

A lethal mtDNA mutation affecting COX is fully rescued by AOX. The mutant genome level remains constant in the somatic tissues along the aging process in heteroplasmic flies. A genetic scheme creates tissue-specific heteroplasmy in otherwise heteroplasmic background and reveals that Ca2+ mishandling contributes to the neurodegeneration.
Abstract
Various human diseases are associated with mitochondrial DNA (mtDNA) mutations, but heteroplasmy—the coexistence of mutant and wild-type mtDNA—complicates their study. We previously isolated a temperature-lethal mtDNA mutation in Drosophila, mt:CoIT300I, which affects the cytochrome c oxidase subunit I (CoI) locus. In the present study, we found that the decrease in cytochrome c oxidase (COX) activity was ascribable to a temperature-dependent destabilization of cytochrome a heme. Consistently, the viability of homoplasmic flies at 29°C was fully restored by expressing an alternative oxidase, which specifically bypasses the cytochrome chains. Heteroplasmic flies are fully viable and were used to explore the age-related and tissue-specific phenotypes of mt:CoIT300I. The proportion of mt:CoIT300I genome remained constant in somatic tissues along the aging process, suggesting a lack of quality control mechanism to remove defective mitochondria containing a deleterious mtDNA mutation. Using a genetic scheme that expresses a mitochondrially targeted restriction enzyme to induce tissue-specific homoplasmy in heteroplasmic flies, we found that mt:CoIT300I homoplasmy in the eye caused severe neurodegeneration at 29°C. Degeneration was suppressed by improving mitochondrial Ca2+ uptake, suggesting that Ca2+ mishandling contributed to mt:CoIT300I pathogenesis. Our results demonstrate a novel approach for Drosophila mtDNA genetics and its application in modeling mtDNA diseases.
INTRODUCTION
Despite its diminutive size (∼17 kb in mammals), mitochondrial DNA (mtDNA) encodes 13 essential subunits of the electron transport complexes (Wallace, 2005) and is vital for life. Various human diseases stem from mutations in mtDNA (Taylor and Turnbull, 2005; Wallace, 2005). mtDNA diseases often affect tissues with high-energy demand, such as muscles and the nervous system (DiMauro and Schon, 2003), which may reflect mitochondria's primary role in energy homeostasis. However, mtDNA diseases also feature great complexity along with a broad spectrum of symptoms that can be manifested in various tissues, suggesting the disruption of pathways other than energy homeostasis. These pathways include reactive oxygen species (ROS) generation and signaling, apoptosis, and calcium homeostasis (Chan, 2006; McBride et al., 2006).
Understanding how mtDNA mutations contribute to mtDNA diseases is complicated by the peculiarities of mtDNA transmission. Each cell contains hundreds to thousands of copies of mtDNA. mtDNA mutations may affect only a subset of the mtDNA copies present in a cell—a situation known as heteroplasmy—or they might affect all mtDNA copies—a situation known as homoplasmy. Whereas some mtDNA diseases are caused by homoplasmic mutations, most pathogenic, mutant mtDNAs are mixed with wild-type (wt) mtDNAs in a cell. The severity of the phenotypes caused by heteroplasmic mtDNA mutations correlates with the proportion of mutant mtDNA in the cells or tissues and often displays a threshold effect (DiMauro and Schon, 2003; Taylor and Turnbull, 2005). Understanding the pathogenic effects of mitochondrial mutations would be greatly facilitated if it were possible to compare cells or tissues with a known proportion of mutant mtDNA. However, random segregation of mtDNAs during cell division makes it extremely difficult to predict or control the mutation load in particular cell or tissue and the consequent phenotypic presentation.
The structure, organization, and gene content are highly conserved between human and Drosophila mtDNAs (Oliveira et al., 2010), which warrants using Drosophila as a model to understand the function and regulation of mtDNA. There is a single XhoI site within mt:CoI locus on Drosophila mtDNA. Expression of a mitochondrially targeted XhoI (MitoXhoI) in female germ line eliminates the wt mtDNA, ablates the germ cells, and leads to female sterility (Xu et al., 2008). However, occasional females give escaper progeny carrying homoplasmic mtDNA mutations on the XhoI site (Xu et al., 2008). Applying this approach, we previously recovered a deleterious mtDNA mutation, mt:CoIT300I, which results in a threonine-to-isoleucine substitution of cytochrome c oxidase subunit I (CoI) protein (Hill, Chen, et al., 2014). Homoplasmic mt:CoIT300I flies developed normally at 18°C but fail to eclose at 29°C (Hill, Chen, et al., 2014). However, the biochemical basis of this temperature sensitivity remains to be elucidated. When shifted to 29°C after eclosion at the permissive temperature, mt:CoIT300I flies only survive up to 5 d (Hill, Chen, et al., 2014), precluding exploration of age-related phenotypes at the restrictive condition. Heteroplasmic flies containing both wt and mutant genomes were generated by germplasm transplantation. Although the mt:CoIT300I level in heteroplasmic flies remains constant over many generations at 18°C, it is dramatically reduced during oogenesis and eventually purged from the population at 29°C (Hill, Chen, et al., 2014). Nonetheless, the segregation and the transmission of mutant genome in somatic tissues of heteroplasmic flies have not been investigated.
In the study reported here, we used mt:CoIT300I flies to ask questions about this mtDNA mutation that would be difficult to address in other systems. The homoplasmic flies provided material for a detailed biochemical characterization of the mt:CoIT300I phenotype. The heteroplasmic flies allowed us to model the age-dependent and tissue-specific phenotypes typically observed in human mtDNA diseases. In particular, heteroplasmic flies provided a healthy background in which we were able to induce tissue-specific homoplasmy, which in turn allowed us to study some tissue-specific phenotypes of the mt:CoIT300I mutation.
RESULTS
mt:CoIT300I disrupts cytochrome c oxidase activity
To understand the biochemical basis of the temperature sensitivity of mt:CoIT300I, we assayed the composition and activity of the respiratory chain complexes of mutant flies at both 25 and 29°C. Although CoI protein levels were comparable in mt:CoIT300I and wt flies (Figure 1A), cytochrome c oxidase (COX) activity in the mutant was decreased to ∼30% of wt activity at 25°C (Supplemental Figure S1A). The mutant COX appears unstable at restrictive condition, as the COX activity of mt:CoIT300I extract quickly diminished to <5% of wild type after incubating at 29°C for 40 min (Hill, Chen, et al., 2014).
FIGURE 1:

mt:CoIT300I disrupts cytochrome c oxidase activity. (A) Western blot analysis of total tissue extracts of mt:CoIT300I and wt flies cultured at 25 or 29°C after eclosion at 25°C, with antibodies against CoI, CoIV, ATP synthase α-subunit (ATPs-α), and tubulin. (B) Spectrophotometric measurements of cytochrome a (cyt. a) and cytochrome c (cyt. c) amounts in isolated mitochondria from 2-d-old mt:CoIT300I and wt flies raised at 25°C (mean ± SD, n = 3). (C) Difference absorbance spectrum of wt and mt:CoIT300I mitochondria at 25°C or after incubation at 29°C for 30 min. Peaks for cytochrome c (cyt. c) and cytochrome a (cyt. a) are labeled. (D) Ubiquitous AOX expression rescues the mt:CoIT300I adult lethality at 29°C. Flies carrying a UAS-AOX transgene were crossed to flies carrying both the homoplasmic mt:CoIT300I mtDNA and a ubiquitous driver actin-Gal4 (ac-Gal4) on the nuclear genome. Because of maternal transmission of mtDNA, only the progeny of mutant females inherited the mt:CoIT300I mutation (right). These flies died as pupae unless they inherited the ac-Gal4 driver, that is, Cy+ (CyO balancer chromosome carries a dominant Cy marker; Cy+ means non-Cy). Overexpression of UAS-AOX under control of ac-Gal4 had no effect on the viability of flies inheriting wide-type (wt) mtDNA from their mothers (left). Asterisk, the CyO escapers died quickly after eclosion.
