

An improvement to the yeast protein-depletion systems using a promoter shutoff is presented. The described combination of tetracycline promoter with N-terminal ubiquitin fusion allows very fast depletion of the target protein in less than an hour. The depleted protein does not require fusion of any large degradation tag.
Abstract
A protein depletion by promoter shutoff or protein destabilization is an important tool in investigation of functions of essential genes. Various approaches using different repressible promoters, inducible degrons, or their combinations were developed. While successful, the current techniques have a drawback in that they require fusion of a large degradation tag to the target protein and/or a change in growth conditions to repress the promoter. We describe efficient protein depletion using the combination of a metabolically inert tetracycline repressible promoter with tetracycline aptamer and constitutive target protein destabilization by means of ubiquitin fusion. The target protein does not require a tag, and its elimination is several fold faster compared with standard promoter shutoff systems. A depletion time of <40 min was sufficient to achieve a robust phenotype.
INTRODUCTION
A protein depletion is a widely used approach for studying functions of essential genes. A protein can be depleted from a cell by, for example, repression of the gene's promoter, degradation or inhibition of its mRNA translation (RNA interference, microRNAs [miRNAs]), or destabilization and degradation of the protein itself. In yeast, the commonly used technique involves replacement of the endogenous promoter with a promoter that can be repressed by a small molecule, usually a metabolite, added to the growth media. Repressible promoters from genes such as GAL1-GAL10, MET3 and MET25, CUP1, and heterologous TetO7 promoter have successfully been used (Johnston and Davis, 1984; Mountain et al., 1991; Dohmen et al., 1994; Bellí et al., 1998). The presence of an appropriate metabolite/small molecule in growth media leads to repression of the promoter and subsequent depletion of the protein of interest from the cell. However, the depletion often takes a long time (12 h or even longer) before the target protein level drops sufficiently enough to exhibit a detectable phenotype. On a promoter repression, the major factors influencing the rate of depletion are the half-life of the protein itself and of its mRNA. In yeast, the median mRNA half-life is ∼20 min (Wang et al., 2002) compared with the median protein half-life of 45 min (Belle et al., 2006). Therefore the stability of the protein is in most cases the major determinant of the time required for a sufficient depletion. Methods to directly destabilize the protein of interest have been developed. A tag containing a degradation signal is fused to the target protein and, upon activation of such a degradation signal, the protein is rapidly degraded. One such approach uses a heat-inducible degron tag fused to the protein of interest. A shift to a higher temperature induces a conformational change of the degron tag and targets the protein for degradation (Dohmen et al., 1994). In a more recently described auxin degradation system, a plant-derived auxin-binding domain directs the fusion protein toward degradation after binding the hormone (Nishimura et al., 2009). While these systems have been successful, they share a disadvantage in the need to fuse a sizable tag to the protein of interest, which might interfere with its function. Furthermore, the change in the growth temperature required to activate the heat-inducible degron can directly affect many cellular processes and thus might not be suitable for the pathways studied.
Almost 30 yr ago, it was shown that protein stability is affected by the N-terminal amino acid (Bachmair et al., 1986). The authors found that the yeast intracellular machinery efficiently recognizes a ubiquitin-coding sequence fused to a reporter protein and precisely cleaves off the ubiquitin moiety. Interestingly, the stability of the remaining reporter part was dependent on the N-terminal amino acid residue remaining after the ubiquitin part was removed. By introduction of different amino acids in the fusion protein between the ubiquitin and the reporter, the authors determined the so-called N-end rule for protein stability (Bachmair et al., 1986; Varshavsky, 1996). Thus fusion of the ubiquitin followed by a destabilizing amino acid residue can be used to constitutively destabilize the target protein. Such ubiquitin fusion systems together with repressible GAL10 or CUP1 gene promoters has been used to improve depletion of target proteins (Cormack and Struhl, 1992; Park et al., 1992). However, in both cases, a change of growth media conditions was required to initiate the depletion, which in turn affected the metabolism of the cells.
We show here that a combination of tetracycline repressible promoter and a ubiquitin fusion system greatly shortens the time required for efficient depletion. The advantage of this combination approach is that the promoter repression is achieved by a metabolically inert molecule (tetracycline/doxycycline), and there is therefore no negative effect on the metabolism of the cell. The ubiquitin moiety is efficiently cleaved off in the cell, leaving the protein to be studied with only one amino acid change at its N-terminus that unlikely affects its function. Using this system, we could reduce the depletion time several fold to <1 h.
