

Abstract
Radiation exposure induces cell and tissue damage causing local and systemic inflammatory responses. Since the inflammasome pathway is triggered by cell death and danger-associated molecular patterns (DAMPs), we hypothesized that the inflammasome may signal acute and chronic immune responses to radiation. Using a mouse radiation model, we show that radiation induces a dose-dependent increase in inflammasome activation in macrophages, dendritic cells, NK cells, T cells, and B cells as judged by cleaved caspase 1 detection in cells. Time course analysis showed the appearance of cleaved caspase 1 in cells by day 1 and sustained expression until day 7 post-radiation. Also, cells showing inflammasome activation co-expressed the cell-surface apoptosis marker, Annexin V. The role of caspase 1 as a trigger for hematopoietic cell losses after radiation was studied in caspase 1 −/− mice. We found less radiation-induced cell apoptosis and immune cell loss in caspase 1 −/− mice than control mice. Next, we tested whether uric acid might mediate inflammasome activation in cells by treating mice with allopurinol and discovered that allopurinol treatment completely blocked caspase 1 activation in cells. Finally, we demonstrate that radiation-induced caspase 1 activation occurs by a Nod-like receptor family protein 3 (NLRP3) independent mechanism since radiation-exposed Nlrp3 −/− mice showed caspase 1 activation profiles that were indistinguishable from wild-type mice. In summary, our data demonstrate that inflammasome activation occurs in many immune cell types following radiation exposure and that allopurinol prevented radiation-induced inflammasome activation. These results suggest that targeting the inflammasome may help control radiation-induced inflammation.
INTRODUCTION
It is well known that radiation exposure occurring by intentional or un-intentional ways causes cell death and damage in a dose-dependent fashion. In mammals, radiation does the most damage to actively dividing cell populations like hematopoietic cells and cells lining mucosal surfaces. Individuals exposed to radiation show significant loss of peripheral immune cells, which predisposes them to a wide range of microbial opportunistic infections. For this reason, a clearer understanding of the cellular and molecular events following radiation injury in humans is of paramount importance if a radionuclear event ever happens. Though radiation therapy is used to treat various types of cancer or for bone marrow transplants in patients, when used medically, radiation exposures are tightly controlled and directed to localized body areas to minimize the damage to healthy tissue or is given at a minimal effective dose for bone marrow transplants. Nevertheless, unwanted side effects of radiation therapy do occur, among them include tissue injury, inflammation and suppressed immune function, resulting in sickness and higher susceptibility to infections in these patients (1). Thus, a better understanding of how radiation exposure influences immune cell activation and function will forward the development of ways to mitigate undesirable effects of radiation exposure on the body.
Cell death after radiation occurs by mitotic catastrophe and by apoptosis. Another cell death modality described after radiation is cell senescence. Cell fate after radiation is mainly determined by the level of DNA damage and by the cell type affected (2). While apoptosis is generally thought to be a relatively silent way for cells to die, dead cells do release molecules which have been shown to activate the immune system (3). These molecules act as endogenous danger signals to alert the immune system and are subsumed under the term alarmins (4, 5). Alarmins and microbial “pathogen-associated molecular patterns” (PAMPs) are together categorized as “danger-associated molecular patterns” (DAMPs) (4). DAMPs are sensed by pattern recognition receptors (PRRs) (4). Currently, four types of PRRs are known and have been classified into: the toll-like receptors (TLRs) and the C-type lectin receptors (CLRs), which are both located on the cell surface; the NOD-like receptors (NLRs) and RIG-I-like receptors (RLRs), which are intracellular PRRs (6).
Though many signaling pathways can be triggered when cells respond to DAMPs, a protein signaling complex called the inflammasome represents a highly-controlled signaling mechanism for acute and chronic inflammatory changes in cells (7). Inflammasome complexes can contain one of the NLRs - NLRP1, NLRP3, NLRP6, IPAF or the HIN-200 family member AIM2 complexed with the adaptor protein called ASC (7, 8). Formation of the inflammasome results in the activation of the caspase 1 enzyme, which results in the formation of the interleukin-1 (IL-1) converting enzyme (ICE). ICE is a homodimer enzyme complex with catalytic domains consisting of two p10 and p20 subunits that mediate the cleavage of the pro-IL-1β and pro-IL-18 into their respective active forms (9).
Caspase 1 activation can also result in caspase 1 dependent cell death in immune cells. This process is also called pyroptosis (10). Pyroptosis can be triggered by a variety of stimuli. These include pathogens like Salmonella and Listeria, but also non-infectious pathologies like stroke and myocardial infarction can induce caspase 1 mediated pyroptosis (11, 12) (13, 14). Interestingly, stroke and myocardial infarction involve a large degree of tissue destruction and cell death, which will result in the release of alarmins. Since radiation also leads to wide-spread cell death with presumed release of alarmins, we hypothesized that: 1) inflammasome activation occurs after radiation; 2) inflammasome activation leads to caspase 1 dependent cell death in immune cells; and 3) inflammasome activation after radiation is mediated by the release of alarmins from dead and damaged cells. To test our hypothesis, we exposed mice to total body irradiation (TBI) from a 137CsCl source irradiator and assessed the state of inflammasome activation in spleen immune cells by intracellular flow cytometry staining for the p10 subunit of activated caspase 1. This flow cytometry method for measuring caspase 1 activation in immune cell subsets has recently been used by our group to demonstrate inflammasome activation after traumatic injury (15). We also tested the influence of caspase 1 deficiency on cell death and immune cell changes after radiation. Moreover, we sought to identify a potential alarmin that might trigger radiation-induced inflammasome activation in immune cells. We found that allopurinol, a drug that inhibits uric acid synthesis, significantly reduced radiation-induced caspase 1 activation in mice. This finding suggests that uric acid or uric acid crystal formation may be a central trigger for radiation-induced inflammasome pathway activation in immune cells (16). Since the Nod-like receptor family protein 3 (NLRP3) is a primary pathway for caspase 1 pathway activation for DAMPs, we tested for radiation-induced caspase 1 activation in Nlrp3−/− mice and discovered that radiation induced caspase 1 activation in immune cells does not depend on the NLRP3 signaling pathway. This report describes the kinetics and dose-dependent effects of whole-body radiation exposure on inflammasome pathway activation in mice and provides evidence to suggest that treating mice with allopurinol, a drug that inhibits the xanthine oxidase enzyme and uric acid synthesis, reduces radiation-induced caspase 1 pathway activation. In addition, we demonstrate that allopurinol treatment reduces radiation-induced inflammation.