COX activity depends on the association of CoI with two heme a cofactors (Babcock and Wikstrom, 1992). Spectral analyses showed that cytochrome a amounts were markedly decreased in mt:CoIT300I flies, whereas the amount of cytochrome c was normal (Figure 1, B and C). In addition, the heme a cofactors further dissociated from COX in mt:CoIT300I mitochondrial extracts after a brief incubation at 29°C (Figure 1C), rendering them spectrally invisible due to the low solubility and high reactivity of free hemes (Severance and Hamza, 2009). These results suggest that the mt:CoIT300I mutation reduces COX activity by weakening the interaction between the a hemes and CoI. Consistent with this hypothesis, the residue mutated in mt:CoIT300I is located in transmembrane helix VIII of the CoI protein, which interacts with the a hemes (Tsukihara et al., 1996).
Although the amounts of individual subunits of complex IV, including CoI and CoIV, were equivalent in mutant and wild type (Figure 1A), the level of whole complex IV was greatly reduced in mt:CoIT300I mitochondria compared with wild type based on the blue native PAGE analysis (Supplemental Figure S1B). This suggests that the mutation might affect the assembly or the stability of the whole complex. We also found that the level of ATP synthase α-subunit, a routinely used mitochondrial marker, was similar in mutant and wild type (Figure 1A). The amounts of complexes I, III, and V were all comparable between wt and mutant on blue native PAGE (Supplemental Figure S1B). In addition, the activities of complexes I–III in mutant flies were normal or slightly higher compared with wt (Supplemental Figure S1C). Taken together, these results show that mt:CoIT300I specifically disrupts COX activity and are consistent with our previous sequence analysis, which did not detect any additional mutation in the mtDNA of mt:CoIT300I mutants (Hill, Chen, et al., 2014).
We showed previously that mt:CoIT300I mutants raised at 29°C failed to eclose from the pupal cases, even though they progressed through embryonic and larval stages normally (Hill, Chen, et al., 2014). To confirm further that pathogenesis of mt:CoIT300I is caused by blockage of the electron transport through COX, we ubiquitously expressed an alternative oxidase, AOX (Fernandez-Ayala, Sanz, Vartiainen, Kemppainen, et al., 2009), under control of an actin-Gal4 (ac-Gal4) driver in the homoplasmic mt:CoIT300I background. AOX catalyzes electron transfer from ubiquinone to molecular oxygen, thereby bypassing the cytochrome chain reactions (Supplemental Figure S2A), and confers the resistance to inhibitors of respiratory chain complexes III and IV in cultured cells (Hakkaart et al., 2006). AOX can also partially suppress nuclear mutations that disrupt COX when expressed in Drosophila (Fernandez-Ayala, Sanz, Vartiainen, Kemppainen, et al., 2009; Kemppainen et al., 2013). Expression of AOX completely restored the viability of mt:CoIT300I flies at 29°C, although it had no obvious effect on flies carrying wt mtDNA (Figure 1D). In addition, ubiquitous expression of AOX at the adult stage partially restored respiration (Supplemental Figure S2, B and C) and extended the lifespan of mt:CoIT300I flies at 25°C (Supplemental Figure S3A) but had no obvious effect on wt flies (Sanz et al., 2010). The genetic rescue by AOX confirmed that pathogenesis of mt:CoIT300I originated from the COX deficiency.
mt:CoIT300I mtDNA is not subject to negative selection in the adult soma of heteroplasmic flies
Homoplasmic mt:CoIT300I flies survived to adult stages when raised at 25°C but lived up to only 2 wk even at this permissive condition (Figure 2A). In addition, their mobility dropped quickly. By the age of 10 d, most mt:CoIT300I flies could hardly move (Figure 2B). mt:CoIT300I flies die within 5 d after being shifted to 29°C (Hill, Chen, et al., 2014), which prevents us from exploring age-related dysfunctions that are normally associated with mtDNA diseases. In addition, most human mtDNA diseases are caused by heteroplasmic mutations and affect specific tissues. We therefore turned to heteroplasmic flies to probe age-related and tissue-specific phenotypes of mt:CoIT300I at the restrictive condition.
FIGURE 2:
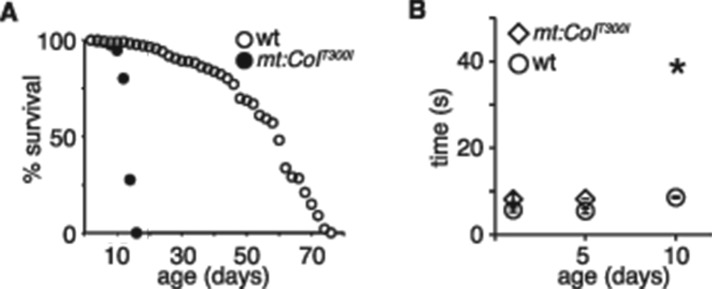
Adult homoplasmic mt:CoIT300I flies show age-associated locomotion defects at 25°C. (A) Lifespan of adult wt and mt:CoIT300I homoplasmic flies at 25°C. (B) Climbing ability of homoplasmic mt:CoIT300I and wt flies at different ages at 25°C, assayed as the time required for 50% of flies in each group to climb 10 cm (means ± SD, n = 3). Asterisk, mutant flies can hardly move at day 10.
We previously generated heteroplasmic flies carrying both wt and mt:CoIT300I genomes using germplasm transplantation (Hill, Chen, et al., 2014). Adult heteroplasmic flies containing as little as 5% of wt genome are able to reach the adult stage (Hill, Chen, et al., 2014). Because heteroplasmic flies fared markedly better than their homoplasmic counterparts even at the restrictive temperature, we wondered whether the mutant mtDNA was eliminated during development or aging. We therefore analyzed the proportion of mutant mtDNA in various tissues of heteroplasmic mt:CoIT300I flies grown at the permissive or restrictive temperature. We cultured heteroplasmic flies at 18°C until eclosion to minimize the detrimental impact and possible negative selection of the mutant mtDNA. We dissected heads, thoraxes, guts, and testes from 10 individual males 2 d after eclosion or aged up to 4 wk at 18, 25, or 29°C and measured heteroplasmic levels in these organs by quantifying the XhoI digestion of mtDNA PCR products (Hill, Chen, et al., 2014).
For flies eclosed and aged at 18°C, the proportion of mutant mtDNA among single individual flies varied from 60 to 90% (Figure 3, A and B), suggesting random segregation of mtDNAs during oogenesis. However, there was no significant difference in the average mutation loads of the various organs (Figure 3E). These results suggest a lack of selection against the mutant genome during development and through the aging process at 18°C for all organs examined. Among the flies cultured at 25°C, the average mutant load in heads, thoraces, and guts was ∼80% and demonstrated little variation (Figure 3E). However, the testes had a much lower mutant load (69 ± 7.4%; Figure 3, C and E). When cultured at 29°C, heteroplasmic flies containing >85% mutant genome were not recovered after 4 wk. Analysis of the survivors revealed that the mutation load in three somatic tissues was similar at ∼70% (Figure 3E) but was significantly reduced in testes (52 ± 11.5%; Figure 3, D and E). Although the spatial resolution is difficult to determine in these experiments, our results suggest that mutant mtDNA is subject to negative selection in testis and only at higher temperatures.