RESULTS AND DISCUSSION
To constitutively destabilize the target protein, we modified the existing TetO7-3HA cassette on a plasmid pMK140 (Alexander et al., 2010) by insertion of the ubiquitin-coding sequence followed by a codon for a destabilizing amino acid (DAA) upstream of the 3× hemagglutinin (HA; 3HA) tag (Figure 1A). The resulting depletion cassette can be integrated in the yeast genome, in frame with the open reading frame of interest. The expression of the resulting fusion protein ubiquitin-DAA-3×HA-proteinX is under the control of the TetO7 promoter. We included the 3×HA tag in the cassettes to allow simple detection of the targeted protein. Although we have not yet observed any adverse effects of the widely used 3×HA tag on the functionality of the several dozen proteins we have studied, an N-terminal tag can negatively affect the function of some fusion proteins. In this case, the integration cassettes can also be amplified using a reverse primer that base pairs upstream of the 3×HA tag and thus fuses only the DAA to the protein of interest. All the experiments were performed in the strain YMK118, in which TetO7 promoter is repressed by doxycycline added to the media (Alexander et al., 2010). Three constructs were created, with leucine, isoleucine, or alanine as DAA to allow titration of the fusion proteins' expression levels and of the rate of depletion. According to the N-end rule, proteins with leucine and isoleucine residues at their N-ends have expected half-lives of 10 and 30 min, respectively. Proteins with alanine as their N-terminal amino acid are considered stable (Bachmair et al., 1986).
FIGURE 1:

Comparison of different depletion systems. (A) Schematic representation of the depletion cassettes used. The promoter is indicated by a broken arrow; DAA, destabilizing amino acid; tc, tetracycline aptamer. (B) Growth of strains with the standard TetO7 promoter and the new TetO7-Ubi-Leu cassette during depletion. Doxycycline or ethanol (solvent) was added to exponentially growing cultures to a final concentration of 2 μg/ml, and optical density was measured at the indicated time points. (C) Reversibility of the depletion in the TetO7-Ubi-Leu-Has1 strain. Doxycycline or ethanol (solvent) was added to exponentially growing yeast to a final concentration of 2 μg/ml at time 0. After 8 h, the doxycycline/ethanol was washed away, and the cultures were monitored until the exponential growth was restored. (D) Viability of cells after 8 h of depletion. Two hundred cells from the TetO7-Ubi-Leu-Has1 strain were grown for 8 h with or without doxycycline and then plated on YPD agar; growing colonies were counted after 2 d. For all experiments, a mean of three independent biological replicas is shown; error bars represent SDs. The y-axes in B and C are in logarithmic scale.
The resulting depletion cassettes were integrated upstream and in frame with the coding sequence of the essential HAS1 gene in the strain YMK118. We compared the growth during depletion of strains with our new TetO7-Leu-DAA-3HA cassette with leucine as the DAA and the strains carrying the standard TetO7-3HA cassette. While ∼6 h of depletion were required until the growth curves of the TetO7-Has1 strain visibly deviated from the nondepleted strain, only 3 h were required for the TetO7-Ubi-Leu-Has1 strain (Figure 1B). The time required for depletion using GAL7 promoter was ∼7 h (unpublished data). The results indicated that Has1p is indeed more rapidly depleted in the TetO7-Ubi-Leu-Has1 strain. The depletion of the Has1p is fully reversible. Cells depleted for 8 h resumed their exponential growth ∼10 h after change into media without doxycycline. As Has1p is a ribosome biogenesis factor, the depleted cells have limiting amounts of functional ribosomes. Therefore the rather slow recommencement of the exponential growth is likely due to a reduced translational capacity of the depleted cells. The viability and colony-forming capabilities of the depleted and nondepleted cells were identical (Figure 1, C and D).