MATERIALS AND METHODS
Mice
Male 5-7 week old CD-1 mice were purchased from Charles River Laboratories (Wilmington, MA). Male caspase 1 gene deficient (caspase 1−/−) mice (strain NOD.129S2(B6)-Casp1tm1Sesh/LtJ) and strain-matched controls (strain NOD/ShiLtJ) were purchased from The Jackson Laboratory (Bar Harbor, ME). Male 7 week old C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, ME) and Nlrp3 −/− mice on the C57BL/6J genetic background were from Dr. Vishva Dixit (Genentech, South San Francisco, CA). All mice were acclimated for at least 5 days in our full-barrier animal facility under a twelve-hour light-dark cycle and were given access to mouse chow and water ad libitum. All animal procedures used in this study were approved by the Harvard Medical School Institutional Animal Care and Use Committee (IACUC) and were performed following NIH guidelines.
Reagents
Fluorescently-labeled antibodies for flow cytometry were purchased from BioLegend (San Diego, CA) or eBioscience (San Diego, CA). The antibody clones used in these experiments were as follows: CD3 (145-2C11), CD11c (N418), CD19 (6D5), CD49b (DX5), and F4/80 (BM8). The caspase 1 p10 specific polyclonal antibody (m315) and the PE-conjugated donkey anti-goat antibody (sc3857) used in these studies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Culture medium, called complete 5 (C5), was RPMI-1640 containing 5 % heat-inactivated fetal bovine serum, HEPES, glutamine, non-essential amino acids, antibiotic-antimycotic, and 2-mercaptoethanol.
Radiation Injury
Mice were exposed to whole body irradiation by timed exposure to 137CsCl radiation delivered by a J.L. Shepherd & Associates Mark I Irradiator (San Fernando, CA). The dose rate for these experiments ranged from 1.63 to 1.65 Gy/minute and doses were delivered by timed exposures to attain the desired radiation dose of 0.5, 1, 2, or 4 Gy.
Allopurinol Treatment
Allopurinol (Cayman Chemical, Ann Arbor, MI), was dissolved in DMSO and injected i.p. (20 mg/kg) 2 hours before and 6 hours after radiation exposure. Control-treated mice were given DMSO diluted in saline at identical concentrations used for allopurinol delivery.
Uric Acid Detection Assay
Uric acid concentrations in heparinized plasma were measured using a colorimetric assay as described by the manufacturer (BioAssay Systems, Hayward, CA). In brief, blood was harvested by cardiac puncture at 2 hours, 6 hours, or 1 day after 4 Gy radiation from individual male, CD-1 mice, immediately following sacrifice by CO2. Blood was centrifuged at slow speed (100 × g) for 15 minutes and plasma was collected for the uric acid assay. The assay was performed using uric acid to generate a standard curve to estimate uric acid levels in mouse plasma samples.
Cell Preparations
Mice were killed by CO2 asphyxiation. Blood was harvested into heparinized 1 cc syringes by cardiac puncture. Spleens were harvested and homogenized under sterile conditions by mincing on sterile 70 μm nylon cell strainers (Falcon, Franklin Lakes, NJ) in C5 medium. Cells were centrifuged at 300 × g for 10 minutes and the pellet was resuspended in 5 ml of red blood cell lysis buffer for 5 minutes followed by neutralization with 5 ml of C5 medium. Following an additional wash by centrifugation, cells were resuspended in C5 medium and filtered through nylon cell strainers (BD Falcon, Bedford, MA) to remove cell debris.
Flow cytometry procedures
Cell suspensions (50 μL) were plated in round-bottom 96-well plates (Costar, NY) for flow cytometry (FACS) staining. All stains were performed in phosphate buffered saline (PBS) with 1% bovine serum albumin (BSA) and 0.1% sodium azide (PBA). Non-specific binding of staining antibodies was blocked by pre-incubation for 10 minutes with TruStain FcX reagent (BioLegend, San Diego, CA). Splenocytes were surface-stained in separate wells with mixtures of fluorescence-tagged anti-F4/80 (macrophages), anti-CD11c (dendritic cells), anti-CD19 (B cells), anti-CD3 (T cells), or anti-CD49b antibodies (NK cells) – all purchased from BioLegend (San Diego, CA). To measure cell apoptosis and pyroptosis by flow cytometry, cells were incubated at 37° C with propidium iodide (PI). After 30 minutes, cells were pelleted by centrifugation at 300 × g for 6 minutes and surface-stained with FITC-labeled Annexin V (BioLegend, San Diego, CA) to identify dead and apoptotic cell populations. When analyzed, cells gated as Annexin V-positive were classified as apoptotic cells, cells gated as Annexin V-positive and PI-positive were identified as pyroptotic cells, and those gated as only PI-positive were classified as necrotic (dead) cells. To stain for caspase 1 activation in cells, spleen cells were added to wells of a round-bottom 96-well plate cells and pelleted by centrifugation at 300 × g for 6 minutes. Cells were fixed with 100 μL of 0.4% paraformaldehyde (PFA) at room temperature. After 10 minutes, cells were pelleted by centrifugation at 300 × g for 6 minutes, and then fixed with 200 μL of ice-cold 100% methanol for 20 minutes on ice. After washing cells twice by centrifugation at 300 × g for 6 minutes in PBA, cells were stained with mixtures of cell subset marker antibodies and antibody specific for activated caspase 1 (p10 subunit) antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). After over-night incubation at 4° C, cells were washed once by centrifugation at 300 × g for 6 minutes and cells were stained with PE-conjugated donkey anti-goat antibody (Santa Cruz Biotechnology, Santa Cruz, CA). After 30 minutes staining, cells were washed twice by centrifugation at 300 × g for 6 minutes, the cells were fixed again with 0.4% PFA. All flow cytometry was performed using a MACSQuant Analyzer (Miltenyi Biotec, Bergisch-Gladbach, Germany). The *.FCS files from flow cytometry were analyzed using FlowJo 7.6.5 software (Tree Star, Ashland, OR). Cells were gated as F4/80+, CD11c+, CD49b+, CD3+ and CD19+ subsets for analysis. Mean fluorescence intensity (MFI) of cleaved caspase 1 p10 staining was calculated for each gated cell subset including Annexin V+ and Annexin V− fractions of each subset. Control stains without anti-p10 antibody addition was used to determine background stain levels and MFI values were calculated by subtracting background MFI from caspase p10-stained samples.
Cytokine Assays
Blood was collected in EDTA anticoagulant by cardiac puncture and plasma was prepared by centrifugation at 12,000 × g for 10 minutes. Splenocytes were prepared from mice and equal cell numbers (5 × 106 cells per well) were plated in round-bottom 96-well tissue-culture treated plates. Cells were cultured for 48 hours at 37° C in the presence of E. coli-derived lipopolysaccharide (LPS 026:B6; Sigma, St. Louis, MO). Plasma and culture supernatants were tested for IL-1α, IL-1β, IL-6, IL-12p40, IL-18, TNFα, and MCP-1 levels using bead-based Luminex assays. Recombinant cytokine standards were used to estimate cytokine concentrations. Data acquisition was performed on a Luminex-200 instrument (Luminex, Austin, TX) with StarStation software (Applied Cytometry, Sheffield, UK).