FIGURE 3:
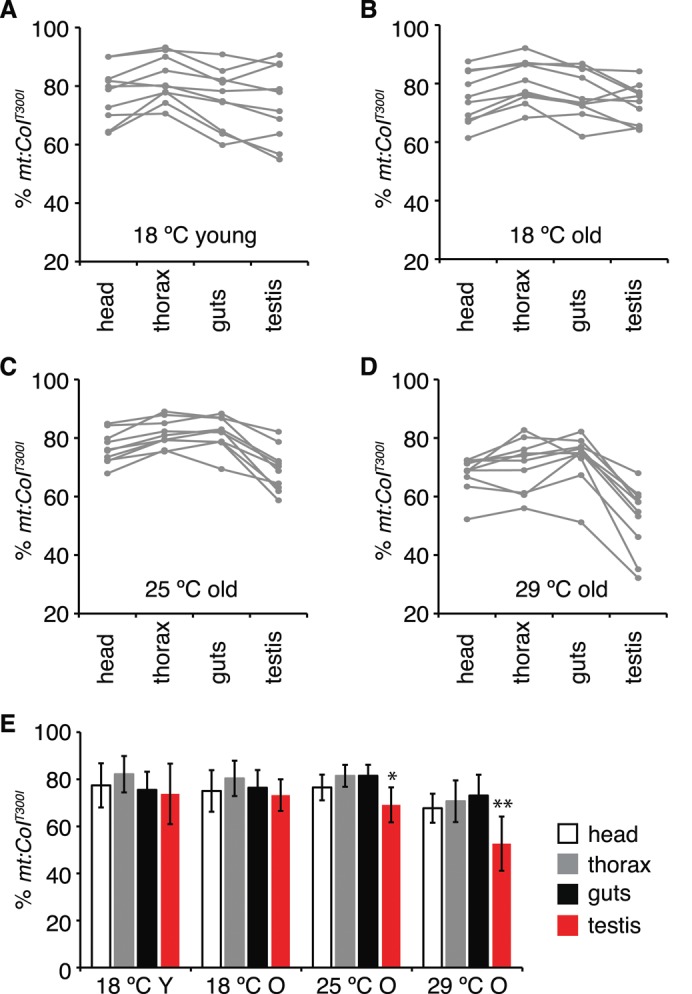
Proportion of mt:CoIT300I mtDNA in different tissues of heteroplasmic flies. The heads, thoraxes, guts, and testes were dissected from 10 individual males eclosed and aged at 18°C for 2 d (A) or 4 wk (B) or shifted to 25°C (C) and 29°C (D) for 4 wk after eclosion at 18°C, and degrees of heteroplasmy were determined by restriction digest analysis. Tissues from a single fly are connected by a gray line. (E) Average mt:CoIT300I levels of each organ from 10 individual flies of each group (means ± SD, n = 10). Y, young, 2-d-old flies; O, old, 4-wk-old flies. Note that at 25 and 29°C, mt:CoIT300I levels are significantly decreased in testes compared with other tissues in the same group (*p < 0.05, **p < 0.005).
Neurological dysfunction plays a major role in the pathology of mt:CoIT300I flies
A genetic scheme generating homoplasmic mutations in specific tissues would overcome the unpredictability of mutation load due to a random segregation and allow us to examine tissue-specific phenotypes with a defined genetic composition. Similar to mosaic analysis for studying essential nuclear genes in heterozygous background (Blair, 2003), this approach would also enable genetic analysis of a lethal mtDNA mutation such as mt:CoIT300I in a particular tissue in an overall healthy heteroplasmic background at a restrictive temperature.
Wild-type mtDNA has an XhoI site, whereas the mt:CoIT300I mitochondrial genome does not and is therefore resistant to XhoI digestion. We reasoned that expression of a mitochondrially targeted form of XhoI, MitoXhoI (Xu et al., 2008), in a heteroplasmic background would destroy wt mtDNA specifically and shift mt:CoIT300I heteroplasmy toward homoplasmy (Supplemental Figure S4A). As a test of principle, we ubiquitously expressed MitoXhoI under control of a tubulin-Gal4 driver (tub-Gal4) in heteroplasmic larvae and quantified mtDNAs based on their sensitivity to XhoI digestion (Supplemental Figure S4B). Expression of MitoXhoI efficiently eliminated wt mtDNA, resulting in nearly 100% mt:CoIT300I. The absolute amount of mtDNA in the heteroplasmic flies expressing MitoXhoI was similar to that in wt flies (Supplemental Figure S4C), suggesting that the copy number of mtDNA remains constant after removal of wt mtDNA. We further tested the efficacy of this approach by generating homoplasmy in specific tissues—larval fat body and eye disk—because of the ease of dissecting homogeneous tissues. Expression of MitoXhoI in fat body driven by Cg-Gal4 or in eye disk by eyeless-Gal4 (ey-Gal4) in the heteroplasmic background completely removed wt mtDNA in the fat body or eye disk, respectively, whereas other tissues remained heteroplasmic (Figure 4A).
FIGURE 4:

Tissue-specific mt:CoIT300I homoplasmy. (A) Tissue-specific expression of MitoXhoI shifted heteroplasmic mt:CoIT300I flies to homoplasmy in fat body and eye disk. A 4.0-kb mtDNA fragment flanking the XhoI site was amplified by PCR from flies with the indicated genotypes (1–6) and further digested by XhoI enzyme. The wt mtDNA carrying the XhoI recognition site could be digested into two fragments (1.6 and 2.4 kb), whereas mutant mtDNA was resistant to XhoI digestion. Here 1–6 denote the genotypes and tissues of different Drosophila lines. Note that tissue-specific expression of MitoXhoI under control of Cg-Gal4 (Cg>mitoXhoI) or eyeless-Gal4 (ey>mitoXhoI) completely eliminated the wt mtDNA and resulted in 100% of mt:CoIT300I in fat body or eye disk, respectively, whereas the whole body remains heteroplasmic. (B–D) mt:CoIT300I homoplasmy in the eye leads to photoreceptor degeneration. (B) Rhabdomeres observed by optical neutralization from 2- or 8-d-old heteroplasmic flies with homoplasmic eyes. Eye tissues were made homoplasmic by eye-specific expression of MitoXhoI under control of eyeless-Gal4 driver (ey>mitoXhoI). Adult flies were kept at 29°C under a 12-h light/12-h dark cycle. (C) Mean number of rhabdomeres per ommatidium, as determined by optical neutralization as a function of age. The numbers of rhabdomeres decreased rapidly in homoplasmic eyes (-o-, ey>mitoXhoI [wt+ mt:CoIT300I]) but were rescued by coexpression of Letm1 (-Δ-, ey >mitoXhoI +letm1 [wt+ mt:CoIT300I]). Expression of MitoXhoI in flies carrying a mtDNA synonymous mutation (mt:CoIsyn) showed no rhabdomere loss (-×-, ey>mitoXhoI [mt:CoIsyn]), demonstrating that overexpression of MitoXhoI per se was not detrimental to photoreceptor cells. Data are presented as means ± SD; n ≥7 flies. (D) Retinal morphology of 2-d or 2-wk-old heteroplasmic flies with or without homoplasmic eyes, as examined by transmission electron microscopy. In addition to the flies used in C, heteroplasmic flies without any nuclear transgene (w1118 [wt+ mt:CoIT300I]) were examined as an additional control. C, cell body; R, rhabdomere. Note the disorganized rhabdomeres (arrowheads) and vesicular structures (arrow) indicating degeneration. Nuclear genotypes are Cg-Gal4/UAS-mitoXhoI (Cg>mitoXhoI), ey-Gal4/UAS-mitoXhoI (ey>mitoXhoI),and ey-Gal4/UAS-mitoXhoI; UAS-letm1/+ (ey >mitoXhoI +letm1).