The rates of Has1p protein depletion in TetO7 and TetO7-Ubi-Leu strains were analyzed by Western blotting (Figure 2A). We observed a rapid drop of Has1p levels to below the 20% level within 2 h of depletion in the TetO7-Ubi-Leu, while ∼75% of the protein remained in the TetO7 strain at the same time point, in agreement with the observed differences in the growth rates of the strains. Next we compared the Has1p depletion using the TetO7-Ubi-DAA with leucine, isoleucine, and alanine as destabilizing residues. The Has1p protein was depleted fastest in the TetO7-Ubi-Leu strain and with an intermediate rate in the TetO7-Ubi-Ile strain. In the TetO7-Ubi-Ala strain, which is expected to produce a stable protein, the depletion rate was very similar to the standard TetO7 strain (Figure 2B). Interestingly, the Has1p protein depletion did not show a simple exponential behavior as would be expected in an ideal case. An initial rapid drop in the Has1p levels was followed by a much slower reduction as the strain growth rate reduces. Furthermore, the Leu-Has1p half-life was ∼60 min, considerably longer than the 10 min predicted by the N-end rule. As a component of large multiprotein complexes, Has1p might not be readily accessible to proteasomes. Therefore only the accessible pool of Has1p is degraded rapidly and the depletion of the remaining Has1p, present in complexes, is much slower and follows duplication of the cells.
FIGURE 2:

Western blot analysis of Has1p levels in strains with different depletion cassettes. Protein samples from strains were collected at the indicated time points following the addition of doxycycline. The proteins were detected using antibodies against the HA tag and actin. The quantification of the blots, normalized to actin levels, is plotted on the right; a mean of two independent experiments is shown. The following strains were used: (A) TetO7-Has1 and TetO7-Ubi-Leu-Has1, (B) TetO7-Ubi-Ile-Has1 and TetO7-Ubi-Ala-Has1, and (C) TetO7-tc-Ubi-Leu-Has1 with added tetracycline aptamer.
The apparent longer than expected half-life could also be caused by an ongoing synthesis of the Has1p due to a delayed shutdown of its gene transcription and/or stability of its mRNA. However, the Northern blot analysis showed that, upon addition of doxycycline, the TetO7 promoter was rapidly repressed and the Has1p mRNA was undetectable after 20 min for all constructs (Figure 3). Therefore the contribution of the ongoing de novo synthesis of Has1p is limited. Nevertheless, we tried to improve the depletion efficiency by introducing a tetracycline-responsive (tc) aptamer into the mRNA upstream of the initiation AUG (Figure 1A). Such an aptamer has been described as blocking translation upon binding of tetracycline (Kötter et al., 2009). This should have allowed us not only to repress the transcription of the target gene but also to directly block its translation by addition of both doxycycline and tetracycline to the media. However, inclusion of the tc aptamer did not further improve the depletion rate, and neither the growth nor the rate of Has1p protein depletion was significantly affected (Figure 2C).
FIGURE 3:
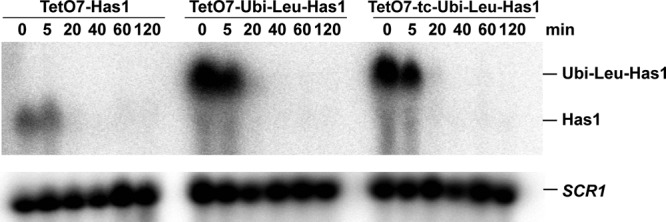
Northern blot analysis of HAS1 mRNA levels during depletion. Total RNA was isolated at indicated time points after addition of doxycycline. The blots were hybridized with probes complementary to HAS1 mRNA and SCR1 RNA (loading control).
To determine whether the TetO7-Ubi-DAA cassettes can be used in general to reduce depletion times, we tested the strategy with six other essential proteins. We selected proteins from different cellular processes and spanning three orders of magnitude in their cellular abundance (Table 1). Depletion of all these proteins in strains carrying the TetO7-Ubi-Leu cassette was significantly faster than in strains with only the TetO7 promoter (Figure 4). In all cases, the depletion time required for an obvious deviation of growth of the depleted versus nondepleted control strains was several hours shorter. The individual protein depletion was also faster in the TetO7-Ubi-Leu strains. The Western blotting analyses revealed that the steady-state levels of Bfr2p, Dbp4p, Prp19, and Rrn3 were reduced already in the absence of doxycycline in TetO7-Ubi-Leu strains, as could be expected, due to the faster turnover of the Ubi-Leu fusion protein. Interestingly, only the TetO7-Ubi-Leu-Dbp4 strain exhibited slightly slower growth in the absence of doxycycline, whereas the other strains were not affected. It is likely that the reduced steady-state levels of Dbp4p in the TetO7-Ubi-Leu strain are already limiting for growth and therefore, upon the start of depletion, the cell growth diminishes more or less immediately, as evidenced by the growth curve (Figure 4A). In this case, use of a less-destabilizing DAA, such as isoleucine, can be used to increase the steady-state (nondepleted) levels of the target protein.