Statistical Analysis
Data was analyzed for statistical significance using GraphPad Prism 5.0.4 software (GraphPad Software, LaJolla, CA). Unpaired student’s t-test or one-way ANOVA with Bonferroni multiple comparisons test were used when appropriate and P values ≤0.05 were considered statistically significant.
RESULTS
Inflammasome activation in response to radiation is dose-dependent
Groups of male, CD-1 outbred mice were exposed to whole-body radiation by timed exposure to 137CsCl. At 1 day after radiation, spleen cells were prepared and stained for caspase 1 activation by measuring the intracellular levels of cleaved-caspase 1 p10 subunit by flow cytometry. Specific immune cell subsets showing increased cleaved caspase 1 p10 were also identified by flow cytometry. The FACS histograms shown in Figure 1A illustrate a detectable increase in caspase 1 p10 expression in spleen cells and all major immune cell subsets examined including macrophages, dendritic cells, T cells, B cells, and NK cells from mice exposed to 1 or 4 Gy radiation. We next performed dose response radiation studies to determine a dose response curve for radiation-induced inflammasome activation. The data plotted in Figure 1B demonstrates that caspase 1 p10 expression is linear and reaches high levels by 4 Gy whole body radiation. A clear dose-dependent increase in inflammasome activation was observed in all immune cell subsets that were analyzed. However, macrophages and dendritic cells showed peak activation by 2 Gy radiation that was sustained at 4 Gy, while NK cells, T cells and B cells showed peak activation at 4 Gy radiation. This observation suggests that macrophages and dendritic cells are comparatively more sensitive to initiators of inflammasome activation following radiation injury.
Figure 1. Inflammasome pathway activation in spleen cell subsets following radiation with increasing whole-body radiation doses.
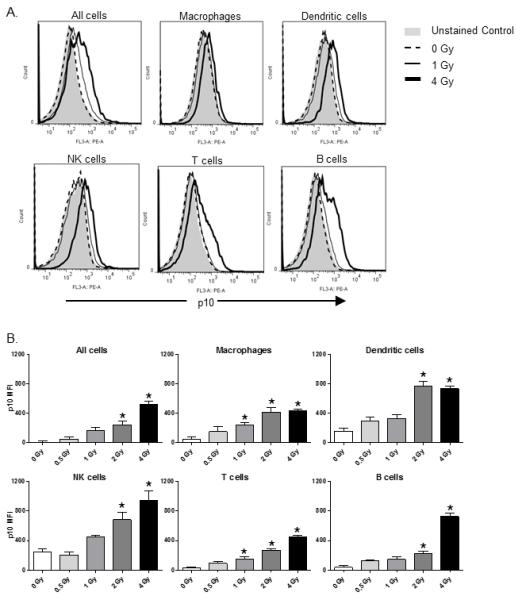
Inflammasome pathway activation was measured by intracellular staining for the p10 subunit of cleaved caspase 1 in spleen immune cell subsets prepared from groups of CD-1 mice (n=5) at 1 day after radiation exposure at the indicated doses. The FACS histograms in (A) are representative of the caspase 1 p10 staining levels seen at 1 day after 1 or 4 Gy whole body radiation exposure by this method. Immune cell subsets were identified by flow cytometry with fluorescently-labeled cell subset specific antibodies as follows: macrophages: F4/80+ cells, dendritic cells: CD11c+ cells, NK cells: NK1.1+ cells, T cells: CD3+ cells, B cells: CD19+ cells. The data plotted in (B) show caspase 1 p10 staining in the indicated spleen immune cell subsets at 1 day after radiation injury with increasing whole-body radiation doses. The levels of caspase 1 p10 staining in cells are plotted as mean fluorescence intensity (MFI). The results indicate a dose-dependent increase in inflammasome pathway activation in all immune cell subsets examined. The * indicates p≤0.05 by one-way ANOVA with Bonferroni multiple comparisons test.
Since radiation is known to induce cell damage and death by apoptosis, we assessed whether cells showing high levels of caspase 1 p10 were also showing signs of apoptosis by measuring caspase 1 p10 expression levels in Annexin V+ versus Annexin V− cells. As expected, we observed that the percentage of apoptotic cells increased in direct relation to whole-body radiation dose (Figure 2A). Macrophages and dendritic cells showed high apoptosis at lower dose radiation exposure than T cells, B cells, and NK cells, similar to what we observed for cleaved caspase 1 p10 expression. When cells were FACS-gated on Annexin V+ cells, we found higher caspase 1 p10 expression in Annexin V+ cells than in Annexin V− cells (Figure 2B). This result indicates that cells showing inflammasome activation are also undergoing apoptosis.
Figure 2. Radiation injury induces a dose-dependent increase in immune cell apoptosis.
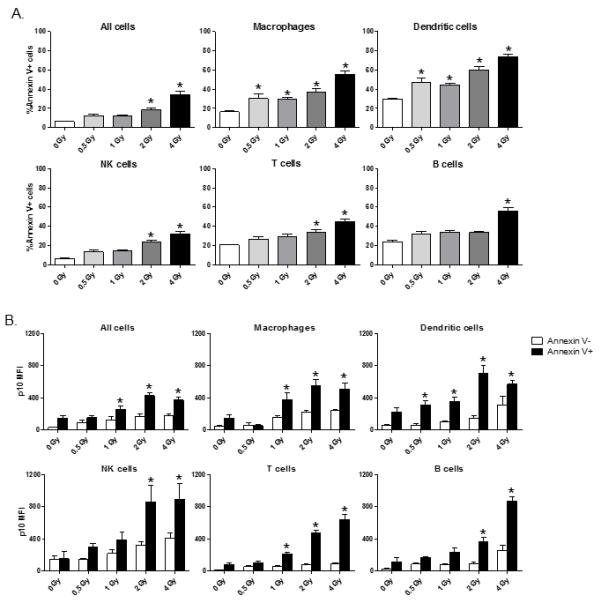
In panel (A), spleen cells were prepared from mice (n=5) at 1 day after 0, 0.5, 1, 2, or 4 Gy whole body radiation injury. Cells were stained with fluorescently-labeled immune cell subset markers and apoptotic cells were identified by Annexin V staining. The data is plotted as mean ± SEM of the percentage of Annexin V+ cells in gated immune cell subsets. The * indicates p≤0.05 for Annexin V+ staining for 0 Gy vs. radiation exposure by one-way ANOVA with Bonferroni multiple comparisons test. The data plots in (B) show inflammasome activation measured by caspase 1 p10 levels in apoptotic cells (Annexin V+) versus non-apoptotic (Annexin V−) spleen cell subsets. Increased inflammasome activation was found in apoptotic cells of all spleen cell subsets. The * indicates p≤0.05 by unpaired t-test, Annexin V+ vs. Annexin V−.