To compare the effect of COX deficiency in different tissues, we drove expression of MitoXhoI with various tissue-specific Gal4 drivers in heteroplasmic flies and evaluated the effect on whole-animal fitness by assaying pupal eclosion rate at 29°C and adult lifespan at 25°C (Table 1). To exclude potential nonspecific damage caused by overproduction of MitoXhoI, we expressed MitoXhoI in a healthy mtDNA mutant background, mt:CoIsyn. We isolated mt:CoIsyn using a MitoXhoI-based selection scheme (Xu et al., 2008); it carries a synonymous mutation within XhoI site at the mt:CoI locus. Heteroplasmic mt:CoIT300I flies expressing MitoXhoI in eye disks, glia, ring glands, hemocytes, or imaginal disks had similar eclosion rates as their respective control flies with the mt:CoIsyn background, which in turn eclosed at rates similar to those of wt pupae (Table 1). However, heteroplasmic mt:CoIT300I flies expressing MitoXhoI in the nervous system had a 25% reduction in eclosion rate and dramatically reduced adult lifespan compared with controls (Table 1). These results suggest that neurological dysfunctions caused by COX deficiency play a major role in reducing the eclosion rate and lifespan of mt:CoIT300I adults. Expression of MitoXhoI in muscles or in salivary glands and amnioserosa was lethal in both mt:CoIsyn and mt:CoIT300I heteroplasmic backgrounds, precluding analysis of mt:CoIT300I homoplasmy in these tissues.
TABLE 1:
Effect of tissue-specific mt:CoIT300I homoplasmy on eclosion rate and lifespan.
| mt:CoIsyn | wt+mt:CoIT300I | ||||
|---|---|---|---|---|---|
| Symbol | Expression pattern | Eclosion rate | Median lifespan | Eclosion rate | Median lifespan |
| No driver | N/A | 0.97 ± 0.03 | 42 ± 6 | 0.98 ± 0.02 | 41 ± 5 |
| ac-Gal4 | Ubiquitous expression | 0.90 ± 0.04 | 45 ± 2 | 0 | N/A |
| tub-Gal4a | Ubiquitous expression | 0 | N/A | 0 | N/A |
| eyeless-Gal4 | Eye | 0.95 ± 0.05 | 43 ± 3 | 0.98 ± 0.02 | 36 ± 2 |
| Cg-Gal4 | Hemocytes, fat body | 0.89 ± 0.10 | 45 ± 4 | 0.92 ± 0.05 | 42 ± 4 |
| repo-Gal4 | Glia | 0.89 ± 0.08 | 39 ± 5 | 0.97 ± 0.02 | 21 ± 2 |
| GawB1407 | Nervous system | 0.94 ± 0.07 | 42 ± 5 | 0.73 ± 0.12 | 8 ± 1 |
| GawB71B | Imaginal disks | 0.99 ± 0.01 | 48 ± 2 | 0.97 ± 0.05 | 14 ± 1 |
| GawBC805 | Gland, guts, and tubes | 0.88 ± 0.21 | 41 ± 9 | 0.91 ± 0.10 | 20 ± 3 |
| Mef2-Gal4a | Muscle | 0 | N/A | 0 | N/A |
| GawB332.3a | Amnioserosa, salivary gland | 0 | N/A | 0 | N/A |
Homoplasmic mt:CoIsyn females or heteroplasmic mt:CoIT300I females (wt+mt:CoIT300I) carrying UAS-mitoXhoI were crossed with different tissue-specific Gal4 lines at 29°C, and their progeny were raised at 25°C. The eclosion rate (number of eclosed adults/number of pupae) and the median lifespan of adult progeny were scored. All of the heteroplasmic virgins, UAS-mitoXhoI (wt+mt:CoIT300I), used in the crosses were collected from the same batch of culture with 60% mutational load in the population. Note that both the eclosion rate and the median lifespan are dramatically reduced in flies with a homoplasmic nervous system (underscored).
aExpression of MitoXhoI ubiquitously (tub-Gal4), in muscle (Mef2-Gal4), or in amnioserosa and salivary gland (GawB332.3) was lethal.
Homoplasmic mt:CoIT300I causes neurodegeneration in the eye
To determine how mt:CoIT300I affects neuronal cells at the restrictive condition, we restricted mt:CoIT300I homoplasmy to the eye by driving MitoXhoI expression with an ey-Gal4 driver (Figure 4A). In newly eclosed heteroplasmic flies that expressed MitoXhoI in their eyes, seven photoreceptor cells with properly organized cell bodies and rhabdomeres were present in each ommatidium (Figure 4, B and D). However, as the flies aged, the number of detectable rhabdomeres decreased (Figure 4C), the rhabdomeres appeared disorganized (Figure 4D; arrowheads), and many vesicular structures appeared in the photoreceptor cell bodies (Figure 4D; arrows), suggesting massive photoreceptor cell degeneration. Neither mt:CoIsyn flies expressing MitoXhoI in the eye nor the heteroplasmic flies displayed any degeneration (Figure 4D). Thus neurodegeneration was indeed caused by homoplasmy of mt:CoIT300I mutation at the restrictive temperature.
Overexpression of a calcium transporter suppresses retinal degeneration in mt:CoIT300I homoplasmic eyes
Disruption of respiratory chain complexes could lead to overproduction of damaging ROS. However, we found no difference in H2O2 level between wt and mutant flies, although there were clearly higher levels of H2O2 in old flies than in young flies (Supplemental Figure S3B). We also assayed the level of protein carbonylation, an alternative approach to evaluate accumulative oxidative damage caused by ROS. Consistent with a previous report (Wehr and Levine, 2012), the levels of protein carbonylation were higher in old flies than in young flies in both total cellular extracts and mitochondrial preparations. However, there was no obvious difference between wt and mutant flies at either age (Supplemental Figure S3C). In addition, overexpression of SOD2 and catalase, two scavenging enzymes that have been shown to successfully suppress phenotypes caused by excess ROS in Drosophila (Anderson et al., 2005), had no effect on the lifespan of mt:CoIT300I flies (Supplemental Figure S3A). Blockage of electron flow could also reduce mitochondrial membrane potential. Indeed, we found that mt:CoIT300I mitochondria had much lower tetramethylrhodamine methylester (TMRM) fluorescence than wt mitochondria (Figure 5, A and B). It is possible that effects of electron blockage on ROS production are offset by the reduction in membrane potential associated with mt:CoIT300I (Lambert and Brand, 2004).
FIGURE 5:

Impaired mitochondrial calcium uptake in the motor neurons of mt:CoIT300I homoplasmic embryos. (A) Membrane potential staining by TMRM in primary motor neurons isolated from wt and mt:CoIT300I homoplasmic embryos. Motor neuron cells were labeled with mitochondrially targeted green fluorescent protein (MitoGFP) driven by D42-Gal4 (D42-Gal4/UAS-mitoGFP). Bar, 10 μm. (B) TMRM fluorescence in wt and mt:CoIT300I motor neurons was measured in arbitrary fluorescence units (AFUs; mean ± SD, wt n = 45, mt:CoIT300I n = 39), p < 0.0005. (C) Mitochondrial Ca2+ dynamics in wt and mt:CoIT300I motor neurons expressing Mitycam in response to 400 μM acetylcholine (arrowhead). Relative changes in Mitycam fluorescence intensity, (F0 − Ft)/F0, are plotted against time. Note that mt:CoIT300I neurons have significantly reduced response to acetylcholine stimulation, indicating an impaired mitochondrial Ca2+ uptake. (D) Average amplitude of the Mitycam responses in wt and mt:CoIT300I neurons upon acetylcholine stimulation (means ± SD, n = 9).
