

Abstract
The nematode Caenorhabditis elegans has two ADF/cofilin isoforms, UNC-60A and UNC-60B, which are expressed from the unc60 gene by alternative splicing. UNC-60A has higher activity to cause net depolymerization, and to inhibit polymerization, than UNC-60B. UNC-60B, on the other hand, shows much stronger severing activity than UNC-60A. To understand the structural basis of their functional differences, we have determined the solution structures of UNC-60A and UNC-60B proteins and characterized backbone dynamics. Both UNC-60A and UNC-60B show conserved ADF/cofilin fold. The G-actin binding regions of the two proteins are structurally and dynamically conserved. Accordingly, UNC-60A and UNC-60B individually bind to rabbit muscle ADP-G-actin with high affinities, with Kd of 32.25 nM and 8.62 nM, respectively. The primary differences between these strong and weak severing proteins were observed in the orientation and dynamics of the F-actin binding loop (F-loop). In the strong severing activity isoform UNC-60B, the orientation of F-loop was towards the recently identified F-loop binding region on F-actin, and the F-loop was relatively more flexible with fourteen residues showing motions on nanosecond-picosecond timescale. In contrast, in the weak severing protein isoform UNC-60A, the orientation of F-loop was away from the F-loop binding region and inclined towards its own C-terminal and strand β6. It was also relatively less flexible with only five residues showing motions on nanosecond-picosecond timescale. These differences in structure and dynamics seem to directly correlate to the differential F-actin site-binding and severing properties of UNC-60A and UNC-60B, and other related ADF/cofilin proteins.
Keywords: G-actin, NMR, F-actin, Protein dynamics, ITC
Introduction
The proteins of the eukaryotic ADF/cofilin family are essential and key regulators of actin filament dynamics [1]. The ADF/cofilin proteins regulate actin filament dynamics through G-actin binding, F-actin binding and depolymerization [2], F-actin severing [3], G-actin monomer sequestering activity [4, 5, 6], and controlling the rate of nucleotide exchange from actin monomers [3, 7, 8, 9]. ADF/cofilin proteins increase the turnover rate of actin filaments by accelerating the dissociation of actin monomers from the pointed ends of filaments. In addition, F-actin filament severing generates new uncapped barded ends and pointed ends where polymerization and depolymerization can occur.
ADF-homology (ADF-H) domains are divided into five classes: ADF/cofilin; glia maturation factor (GMF); coactosin; twinfilin; and Abp1/debrins [10]. The ADF/cofilins represent the highly conserved ADF-H fold, which is also observed in destrin, actophorin, and gelsolin [11]. This canonical fold, which is well represented by the structure of 143 residues cofilin from Saccharomyces cerevisiae (ScCof), consists of a six stranded mixed β-sheet in which the four central strands are anti-parallel, while the two edge strands run parallel to the neighbouring strands. The β-sheet is sandwiched between a pair of α-helices on each face. The longest helix (α3) is kinked at the position of a serine residue in a conserved segment [12].
ADF/cofilins have two distinct actin-binding sites. These have been named as the G/F-site and the F-site. The G/F-site is required for binding to both, the G-actin and the F-actin. This site corresponds to the N-terminal flexible region, the long kinked helix (α3), the last β-strand (β5 or β6), and the loop before the C-terminal helix (α4) [2, 10]. The long-kinked helix of ADF/cofilins binds at the groove between subdomain 1 and subdomain 3 of actin, which is also a site for the binding of many other actin binding proteins [2, 10]. F-site is responsible for binding to F-actin and is comprised of the ‘F-loop’, which is a long loop between the strand β4 and β5 (or β3 and β4) that typically protrudes out of the structure; C-terminal α-helix; and the C-terminal residues, all of which contain several basic or charged residues [2, 10].
In the regular actin filament, residues of SD2 and SD4 of protomer n, interact with SD1 and SD3 of protomer n+2, respectively, which are longitudinally connected with each other [13]. ADF/cofilin molecule contacts with two longitudinally connected actin protomers (n and n+2) in an actin filament by binding in a cleft between them [13]. The F-site interacts with the protomer n, while G/F-site interacts with longitudinally succeeding protomer n+2. Rotation of outer domain (SD1 and SD2) of actin protomer in actin filament, upon binding of ADF/cofilin, leads to breaking of all longitudinal contacts between longitudinally adjacent protomers. This change cooperatively continues to the bare region of actin filament. As a result, severing of actin filament takes place at the junction of bare and ADF/cofilin decorated region [13].
Actin dynamics has been shown to play an important role in oocyte development, fertilization, and a wide range of important events during early embryogenesis including proper chromosome segregation and cytokinesis, in the nematode Caenorhabditis elegans [14]. C. elegans has been used as the model organism to study the organization and function of myofibrils, because the body wall muscles of this organism are obliquely striated muscle and are structurally and biochemically related to vertebrate striated muscles [15]. Among the several actin dynamics and modulatory proteins that have been characterized for C. elegans, two important proteins are UNC-60A and UNC-60B, which belong to the ADF/cofilin family and are expressed from the unc60 gene by alternative splicing [14]. Mutation in the unc60 gene results in slow movement and paralyzed nematodes [14]. UNC-60A has been shown to be essential for organized assembly of contractile actin network in the myoepithelial sheath of C. elegans somatic gonads, while UNC-60B is expressed in body wall muscles, and is required for the proper assembly of actin in myofibrils [15, 16].
UNC-60A is 165 amino acids in length and exhibits strong pointed end depolymerzation on C. elegans actin (Ce-actin), strong inhibition of polymerization, strong monomer sequestering activity, weak severing activity, and low affinity for F-actin binding [9]. UNC-60B is 152 amino acids in length and exhibits strong pointed end depolymerization on rabbit muscle actin, strong severing activity, and high affinity for F-actin binding [9]. UNC-78, an AIP-1 isoform, disassembles UNC-60B-bound actin filaments, prevents formation of aggregates, and maintains the dynamic state of their interactions. Both UNC-60A and UNC-60B isoforms show only slight effect of pH on their activity [9]. On the basis of activity, UNC-60A is classified in the ADF subgroup and UNC-60B is classified in cofilin subgroup. UNC-60B has greater impact on actin filament dynamics because of strong severing activity, while UNC-60A maintains pool of monomeric form of actin because of its high monomer sequestering activity [9, 16].
To understand the structure activity relationship of these two ADF/cofilins, we have determined the solution structures of UNC-60A and UNC-60B using NMR spectroscopy. We have also characterized the dynamics of these proteins by measuring the 15N-relaxation rates and steady state heteronuclear {1H}-15N NOE, and analysing these through the Lipari-Szabo formalism. The dynamics were also characterized by relaxation dispersion studies. Further, we have performed NMR chemical shift perturbation analysis of the effect of F-loop and C-terminal mutation on UNC-60A and UNC-60B structure. The UNC-60A and UNC-60B structures were docked on the crystal structure of rabbit muscle G-actin monomer. The binding affinities, characterizing the interaction of UNC-60A and UNC-60B with rabbit muscle ADP-G-actin, were determined by isothermal titration calorimetry. Our results demonstrate that specific combinations of the F-loop orientation, flexibility, and charge are the determining factors for both actin sequestering and F-actin binding.
Experimental procedures
Sequence alignment of UNC-60B with UNC-60A and other ADF/cofilin proteins
Protein-protein BLAST was carried out at the NCBI server (http://www.ncbi.nlm.nih.gov/BLAST) by taking UNC-60B sequence as a template. A multiple sequence alignment, including UNC-60A and other ADF/cofilin proteins described below, was generated using ClustalW. The following protein sequences (with the GenBankTM sequence ID nos.) were retrieved from NCBI and used in the alignment: AtADF1, Arabidopsis thaliana ADF1 (AAC72407); AcActophorin, Acanthamoeba castellani actophorin (AAA2909); ScCOF, Saccharomyces cerevisiae cofilin (AAA13256); C. elegans UNC-60A (AAL02461); C. elegans UNC-60B (AAC14457),; Ldcof, Leishmania donovani ADF/cofilin (AAY99389), PfADF2, Plasmodium falciparum ADF2 (NP705497); PfADF1, P. falciparum ADF1(NP703379), and TgADF, Toxoplasma gondii ADF (AAC47717). The alignment file was used as the input file for the program ESPript (http://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi) which was used to prepare the figure of alignment.
Cloning and Preparation of NMR samples
Clones of UNC-60A and UNC-60B were obtained from Dr. Shoichiro Ono, in expression vector pET-3d [15]. The Caenorhabditis elegans unc60a and unc60b genes were subcloned into pETNH6 vector using restriction sites NcoI and BamHI. The clones were over-expressed in BL21 (λDE3) strain of E. coli. The cloning procedure added extra residues at the N-terminal that included a hexa-histidine tag and a TEV-protease site. Conditions for optimal over-expression and purification were standardized. The yield of purified UNC-60A protein was 30 mg/L and UNC-60B was 35mg/ L of culture medium. For isotopic labelling, over-expressions of UNC-60A and UNC-60B were standardized in minimal media containing 15N-ammonium sulphate and 13C-glucose (CIL, MA, USA) as the sole nitrogen and carbon sources, respectively.