Inflammasome pathway activation and cell death is a delayed event after radiation injury
The kinetics of radiation-induced inflammasome activation was defined by exposing mice to 2 Gy whole-body radiation and measuring caspase 1 p10 levels in cells at 1 hour, 4 hours, 1 day, 3, 7, and 14 days after radiation. We were surprised to find that inflammasome activation was not significantly induced at 1 or 4 hours after radiation injury, but was easily detected at day 1 after radiation in all immune cell subsets indicating that inflammasome activation by radiation injury is a delayed host response (Figure 3A). Moreover, we found that inflammasome activation was sustained for 7 days after radiation injury in all immune cell subsets and returned to baseline levels by 14 days after radiation. Cell death as measured by Annexin V expression on cells followed the exact kinetics of inflammasome activation (Figure 3B) for most immune cell subsets examined. The exceptions were that T cells showed increased apoptosis at 4 hours after radiation and that B cells did not show signs of apoptosis until 3 days after radiation. These results suggest that radiation injury causes prolonged inflammasome activation in immune cells and that the kinetics of inflammasome activation coincides with increased Annexin V expression on most immune cell types.
Figure 3. Time course of radiation-induced inflammasome pathway activation and immune cell pyroptosis.
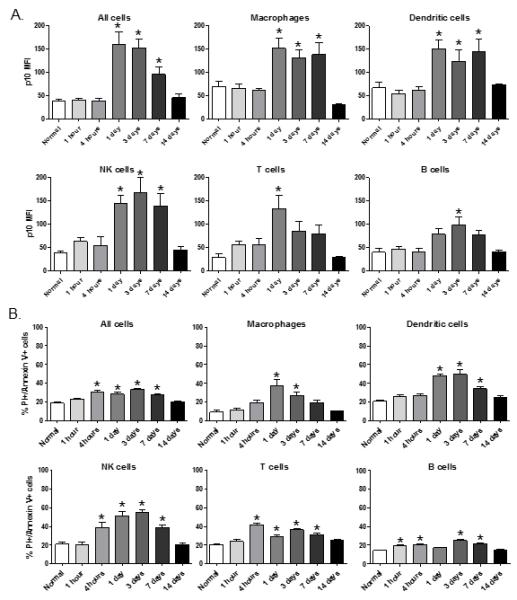
Groups of mice (n=5) were exposed to 2 Gy whole-body radiation and spleen cells were harvested at 1 hour, 4 hours, 1 day, 3, 7, or 14 days after radiation. In (A), spleen cells were stained with immune cell subset specific antibodies and counterstained for caspase 1 p10 to measure inflammasome pathway activation. In (B), the same cell preparations were stained for Annexin V and propidium iodide (PI) to measure cells undergoing cell death by pyroptosis. The * indicates p≤0.05 by one-way ANOVA with Bonferroni test for multiple comparisons.
Enhanced immune cell survival following radiation injury in caspase 1 gene deficient mice
To identify a functional role for inflammasome activation in radiation injury, we tested the effects of radiation injury on cell death and apoptosis in caspase 1 gene deficient (−/−) mice. Caspase 1 −/− and wild-type (WT) mice were exposed to 2 Gy whole-body radiation and spleen cells were compared at 1 day after radiation for changes in cell numbers, and cell-surface Annexin V/PI staining as a measure for pyroptosis. As shown in Figure 4A, caspase 1 −/− mice exposed to 2 Gy radiation had significantly higher immune cell numbers than WT mice. Accordingly, the percentages of cells expressing Annexin V and PI were lower in caspase 1 −/− mice as compared to WT mice (Figure 4B). These results indicate that caspase 1 activation plays an active role in controlling cell death by radiation injury.
Figure 4. Reduced radiation-induced immune cell loss in caspase 1−/− mice.
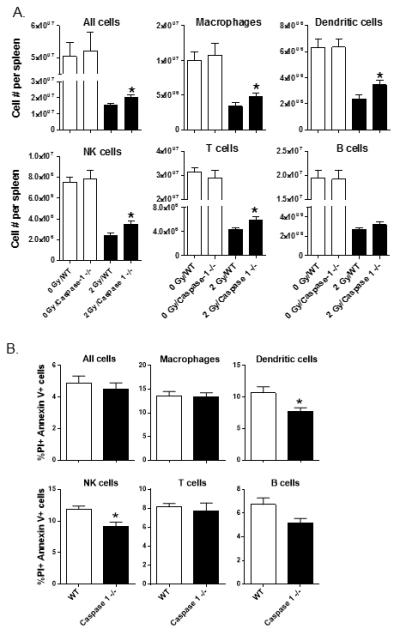
Groups of WT and caspase 1−/− mice (n=5 per group) were exposed to 2 Gy whole-body radiation One day later, spleen cells were prepared and tested for radiation-induced immune cell subset losses by FACS (A). The * indicates p≤0.05 for WT vs. caspase 1−/− by a one-tailed unpaired t test. In (B), the indicated immune cell subsets were stained with Annexin V and PI to measure the percentages of cells undergoing pyroptosis. The * indicates p≤0.05 for 2 Gy WT vs. 2 Gy caspase 1−/− by a one-tailed unpaired t test.
Uric acid synthesis plays a role in caspase 1 activation following radiation
Uric acid is known to potently activate the inflammasome pathway in cells by acting as a danger associated molecular pattern (DAMP) molecule (16, 17). Given the high level of cell death caused by radiation injury, we suspected that uric acid might be a trigger for inflammasome activation in cells. To test this idea, we first measured systemic levels in uric acid in mice that were exposed to 4 Gy whole-body radiation. As shown in Figure 5, we observed increased levels of uric acid in the plasma of mice at 2 hours, 6 hours, and 1 day after radiation exposure. Next, we tested the functional effects of blocking uric acid generation by treating mice with the xanthine oxidase inhibitor drug, allopurinol, before and after 2 Gy whole-body radiation. We observed that allopurinol treatment significantly decreased inflammasome activation in macrophages, dendritic cells, and NK cells to levels observed in sham mice (Figure. 6A). Allopurinol treatment also reduced inflammasome activation in T and B cells, but the reduction was not statistically significant. However, allopurinol treatment did not have a major effect on immune cell losses from radiation injury, but did cause significant increases in spleen cell numbers in sham mice (Figure 6B). Importantly, allopurinol treatment did significantly suppress radiation-induced increases in LPS-stimulated IL-1β production by spleen cells and also had a generalized suppressive effect on LPS-induced inflammatory cytokine production as shown by reduced IL-1α, IL-6, IL-12p40, MCP-1, and TNFα production (Figure 7). These data support that uric acid generation may be a primary stimulus for inflammasome pathway activation and might act to control the inflammatory behavior of the immune system following radiation.