Because membrane potential drives mitochondrial Ca2+ uptake (Rizzuto et al., 2000), we next assessed the effect of mt:CoIT300I on mitochondrial Ca2+ homeostasis. We expressed a mitochondrially targeted fluorescent Ca2+ reporter, Mitycam (Terhzaz, Southall, et al., 2006), in the motor neurons of both wt and homoplasmic mt:CoIT300I embryos using a UAS-mitycam transgene and the P{GawD42} Gal4 (D42-Gal4) driver (Pilling et al., 2006). We imaged mitochondrial Ca2+ dynamics in cultured primary motor neurons after stimulation with acetylcholine, a potent neurotransmitter that elicits Ca2+ influx into the cytoplasm and triggers mitochondrial Ca2+ uptake (Rohrbough and Broadie, 2002). Compared to wt mitochondria, mt:CoIT300I mitochondria took up Ca2+ more slowly and reached lower Ca2+ concentrations, demonstrating an impaired capacity for Ca2+ uptake (Figure 5, C and D, and Supplemental Movies S1 and S2).
We then applied the genetic mosaic scheme described earlier to test the potential role of Ca2+ mishandling in photoreceptor degeneration caused by mt:CoIT300I homoplasmy in the eye. We coexpressed Letm1, a Ca2+ transporter on the mitochondrial inner membrane (Jiang et al., 2009; Tsai et al., 2014), with MitoXhoI in the eye of heteroplasmic flies using the ey-Gal4 driver. We found that overexpression of Letm1 significantly suppressed retinal degeneration in homoplasmic mt:CoIT300I eyes (Figure 4, C and D). Many vacuoles were present in both young and old Letm1-overexpressing photoreceptors (Figure 4D), but the origin of these structures remains to be determined. Nonetheless, considering the role of Letm1 in mitochondrial Ca2+ uptake, this genetic suppression suggests that impaired mitochondrial Ca2+ uptake contributes to neurodegeneration in homoplasmic mt:CoIT300I eyes.
DISCUSSION
One of the problems hindering our understanding of the pathogenic mechanism of mtDNA mutations is the difficulty in obtaining homoplasmic mutant tissues, without which it is difficult to readily ascribe a phenotype to a given mtDNA mutation. We demonstrated that mitochondrially targeted restriction enzymes provide an effective means for selecting inheritable, homoplasmic mtDNA mutations in Drosophila (Xu et al., 2008). Here we report the genetic and biochemical analyses of mt:CoIT300I, a deleterious mutation we previously obtained through such selection that can propagate in both homoplasmic and heteroplasmic states (Hill, Chen, et al., 2014).
The mt:CoIT300I mutation disrupts the cytochrome c oxidase locus and is lethal at 29°C (Hill, Chen, et al., 2014). We show here that even at a permissive temperature (25°C), this mutation causes reduced lifespan and impaired mobility in adults. Strikingly, ectopic expression of a nucleus-encoded oxidase, AOX, which restores mitochondrial electron transport, could fully rescue the viability of homoplasmic mutant flies. Consistent with the proposed role of AOX in bypassing complex III and IV that pump protons into the intermembrane space, expression of AOX did not restore membrane potential or Ca2+ uptake in mt:CoIT300I motor neurons, which might explain why AOX expression only moderately extended the lifespan of adult mt:CoIT300I flies. Nonetheless, genetic rescue of mt:CoIT300I viability by AOX confirms that mortality at the pupal stage is caused by blockage of electron transport. Of greater importance, it validates ectopic expression of AOX as a potential therapeutic approach for disorders caused by mitochondrial mutations affecting cytochrome chains.
Besides its direct effect on energy production, disruption of electron transport by the mt:CoIT300I mutation could trigger a myriad of cellular deficiencies. As an example, we found that membrane potential was reduced in mt:CoIT300I mitochondria. We also found that Ca2+ uptake was impaired in mitochondria of mutant motor neurons, a likely consequence of reduced membrane potential (Rizzuto et al., 2000). Overexpression of Letm1, a mitochondrial Ca2+ transporter, partially suppressed the neurodegenerative phenotype caused by the mt:CoIT300I mutation, confirming that defective mitochondrial Ca2+ homeostasis contributes to the pathogenesis of mt:CoIT300I flies. Mitochondrial Ca2+ uptake plays an important role in attenuating intracellular Ca2+ signaling. Ca2+ also stimulates ATP production because it up-regulates a few key enzymes in the Krebs cycle and electron transport chain complexes (Wan et al., 1989; Glancy and Balaban, 2012). Of interest, impaired mitochondrial Ca2+ uptake and defective cytosolic Ca2+ regulation have been reported in cultured cells carrying other mtDNA mutations that disrupt electron transfer (Brini et al., 1999; Trevelyan, Kirby, et al., 2010). Disrupted mitochondrial electron transport would impede mitochondrial Ca2+ uptake and ATP production, which in turn compromise ATP-dependent cytosolic Ca2+ extrusion. It is likely that these two interconnected processes—Ca2+ mishandling and impaired energy homeostasis—play important roles in the pathogenesis of mitochondrial mutations, especially in tissues that are highly energy demanding and dependent on Ca2+ signaling, such as neurons and muscles.
Random segregation of mtDNA leads to variable mutational loads in different tissues. Although there is generally some correlation between mutation load and the severity of symptoms, symptoms often show a threshold effect. Thus it is extremely difficult, if not impossible, to establish a strict correlation between mutational load in a given tissue and its phenotypic presentation. Our ability to generate homoplasmic tissues in a heteroplasmic background allowed us to circumvent this limitation. Using this technique, we found that heteroplasmic flies harboring a homoplasmic mt:CoIT300I nervous system had a reduced eclosion rate and significantly reduced lifespan, similar to those of purely homoplasmic flies. These results suggest that bioenergetic deficiency in the nervous system contributes most to the demise of the whole organism. Our results do not exclude the possibility that COX disruption in other tissues also contributes to the eclosion and lifespan phenotypes. Nevertheless, they show that the effect of mt:CoIT300I on the nervous system is strong. To validate use of our tissue-mosaic approach for the study of mitochondrial mutations, we induced mt:CoIT300I homoplasmy in the eye, a nonessential organ that is widely used to model human neurodegenerative diseases. We found that homoplasmy caused severe retinal degeneration over time, highlighting the utility of this mosaic analysis in examining age-related phenotypes, as well as for future genetic dissection of the underlying pathogenic mechanisms.
Although mtDNAs are usually randomly segregated during cell division (Taylor and Turnbull, 2005), directional segregation of mutant mtDNAs in different tissues has been reported (Wallace and Chalkia, 2013), although the underlying mechanisms remain largely unknown. We found that mt:CoIT300I load remained constant at the restrictive temperature in most postmitotic tissues, including neurons and muscles, demonstrating a lack of selection mechanism against mtDNA mutations in these tissues. Mitophagy has been proposed to be a quality control mechanism to clear dysfunctional mitochondria through autophagic engulfment and degradation of the whole organelle, including mtDNA (Tolkovsky, 2009). Our results suggest a lack of such a potent mechanism to remove defective mitochondria containing this deleterious mtDNA mutation in Drosophila somatic tissues. The mt:CoIT300I level was significantly reduced in testes of aged heteroplasmic flies. We previously showed that replication competition contributed to the decline of the mt:CoIT300I genome in the female germline of heteroplasmic flies (Hill, Chen, et al., 2014). The same mechanism is probably at play in testes, which are highly proliferative compared with somatic tissues. It would be intriguing to test whether promoting mtDNA replication in other postmitotic cells could reduce the mt:CoIT300I load.
In conclusion, we anticipate that the genetic schemes we developed to mutate mtDNA, together with the powerful genetic tools available to manipulate the nuclear genome in Drosophila, will prove handy to dissect the pathological processes of other deleterious mtDNA mutations and understand the principles guiding their segregation and inheritance.