NMR samples of 13C/15N-labeled UNC-60A and UNC-60B were prepared at concentration of approximately 0.65 mM and 1.0 mM, respectively in NMR buffer (20 mM sodium phosphate pH 6.5, 50 mM NaCl, and 0.1% NaN3) containing 93% H2O/7% 2H2O. For backbone dynamics study, NMR sample of 15N-labeled UNC-60A and UNC-60B were prepared at concentration of approximately 0.7 mM in NMR buffer containing 93% H2O/7% 2H2O.
For chemical shift perturbation (CSP) analysis, mutants K100T (F-loop) and K161N (C-terminal) were constructed for UNC-60A, and R80A, K91A and R80A-K91A (F-loop), and ΔRI, I152A, R151A (C-terminal) were constructed for UNC-60B and sequences were verified by DNA sequencing. Proteins were expressed and purified as described above for the wtUNC-60A and wtUNC-60B.
Structure calculations
Distance restraints were obtained from 3D 15N-edited NOESY-HSQC spectra (τmix-150 ms), 13C-edited NOESY-HSQC (τmix-160 ms), and 13C (aromatic)-edited NOESY-HSQC spectra (τmix-160 ms) recorded on a Bruker 800MHz NMR spectrometer. All the spectra were processed using Topspin3.0 and all NOE were assigned manually using CARA-1.8.4 [17]. Integrated NOE peaks were calibrated and converted to distance restraints with the program CALIBA [18]. In the final structural determination, the program CYANA-3.0 [19] was used. The torsion angle restraints were obtained from assigned backbone chemical shifts using the program TALOS+ [20]. In total 2827 NOE and 2400 NOE distance restrains were identified, to which 74 and 60 hydrogen bond restrains were added for determination of UNC-60A and UNC-60B structures, respectively. In total, for the structures of UNC-60A and UNC-60B, 200 randomized conformers were generated and 10 conformers with lowest target function with no distance violation > 0.5 Å and no angle violations were selected for each. These 10 structures with lowest target functions were further subjected to molecular dynamics simulation in explicit water with NMR derived distance restraints and angle restraints using the CNS 1.21 program [21] and the standard water shell refinement protocol [22, 23]. At this stage the distances were relaxed. This step improved the Ramachandran plot statistics and also the Z-score for the Procheck (phi-psi) and Procheck (all) for the ordered residues. The programs CING and PSVS v1.4 (www.psvs-1_4.nesg.org) were used to analyze the quality of the structures. The program PYMOL (http://pymol.sourceforge.net/) was used for generating figures for structures.
Effect of point mutation by chemical shift perturbation analysis
15N-labelled proteins of different mutants of UNC-60A and UNC-60B were prepared as described for wtUNC-60A and wtUNC-60B, and 15N-HSQC spectra were recorded. CSPs were analysed to check the effect of point mutation on each residue. Combined chemical shift perturbation ΔTOTAL of 1HN and 15N nuclei were weighted according to ΔTotal = [(Δ1H)2 + (0.2*Δ15N)2]1/2 to normalize the larger chemical shift range of 15N [24]. The resonance frequencies at pH 6.5 of wtUNC-60A and wtUNC-60B were taken as reference points for their respective mutant.
Docking of rabbit muscle actin monomer with UNC-60A, UNC-60B and N-terminal mutant (G4K) of UNC-60B
The Crystal structure of C-terminal ADF-H domain of Twinfilin (Twf-C) in complex with G-actin monomer (PDB ID: 3DAW) was taken as a template to build a docked model of UNC-60A and UNC-60B with G-actin monomer [10]. G/F-site required for binding of ADF/cofilins with G-actin is highly conserved in UNC-60A and UNC-60B. We used ClusPro protein-protein docking web server which is publicly available (http://cluspro.bu.edu/login.php) [25] for docking studies. UNC-60B was taken as ligand and the G-actin monomer from the crystal structure of PDB ID: 3DAW, after removing the Twf-C from it, was taken as receptor and docked using the ClusPro protein-protein docking server. The details of the interface area were analyzed by using the web based server PDBePISA (http://www.ebi.ac.uk/msdsrv/prot_int/pistart.html).
Isothermal Titration Calorimetry
ITC experiments were performed at 25°C on a VP-ITC calorimeter from MicroCal™ (Northampton, MA, USA). The calorimeter was calibrated according to the user manual of the instrument. All the samples were briefly centrifuged and then degassed for 20 min before each of the ITC experiments. Titrations were performed at least in duplicate using the same set of stock solutions. The ITC experiments were performed by adding aliquots of ligands to rabbit muscle actin. The rabbit muscle actin for the experiments and samples for ITC (Stock solution of UNC-60A, UNC-60B and rabbit muscle ADP-G-actin) were prepared as described earlier [26, 27]. The sample cell was filled with 1.459 mL of 0.01 mM of rabbit muscle actin (titrand) and titrated against UNC-60A which was filled in the syringe of 290 μL at a concentration of 0.100 mM. For UNC-60B 0.005 mM of actin was titrated with 0.05 mM of UNC-60B. The injectant volume was set at 10 μL per injection, and the duration of the injection was 20 s, with an interval of 180 s between injections. During the titration, the reaction mixture was continuously stirred at 351 rpm. Control experiments were performed by injecting UNC-60B into G-actin buffer under conditions exactly similar to the rabbit muscle actin/titrand titration, to take into account heats of dilution and viscous mixing. The heats of injection of the control experiment were subtracted from the raw data of rabbit muscle actin and titrand titration.
The ITC data were analyzed using the ORIGIN version 7.0 software provided by MicroCal™. The heats of binding were normalized with respect to the titrand concentration, and a volume correction was performed to take into account dilution of titrand during each injection. The amount of heat produced per injection was calculated by integration of the area under each peak using a baseline selected by the ORIGIN program, assuming a one site binding model. The dissociation constant (Kd) and molar enthalpy (ΔH) for the binding of titrand to actin was determined by non-linear least square fitting to the data.
Relaxation measurements
To study backbone dynamics of UNC-60A and UNC-60B, experiments for measurement of 15N longitudinal and transverse relaxation rate constants and {1H}-15N steady state NOE were recorded as described previously [28]. For backbone amide 15N longitudinal relaxation rate constants, R1, two-dimensional correlation spectra were measured for 10 different relaxation delays: 0, 10, 70, 110, 190, 280, 450, 610, 890, and 1330 ms. Transverse relaxation rate constants, R2, were determined from correlation spectra measured with 9 different relaxation delays: 10, 30, 50, 70, 90, 130, 170, 190, 230 ms. {1H}-15N steady state NOE experiments were recorded in an interleaved manner with and without 3 second 1H saturation during the 5 second recycle delay. All of the relaxation parameters of UNC-60A and UNC-60B, along with the {1H}-15N nuclear Overhauser effect (NOE) intensities for backbone 15N nuclei, were measured at 25°C, at a magnetic field strength of 14.1 T (corresponding to the resonance frequency of 599.721 MHz for 1H). All the relaxation data were acquired with 96 Χ 1024 (t1 Χ t2) complex points and spectral width of 2309.494 Hz and 8396.300 Hz, respectively. Relaxation rate constants of UNC-60A and UNC-60B were determined from the single exponential decay of peak intensities in correlation spectra using the software Curvefit, while steady-state NOE values of UNC-60A and UNC-60B were determined from the ratio of peak-heights for spectra recorded with and without 1H saturation.
The heteronuclear 15N relaxation parameters R1, R2, and the steady state heteronuclear {1H}-15N NOE values were analyzed by Modelfree formalism as described earlier [28, 29, 30, 31]. For Modelfree analysis of UNC-60A and UNC-60B, the N-H bond length was assumed to be 1.01 Å and 15N chemical shift anisotropy value of 170 ppm was considered. The uncertainties in R1 and R2 of UNC-60A were set to an upper limit of 3% and 5%, respectively. While the uncertainties in steady state heteronuclear {1H}-15N NOE of UNC-60A was fixed at 0.05. In UNC-60B, the uncertainties of R1 and R2 were set to 3.5% and 5%, respectively, and the uncertainties in steady state heteronuclear {1H}-15N NOE of UNC-60B was fixed at 0.04. R1, R2, and steady state heteronuclear {1H}-15N NOE were subjected to Modelfree analysis [30, 31 32, 33] and Fast-Modelfree interface [33]. The dynamic parameters were extracted by using five simple models as proposed by Palmer and co-workers using Modelfree 4.1 program [34]. The models considered were : (1) S2 only, τe & Rex are negligible; (2) S2 and τe only, Rex is negligible; (3) model 1 and Rex term; (4) model 2 and Rex term; (5) incorporation of an additional order parameter for anisotropic rotational diffusion. The 15N-1H vectors with relaxation data that can be fitted to model 1 usually have large S2 values and are more rigid. 15N-1H vectors with relaxation data that must be fit to models 3 and 4 are those displaying conformational exchange processes on the millisecond to microsecond timescale. 15N-1H vectors with relaxation data fit to models 2, 4, or 5 display internal motions on the picosecond to nanosecond timescale.