TABLE 1:
Function and abundance of proteins used in this study.
| Name | Molecular function, process | Abundance [molecules/cell] ( Ghaemmaghami et al., 2003) |
|---|---|---|
| Bfr2p | Ribosome biogenesis | 15,000 |
| Dbp4p | RNA helicase, ribosome biogenesis | 8500 |
| Erg1p | Squalene epoxidase, ergosterol biosynthesis | 65,000 |
| Has1p | RNA helicase, ribosome biogenesis | — |
| Prp19p | RNA splicing factor | 11,700 |
| Rrn3p | Transcription factor for RNA polymerase I | 138 |
| Sec62p | Protein transporter, translocation into endoplasmic reticulum | 16,500 |
FIGURE 4:

Comparison of depletion of various proteins in TetO7 and Tet07-Ubi-Leu strains. Growth curves (left) and Western blot analysis (middle and right) of strains depleted of essential proteins Dbp4p (A), Bfr2p (B) Erg1 (C), Sec62 (D), Prp19 (E), and Rrn3 (F) using different depletion cassettes. Samples were collected at the indicated time points following the addition of doxycycline. The individual proteins were visualized using antibodies against the HA tag and actin. The quantification of the blots, normalized to actin levels, is shown on the right; a mean of two independent experiments is shown. For growth curves (left charts), the mean of three independent experiments is plotted; vertical y-axes are in logarithmic scale.
Figures 1 and 4 show that the depletion rates (and subsequent growth defects) are to a certain extent protein dependent. This is not surprising, as various proteins play roles in different cellular processes, and cells might continue growing for some time even after a protein is eliminated. For example, while depletion of a ribosome biogenesis factor prevents synthesis of new ribosomes, the cells can use the already existing ribosomes and continue dividing, albeit more slowly, for a few generations. With regard to the use of constitutive protein destabilization, an important factor to consider is protein stability. Depletion of inherently unstable proteins with short half-lives might benefit little or not at all from the constitutive destabilization system presented here. On the other hand, the approach should work well for proteins with longer half-lives.
The results presented so far show that growth in the strains carrying the TetO7-Ubi-Leu cassette slows several hours before growth in the strains carrying the standard TetO7 promoter. While convenient, monitoring of cell growth allows only a gross measure of the defects resulting from the reduced levels of the protein studied. The change in the growth rate cannot be used to reliably time the first appearance of a phenotype resulting from the target protein depletion. Therefore we took advantage of the easily detectable RNA-processing phenotypes of Has1p and Prp19p and determined the time of appearance of first RNA-processing defects in the depleted strains. The Has1p protein is essential for rRNA processing, in particular for production of the 20S pre-rRNA intermediate (Emery et al., 2004). Therefore we used Northern blotting to determine the shortest depletion time required to abolish 20S pre-rRNA synthesis. Surprisingly, only 40 min after the start of depletion, the 20S pre-rRNA levels were clearly reduced and synthesis of 20S was barely detectable after 60 min in the TetO7-Ubi-Leu-Has1 strain (Figure 5A). The inclusion of the tc aptamer had only a mild positive effect on the depletion (Figure 5A, bottom panel). In comparison, in the standard TetO7-Has1 strain, the reduction in the 20S pre-rRNA levels was noticeable after only 240 min, with the 20S pre-rRNA signal comparable with the 40-min time point in the TetO7-Ubi-Leu-Has1 strain. Therefore the depletion time required to achieve a robust measurable phenotype was reduced sixfold in the TetO7-Ubi-Leu-Has1 strain. The Prp19p is an essential splicing factor. Northern blotting was used to detect unspliced, intron-containing U3 small nucleolar RNA (snoRNA) at different time points during depletion (Figure 5B). It is important to note that the unspliced U3 snoRNA (similar to unspliced pre-mRNAs) can also be detected in normally growing wild-type cells, explaining the weak signal present in all the analyzed time points. Increased levels of the unspliced U3 were detected after 1–2 h in the TetO7-Ubi-Leu-Prp19 strain compared with 8 h in TetO7-Prp19 strain. Thus, also in the case of Prp19p, the appearance of the first processing defects resulting from the protein depletion is largely accelerated.