MATERIALS AND METHODS
Fly genetics and maintenance
Fly stocks were maintained on cornmeal/agar/molasses medium at 25°C under ambient light condition unless otherwise specified. mt:CoIT300I and mt:CoIsyn flies were selected as described previously (Xu et al., 2008) and backcrossed with w1118 male for more than five generations to clean up the nuclear background. All nuclear and mitochondrial genome combinations were obtained by crossing female flies carrying the desired mtDNA with males carrying desired nuclear mutations or transgenes. In all experiments described in this study, wt refers to wild-type mitochondrial genome, and w1118 was used as wild-type nuclear genome. When describing genotypes, mitochondrial genotypes are specified in parentheses, following nuclear genotypes. mt:CoIT300I heteroplasmic flies were generated as described previously (Hill, Chen, et al., 2014). mtDNA genotypes and levels of heteroplasmy were determined by molecular analyses described previously (Xu et al., 2008). Transgenic flies expressing AOX and Letm1 were generated through standard germline transformation procedures. Fly strains carrying UAS-mitoGFP, UAS-mitycam, UAS-SOD2, and UAS-catalase were described previously (Anderson et al., 2005; Terhzaz, Southall, et al., 2006; Hill, Chen, et al., 2014). Two Gal4 lines, actin-Gal4 (P{Act5C-GAL4}25FO1; Ekengren et al., 2001) and tubulin-Gal4 (P{tubP-GAL4}LL7; Lee and Luo 1999), were used to drive ubiquitous expressions of UAS-AOX and UAS-mitoXhoI. The following Gal4 lines were used to drive tissue-specific expression: D42-Gal4 (P{GawB}D42) in embryonic motor neurons (Pilling et al., 2006); eyeless-Gal4 (P{GAL4-ey.H}3-8) in eye (Stowers and Schwarz, 1999); Cg-Gal4(P{Cg-GAL4.A}2) in hemocytes and fat body (Henning et al., 2006); repo-Gal4(P{GAL4}repo) in glia cells (Sepp et al., 2001); Mef2-Gal4(P{GAL4-Mef2.R}3) in somatic muscles (Ranganayakulu et al., 1998); P{GawB}inscMz1407 in all neurons (Sweeney, Broadie, et al., 1995); P{GawB}71B in imaginal disks (Brand and Perrimon 1993); P{GawB}C805 in ring glands, larva guts, and tubules (Hrdlicka et al., 2002); and P{GawB}332.3 in amnioserosa and salivary glands (Wodarz et al., 1995). Unless otherwise specified, all fly stocks were obtained from Bloomington Drosophila Stock Center (Bloomington, IN).
Viability and lifespan analyses
Adult virgin females were allowed to mate with adult males in a mating cage. After 12 h, 50 embryos were collected from Petri dishes and transferred to fresh vials and cultured at 25 or 29°C. The numbers of pupae and emerging adult flies were recorded for each vial. To assay the lifespan, newly eclosed flies were separated by sex, and ∼20 flies were placed in one vial. Flies were transferred to fresh vials, and survivorship was recorded every 2 d. At least 10 vials of each genotype were used to determine the mean 50% survivor rate and SD.
Climbing assay
Twenty wt or mt:CoIT300I flies were randomly selected and transferred to a glass tube (15 cm long, 1.5 cm in diameter) with cotton covering the opening. After acclimation for 1 h, the flies were knocked to the bottom of tube by gently tapping the tubes. The time required for 50% of flies to climb to a 10-cm line was recorded. Three trials were used for each group, and three groups were tested for each genotype.
Cytochrome c oxidase activity assay
Ten male flies of each genotype were homogenized in 100 μl of sodium phosphate buffer containing 0.05% Tween-80. Supernatants were collected by centrifugation at 4000 g for 1 min. COX activity was determined using a COX assay kit (CYTOCOXI; Sigma-Aldrich, St. Louis, MO) and normalized with the protein concentration determined by Bradford assay. Data shown represent the average of three independent experiments.
Electron transport chain complex activity
Mitochondrial complex I (NADH dehydrogenase) activity was measured as the decrease in absorbance due to oxidation of NADH at 340 nm (extinction coefficient is 6220 M−1 cm−1). The reaction mixture (50 mM sodium phosphate, pH 7.4, 5 mM MgCl2, 2 mM KCN, 2.5 mg/ml bovine serum albumin, 2 μg/ml antimycin A, 130 μM NADH, and 100 μM decylubiquinone) was equilibrated at 25°C. The reaction was initiated by addition of isolated mitochondria (20 μg of protein), the linear decrease in absorbance was monitored for 2 min, rotenone (5 μM) was added, and any rotenone-insensitive activity was measured for 2 min. Complex I activity reported here was the rotenone-sensitive activity.
The activity of NADH-cytochrome c oxidoreductase (complexes I/III) or succinate-cytochrome c oxidoreductase (complexes II/III) was measured as the increase in absorbance due to the reduction of cytochrome c at 550 nm (extinction coefficient is 27.8 mM−1 cm−1). The reaction mixture consisted of 50 mM sodium phosphate buffer (pH 7.4), 80 μM horse heart cytochrome c, 2 mM KCN, and either 200 μM NADH (complexes I/III) or 5 mM succinate (complexes II/III). The reaction was initiated by adding isolated mitochondria (20 μg of protein), and the linear increase in absorbance was measured for 2 min, after which 5 μM rotenone was added and the absorbance was monitored for another 2 min. The activity reported for each complex was the rotenone-sensitive rate.
Isolation of mitochondria
Around 300 male flies were immobilized on ice and then ground by plastic pestles in 500 μl of ice-cold isolation buffer (250 mM sucrose, 5 mM Tris-HCl, 2 mM ethylene glycol tetraacetic acid, 1% [wt/vol] fatty acid–free bovine serum albumin [BSA], pH 7.4, at 4°C). The liquid was passed through two layers of gauze pads (Johnson & Johnson, New Brunswick, NJ) and immediately centrifuged at 500 × g for 3 min at 4°C. Supernatants were filtered by one layer of gauze pads and centrifuged at 9000 × g for 10 min. Supernatants were discarded, and pellets were washed twice in the isolation buffer without BSA. Pellets were carefully resuspended in 100 μl of isolation buffer without BSA. Protein concentrations of mitochondrial samples were determined by Bradford assay.
Mitochondrial cytochrome a and c measurement
Mitochondrial cytochrome a and c contents were measured spectrophotometrically with modifications described previously (Chess et al., 2013). Briefly, isolated mitochondria were solubilized with a 4% solution of dodecyl-β-maltoside (DDM) in 100 mM sodium phosphate buffer (pH 7.0). After mixing, the suspension was centrifuged to remove any residual solid material. Supernatants were used to measure the difference spectra by scanning from 500 to 650 nm on a spectrophotometer (Lambda 3B; PerkinElmer-Cetus, Waltham, MA) between oxidized and reduced states. The reduced state was obtained by incubating mitochondrial solution with 10 mM potassium cyanide and sodium ascorbate. Cytochrome a and c contents were determined using the 605- and 550-nm wavelengths, respectively. The molar extinction coefficients used for cytochrome a and c were 12 and 20.8 mM−1, respectively.
Blue native PAGE
Blue native PAGE was performed with isolated mitochondria using the NativePAGE Novex Bis-Tris Gel system (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol.