Results
Sequence comparison of UNC-60A and UNC-60B proteins with other members of ADF/cofilin family
UNC-60A protein is 36% identical to UNC-60B [Fig. 1, bit score 110 from BLAST] and this intra-species identity is low in comparison to the three mammalian ADF/cofilins, which are all approximately 70% identical to each other. Among nematodes, ADF like UNC-60A shows high sequence identity (67–75%) with the ADF like isoforms in Ascaris suum, Loa loa, and Brugia malayi, while cofilin like isoform UNC-60B shows high sequence identity with cofilin like isoform in Ascaris suum, Loa loa and Brugia malayi (77%). However, both UNC-60A and UNC-60B show lower sequence identity (<40%) with ADF/cofilins from other organisms. In comparison to the canonical 143 residue long yeast cofilin sequence, UNC-60A and UNC-60B have extra residues after β3, in α2, and in the F-loop. Therefore, both proteins have longer F-loop and α2 in comparison to other ADF/cofilins [12]. In comparison to yeast cofilin sequence, the vertebrate cofilins, which are all approximately 165 amino acid in length, have two inserts and an extended C-terminal. The first insert forms a helix between α1 and β2, which contains a nuclear localization signal (NLS). The second insert follows β3 and forms a bulge and β strand, which pairs with the N-terminal of β2 [35]. In comparison to UNC-60B, UNC-60A protein is 13 amino acids longer. These extra residues in UNC-60A align in the form of two inserts. The first insert is of four residues from K36 to V39, while the second insert is from I50 to D57, which, incidentally, includes four acidic residues.
Figure 1. Sequence alignment of ADF/cofilin proteins.

A sequence alignment was constructed for UNC-60A, UNC-60B and other eukaryotic ADF/cofilins, for which three-dimensional structures are known. Secondary structure elements are indicated for UNC-60B. Strictly conserved residues are shown in Black boxes, and regions of residues with similar properties are indicated with a thin Black lining. Sequences of ADF/cofilins are shown for C. elegans, T. gondii (TgADF), P. falciparum (PfADF1; PfADF2), Acanthamoeba Actophorin, S. cerevisiae (Yeast cofilin), A. thaliana (AtADF) and L. donovani (LdCof). The gaps in the alignment are represented as dots. The alignment figures were made using program ESPRIPT.
Solution structure of UNC-60A and UNC-60B
The ribbon representations of the lowest energy structures and the wire representations of the final ensemble of 10 structures of UNC-60A and UNC-60B are shown in Fig. 2. The pair wise RMSDs for the secondary structure regions of the final ensemble of 10 lowest energy structures of UNC-60A and UNC-60B were 0.7 Å, and 0.6 Å, respectively. The atomic coordinates for all 10 structures of both proteins have been deposited in the Protein Data Bank (UNC-60A; PDB ID: 2MP4 and UNC-60B; PDB ID: 2LXX). Structural parameters for the solution structures of UNC-60A and UNC-60B are summarized in Table 1.
Figure 2. Solution structures of UNC-60A and UNC-60B.

(A) & (C) Ribbon diagram of structure of UNC-60A and UNC-60B, respectively. The individual β strands and α helices are labelled. In UNC-60A, the β-strands are β1 (M6-V7), β2 (I29-I32), β3 (I38-A42), β4 (C80-C91), β5 (S99-I109) and β6 (L139-V143), and the α-helices are α1 (D10-E20), α2 (K60-T74), α3 (I116-L133) and α4 (H151-K161). In UNC-60B, the β-strands are β1 (K6-V7), β2 (S25-D32), β3 (T36-G44), β4 (R68-V78), β5 (T86-V97) and β6 (Q128-A131), and the four helices correspond to α1 (P10-K21), α2 (Y50-E62), α3 (V104-L121), and α4 (E139-Q150) (B) & (D) Superimposition of backbone traces from final ensemble of 10 structures of UNC-60A and UNC-60B, respectively.
Table 1.
Experimental restraints and structural statistics for final ensemble of 10 structures of UNC-60A and UNC-60B.
| Distance Restraint List | ||
| Classes | UNC-60A | UNC-60B |
| Sequential | 548 | 456 |
| intra-residual | 808 | 696 |
| medium-range | 800 | 656 |
| long-range | 671 | 582 |
| Hydrogen bonds | 74 | 60 |
| Dihedral angle restrains (ϕ and φ) | 266 | 241 |
| RMSD values (Å) | ||
| All backbone atoms | 0.7 Å | 0.6 |
| All heavy atoms | 1.1 Å | 0.9 |
| Ramachandran plot statisticsa | ||
| Most favored region (%) | 94.1% | 94.7% |
| Allowed region (%) | 5.9% | 5.3% |
| Additionally allowed region (%) | 0.0% | 0.0% |
| Disallowed region (%) | 0.0% | 0.0% |
| CING ROG analysisa | ||
| Red | 13(12%) | 6(6%) |
| Orange | 24(21%) | 35(38%) |
| Green | 76(67%) | 72(56%) |
| WHAT IF Summarya | ||
| Structure Z-scores | ||
| 1st generation packing quality | 2.349+/− 1.015 | 2.344+/−1.175 |
| 2nd generation packing quality | 4.417+/− 1.932 | 5.478+/−1.961 |
| Ramachandran plot appearance | −2.555+/− 0.343 | −2.624+/−0.529 |
| chi-1/chi-2 rotamer normality | −5.135+/− 0.256 | −4.122+/−0.273 |
| Backbone conformation | 0.124+/− 0.603 | 0.694+/−0.601 |
| RMS Z-scores | ||
| Bond lengths | 1.035+/− 0.001 | 1.031+/−0.001 |
| Bond angles | 0.283+/− 0.008 | 0.288+/−0.005 |
| Omega angle restraints | 0.745+/− 0.053 | 0.663+/−0.067 |
| Side chain planarity | 0.393+/− 0.039 | 0.381+/−0.041 |
| Improper dihedral distribution | 0.453+/− 0.011 | 0.439+/−0.017 |
| Inside/Outside distribution | 1.026+/− 0.014 | 1.097+/−0.022 |
region, UNC-60A: 6M-7V,10D-20E,27I-32I,37V-42A,45Q-48L,54D-56D,60K-72S,80C-93R,96A-109I,116I-132S,138I-143V,151H-161K
region, UNC-60B: 6K-7V, 9P-18L, 26Y-30K, 37A-44G, 50K-60L, 69Y −79Q, 88T-95V, 104V-125S, 134M-136D, 139E-148S
The solution structures of both UNC-60A and UNC-60B possess typical ADF/cofilin fold, the core of which consists of a central six-stranded mixed β-sheet. The strand ordering is β1-β3-β2-β4-β5-β6, in which the four central strands are antiparallel, while the strands β1-β3 and β5-β6 run parallel to each other. The central β-sheet core is surrounded by helices α1 and α3 on one face, and by helices α2 and α4 on the opposite face. Solution structures of UNC-60A and UNC-60B show conserved features of both G/F-site and F-site, which include the characteristic long kinked helix α3 (G/F-site), the β4-β5 loop (F-loop, F-site), and the C-terminal helix (F-site). The G-actin binding long helix α3 retains the structurally conserved kink in both of these structures.
As discussed above, UNC-60A protein possesses 13 extra residues subsequent to the strand β3 (post β3 insert), in comparison to UNC-60B. This post β3 insert forms two helical turns, T44-L48 and D54-D56. The first helical turn (T44-L48) is in close proximity with N-terminal of α2, while the second helical turn (D54-D56) is in close proximity with β4 and β5 [Fig. 3]. This kind of structural feature has not been observed in the structure of any other ADF/cofilin. Residues D53 and D56 of this region form salt bridges with K88, and K100 of β4 and β5, respectively [Fig. 3]. In addition to this unique feature, UNC-60A has longer α2 in comparison to UNC-60B and other ADF/cofilins.
Figure 3. Structure of UNC-60A protein showing unique structural features.
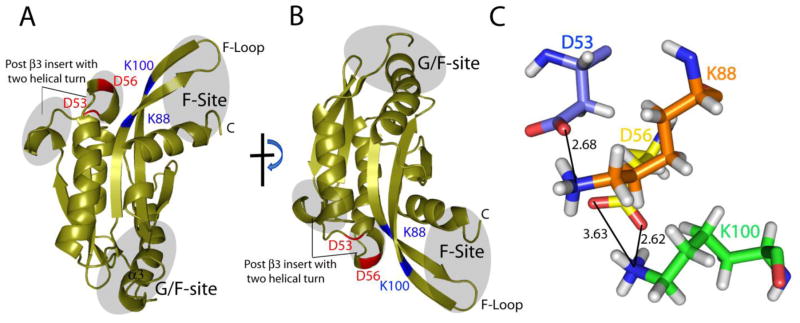
(A) & (B) Ribbon diagram of structure of UNC-60A at two different orientations showing unique post β3 insert, G/F-site and F-site. (C) Salt bridges formed between the residues of post β3 insert and F-loop.
