44938–44944. doi: 10.1074/jbc.M408507200. [DOI] [PubMed] [Google Scholar]
- Liao W, Chang BH, Mancini M, Chan L. Ubiquitin-dependent and -independent proteasomal degradation of apoB associated with endoplasmic reticulum and Golgi apparatus, respectively, in HepG2 cells. J Cell Biochem. 2003;89:1019–1029. doi: 10.1002/jcb.10538. [DOI] [PubMed] [Google Scholar]
- Moore P, Bernardi KM, Tsai B. The Ero1alpha-PDI redox cycle regulates retro-translocation of cholera toxin. Mol Biol Cell. 2010;21:1305–1313. doi: 10.1091/mbc.E09-09-0826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota T, Gayet C, Ginsberg HN. Inhibition of apolipoprotein B100 secretion by lipid-induced hepatic endoplasmic reticulum stress in rodents. J Clin Invest. 2008;118:316–332. doi: 10.1172/JCI32752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan M, Liang JS, Fisher EA, Ginsberg HN. The late addition of core lipids to nascent apolipoprotein B100, resulting in the assembly and secretion of triglyceride-rich lipoproteins, is independent of both microsomal triglyceride transfer protein activity and new triglyceride synthesis. J Biol Chem. 2002;277:4413–4421. doi: 10.1074/jbc.M107460200. [DOI] [PubMed] [Google Scholar]
- Pan M, Maitin V, Parathath S, Andreo U, Lin SX, St Germain C, Yao Z, Maxfield FR, Williams KJ, Fisher EA. Presecretory oxidation, aggregation, and autophagic destruction of apoprotein-B: a pathway for late-stage quality control. Proc Natl Acad Sci USA. 2008;105:5862–5867. doi: 10.1073/pnas.0707460104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primm TP, Walker KW, Gilbert HF. Facilitated protein aggregation. Effects of calcium on the chaperone and anti-chaperone activity of protein disulfide-isomerase. J Biol Chem. 1996;271:33664–33669. doi: 10.1074/jbc.271.52.33664. [DOI] [PubMed] [Google Scholar]
- Puig A, Gilbert HF. Protein disulfide isomerase exhibits chaperone and anti-chaperone activity in the oxidative refolding of lysozyme. J Biol Chem. 1994;269:7764–7771. [PubMed] [Google Scholar]
- Raabe M, Veniant MM, Sullivan MA, Zlot CH, Bjorkegren J, Nielsen LB, Wong JS, Hamilton RL, Young SG. Analysis of the role of microsomal triglyceride transfer protein in the liver of tissue-specific knockout mice. J Clin Invest. 1999;103:1287–1298. doi: 10.1172/JCI6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajpal G, Schuiki I, Liu M, Volchuk A, Arvan P. Action of protein disulfide isomerase on proinsulin exit from endoplasmic reticulum of pancreatic beta-cells. J Biol Chem. 2012;287:43–47. doi: 10.1074/jbc.C111.279927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusinol A, Verkade H, Vance JE. Assembly of rat hepatic very low density lipoproteins in the endoplasmic reticulum. J Biol Chem. 1993;268:3555–3562. [PubMed] [Google Scholar]
- Rutkevich LA, Cohen-Doyle MF, Brockmeier U, Williams DB. Functional relationship between protein disulfide isomerase family members during the oxidative folding of human secretory proteins. Mol Biol Cell. 2010;21:3093–3105. doi: 10.1091/mbc.E10-04-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge AC, Su Q, Adeli K. Apolipoprotein B100 biogenesis: a complex array of intracellular mechanisms regulating folding, stability, and lipoprotein assembly. Biochem Cell Biol. 2010;88:251–267. doi: 10.1139/o09-168. [DOI] [PubMed] [Google Scholar]
- Shelness GS, Thornburg JT. Role of intramolecular disulfide bond formation in the assembly and secretion of apolipoprotein B-100-containing lipoproteins. J Lipid Res. 1996;37:408–419. [PubMed] [Google Scholar]
- Sideraki V, Gilbert HF. Mechanism of the antichaperone activity of protein disulfide isomerase: facilitated assembly of large, insoluble aggregates of denatured lysozyme and PDI. Biochemistry. 2000;39:1180–1188. doi: 10.1021/bi992246q. [DOI] [PubMed] [Google Scholar]
- Sundaram M, Yao Z. Recent progress in understanding protein and lipid factors affecting hepatic VLDL assembly and secretion. Nutr Metab (Lond) 2010;7:35. doi: 10.1186/1743-7075-7-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran K, Boren J, Macri J, Wang Y, McLeod R, Avramoglu RK, Adeli K, Yao Z. Functional analysis of disulfide linkages clustered within the amino terminus of human apolipoprotein B. J Biol Chem. 1998;273:7244–7251. doi: 10.1074/jbc.273.13.7244. [DOI] [PubMed] [Google Scholar]
- Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- Wang S, Chen Z, Lam V, Han J, Hassler J, Finck BN, Davidson NO, Kaufman RJ. IRE1alpha-XBP1s induces PDI expression to increase MTP activity for hepatic VLDL assembly and lipid homeostasis. Cell Metab. 2012;16:473–486. doi: 10.1016/j.cmet.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Fast DG, Attie AD. The enzymatic and non-enzymatic roles of protein-disulfide isomerase in apolipoprotein B secretion. J Biol Chem. 1997;272:27644–27651. doi: 10.1074/jbc.272.44.27644. [DOI] [PubMed] [Google Scholar]
- Wang S, Kaufman RJ. How does protein misfolding in the endoplasmic reticulum affect lipid metabolism in the liver. Curr Opin Lipidol. 2014;25:125–132. doi: 10.1097/MOL.0000000000000056. [DOI] [PubMed] [Google Scholar]
- Wetterau JR, Combs KA, McLean LR, Spinner SN, Aggerbeck LP. Protein disulfide isomerase appears necessary to maintain the catalytically active structure of the microsomal triglyceride transfer protein. Biochemistry. 1991;30:9728–9735. doi: 10.1021/bi00104a023. [DOI] [PubMed] [Google Scholar]
- Yamaguchi J, Gamble MV, Conlon D, Liang JS, Ginsberg HN. The conversion of apoB100 low density lipoprotein/high density lipoprotein particles to apoB100 very low density lipoproteins in response to oleic acid occurs in the endoplasmic reticulum and not in the Golgi in McA RH7777 cells. J Biol Chem. 2003;278:42643–42651. doi: 10.1074/jbc.M306920200. [DOI] [PubMed] [Google Scholar]
- Yang LY, Kuksis A, Myher JJ, Steiner G. Origin of triacylglycerol moiety of plasma very low density lipoproteins in the rat: structural studies. J Lipid Res. 1995;36:125–136. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.