Figure 5. Radiation exposure increases systemic uric acid levels in mice.
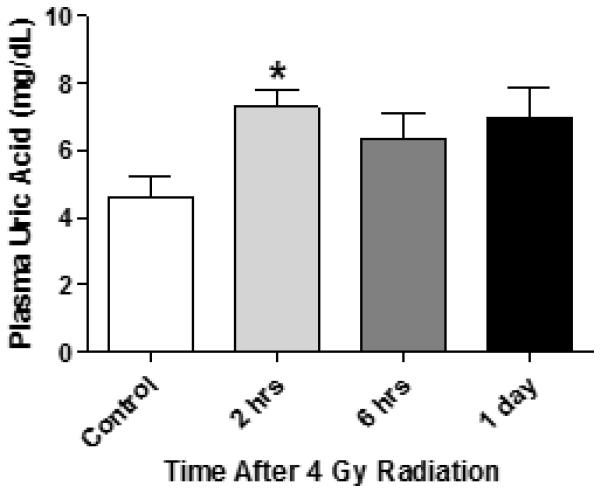
Groups of outbred CD-1 mice (n=5) were exposed to 4 Gy whole-body radiation. Blood was harvested in heparin anti-coagulant at 2 hours, 6 hours, or 1 day after radiation. Plasma was tested for uric acid levels by a colorimetric assay using uric acid as a standard as described in materials and methods. The * indicates p≤0.05 vs control plasma by one-way ANOVA and the Bonferroni test for multiple comparisons.
Figure 6. Allopurinol treatment prevents radiation-induced inflammasome pathway activation in immune cells.
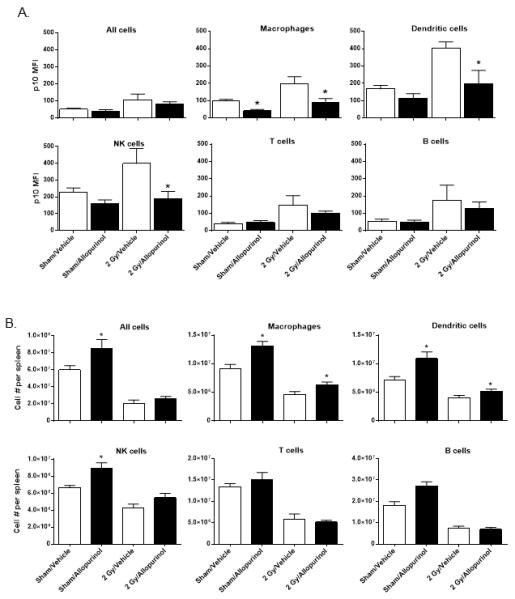
Mice were given 20 mg/kg allopurinol or vehicle by i.p. injection at 2 hours before and 6 hours after 2 Gy whole-body radiation. Spleen cells were prepared and FACS analysis was performed to detect p10 cleaved caspase 1 levels in the indicated FACS-gated immune cell subsets (A). In (B), spleen cells were analyzed by FACS to determine allopurinol treatment effects on the indicated immune cell subset numbers. The data is plotted as the mean ± SEM for n=6 mice per group. The * indicates p≤0.05 for allopurinol vs vehicle-treated mice by one-way ANOVA with Bonferroni multiple comparisons test.
Figure 7. Allopurinol treatment blocks the radiation-induced pro-inflammatory phenotype of immune cells.
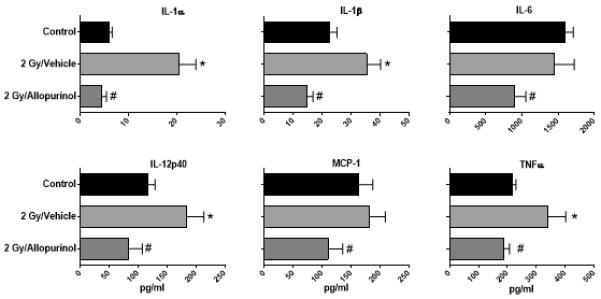
Spleen cells were prepared from mice exposed to 2 Gy radiation and treated with 20 mg/kg of allopurinol or vehicle at 2 hours before and 6 hours after radiation or control mice that were not exposed to radiation. Spleen cells were cultured with 1 μg/ml LPS and48-hour culture supernatants were tested for cytokine levels using a Luminex multiplex cytokine assay platform. The data is plotted as the mean ± SEM for n=6 mice per group. The * indicates p≤0.05 for control vs. 2 Gy + Vehicle and the # indicates p≤0.05 for 2 Gy + Vehicle vs. 2 Gy + Allopurinol by one-way ANOVA with Bonferroni multiple comparisons test.
The NLRP3 signaling pathway does not influence radiation-induced caspase 1 activation in immune cells
The observation that allopurinol could block radiation-induced caspase 1 activation suggested to us that uric acid synthesis, uric acid, or uric acid crystal formation in tissue could be mediating radiation-induced inflammasome pathway activation in immune cells. Though multiple pathways can trigger assembly of the inflammasome and caspase 1 activation, the NLRP3 signaling pathway is a promiscuous sensor of DAMPs and has been shown to be involved in uric acid mediated caspase 1 activation(18, 19). Thus, we tested whether Nlrp3 gene deficient (Nlrp3 −/−) mice might show differences in radiation-induced inflammasome pathway activation and cell loss. Nlrp3 −/− or C57BL/6J (WT) mice were exposed to 2 Gy TBI and caspase 1 activation was measured in spleen immune cell subsets by p10 staining using flow cytometry at 1 day after radiation. Changes in immune cell numbers were also measured by flow cytometry. As shown in Figure 8A, immune cell subsets from Nlrp3 −/− mice showed levels of caspase 1 activation that were indistinguishable from WT mice. In addition, radiation-induced immune cell loss in Nlrp3 −/− mice was not significantly different from WT mice (Figure 8B). However, we did observe that Nlrp3 −/− intrinsically have higher numbers of total spleen cells and macrophages than age- and sex-matched WT mice. Given that caspase 1 −/− and Nlrp3 −/− mice showed differences in radiation responses, we compared their inflammatory response to radiation exposure by measuring circulating cytokine levels at 1 day after 2 Gy radiation exposure using multiplex Luminex bead cytokine assays. The plasma cytokine data shown in Figure 9 demonstrate that caspase 1 −/− mice produce significantly lower levels of circulating IL-1β, IL-18, IL-6, and IL-12p40 than WT mice. Interestingly, Nlrp3 −/− mice also showed significantly lower levels of plasma IL-1β, IL-18, and IL-6 than WT mice, but IL-18 and IL-12p40 levels were higher than those observed in caspase 1 −/− mice. Thus, both caspase 1−/− and Nlrp3 −/− mice showed lower inflammatory-type responses to radiation exposure than genetically normal mice suggesting that these pathways are involved in triggering the host response to radiation injuries. However, it appears that radiation-induced caspase 1 activation does not depend on the NLRP3 inflammasome activation path-way.