Analysis of retinal degeneration
Photoreceptor cell numbers were assessed by optical neutralization (Xu et al., 2004). Flies were allowed to eclose at 29°C and aged at 29°C under a 12-h light/12-h dark cycle. To assay the time course of photoreceptor loss, fly heads were dissected at different time points and immersed in immersion oil, and the mean number of rhabdomeres per ommatidium was determined. Each data point was based on examination of ≥90 ommatidia from at least seven flies. Photoreceptor morphology was assessed by electron microscopy (Xu et al., 2008). Hemisected fly heads were fixed in fixation solution (0.1 M sodium phosphate buffer, pH 7.4, 2% paraformdehyde, and 2% gluteraldehyde) overnight and postfixed with 1% Os2O4 for 2 h. After dehydration with ethanol series, the samples were embedded in LR white embedding resin (Polysciences, Warrington, PA). The 8-nm thin sections were stained with uranyl acetate and lead citrate and viewed on a Tecnai T12 (FEI, Hillsboro, OR) transmission electron microscope.
Western blot
Immunoblotting was carried out according to the standard protocol. Primary antibodies against CoI (Molecular Probes, Eugene, OR), CoIV (Abcam, Cambridge, MA), ATP synthase α-subunit (MitoSciences, Eugene, OR), and tubulin (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA) were used with 1:1000 dilutions. Horseradish peroxidase–labeled anti-mouse or anti-rabbit secondary antibodies (GE Healthcare, Pittsburgh, PA) were used at a 1:10,000 dilution. The immunoreactivity was revealed with Supersignal West Dura Chemiluminescent Substrate (Pierce Biotechnology, Rockford, IL).
Imaging of mitochondrial calcium
Mitycam imaging was conducted with cultured neurons isolated from embryos of wt and mt:CoIT300I flies. Male UAS-mitycam transgenic flies were crossed with female D42-Gal4 (mt:CoIT300I) flies. Embryos were collected 6 h after fertilization. Cells were dissociated by crushing embryos and cultured on a chambered coverglass in Schneider's Drosophila medium (Invitrogen) with 2 μg/ml cytochalasin D (Sigma-Aldrich; Pilling et al., 2006). Progeny from the cross between female UAS-mitycam transgenic flies and male D42-Gal4/CyO (mt:CoIT300I) flies, which have the same nuclear genotype but wt mtDNA, were used as control. Because Mitycam expression was driven by D42-Gal4, neuronal cells could be identified by the expression of Mitycam. The 2-d cultures were washed with Ringer's solution (130 mM NaCl, 5 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 36 mM sucrose, 5 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) three times and then incubated in Ringer's solution with 2 mM 2-deoxy-d-glucose (2-DG; Sigma-Aldrich). Neuronal cells expressing Mitycam were imaged live (excitation 488 nm/emission 520 nm) on a PerkinElmer Ultraview Vox system. Acetylcholine chloride (final concentration 400 μM; Sigma-Aldrich) was added at indicated time points.
Membrane potential measurements
TMRM staining was used for measuring membrane potential. mt:CoIT300I or wt females were crossed with UAS-mitoGFP;D42-Gal4 males, and embryonic primary cells were isolated from their progeny. After 2 d in culture, primary cells were washed with Ringer's solution (with 2 mM 2-DG) three times and loaded with 10 nM TMRM for 20 min at room temperature. Motor neuron cells expressing MitoGFP were imaged on an Ultraview Vox system. Fluorescence intensities of each region of interest were obtained and analyzed with Volocity 6.1.1 software (PerkinElmer).
Quantification of heteroplasmy
The heteroplasmic level was measured as described previously (Hill, Chen, et al., 2014). Briefly, total DNA was extracted from tissues or whole bodies of Drosophila. A 4-kb mtDNA fragment spanning the XhoI site on mtDNA was PCR amplified with primers 5′TGGAGCTATTGGAGGACTAAATCA3′ and 5′GCTCCTGTTAATGGTCATGGACT3′ and gel purified. A 500-ng amount of PCR product was digested with XhoI at 37°C overnight. The digested DNA was analyzed on an Agilent 2100 Bioanalyzer using DNA 7500 kit (Agilent, Santa Clara, CA). The proportion of mutant DNA was calculated by dividing the amount of undigested 4-kb fragment with the total amount of undigested 4 kb plus 2.4-kb and 1.6-kb XhoI-digested fragments.
Determination of ROS level
ROS levels were measured with the chemical probe H2DCF (Molecular Probes), which becomes fluorescent upon reacting with ROS.
For each group, 10 males were grounded in 200 μl of PBST (PBS, 0.1% Tween-20). After centrifugation at 13,000 × g, 4°C, for 10 min, the supernatant was collected and incubated with 50 μM H2DCF, and fluorescence intensities (excitation 490 nm/emission 520 nm) were measured and normalized to protein concentrations determined by Bradford method.
Protein carbonylation assay
Protein carbonylation assay was performed as described previously (Wehr and Levine, 2012). For preparation of total protein, 10 male flies were homogenized in 50 μl of RIPA buffer (Pierce Biotechnology) with protease inhibitor cocktail (Roche, Indianapolis, IN). After centrifugation at 12,000 × g for 10 min at 4°C, the supernatant was collected, and protein concentration was determined. Total or mitochondrial protein samples were adjusted to the same protein concentration and dissolved with a final concentration of 6% SDS and then derivatized by adding 20 mM 2,4-dinitrophenylhydrazine (DNPH; Sigma-Aldrich) in 10% trifluoroacetic acid (Pierce Biotechnology). Samples were neutralized with 2 M Tris and glycerol and then reduced in 2% β-mercaptoethanol. Western blot assay was performed to analyze the protein carbonylation. Goat anti-DNPH primary antibody (Bethyl, Montgomery, TX) was used at a 1:2000 dilution.
Drosophila respiration measurement
Drosophila respiration was performed at room temperature with Q-Box RP1LP low-range respiratory system (Qubit, Kingston, Canada). CO2 produced by 20 male flies in an open flow system was monitored and recorded for 15 min. The rate of CO2 produced per unit time by each fly was calculated, assuming the value of RQ (=VCO2/VO2) is 0.85.
Statistical analysis
Data were analyzed using Student's t test or one-way analysis of variance. The difference was considered statistically significant when p < 0.05.
Supplementary Material
Acknowledgments
We thank F. Chanut, K, Delaney, T. Finkel, and P. O'Farrell for their comments and editing of the manuscript and J. Phillips, J. Dows, and the Bloomington Drosophila Stock Center for fly stocks. This work is supported by the National Heart, Lung, and Blood Institute Intramural Program.
Abbreviations used:
- AOX
alternative oxidase
- ATPs-α
ATP synthase α
- CoI
cytochrome c oxidase subunit I
- COX
cytochrome c oxidase
- Mito-GFP
mitochondrially targeted green fluorescent protein
- MitoXhoI
mitochondrially targeted XhoI
- mtDNA
mitochondrial DNA
- ROS
reactive oxygen species.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-11-1513) on December 10, 2014.
REFERENCES
Boldface names denote co–first authors.