Upon alignment of structures of these two proteins, finer structural differences can be distinguished in the F-sites of UNC-60A and UNC-60B [Fig. 4A]. In UNC-60A, the inclination of the F-loop towards the C-terminal is ~570, while in UNC-60B, the inclination of the F-loop towards the C-terminal is ~430, as shown in Fig. 4B and Fig. 4C. Therefore, F-loop is closer to the C-terminal in the structure of UNC-60A than UNC-60B. Residues F89 (β4) and L154 (α4) of UNC-60A are positionally correlated to the residues V76 (β4) and V142 (α4), respectively, of UNC-60B. The distance between Cα of F89 and L154 is 8.9 Å in UNC-60A [Fig. 4B]. However, as a consequence of difference in the inclination of F-loop towards the C-terminal in UNC-60B, the distance between Cα of V76 and V142 is increased to 11.12 Å [Fig. 4C]. In addition to the above, slight change is also observed in the orientations of the C-terminals of these proteins. In UNC-60B, the C-terminal is almost parallel to β6, while in UNC-60A, it forms an angle of ~200 with respect to β6. Further, the composite F-site (F-loop and C-terminal helix) is more inclined towards and closer to β6 in the case of UNC-60A, in comparison to UNC-60B [Fig. 4A]. Similar differences in the F-sites are also observed upon comparison of structures of yeast cofilin and LdCof [Fig. 4D]. The F-loop is inclined by 520 towards the C-terminal in LdCof [PDB ID: 2KVK], while this inclination is 420 in yeast cofilin [PDB ID: 1COF] [Fig. 4D]. Similarly, C-terminal is almost parallel to β6 in yeast cofilin, while it forms an angle of ~200 with β6 in LdCof [12, 26].
Figure 4. Comparison of UNC-60A protein with UNC-60B, yeast cofilin and LdCof.

(A) Overlap of UNC-60A with UNC-60B showing different orientation of F-site (F-loop and C-terminal) (B) & (C) Structure of UNC-60A and UNC-60B showing detailed description of difference in the F-site in term of the angle and distance (D) Overlap of yeast cofilin with Leishmania cofilin showing different orientation of F-site (E) Overlap of UNC-60B with Yeast cofilin showing same orientation of F-site (F) Overlap of UNC-60A with Leishmania cofilin showing same orientation of F-site. Structures of UNC-60A, UNC-60B, Yeast cofilin, and Leishmania cofilin, are shown in cyan, purple, green, and magenta in color, respectively.
Overall, on the basis of the comparison of the F-site, various ADF/cofilin structures can be divided into two categories. The inclination of F-loop towards the C-terminal is ~400 in the case of UNC-60B, yeast cofilin [PDB ID: 1COF], Acanthamoeba actophorin [PDB ID: 1CNU], human cofilin [PDB ID: 4BEX], and chick cofilin [PDB ID: 1TVJ]. On the other hand, the inclination of F-loop towards the C-terminal is ~550 in the case of UNC-60A and LdCof [PDB ID: 2KVK] [12, 26, 35, 36, 37]. In UNC-60B, yeast cofilin, Acanthamoeba actophorin, human cofilin and chick cofilin, the C-terminal is almost parallel to β6, while in UNC-60A, and LdCof, it forms an angle of approximately ~200 with respect to β6 [12, 26, 35, 36, 37]. Superposition of UNC-60B and yeast cofilin structure, and superposition of UNC-60A and LdCof structures showing similar orientation of F-site are shown in Fig. 4E and Fig. 4F, respectively. In UNC-60A, F-loop residues are involved in seven salt bridges, while in UNC-60B F-loop residues are involved in only two salt bridges [Table 2]. In LdCof, the residues of F-loop are involved in nine salt bridges. On the other hand, F-loop residues are involved in one to three salt bridges in yeast cofilin (ScCof), Acanthamoeba actophorin, human cofilin, and chick cofilin [12, 26, 35, 36, 37] [Table 2].
Table 2.
Salt bridges formed from the residues of F-loop with the other region in different ADF/cofilins.
| Salt Bridge from residues of F-loop in UNC-60A | Salt Bridge from residues of F-loop in LdCof | |||||
|---|---|---|---|---|---|---|
| 1 | LYS23 (NZ) | 2.67Å | ASP 102 (OD1) | ASP68(OD1) | 2.66 Å | LYS22(NZ) |
| 2 | LYS88 (NZ) | 2.68 Å | ASP53(OD2) | ASP68(OD2) | 2.80Å | LYS22(NZ) |
| 3 | LYS 100 (NZ) | 2.62 Å | ASP56(OD1) | GLU79 (OE1) | 2.63Å | LYS22(NZ) |
| 4 | LYS100 (NZ) | 3.63Å | ASP56(OD2) | ASP68 (OD2) | 2.66 Å | ARG 25(NH1) |
| 5 | LYS103 (NZ) | 3.36 Å | ASP86 (OD1) | GLU70 (OE1) | 2.73 Å | LYS77(NZ) |
| 6 | LYS103(NZ) | 2.62 Å | ASP86(OD2) | ASP73 (OD1) | 2.60 Å | ARG134 (NH1) |
| 7 | LYS103(NZ) | 3.85 Å | ASP102 (OD1) | ASP73 (OD1) | 3.67 Å | ARG134(NH2) |
| 8 | ASP73(OD1) | 3.40 Å | ARG137(NH2) | |||
| 9 | ASP73 (OD2) | 2.76 Å | ARG137(NH2) | |||
| Salt Bridge from residues of F-loop in UNC-60B | Salt Bridge from residues of F-loop in Yeast cofilin | |||||
| 1 | ASP73 (OD1) | 2.55 Å | HIS19 (ND1) | ASP 68(OD1) | 3.85 Å | LYS 23(NZ) |
| 2 | GLU75 (OE1) | 3.57 Å | LYS 46(NZ) | ASP 68(OD2) | 3.01 Å | LYS 23 (NZ) |
UNC-60A and UNC-60B display high G-actin binding affinities
In order to understand the structural basis of interaction of UNC-60A and UNC-60B with actin, the structures of these proteins were docked on the rabbit muscle G-actin crystal structure [10]. In both UNC-60A/G-Actin and UNC-60B/G-Actin docked models, several hydrogen bonds and salt bridges were observed between the residues of the proteins forming the pairs. The details of interacting residues of UNC-60A/G-Actin and UNC-60B/G-Actin pairs in the docked models are given in Supplementary Table 1. In the UNC-60B/G-Actin docked model, the orientation of α3 of UNC-60B, in the cleft between subdomain1 and subdomain3 of actin, is similar to the orientation of the long helix of Twf-C in the crystal structure of G-actin and Twf-C complex [Fig. 5C]. However, in the UNC-60A/G-Actin model, the orientation of α3 is slightly different from that of Twf-C [Fig. 5A]. The interacting regions of the actin monomer are almost similar for the UNC-60A/G-Actin and UNC-60B/G-Actin models.
Figure 5. Molecular docking of UNC-60A/UNC-60B with G-actin and comparison of interfacial residues with G/F-site residues displaying motion on NMR timescale.

(A) & (C) UNC-60A/G-actin and UNC-60B/G-actin docked model showing residues of UNC-60A and UNC-60B involved in interaction with G-actin, respectively (B) & (D) UNC-60A/G-actin and UNC-60B/G-actin docked model showing dynamic residues of UNC-60A and UNC-60B at NMR time scale, respectively. This figure shows that the interacting orientation of the UNC-60A and UNC-60B at the cleft between the subdomain 1 and subdomain 3 is slightly different. This Figure also displays that backbone dynamics of G/F-site correlate well with the docking study as residues involved in interaction with G-actin also show dynamic flexibility at NMR timescale. The F-loop region is highlighted in these Figures.
In UNC-60B/G-Actin model, residues R104, R105, R107 of α3 show interaction with D25 (subdomain1), S125, R147 (helix connecting subdomain1 and subdomain3), and E167 (subdomain3) of actin monomer. In addition to this, N34 and T36 (loop connecting β2-β3) of UNC-60B form H-bonds with S350 and T351 (subdomain3) of actin. Residue D100 (β5-α3 loop) of UNC-60B forms many H-bonds and salt bridges with R147 (helix connecting subdomain1 and subdomain3), K328 (subdomain3) of actin. Details of H-bonds and salt bridges in UNC-60A/G-Actin docked model and in UNC-60B/G-Actin docked model are given in Supplementary Table 1.
The structural features of UNC-60A and UNC-60B are well supported by a direct demonstration of their binding with rabbit muscle G-actin in the presence of ADP. Isothermal titration calorimetry (ITC) titration of UNC-60A with ADP-G-actin reveals 1:1 stoichiometry and a dissociation constant Kd of 32.25 nM, with ΔG of −7.899 X 103 cal/mole, ΔH of −2.305 X 103 ± 71.14 cal/mole and ΔS of 26.5 cal/mole K, while titration of UNC-60B with ADP-G-actin reveals Kd of 8.62 nM with ΔG of −1.1484 X 104 cal/mole, ΔH of −6.716 X 103 ± 116.9 cal/mole and ΔS of 16.0 cal/mole K. The ITC curves are shown in Fig. 6A; 6B.
Figure 6. ITC characterization of binding of UNC-60A/UNC-60B with G-actin.

(A) Probing UNC-60A/ADP-G-Actin interactions by ITC. (B) Probing UNC-60B/ADP-G-Actin interactions by ITC. The negative peaks indicate an exothermic reaction. The area under each peak represents the heat released after an injection. (Lower) Binding isotherms obtained by plotting peak areas against the molar ratio of titrands. The lines represent the best-fit curves obtained from least-squares regression analyses assuming a one-site binding model.