Figure 8. Nlrp3 −/− mice demonstrate normal radiation-induced caspase 1 activation and cell loss profiles.
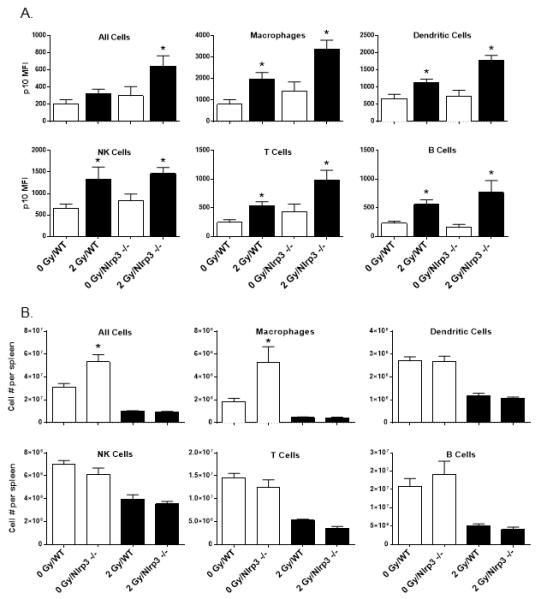
Groups of C57BL/6J (WT) and Nlrp3 −/− mice (n=8 per group) were exposed to 2 Gy whole-body radiation One day later, spleen cells were prepared and tested for radiation-induced caspase 1 activation as judged by FACS detection of cleaved caspase 1 p10 levels (A). The data plots in (B) show the comparative levels of immune cell loss in the spleens of WT versus Nlrp3 −/− mice (n=8 per group) exposed to 2 Gy whole-body radiation. The * indicates p≤0.05 for WT vs. Nlrp3 −/− mice by one-way ANOVA with Bonferroni multiple comparisons test.
Figure 9. Circulating cytokine levels at 1 day after 2 Gy radiation exposure in WT, caspase 1 −/−, and Nlrp3 −/− mice.
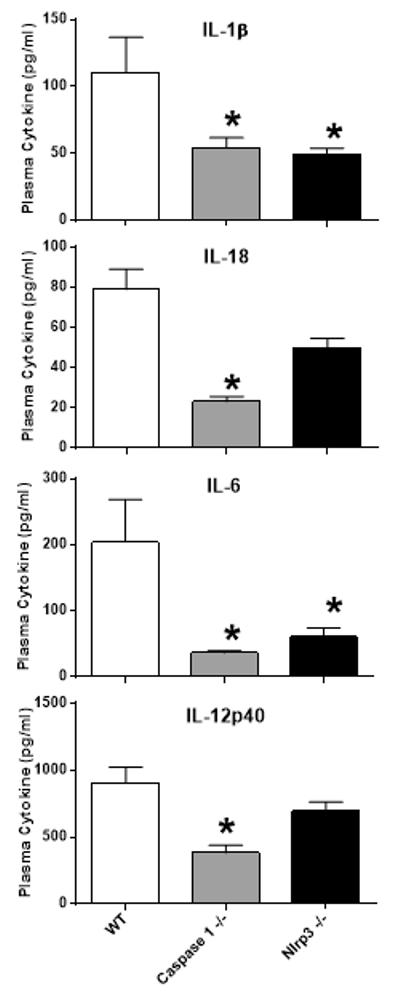
Mice were exposed to 2 Gy radiation and plasma samples were tested for cytokine levels using a multiplex Luminex assay panel for 17 cytokines. The cytokine data shown is plotted as the mean ± SEM for n=8 mice per group and include only those cytokines with detectable levels in the plasma of WT, caspase 1 −/−, or Nlrp3 −/− mice. The * indicates p≤0.05 for plasma cytokine level differences between WT mice and caspase 1 −/− or Nlrp3 −/− mice by one-way ANOVA with Bonferroni multiple comparisons test.
DISCUSSION
A primary goal of this study was to contribute an improved basic understanding of how radiation exposure activates the immune system. Though it is known that radiation causes a dose-dependent reduction in hematopoietic and peripheral immune cells, less is known about the specific phenotypic changes in the cellular and molecular pathways in immune cells that remain after radiation injury. In this study, we focused our efforts on studying the influence of radiation on activating the inflammasome activation pathway in immune cells for several reasons. First, we believed that the extensive cell death and damage that occurs after radiation exposure might provide an early stimulus for inflammasome pathway activation by the immune system. Second, the physiological consequences of radiation injury suggest that radiation injury induces clinical symptoms similar to severe infections and trauma, which include fever, fatigue, loss of appetite, vomiting, and diarrhea. Third, radiation injury promotes chronic inflammatory conditions that can predispose exposed individuals to inflammatory diseases such as pulmonary fibrosis and cancer. The inflammasome pathway has been shown to be a central feature of these diseases (20-23).