- Anderson PR, Kirby K, Hilliker AJ, Phillips JP. RNAi-mediated suppression of the mitochondrial iron chaperone, frataxin, in Drosophila. Hum Mol Genet. 2005;14:3397–3405. doi: 10.1093/hmg/ddi367. [DOI] [PubMed] [Google Scholar]
- Babcock GT, Wikström M. Oxygen activation and the conservation of energy in cell respiration. Nature. 1992;356:301–309. doi: 10.1038/356301a0. [DOI] [PubMed] [Google Scholar]
- Blair SS. Genetic mosaic techniques for studying Drosophila development. Development. 2003;130:5065–5072. doi: 10.1242/dev.00774. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Brini M, Pinton P, King MP, Davidson M, Schon EA, Rizzuto R. A calcium signaling defect in the pathogenesis of a mitochondrial DNA inherited oxidative phosphorylation deficiency. Nat Med. 1999;5:951–954. doi: 10.1038/11396. [DOI] [PubMed] [Google Scholar]
- Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Chess DJ, Billings E, Covian R, Glancy B, French S, Taylor J, de Bari H, Murphy E, Balaban RS. Optical spectroscopy in turbid media using an integrating sphere: mitochondrial chromophore analysis during metabolic transitions. Anal Biochem. 2013;439:161–172. doi: 10.1016/j.ab.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- Ekengren S, Tryselius Y, Dushay MS, Liu G, Steiner H, Hultmark D. A humoral stress response in Drosophila. Curr Biol. 2001;11:714–718. doi: 10.1016/s0960-9822(01)00203-2. [DOI] [PubMed] [Google Scholar]
- Fernandez-Ayala DJ, Sanz A, Vartiainen S, Kemppainen KK, Babusiak M, Mustalahti E, Costa R, Tuomela T, Zeviani M, Chung J, et al. Expression of the Ciona intestinalis alternative oxidase (AOX) in Drosophila complements defects in mitochondrial oxidative phosphorylation. Cell Metab. 2009;9:449–460. doi: 10.1016/j.cmet.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry. 2012;51:2959–2973. doi: 10.1021/bi2018909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakkaart GAJ, Dassa EP, Jacobs HT, Rustin P. Allotopic expression of a mitochondrial alternative oxidase confers cyanide resistance to human cell respiration. EMBO Rep. 2006;7:341–345. doi: 10.1038/sj.embor.7400601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig KM, Colombani J, Neufeld TP. TOR coordinates bulk and targeted endocytosis in the Drosophila melanogaster fat body to regulate cell growth. J Cell Biol. 2006;173:963–974. doi: 10.1083/jcb.200511140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JH, Chen Z, Xu H. Selective propagation of functional mitochondrial DNA during oogenesis restricts the transmission of a deleterious mitochondrial variant. Nat Genet. 2014;46:389–392. doi: 10.1038/ng.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrdlicka L, Gibson M, Kiger A, Micchelli C, Schober M, Schöck F, Perrimon N. Analysis of twenty-four Gal4 lines in Drosophila melanogaster. Genesis. 2002;34:51–57. doi: 10.1002/gene.10125. [DOI] [PubMed] [Google Scholar]
- Jiang D, Zhao L, Clapham DE. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science. 2009;326:144–147. doi: 10.1126/science.1175145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemppainen KK, Rinne J, Sriram A, Lakanmaa M, Zeb A, Tuomela T, Popplestone A, Singh S, Sanz A, Rustin P, Jacobs HT. Expression of alternative oxidase in Drosophila ameliorates diverse phenotypes due to cytochrome oxidase deficiency. Hum Mol Genet. 2013;23:2078–2093. doi: 10.1093/hmg/ddt601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert AJ, Brand MD. Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem J. 2004;382:511–517. doi: 10.1042/BJ20040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Luo L. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron. 1999;22:451–461. doi: 10.1016/s0896-6273(00)80701-1. [DOI] [PubMed] [Google Scholar]
- McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol. 2006;16:R551–560. doi: 10.1016/j.cub.2006.06.054. [DOI] [PubMed] [Google Scholar]
- Oliveira MT, Garesse R, Kaguni LS. Animal models of mitochondrial DNA transactions in disease and ageing. Exp Gerontol. 2010;45:489–502. doi: 10.1016/j.exger.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilling AD, Horiuchi D, Lively CM, Saxton WM. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol Biol Cell. 2006;17:2057–2068. doi: 10.1091/mbc.E05-06-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganayakulu G, Elliott DA, Harvey RP, Olson EN. Divergent roles for NK-2 class homeobox genes in cardiogenesis in flies and mice. Development. 1998;125:3037–3048. doi: 10.1242/dev.125.16.3037. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Bernardi P, Pozzan T. Mitochondria as all-round players of the calcium game. J Physiol. 2000;529:37–47. doi: 10.1111/j.1469-7793.2000.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrbough J, Broadie K. Electrophysiological analysis of synaptic transmission in central neurons of Drosophila larvae. J Neurophysiol. 2002;88:847–860. doi: 10.1152/jn.2002.88.2.847. [DOI] [PubMed] [Google Scholar]
- Sanz A, Fernández-Ayala DJM, Stefanatos RK, Jacobs HT. Mitochondrial ROS production correlates with, but does not directly regulate lifespan in Drosophila. Aging (Albany NY) 2010;2:200–223. doi: 10.18632/aging.100137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepp KJ, Schulte J, Auld VJ. Peripheral glia direct axon guidance across the CNS/PNS transition zone. Dev Biol. 2001;238:47–63. doi: 10.1006/dbio.2001.0411. [DOI] [PubMed] [Google Scholar]
- Severance S, Hamza I. Trafficking of heme and porphyrins in metazoa. Chem Rev. 2009;109:4596–4616. doi: 10.1021/cr9001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowers RS, Schwarz TL. A genetic method for generating Drosophila eyes composed exclusively of mitotic clones of a single genotype. Genetics. 1999;152:1631–1639. doi: 10.1093/genetics/152.4.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney ST, Broadie K, Keane J, Niemann H, O'Kane CJ. Targeted expression of tetanus toxin light chain in Drosophila specifically eliminates synaptic transmission and causes behavioral defects. Neuron. 1995;14:341–351. doi: 10.1016/0896-6273(95)90290-2. [DOI] [PubMed] [Google Scholar]
- Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terhzaz S, Southall TD, Lilley KS, Kean L, Allan AK, Davies SA, Dow JA. Differential gel electrophoresis and transgenic mitochondrial calcium reporters demonstrate spatiotemporal filtering in calcium control of mitochondria. J Biol Chem. 2006;281:18849–18858. doi: 10.1074/jbc.M603002200. [DOI] [PubMed] [Google Scholar]
- Tolkovsky AM. Mitophagy. Biochim Biophys Acta. 2009;1793:1508–1515. doi: 10.1016/j.bbamcr.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Trevelyan AJ, Kirby DM, Smulders-Srinivasan TK, Nooteboom M, Acin-Perez R, Enriquez JA, Whittington MA, Lightowlers RN, Turnbull DM. Mitochondrial DNA mutations affect calcium handling in differentiated neurons. Brain. 2010;133:787–796. doi: 10.1093/brain/awq023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai M-F, Jiang D, Zhao L, Clapham D, Miller C. Functional reconstitution of the mitochondrial Ca2+/H+ antiporter Letm1. J Gen Physiol. 2014;143:67–73. doi: 10.1085/jgp.201311096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Chalkia D. Mitochondrial DNA Genetics and the Heteroplasmy Conundrum in Evolution and Disease. Cold Spring Harb Perspect Biol. 2013;5:a021220. doi: 10.1101/cshperspect.a021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan B, LaNoue KF, Cheung JY, Scaduto RC. Regulation of citric acid cycle by calcium. J Biol Chem. 1989;264:13430–13439. [PubMed] [Google Scholar]
- Wehr NB, Levine RL. Quantitation of protein carbonylation by dot blot. Anal Biochem. 2012;423:241–245. doi: 10.1016/j.ab.2012.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wodarz A, Hinz U, Engelbert M, Knust E. Expression of crumbs confers apical character on plasma membrane domains of ectodermal epithelia of Drosophila. Cell. 1995;82:67–76. doi: 10.1016/0092-8674(95)90053-5. [DOI] [PubMed] [Google Scholar]
- Xu H, DeLuca SZ, O'Farrell PH. Manipulating the metazoan mitochondrial genome with targeted restriction enzymes. Science. 2008;321:575–577. doi: 10.1126/science.1160226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Lee S-J, Suzuki E, Dugan KD, Stoddard A, Li H-S, Chodosh LA, Montell C. A lysosomal tetraspanin associated with retinal degeneration identified via a genome-wide screen. EMBO J. 2004;23:811–822. doi: 10.1038/sj.emboj.7600112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.