15N Relaxation and Modelfree analysis for UNC-60A and UNC-60B
The backbone dynamics of UNC-60A and UNC-60B were characterized by measurements of the backbone 15N longitudinal relaxation rates (R1), transverse relaxation rates (R2), and steady state heteronuclear {1H}-15N NOEs. The longitudinal relaxation rates, R1, are sensitive to nanosecond-to-picosecond timescale. The transverse relaxation (R2) rates are more sensitive to nanosecond motions, but they also reflect contributions from slower millisecond-to-microsecond exchange processes [38, 39, 40]. The heteronuclear [1H] 15N NOEs are typically most sensitive to picosecond motions with lower values indicating increased local flexibility of the polypeptide [38, 39, 40]. R2/R1 ratios characterize the different time-scale local motions. Residues with low value of R2/R1 reflect fast fluctuations on the nanosecond-to-picosecond timescale, while residues with high value of R2/R1 reflect contributions from slower millisecond-to-microsecond exchange processes [38, 39, 40].
The backbone relaxation rates were determined for 137 out of 163 non proline residues of UNC-60A, and 116 out of 149 non proline residues of UNC-60B, by using the program Curvefit. The average values of relaxation parameters for residues of UNC-60A are R1 = 1.35 s−1, R2 = 14.29 s−1, {1H}-15N NOE = 0.70. For residues of UNC-60B, R1 = 1.32 s−1, R2 = 13.68 s−1, {1H}-15N NOE = 0.59. The residue specific R1, R2 and steady state heteronuclear {1H}-15N NOE values obtained for UNC-60A and UNC-60B at 600 MHz are shown in Fig. 7A and 7B.
Figure 7. Sequence dependent variations of backbone 15N relaxation parameters and Modelfree internal mobility parameters of UNC-60A and UNC-60B.

(A) & (B) R1; R2; steady state {1H}-15N NOE and R2/R1 ratio values versus amino acid sequence of UNC-60A and UNC-60B, respectively. All experiments of UNC-60A and UNC-60B were performed on 0.7mM 15N labelled sample at 25°C on a Varian Inova spectrometer at 600 MHz.(C) & (D) Plots of Modelfree parameters like generalized order parameter (S2), the effective correlation time (τe) and the chemical exchange parameter (Rex) as a function of residue number for UNC-60A and UNC-60B, respectively.
The relaxation parameters were analysed using the Modelfree formalism with isotropic model for both proteins [28, 29, 30, 31], which yielded optimized value of molecular rotational correlation times, τm, of 10.098 ns and 10.088 ns for UNC-60A and UNC-60B, respectively, both of which correspond to the correlation time expected for a monomeric protein of ~20KDa. All 137 residues of UNC-60A, for which the relaxation rates were determined, could be fitted to one of the five models of Modelfree. However, for UNC-60B, out of 116 residues, only 108 residues could be fitted, while eight residues, M1, A2, S3, E55, D63, A71, N101, and A102, could not be fitted to any of the five models.
Three dynamic parameters were extracted from Modelfree analysis i.e. generalized order parameter (S2), effective correlation time (τe), and chemical exchange rate (Rex) [30, 31, 32, 33]. S2 and τe are the measure of the amplitude and correlation time of wobbling of N-H vectors in space, with respect to overall molecular tumbling, on picosecond to nanosecond time scales. Motions on this timescale reflect local flexibility and entropic contributions to protein-ligand interactions. Rex provides a measure of the rate of local exchange processes on a millisecond-to-microsecond timescale for the residues involved in such processes [30, 31, 32, 33, 41, 42], and is particularly valuable in understanding how dynamics drives enzyme catalysis. The generalized order parameter (S2), the effective internal correlation time (τe), and a conformational exchange broadening parameter (Rex) for each backbone amide NH vector that could be determined using the Modelfree formalism for UNC-60A and UNC-60B and are shown in Fig. 7C and 7D. The ribbon representations of UNC-60A and UNC-60B structures shaded according to residue specific generalized order parameters (S2), effective correlation time (τe), and conformational exchange term (Rex) are shown in Fig. 8A-8D and details of residues showing chemical exchange (Rex) and effective correlation time (τe) are given in Table 3.
Figure 8. Ribbon diagram representing the dynamics properties of UNC-60A and UNC-60B.

(A) & (C) The ribbon representations of UNC-60A and UNC-60B solution structures shaded according to the S2 values derived from Modelfree analysis, respectively. The color coding is from blue for S2=1, to red for S2=0.2. Prolines and residues that are not included in Modelfree analysis are colored gray. (B) & (D) The ribbon representations of UNC-60A and UNC-60B shaded according to chemical exchange (Rex) terms, effective correlation (τe) times from Modelfree analysis, respectively. The residues displaying motion on picosecond to nanosecond timescale (τe) are represented in blue, residues displaying conformational exchange (Rex) from Modelfree analysis are represented in green.
Table 3.
Residues of UNC-60A and UNC-60B showing effective correlation time (τe) and chemical exchange (Rex) from Modelfree analysis and relaxation dispersion analysis
| Modelfree | Residues of UNC-60A | Residues of UNC-60B |
|---|---|---|
| Residues in effective correlation time (τe) | G4-V7 (N-terminal and β1), Q12 (α1), G21, K23-E24 (loop after α1), G52-D53 (insert after β3), R93-G97 (F-loop), K117(α3), T135(loop after α3), S150 (loop before C-terminal helix), and Y162-H165 (C-terminal region) | G4-K6 (N-terminal and β1), N34 (loop between β2 and β3), V61(α2), E66 (loop after α2), T77-Q79 (β4), Q81-N90, V92 (F-loop), C98, D100 (loop after β5), S148, and Q150-I152 (C-terminal) |
| Residues in chemical exchange(Rex) | D10-F15, K17-S19 (α1), S59-A61(second helical turn of extra region), F63-K65, V67-T74 (α2 and loop after α2), C80 (β4), C110 (loop after β5), K130 (loop after α3), V143 (loop after β6), and H165 (C-terminal) | K12-N13, Y15, L17-L18 (α1), K21(loop after α1), F29 (β2), V43-G44 (β3), Y50-A51, F53-V54, M57-K58, V61(α2), V74 (β4), C98, D100 (loop after β5), R105 (α3), G122-L123, L126 (loop after α3), N130 (β6), D136 (helical turn), and S144 (α4) |
| Relaxation Dispersion | Residues of UNC-60A | Residues of UNC-60B |
| Residues in chemical exchange(Rex) | Q12-S14, K17, S19, E20 (α1), E40, T44, T51, G52, D54, D57 (insert after β3), A61, K65, V67-V70 (α2 ), D75-L77 (loop after α2), G95-T98, K100-D102 (F-loop), C110, D112, G113 (β5-α3 loop), K119, Y122, S125, T131 (α3), L133, T135 (α3-β6 loop), D145, E146, E148 (helical turn), K152-L154, N156, L158, K161 (C-terminal) | S10, K12-A14, H19 (α1), N34-D35 (loop between β2-β3), E52-E55, M57, K59 (α2), E62, G64, E66 (loop after α2), R105, L109, Y110, L117, K118, L121 (α3), L123, L126 (α3-β6 loop), D138 (loop after β6), V142-S144, L146 (C-terminal) |
Overall, for both UNC-60A and UNC-60B, residues in secondary structure elements display high order parameter (S2) [Fig. 8A; 8C], while the N-terminal, the C-terminal, and the F-loop residues are flexible. Interestingly, the helix α1 and α2 are comparatively more flexible than the remaining secondary structure elements, as evidenced by their lower values of S2 in comparison to the average value for the regions with secondary structure.
In UNC-60A, residues showing motion on nanosecond to picosecond (τe) are present in the five main clusters i.e. N-terminal, loop after α1, post β3 insert, F-loop, and C-terminal. In UNC-60B, residues showing motion on nanosecond to picosecond (τe) are present in the four main clusters i.e. N-terminal, β2-β3 loop, F-loop and C-terminal. A clear difference in the dynamics of F-loop is seen between UNC-60A and UNC-60B. In UNC-60A, five residues (R93-G97) of F-loop show fluctuations on fast picosecond-to-nanosecond time scale, while in UNC-60B, fourteen residues (T77-Q79, Q81-N90, V92) show fluctuations on fast picosecond-to-nanosecond time scale. Therefore, the F-loop of UNC-60B is more dynamic in comparison to the F-loop of UNC-60A.