The results presented in this report demonstrate that whole-body radiation, also referred to as total body irradiation (TBI), induces substantial inflammasome pathway activation in immune cell populations, especially macrophages, dendritic cells, and NK cells. The activation was found to be radiation dose-dependent, sustained, and partially inhibited by treatment with allopurinol, a xanthine oxidase inhibitor drug that inhibits uric acid synthesis by altering purine metabolism. To our knowledge, this may be the first report to demonstrate that whole-body ionizing radiation exposure causes significant inflammasome pathway activation in immune cells. Though not investigated in this study, it is also possible that the inflammasome pathway is activated in non-immune cell types by radiation exposure, which could contribute to prolonged tissue inflammation or damage. The results reported here were greatly facilitated by a flow cytometry technique that detects cleaved caspase 1 p10 as a measure for caspase 1 activation in immune cells. Since caspase 1 activation is the final step of inflammasome pathway activation, we made the assumption that increased levels of cleaved caspase 1 p10 detection in cells should indicate assembly of the inflammasome signaling complex (7). When this approach was developed, we compared the p10 staining profiles in cells to those measured using a fluorescent caspase 1 detection reagent called fluorochrome-labeled inhibitor of caspases (FLICA), a FAM-tagged cleaved caspase 1 inhibitor – FAM-YVAD-fmk(15). Comparative flow cytometry analysis showed that the p10 staining results in cells were directly comparable to those measured by FLICA(15). The primary advantage of using flow cytometry to measure caspase 1 activation was that it allowed us to specifically identify which cell subsets showed the highest levels of inflammasome pathway activation. Furthermore, this technique allowed us to efficiently follow the kinetics of inflammasome pathway activation in immune cells. We also used anti-p10 antibody staining to measure caspase 1 activation because staining cells for caspase activation by flow cytometry using FLICA has been criticized as being non-specific (24). Accordingly, we discovered an anticipated dose-dependent effect of radiation exposure on inflammasome pathway activation. However, we did not foresee that radiation exposure would induce sustained inflammasome pathway activation in immune cells. This prolonged effect of radiation on the immune system suggests that radiation has both acute and chronic pro-inflammatory influences on the immune system that are mediated in part by the inflammasome. The sustained activation observed in these experiments also supports the idea that inflammasome activating molecules remain active in the body for long periods of time after radiation injury.
The sustained inflammasome pathway activation raises the question if radiation directly activates the inflammasome pathway, which has been described in response to ultraviolet radiation in the skin or if alarmins released by damaged cells may be the trigger for activation (25). One way to answer this question was to assess the time course for inflammasome activation after radiation. Since we found that radiation-induced inflammasome activation was delayed until day 1, direct activation of the inflammasome by radiation seems unlikely. Furthermore, the activation was detectable in cells even at 7 days after radiation exposure suggesting that inflammasome activation occurs secondarily to radiation injuries. Based on these results, we favor the hypothesis that inflammasome activation is induced by the release of DAMPs, in response to radiation-induced cell death or damage. This hypothesis is supported by findings in other related studies, which showed that trauma, osteoarthritis, experimental autoimmune encephalitis, and lung injury induced by ventilation or anthrax could induce delayed and sustained inflammasome pathway activation (15, 26-28).
Because inflammasome activation can lead to caspase 1-mediated cell death, referred to as pyroptosis, we wished to determine if immune cell types in mice showing inflammasome activation by radiation also show signs of undergoing cell death by pyroptosis (10). Using a combination of cell-surface Annexin V binding as a marker to detect phosphatidylserine residues that appear on the outer membrane leaflet of stressed or dying cells and propidium iodide (PI) DNA intercalation staining, we discovered that cells showing high caspase 1 activation showed evidence of dying by pyroptosis. Based on this observation, it appears that caspase 1 activation after radiation is linked to cell death and establishes that many types of immune cells can undergo caspase 1-dependent cell death following radiation. In addition, though the early cell loss that is seen after radiation may occur by apoptosis and mitotic catastrophe, caspase 1-mediated cell death by pyroptosis appears to be an ongoing process after radiation exposure (2). In relation to this observation, links between activation of tumor suppressor p53 and the activation of caspases in cells have been described (29-31). Moreover, it is known that radiation exposure increases p53 expression in cells that undergo radiation-induced apoptosis but p53 can also protect cells from death by acting as a DNA repair enzyme (32). The overlapping activation pattern between p53 and caspases suggest that there may also be a link between p53 and caspase 1. If so, then modulating caspase 1 or p53 activation may have similar effects on cell survival and inflammation after radiation exposure. Given the importance of tumor suppressor p53 for regulating radiation-induced cell death, future studies could address whether allopurinol might have cell protective effects by influencing p53 expression in cells showing inflammasome activation.
To further test the idea that cells undergo caspase 1 initiated cell death following radiation exposure, we tested the influence of caspase 1 gene deficiency on radiation-induced cell death. We discovered that caspase 1 gene deficiency results in higher cell numbers and decreased percentages of dying cells in the spleen. These findings further support the hypothesis that the caspase 1 pathway contributes to some of the cell death after whole-body radiation exposure. This finding is line with a host of other publications demonstrating that cell death by caspase 1 mediated pyroptosis occurs in sterile inflammatory conditions like stroke or hypoxia-induced renal damage (33, 34). Other findings also suggest that caspase 1 activation plays a direct role in cell death which goes beyond initiating pyroptosis (35). Nevertheless, we found that ablating caspase 1 activity in vivo rescued only a fraction of the cells dying after radiation, which supports the conclusion that radiation-induced cell death is not wholly caspase 1 dependent. However, in contrast, we found that Nlrp3 −/− mice did not show any difference in radiation-induced cell loss as compared to wild-type mice. This finding is novel and further supports that caspase 1 and inflammasome pathway activation may occur independent of the NLRP3 signaling pathway. Several other reports also demonstrated caspase 1 independent NLRP3 signaling (36, 37).
Next, we sought to identify possible signaling triggers for inflammasome activation following radiation. We suspected that uric acid or uric acid synthesis may be involved due to the wide-spread cell destruction that occurs after radiation exposure (38). This is because cells contain intracellular uric acid and extracellular uric acid levels rise significantly due to the release and degradation of DNA and RNA from dying cells (39). Extracellular uric acid, especially uric acid crystals, are recognized by the immune system as DAMPs and trigger activation of the inflammasome (40). Accordingly, we examined circulating levels of uric acid in mice exosed to 4 Gy whole-body radiation and found significantly higher uric acid levels as compared to control mice. Thus, we suspected that targeting uric acid might attenuate radiation-induced inflammasome activation. To test this hypothesis, mice were treated with the xanthine oxidase inhibitor drug, allopurinol, to investigate if reducing in vivo uric acid synthesis might reduce inflammasome pathway activation in immune cells. We discovered that allopurinol treatment blocked radiation-induced inflammasome activation in immune cells and also increased immune cell numbers in the spleen. Thus, it appears that uric acid or uric acid crystals may act as a central trigger for radiation-induced inflammasome pathway activation by the immune system. Allopurinol is a xanthine oxidase inhibitor and may have other effects that are independent of reducing uric acid production that could contribute to its effects on suppressing inflammasome activation during radiation exposure. For example, allopurinol could affect reactive oxygen and nitrogen generation by acting as a xanthine oxidase enzyme inhibitor (41, 42) (43).