FIGURE 5:

Analysis of RNA processing and splicing during depletion of Has1p and Prp19p using different cassettes. Total RNA was isolated at the indicated time points following the addition of doxycycline. (A) Analysis of 20S pre-rRNA synthesis in TetO7-Has1, TetO7-Ubi-Leu-Has1, and TetO7-tc-Ubi-Leu-Has1 strains. Total RNA was separated in a 1% glyoxal agarose gel and blotted, and the membrane was hybridized with oligonucleotides complementary to 20S pre-rRNA and SCR1 RNA, which was used as a loading control. (B) Analysis of U3 snoRNA splicing in TetO7-Prp19 and TetO7-Ubi-Leu-Prp19 strains. The total RNA was separated in 6% denaturing polyacrylamide gel and blotted, and the membranes were hybridized with oligonucleotides complementary to the U3 snoRNA intron and 5S rRNA (loading control).
In summary, the combination of tetracycline promoter shutoff with a constitutive protein destabilization described here represents a simple and valuable improvement on the existing depletion systems. It greatly reduces the time required for an efficient depletion of a studied target protein, potentially to <1 h. In addition, the use of TetO7 promoter does not require a change of growth conditions that could affect the process under investigation. Furthermore, the integration cassettes described here can be used to create fusion protein with or without 3×HA tag at the N-terminus. The protein of interest carries only a single additional amino acid at its N-terminus that is unlikely to affect its normal function. The faster elimination of the protein from the cell helps to avoid the appearance of confounding secondary phenotypic effects due to prolonged depletion. The approach is simple and compatible with the existing tetracycline promoter–depletion strains and can therefore be readily used in any yeast lab.
MATERIALS AND METHODS
Plasmids and strains
The PTetO7-ubiquitin destabilizing cassettes were generated by cloning the ubiquitin-coding sequence (PCR-amplified from yeast UBI3 gene) along with the codon for the destabilizing amino acid residue (leucine, isoleucine, or alanine) into the HindIII site of the plasmid pMK140 containing the PTetO7-3HA cassette (Alexander et al., 2010). The tetracycline aptamer (Kötter et al., 2009) was inserted by PCR into the NsiI and ClaI sites of the plasmid-containing TetO7-Ubi-Leu-3HA cassette, upstream of the ubiquitin sequence. Oligonucleotides used are listed in Supplemental Table S4.
The Saccharomyces cerevisiae strain YMK118 was used for all experiments (Alexander et al., 2010). This strain carries both the tetracycline transcriptional activator and repressor integrated in the genome and allows fast shutoff of the TetO7 promoter by doxycycline. All strains and plasmids used are listed in Supplemental Tables S1–S3. Cells were grown in yeast–peptone–dextrose (YPD) media, and depletion was initiated by addition of doxycycline to the growth media in the final concentration 2 μg/ml. For nondepleted strains, the same volume of ethanol (solvent) was added.
The yeast strain YMK118 and plasmids used to generate the depletion strains were deposited in the Euroscarf collection (Euroscarf.de).
Western blotting
Total protein extract from equal numbers of cells was prepared using the TCA extraction method. An equal amount of proteins was resolved in an 8% SDS–PAGE gel and transferred to an Immobilon-FL (Millipore, Darmstadt, Germany) membrane using a wet electrotransfer system (Bio-Rad, Munich, Germany). The membranes were incubated with an anti-HA (Abcam, Cambridge, UK) antibody followed by anti-rabbit immunoglobulin G antibody coupled with Alexa Fluor 680 (Molecular Probes, Life Technologies, Darmstadt, Germany) and scanned on the Odyssey Clx imager (Li-Cor, Bad Homburg, Germany). The Western blots were quantified using AIDA software (Raytek, Berlin, Germany).
RNA isolation and Northern blotting
The total RNA isolation and Northern blotting were performed as previously described (Boon and Koš, 2010). Briefly, total RNA from 0.2 ODs of cells (loading by OD units) was resolved on either 1% agarose or 6% denaturing polyacrylamide gels and transferred to a nylon membrane using wet electroblotting. The blots were hybridized with random primed [32P]-labeled probes (High Prime, Roche) complementary to the HAS1 gene or [32P]-labeled oligonucleotide complementary to the U3 snoRNA intron sequence (5′-AAGCTGCTGCAATGGTT-3′), 20S pre-rRNA (5′-CGGTTTTAATTGTCCTA-3′), 5S rRNA (5′-GATTGCAGCACCTGAGT-3′), or SCR1 RNA(5′-ATCCCGGCCGCCTCCATCAC-3′) (Boon and Koš, 2010). The blots were exposed to phosphoimager plates and scanned on the FLA-7000 imager (Fuji).