Residues involved in millisecond to microsecond time scale exchange processes characterized by Rex were also identified from the Modelfree analysis. In UNC-60A, residues displaying Rex were present in four main clusters i.e. α1, second helical turn of the post β3 insert, α2, and loop after α2. In UNC-60B, residues displaying Rex were present in five main clusters i.e. α1, β3, α2, β5-α3 loop, and α3-β6 loop. Millisecond to microsecond timescale motions of UNC-60A and UNC-60B were subsequently examined quantitatively by using 15N-CPMG relaxation dispersion NMR method, which is able to probe the chemical exchange phenomena in proteins [43, 44, 45]. Details of 15N-CPMG relaxation dispersion NMR method are given in supplementary text. In UNC-60A, residues displaying chemical exchange rate analysed from relaxation dispersion [Rex (RD)] were mainly present in one cluster of G/F-site (α3), two clusters of F-site (F-loop, C-terminal), and five other clusters (α1, post β3 insert, α2, β5-α3 loop, α3-β6 loop) [Fig. 9A]. In UNC-60B, residues displaying Rex (RD) were mainly present in one cluster of G/F-site (α3), one cluster of F-site (C-terminal) and four other clusters (α1, α2, β5-α3 loop, α3-β6 loop) [Fig. 9B]. In UNC-60A, L133 and T135 of α3-β6 loop show conformational exchange at NMR timescale. Similarly, the residues G122, L123 and L126 of this loop in UNC-60B are also in conformational exchange. Backbone dynamics of G/F-site correlate well with the docking study as residues involved in interaction with G-actin also show dynamic flexibility at NMR timescale. To illustrate this point, interfacial residues of UNC-60A and UNC-60B (shown in red) are labelled in Fig. 5A and 5C, respectively, while the residues, displaying dynamics on the NMR timescale of the G/F-site of UNC-60A and UNC-60B are labelled in Fig. 5B and 5D, respectively (red for τe and yellow for Rex).
Figure 9. Slow time scale motion characterization for UNC-60A and UNC-60B by Relaxation Dispersion method.

(A) & (B) Plot of residue wise Rex (sec−1) values for UNC-60A and UNC-60B, respectively, determined by fitting the relaxation dispersion data to Eqn. 2. The residues showing significantly higher than the average Rex Values have been labeled.
Comparison of slow time scale motions for UNC-60A and UNC-60B, which were derived from CPMG-based Relaxation Dispersion experiments, shows that almost similar clusters of residues, which are spatially comparable, are involved in exchange process. Except for the post β3 insert, which is unique for UNC-60A, the prominent difference between these two proteins is in the F-loop. Residues of F-loop (G95-T98, K100-D102) in UNC-60A were found to be in chemical exchange, while no residues of F-loop of UNC-60B were in chemical exchange [Fig. 9A; 9B]. This further shows that the F-loop is more dynamic in UNC-60B where fast nanosecond to picosecond timescale motions dominate, while slow millisecond to microsecond timescale motions are observed for the F-loop of UNC-60A.
Discussion
Structure and dynamics rationale for F-actin binding and different activities of ADF/cofilins
In this study, we have presented the solution structures and dynamics of the two ADF/cofilin isoforms of C. elegans, UNC-60A and UNC-60B, which differ significantly in their F-actin severing activities, monomer sequestering activities, and co-sedimentation properties.
Structurally, both UNC-60A and UNC-60B proteins have similar F-site features, characterized by the presence of long F-loop, which protrudes beyond the core globular structure, C-terminal helix, and presence of conserved basic residue on F-loop (K103 in UNC-60A and K91 in UNC-60B) [12]. Amongst all the known ADF/cofilin structures, structures of PfADF1 and TgADF display the shortest F-loop that does not protrude out of the structure [27, 46]. In both these proteins, there is a single basic residue (K72 in PfADF1; K68 in TgADF), which is essential for their weak severing activity. The K72A mutation leads to complete loss of severing activity for PfADF1 [46]. The orientation of this basic residue is conserved in the structures of various ADF/cofilins, and it represents the minimal structural feature required for weak severing activity. In the case of human cofilin, point mutation of corresponding K96 residue in the F-loop leads to a loss of severing activity and increased depolymerizing activity [47]. As mentioned above, while this feature is conserved in the structures of both UNC-60A and UNC-60B, it is not sufficient to explain the stronger severing activity of UNC-60B as well as the weaker severing activity of UNC-60A. In UNC-60A and LdCof, both of which show weak severing activities [26, 48], the F-loop has three basic residues and the C-terminal has five basic residues. On the other hand, in UNC-60B, which shows strong severing activity, the F-loop has only two basic residues and the C-terminal has only three basic residues. This suggests that severing activity does not linearly correlate with the number of positively charged residues on the F-Loop and the C-terminal.
Since the finer structure and dynamics differences between these two isoforms have been mapped in this study, it is pertinent to overlay these structures with the structures of ADF/cofilins having strong and weak severing activities, viz. yeast cofilin and LdCof, respectively [2, 12, 26, 48]. Structural overlays of various combinations of pairs of these four proteins are shown in Fig. 4. It is seen that the structural features of the F-site (F-loop and C-terminal) of UNC-60A, i.e. inclination of F-loop towards C-terminal, and inclination of F-site towards β6, match closely to that of LdCof [Fig. 4F] [26], and are different from the F-site of UNC-60B and yeast cofilin [Fig. 4A] [12]. Concurrently, the orientations of F-sites of UNC-60B and yeast cofilin match closely to each other [Fig. 4E], and to other strong severing proteins like Acanthamoeba actophorin, human cofilin, and chick cofilin [12, 35, 36, 37]. Further, the F-sites of LdCof and yeast cofilin differ structurally in the same manner as they differ for UNC-60A and UNC-60B [Fig. 4A; 4D].
In order to extend this understanding, we have individually superimposed the structures of UNC-60A, UNC-60B, and LdCof on the structure of HsCof in the HsCof-F-actin structure determined from cryo-electron microscopy [PDB ID: 3J0S] [13]. These overlays are shown in Fig 10. From the overlays, it is seen that the F-loops of UNC-60B and yeast cofilin have similar orientations as the F-loop of human cofilin in its bound form. This orientation enables the interaction of F-loop with residues 21–28 and 90–96 of actin, which have been identified as the putative F-loop binding site on the actin surface [Fig. 10A; 10B]. On the other hand, because of more inclination, F-loops of UNC-60A and LdCof are oriented away from this F-loop binding site, and are oriented towards the interface of actin protomers n and n+2 [Fig. 10A; 10B]. This suggests that correct positioning of F-loop, as in the case of UNC-60B and HsCof, is necessary for stable association of ADF/cofilin with the filamentous form of actin, which further leads to efficient F-actin severing.
Figure 10. Superimposition of UNC-60A, UNC-60B and LdCof on HsCof in the structure of HsCof/F-actin.
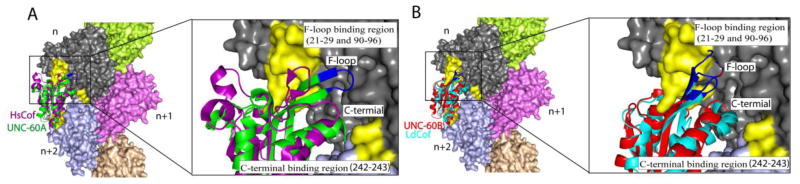
To model the binding of different ADF/cofilins with F-actin, the structures of UNC-60A, UNC-60B and LdCof were superimposed on the HsCOF in the cryo-electron microscopy structure of human cofilin decorated F-actin to observe the binding of these proteins on F-actin. (A) Overlays of structure of unc-60a with cryoelectron microscopy structure of HsCof with F-actin. (B) Overlay of structure of UNC-60B and LdCOF on HsCof/F-actin. In overlays, actin protomers are shown in different colors. Structure of HsCof, UNC-60A, UNC-60B and LdCof are shown in magenta, green, red and cyan colors, respectively. ADF/cofilins are shown in cyan color, residues of F-loop showing motion on nanaosecond-picoseconds are shown in blue color. Structure of HsCof was omitted from overlays of UNC-60B and LdCOF for clearity.
Comparison of dynamics of UNC-60A and UNC-60B with other ADF/cofilins reveals that the dynamic behaviour of UNC-60A is similar to LdCof in terms of number of flexible residues present in the F-loop [26]. In UNC-60A five residues (R93-G97), and in LdCof four residues (D73-S76), of F-loop show motion on nanosecond-picosecond timescale [26]. In UNC-60B fourteen residues of F-loop show motion on nanosecond-picosecond timescale. In weak severing proteins like UNC-60A and LdCof, seven and nine salt bridges are formed by the residues of F-loop with the residues of other regions, respectively [27]. In strong severing proteins like UNC-60B, yeast cofilin, Acanthamoeba actophorin, human cofilin and chick cofilin only one to three salt bridges are formed by the residues of F-loop with the residues of other regions [2, 12, 35, 36, 37] [Table 2]. It is possible that the presence of higher number of salt bridges in weak severing proteins UNC-60A and LdCof makes the F-loop relatively rigid in comparison to the strong severing proteins UNC-60B, Acanthamoeba actophorin, human cofilin and chick cofilin. It seems that the weaker severing activity conformation of F-loop, which is oriented towards the interface of two actin protomers in F-actin, is relatively more rigid. On the other hand, sronger severing conformation of the F-loop, which is oriented towards the binding region of F-loop in F-actin, is relatively more flexible. This orientation and flexibility appears to facilitate stable association with F-actin and stronger severing activity towards actin filaments. Overall, these comparisons suggest that both F-loop orientation and flexibility are important determinants of the severing activities and co-sedimentation properties of UNC-60A and UNC-60B.