Supplementary Material
Acknowledgments
We thank all the members of our lab for helpful discussions and advice. We also thank Emma Thomson for advice and help with the manuscript. M.K.'s group is funded by the Cluster of Excellence Cell Networks (grant number EXC 81).
Abbreviations used:
- DAA
destabilizing amino acid
- tc
tetracycline-responsive
- YPD
yeast–peptone–dextrose.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-07-1186) on December 24, 2014.
REFERENCES
- Alexander RD, Barrass JD, Dichtl B, Kos M, Obtulowicz T, Robert M-C, Koper M, Karkusiewicz I, Mariconti L, Tollervey D, et al. RiboSys, a high-resolution, quantitative approach to measure the in vivo kinetics of pre-mRNA splicing and 3′-end processing in Saccharomyces cerevisiae. RNA. 2010;16:2570–2580. doi: 10.1261/rna.2162610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmair A, Finley D, Varshavsky A. In vivo half-life of a protein is a function of its amino-terminal residue. Science. 1986;234:179. doi: 10.1126/science.3018930. [DOI] [PubMed] [Google Scholar]
- Belle A, Tanay A, Bitincka L, Shamir R, O'shea EK. Quantification of protein half-lives in the budding yeast proteome. Proc Natl Acad Sci USA. 2006;103:13004–13009. doi: 10.1073/pnas.0605420103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellí G, Garí E, Aldea M, Herrero E. Functional analysis of yeast essential genes using a promoter-substitution cassette and the tetracycline-regulatable dual expression system. Yeast. 1998;14:1127–1138. doi: 10.1002/(SICI)1097-0061(19980915)14:12<1127::AID-YEA300>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Boon K-L, Koš M. Deletion of Swm2p selectively impairs trimethylation of snRNAs by trimethylguanosine synthase (Tgs1p) FEBS Lett. 2010;584:3299–3304. doi: 10.1016/j.febslet.2010.07.001. [DOI] [PubMed] [Google Scholar]
- Cormack BP, Struhl K. The TATA-binding protein is required for transcription by all three nuclear RNA polymerases in yeast cells. Cell. 1992;69:685–696. doi: 10.1016/0092-8674(92)90232-2. [DOI] [PubMed] [Google Scholar]
- Dohmen RJ, Wu P, Varshavsky A. Heat-inducible degron: a method for constructing temperature-sensitive mutants. Science. 1994;263:1273–1276. doi: 10.1126/science.8122109. [DOI] [PubMed] [Google Scholar]
- Emery B, De La Cruz J, Rocak S, Deloche O, Linder P. Has1p, a member of the DEAD-box family, is required for 40S ribosomal subunit biogenesis in Saccharomyces cerevisiae. Mol Microbiol. 2004;52:141–158. doi: 10.1111/j.1365-2958.2003.03973.x. [DOI] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh W-K, Bower K, Howson RW, Belle A, Dephoure N, O'shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- Johnston M, Davis RW. Sequences that regulate the divergent GAL1-GAL10 promoter in Saccharomyces cerevisiae. Mol Cell Biol. 1984;4:1440–1448. doi: 10.1128/mcb.4.8.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kötter P, Weigand JE, Meyer B, Entian K-D, Suess B. A fast and efficient translational control system for conditional expression of yeast genes. Nucleic Acids Res. 2009;37:e120. doi: 10.1093/nar/gkp578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mountain HA, Byström AS, Larsen JT, Korch C. Four major transcriptional responses in the methionine/threonine biosynthetic pathway of Saccharomyces cerevisiae. Yeast. 1991;7:781–803. doi: 10.1002/yea.320070804. [DOI] [PubMed] [Google Scholar]
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods. 2009;6:917–922. doi: 10.1038/nmeth.1401. [DOI] [PubMed] [Google Scholar]
- Park EC, Finley D, Szostak JW. A strategy for the generation of conditional mutations by protein destabilization. Proc Natl Acad Sci USA. 1992;89:1249–1252. doi: 10.1073/pnas.89.4.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A. The N-end rule: functions, mysteries, uses. Proc Natl Acad Sci USA. 1996;93:12142–12149. doi: 10.1073/pnas.93.22.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Liu CL, Storey JD, Tibshirani RJ, Herschlag D, Brown PO. Precision and functional specificity in mRNA decay. Proc Natl Acad Sci USA. 2002;99:5860–5865. doi: 10.1073/pnas.092538799. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.