G-actin sequestration property of ADF/cofilins may be dependent on their two activities, viz strong pointed end depolymerization and strong inhibition of F-actin polymerization. Inhibition of polymerization/nucleation does not directly correlate to the binding affinity of ADF/cofilins with G-actin, as this affinity is lower for UNC-60A in comparison to UNC-60B. It is possible that F-loop plays an important role in inhibition of polymerization/nucleation activity as observed in the case of Schizosaccharomyces pombe cofilin (SpCof) where point mutation of R78 in the F-loop resulted in a loss of nucleating activity [3]. As described above, juxtaposition of the F-site of UNC-60A at the interface of two longitudinally attached protomers may obstruct the interaction of helix (I287-L293) of subdomain 3 of one protomer with the helix (T203-K215) of subdomain 4 of other protomer of actin, as shown schematically in Fig 11. This implies that the F-loop and C-terminal of UNC-60A bound to G-actin may present a steric restriction for the approach of a second G-actin monomer longitudinally, which is necessary for polymerization and/or nucleation. Additionally, this region is also more rigid in UNC-60A in comparison to UNC-60B, and this higher rigidity may also contribute towards the inhibition of polymerization and/or nucleation. Therefore, it seems that the properties of weak severing activity and inhibition of polymerization and/or nucleation may share a common basis in terms of F-loop structure and dynamics.
Figure 11. Schematic representation of the role of F-loop of UNC-60A in inhibiting polymerization/nucleation.

An actin monomer is placed on the surface of UNC-60A/G-actin model, the F-loop of UNC-60A gets in close proximity with the helix (T203-K215) of subdomain 4 of actin (yellow), which interacts with the helix (I287-L293) of subdomain 3 of adjacent protomer (blue) in co-filament structure of ADF and F-actin.
Functional importance of α3-β6 loop
Residues of α3-β6 loop (L133, T135) of UNC-60A and (G122, L123, L126) of UNC-60B show conformational flexibility on the millisecond to microsecond time scale in both proteins, and similar dynamic behaviour for this region was also observed for TgADF [27]. In the F-loop mutant K91A of UNC-60B, in addition to residues of F-site, some residues of α3-β6 loop also show CSPs. In K96Q mutant of Human cofilin, in addition to residues of F-loop and C-terminal, CSPs were also observed for the residues of α3-β6 loop (L132, H133) [35]. Residues of α3-β6 loop of human cofilin are close to the residues K50, E57 and N92 of actin protomer n, in the co-filament structure as observed in the cryo-electron microscopy image reconstruction [Supplementary Fig S5]. This suggests that in addition to the previously recognised F-actin binding site, the α3-β6 loop of ADF/cofilin may be involved in interaction with F-actin.
Structure and dynamics rationale for ADP-G-actin binding affinity of ADF/cofilins
Structures of many ADF/cofilins and other related ADF-H domains have been characterized and they show conserved common features for G-actin binding, but with different affinities. It indicates that besides conserved biochemical and structural features, protein dynamics also contributes to binding affinity. G-actin binding sites in UNC-60A and UNC-60B are well formed and 15N-relaxation dynamics of residues of G-actin binding site show desired interfacial flexibility as having flexible N-terminal (G4-V7 in UNC-60A and G4-K6 in UNC-60B) and N-terminal of α3 (K117, K119, Y122 in UNC-60A and R105, L109, Y110 in UNC-60B) to facilitate the interaction with G-actin. While the N-terminal is flexible in almost all of the ADF/cofilins, the flexibility at the N-terminal of α3 appears to enhance the binding affinity [27]. As described above, backbone dynamics of G/F-site correlate well with the docking study [Fig 5]. In UNC-60A/G-actin and UNC-60B/G-actin docking models, one acidic residue (D112 in UNC-60A and D100 in UNC-60B) of β5-α3 loop is involved in interaction with the G-actin by forming H-bond and salt-bridge, and this residue is dynamic in nature at NMR timescale. Presence of acidic residue at corresponding position is highly conserved in all ADF/cofilins, which strongly suggests that this residue may be involved in binding with G-actin.
In conclusion, we have presented the first solution structures of C. elegans ADF/cofilins, UNC-60A and UNC-60B, which are also the first representatives from the nematode phylum. The main finding of this study is that, with reference to their putative binding region on F-actin, the relatively flexible vertical orientation of F-site, as observed for UNC-60B, is associated with stronger severing activity and co-sedimentation property, while the relatively rigid, inclined orientation of F-site, as observed for UNC-60A, is associated with the weak severing activity. This conclusion is further corroborated by structural comparisons with other strong and weak severing ADF/cofilin proteins like yeast cofilin, Actophorin, human cofilin, chick cofilins and LdCof. UNC-60A and UNC-60B display strong affinity with ADP-G-actin, which results from structurally and dynamically conserved G/F-site, specially the flexibility of the N-terminal of the long-kinked helix, α3.
Supplementary Material
Acknowledgments
We are thankful to Dr. C.L. Khetrapal for usage of 800 MHz NMR spectrometer at the Centre for Biomedical Research, Lucknow. This work was supported by Council of Scientific and Industrial Research (CSIR) Network Project UNSEEN, and National Biosciences Award Grant from Department of Biotechnology (DBT), to AA. V.K.S. and D.M. are recipients of research fellowships from CSIR, New Delhi, India. R.Y. and A.K. are recipients of research fellowship from DBT and Indian Council for Medical Research, New Delhi, India, respectively. Work relating to unc-60a and unc-60b clones was supported by a grant from National Institutes of Health (AR048615) to S.O.
Footnotes
Author Contributions: Solution structures of UNC-60A and UNC-60B, molecular docking study, mutational analysis, and relaxation dispersion analysis were done by V. K. Shukla. A. Kabra and D. Maheshwari assisted in sample preparation and data analysis. Subcloning of unc60a and unc60b gene was done by R. Yadav. Modelfree analysis and ITC experiments were done by A. Jain and S. Tripathi, respectively. S. Ono provided the clones of unc60a and unc60b in pET3d vector, and gave useful suggestions. Acquisition of relaxation dispersion data and NOESY-HSQC data was done by D. Kumar. A. Arora conceptualized, designed and directed the study, acquired NMR data, analyzed the results, and wrote the manuscript.
ACCESSION NUMBERS: Coordinates and structure factors of UNC-60A and UNC-60B have been deposited in the Protein Data Bank with accession number 2MP4 and 2LXX, respectively.
References
- 1.Ono S. The mechanism of depolymerization and severing of actin filaments and its significance in cytoskeletal dynamics. Int Rev Cytol. 2007;258:1–82. doi: 10.1016/S0074-7696(07)58001-0. [DOI] [PubMed] [Google Scholar]
- 2.Lappalainen P, Fedorov EV, Fedorov AA, Almo SC, Drubin DG. Essential functions and actin-binding surfaces of yeast cofilin revealed by systematic mutagenesis. EMBO J. 1997;16:5520–5530. doi: 10.1093/emboj/16.18.5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andrianantoandro E, Pollard TD. Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol Cell. 2006;24:13–23. doi: 10.1016/j.molcel.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 4.Chen H, Bernstein BW, Sneider JM, Boyle JA, Minamide LS, Bamburg JR. In vitro activity differences between proteins of the ADF/cofilin family define two distinct subgroups. Biochemistry. 2004;43:7127–7142. doi: 10.1021/bi049797n. [DOI] [PubMed] [Google Scholar]
- 5.Mehta S, Sibley LD. Toxoplasma gondii actin depolymerizing factor acts primarily to sequester G-actin. J Biol Chem. 2010;285:6835–6847. doi: 10.1074/jbc.M109.068155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nachmias VT. Small actin-binding proteins: the beta-thymosin family. Curr Opin Cell Biol. 1993;5:56–62. doi: 10.1016/s0955-0674(05)80008-0. [DOI] [PubMed] [Google Scholar]
- 7.Hawkins M, Pope B, Maciver SK, Weeds AG. Human actin depolymerizing factor mediates a pH sensitive destruction of actin filaments. Biochemistry. 1993;32:9985–9993. doi: 10.1021/bi00089a014. [DOI] [PubMed] [Google Scholar]
- 8.Hayden SM, Miller PS, Brauweiler A, Bamburg JR. Analysis of the interactions of actin depolymerizing factor with G- and F-actin. Biochemistry. 1993;32:9994–10004. doi: 10.1021/bi00089a015. [DOI] [PubMed] [Google Scholar]
- 9.Yamashiro S, Mohri K, Ono S. The Two Caenorhabditis elegans actin depolymerizing factor/cofilin proteins differently enhance actin filament severing and depolymerization. Biochemistry. 2005;44:14238–14247. doi: 10.1021/bi050933d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paavilainen VO, Oksanen E, Goldman A, Lappalainen P. Structure of the actin-depolymerizing factor homology domain in complex with actin. J Cell Biol. 2008;182:51–59. doi: 10.1083/jcb.200803100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paavilainen VO, Hellman M, Helfer E, Bovellan M, Annila A, Carlier MF, Permi P, Lappalainen P. Structural basis and evolutionary origin of actin filament capping by twinfilin. Proc Natl Acad Sci USA. 2007;104 :3113–3118. doi: 10.1073/pnas.0608725104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fedorov AA, Lappalainen P, Fedorov EV, Drubin DG, Almo SC. Structure determination of yeast cofilin. Nat Struct Biol. 1997;4(5):366–9. doi: 10.1038/nsb0597-366. [DOI] [PubMed] [Google Scholar]
- 13.Galkin VE, Orlova A, Kudryashov DS, Solodukhin A, Reisler E, Schröder GF, Egelman EH. Remodeling of actin filaments by ADF/cofilin proteins. 2011;108:20568–20572. doi: 10.1073/pnas.1110109108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ono S, Benian GM. Two Caenorhabditis elegans actin depolymerizing factor/cofilin proteins, encoded by the unc-60 gene, differentially regulate actin filament dynamics. J Biol Chem. 1998;273:3778–3783. doi: 10.1074/jbc.273.6.3778. [DOI] [PubMed] [Google Scholar]
- 15.Ono S, Baillie DL, Benian GM. UNC-60B, an ADF/cofilin family protein, is required for proper assembly of actin into myofibrils in Caenorhabditis elegans body wall muscle. J Cell Biol. 1999;145:491–502. doi: 10.1083/jcb.145.3.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ono K, Parast M, Alberico C, Benian GM, Ono S. Specific requirement for two ADF/cofilin isoforms in distinct actin-dependent processes in Caenorhabditis elegans. J Cell Sci. 2003;116:2073–2085. doi: 10.1242/jcs.00421. [DOI] [PubMed] [Google Scholar]
- 17.Keller R. Diss ETH No 15947. Swiss federal Institute of Technology; Zurich: 2005. Optimizing the process of nuclear magnetic resonance spectrum analysis and computer aided resonance assignment. [Google Scholar]
- 18.Guntert P, Qian YQ, Otting G, Muller M, Gehring W, Wuthrich K. Structure determination of the Antp (C39----S) homeodomain from nuclear magnetic resonance data in solution using a novel strategy for the structure calculation with the programs DIANA, CALIBA, HABAS and GLOMSA. J Mol Biol. 1991;217:531–40. doi: 10.1016/0022-2836(91)90755-u. [DOI] [PubMed] [Google Scholar]
- 19.Guntert P, Mumenthaler C, Wuthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol. 1997;271:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 20.Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 22.Jung JW, Yee A, Wu B, Arrowsmith CH, Lee W. Solution structure of YKR049C, a putative redox protein from Saccharomyces cerevisiae. J Biochem Mol Biol. 2005;38:550–554. doi: 10.5483/bmbrep.2005.38.5.550. [DOI] [PubMed] [Google Scholar]
- 23.Linge JP, Williams MA, Spronk AEM, Bonvin AM, Nilges M. Refinement of protein structures in explicit solvent. Proteins. 2003;50:496–506. doi: 10.1002/prot.10299. [DOI] [PubMed] [Google Scholar]
- 24.Wishart DS, Sykes BD, Richards FM. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J Mol Biol. 1991;222:311–333. doi: 10.1016/0022-2836(91)90214-q. [DOI] [PubMed] [Google Scholar]
- 25.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: a fully automated algorithm for protein protein docking. Nucleic Acids Research. 2004;32:W96–W99. doi: 10.1093/nar/gkh354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pathak PP, Pulavarti SVSRK, Jain A, Sahasrabuddhe AA, Gupta CM, Arora A. Solution structure and dynamics of ADF/cofilin from Leishmania donovani. J Struc Biol. 2010;172:219–224. doi: 10.1016/j.jsb.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 27.Yadav R, Pathak PP, Shukla VK, Jain A, Srivastava S, Tripathi S, Krishna Pulavarti SV, Mehta S, Sibley LD, Arora A. Solution structure and dynamics of ADF/cofilin from Toxoplasma gondii. J Struc Biol. 2011;176(1):97–111. doi: 10.1016/j.jsb.2011.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pulavarti SVSRK, Jain A, Pathak PP, Mahmood A, Arora A. Solution structure and dynamics of peptidyl-tRNA hydrolase from Mycobacterium tuberculosis H37Rv. J Mol Biol. 2008;378:165–177. doi: 10.1016/j.jmb.2008.02.027. [DOI] [PubMed] [Google Scholar]
- 29.Lipari G, Szabo A. Modelfree approach to the interpretation of nuclear magnetic-resonance relaxation in macromolecules I. Theory and range of validity. J Am Chem Soc. 1982a;104:4546–4559. [Google Scholar]
- 30.Lipari G, Szabo A. Modelfree approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules II. Analysis of experimental results. J Am Chem Soc. 1982b;104:4559–4570. [Google Scholar]
- 31.Clore GM, Szabo A, Bax A, Kay LE, Driscoll PC, Gronenborn AM. Deviations from the simple two parameter model free approach to the interpretation of 15N nuclear magnetic relaxation of proteins. J Am Chem Soc. 1990;112:4989–4991. [Google Scholar]
- 32.Nicholls A, Sharp KA, Honig B. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins Struct Funct Genet. 1991;11:281–285. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 33.Roger C, Loria JP. FAST-Modelfree: a program for rapid automated analysis of solution NMR spin-relaxation data. J Biomol NMR. 2003;26:203–213. doi: 10.1023/a:1023808801134. [DOI] [PubMed] [Google Scholar]
- 34.Palmer AG, Rance M, Wright PE. Intramolecular motions of a zinc finger DNA-binding domain from Xfin characterized by proton-detected natural abundance 13C heteronuclear NMR spectroscopy. J Am Chem Soc. 1991;113:4371–4380. [Google Scholar]
- 35.Pope BJ, Ziegler-Gould KM, Kuhne R, Weeds AG, Ball LJ. Solution structure of human cofilin: actin binding, pH sensitivity, and relationship to actin-deploymerizing factor. J Biol Chem. 2004;279:4840–4848. doi: 10.1074/jbc.M310148200. [DOI] [PubMed] [Google Scholar]
- 36.Maciver SK, Zot HG, Pollard TD. Characterization of actin filament severing by actophorin from Acanthamoeba castellanii. J Cell Biol. 1991;115(6):1611–20. doi: 10.1083/jcb.115.6.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gorbatyuk Vitaliy Y, Nosworthy Neil J, et al. Mapping the Phosphoinositide-Binding Site on Chick Cofilin Explains How PIP2 Regulates the Cofilin-Actin Interaction. Molecular Cell. 2006;24:511–522. doi: 10.1016/j.molcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 38.Palmer AG., III NMR characterization of the dynamics of biomacromolecules. Chem Rev. 2004;104:3623–3640. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- 39.Kay LE, Torchia DA, Bax A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry. 1989;28:8972–8979. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- 40.Cavanagh J, Fairbrother WJ, Palmer AG, Skelton NJ. Protein NMR Spectroscopy. Academic Press; San Diego: 1996. [Google Scholar]
- 41.Palmer AG., III NMR characterization of the dynamics of biomacromolecules. Chem Rev. 2004;104:3623–3640. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- 42.Boehr DD, Dyson HJ, Wright PE. An NMR perspective on Enzyme dynamics. Chem Rev. 2006;106(8):3055–3079. doi: 10.1021/cr050312q. [DOI] [PubMed] [Google Scholar]
- 43.Palmer AG, Kroenke CD, Loria JP. Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Methods Enzymol. 2001;339:204–238. doi: 10.1016/s0076-6879(01)39315-1. [DOI] [PubMed] [Google Scholar]
- 44.Mukherjee S, Pondaven SP, Jaroniec CP. Conformational Flexibility of a Human Immunoglobulin Light Chain Variable Domain by Relaxation Dispersion Nuclear Magnetic Resonance Spectroscopy: Implications for Protein Misfolding and Amyloid Assembly. 2011;50:5845–5857. doi: 10.1021/bi200410c. [DOI] [PubMed] [Google Scholar]
- 45.Rawat A, Kumar D. NMR investigations of structural and dynamics features of natively unstructured drug peptide salmon calcitonin: implication to rational design of potent sCT analogs. 2012;19(1):33–45. doi: 10.1002/psc.2471. [DOI] [PubMed] [Google Scholar]
- 46.Wong W, Skau CT, Marapana DS, Hanssen E, Taylor NL, Riglar DT, Zuccala ES, Angrisano F, Lewis H, Catimel B, Clarke OB, Kershaw NJ, Perugini MA, Kovar DR, Gulbis JM, Baum J. Minimal requirements for actin filament disassembly revealed by structural analysis of malaria parasite actin-depolymerizing factor 1. PNAS. 2011;108:9869–9874. doi: 10.1073/pnas.1018927108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pope BJ, Gonsior SM, Yeoh S, McGough A, Weeds AG. Uncoupling actin filament fragmentation by cofilin from increased subunit turnover. J Mol Biol. 2000;298:649–661. doi: 10.1006/jmbi.2000.3688. [DOI] [PubMed] [Google Scholar]
- 48.Tammana TVS, Sahasrabuddhe AA, Mitra K, Bajpai VK, Gupta CM. Actin-depolymerizing factor, ADF/cofilin, is essentially required in assembly of Leishmania flagellum. Mol Microbiol. 2008;70:837–852. doi: 10.1111/j.1365-2958.2008.06448.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.