We believe our finding that uric acid or uric acid synthesis may mediate radiation-induced inflammasome activation in vivo is clinically significant for several reasons. First, we demonstrate that the inflammasome pathway is activated in immune cells for up to seven days following radiation exposure. Sustained inflammatory conditions are believed to promote cancer development in tissues (44). Thus, it is possible that blunting uric acid production in tissues following radiation may reduce the risk of developing inflammatory complications or cancer after accidental or therapeutic radiation exposure. Second, our findings suggest that reducing uric acid production by allopurinol or other uric acid lowering drugs like rasburicase, a uricase drug, may improve immune cell survival and recovery. We suspect that modulating uric acid levels or reducing uric acid crystal formation could in turn improve immune function in people after radiation exposure and reduce the development of opportunistic infections, a common cause of morbidity and mortality following radiation injuries. We speculate that radiation effects on anti-microbial immunity might be mediated in part by chronic stimulation of the immune system by uric acid or uric acid crystals, which may promote immune system exhaustion in a manner similar to what has been reported to occur in chronic diseases like cancer and HIV infection (45, 46). In support of this idea, a recent report showed that ultraviolet radiation increased uric acid levels in the skin (47). The increased uric acid levels in the skin was found to be associated with decreased contact hypersensitivity responsiveness since allopurinol treatment reduced skin uric acid levels and restored contact hypersensitivity responses in mice following ultraviolet radiation injury (47). This observation suggests that uric acid levels affect cell-mediated immune responses, which supports the idea that inflammasome activation by radiation can suppress immune function by a uric acid dependent mechanism. In addition, it has been reported that blocking xanthine oxidase after radiation protected the vascular endothelium integrity in the aorta of rats (48). This provides evidence that blocking uric acid production after radiation injury might have additional beneficial effects which extend beyond the immune system and may also be mediated by inflammasome-independent mechansims.
In summary, we report for the first time that whole-body radiation exposure causes sustained inflammasome activation in most major immune cell subsets and that radiation-induced cell death is partly dependent on caspase 1 activation. Furthermore, we provide evidence that inflammasome activation by radiation can be modulated by allopurinol, a drug that is used to reduce uric acid production and reduce uric acid mediated inflammation. Thus, therapeutic reduction of uric acid synthesis following radiation exposure could help reduce radiation-induced damage to radiation-sensitive tissues and cells and could also help reduce inflammatory complication of radiation injury and immune system functions. Thus, future studies will need to address the potential of using drugs to modulate uric acid production or levels as a way to protect individuals from the damaging effects of radiation on the body if a radionuclear accident or event does occur.
Footnotes
This research was supported by funding from NIH grant # R21/R33 AI080565 as part of Project Bioshield and the Medical Countermeasures for Radiological and Nuclear Threats Program.
REFERENCES
- 1.Redding SW, Zellars RC, Kirkpatrick WR, McAtee RK, Caceres MA, Fothergill AW, Lopez-Ribot JL, Bailey CW, Rinaldi MG, Patterson TF. Epidemiology of Oropharyngeal Candida Colonization and Infection in Patients Receiving Radiation for Head and Neck Cancer. Journal of Clinical Microbiology. 1999;37:3896–3900. doi: 10.1128/jcm.37.12.3896-3900.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eriksson D, Stigbrand T. Radiation-induced cell death mechanisms. Tumor Biology. 2010;31:363–372. doi: 10.1007/s13277-010-0042-8. [DOI] [PubMed] [Google Scholar]
- 3.Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425:516–521. doi: 10.1038/nature01991. [DOI] [PubMed] [Google Scholar]
- 4.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. Journal of Leukocyte Biology. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 5.Oppenheim JJ, Yang D. Alarmins: chemotactic activators of immune responses. Curr Opin Immunol. 2005;17:359–365. doi: 10.1016/j.coi.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 6.Takeuchi O, Akira S. Pattern Recognition Receptors and Inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 7.Schroder K, Tschopp J. The Inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 8.Anand PK, Malireddi RKS, Lukens JR, Vogel P, Bertin J, Lamkanfi M, Kanneganti T-D. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature. 2012 doi: 10.1038/nature11250. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walker NPC, Talanian RV, Brady KD, Dang LC, Bump NJ, Ferenza CR, Franklin S, Ghayur T, Hackett MC, Hammill LD, Herzog L, Hugunin M, Houy W, Mankovich JA, McGuiness L, Orlewicz E, Paskind M, Pratt CA, Reis P, Summani A, Terranova M, Welch JP, Xiong L, Möller A, Tracey DE, Kamen R, Wong WW. Crystal structure of the cysteine protease interleukin-1β-converting enzyme: A (p20/p10)2 homodimer. Cell. 1994;78:343–352. doi: 10.1016/0092-8674(94)90303-4. [DOI] [PubMed] [Google Scholar]
- 10.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Micro. 2009;7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cervantes J, Nagata T, Uchijima M, Shibata K, Koide Y. Intracytosolic Listeria monocytogenes induces cell death through caspase-1 activation in murine macrophages. Cellular Microbiology. 2008;10:41–52. doi: 10.1111/j.1462-5822.2007.01012.x. [DOI] [PubMed] [Google Scholar]
- 12.Brennan MA, Cookson BT. Salmonella induces macrophage death by caspase-1-dependent necrosis. Molecular Microbiology. 2000;38:31–40. doi: 10.1046/j.1365-2958.2000.02103.x. [DOI] [PubMed] [Google Scholar]
- 13.Frantz S, Ducharme A, Sawyer D, Rohde LE, Kobzik L, Fukazawa R, Tracey D, Allen H, Lee RT, Kelly RA. Targeted deletion of caspase-1 reduces early mortality and left ventricular dilatation following myocardial infarction. Journal of Molecular and Cellular Cardiology. 2003;35:685–694. doi: 10.1016/s0022-2828(03)00113-5. [DOI] [PubMed] [Google Scholar]
- 14.Rabuffetti M, Sciorati C, Tarozzo G, Clementi E, Manfredi AA, Beltramo M. Inhibition of Caspase-1-Like Activity by Ac-Tyr-Val-Ala-Asp-Chloromethyl Ketone Induces Long-Lasting Neuroprotection in Cerebral Ischemia through Apoptosis Reduction and Decrease of Proinflammatory Cytokines. The Journal of Neuroscience. 2000;20:4398–4404. doi: 10.1523/JNEUROSCI.20-12-04398.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Osuka A, Hanschen M, Stoecklein V, Lederer JA. A Protective Role for Inflammasome Activation Following Injury. Shock. 2012;37:47–55. doi: 10.1097/SHK.0b013e318234f7ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi Y, Mucsi AD, Ng G. Monosodium urate crystals in inflammation and immunity. Immunol Rev. 2010;233:203–217. doi: 10.1111/j.0105-2896.2009.00851.x. [DOI] [PubMed] [Google Scholar]
- 17.Ghaemi-Oskouie F, Y Shi. The role of uric acid as an endogenous danger signal in immunity and inflammation. Curr Rheumatol Rep. 2011;13:160–166. doi: 10.1007/s11926-011-0162-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Goutassociated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 19.Giamarellos-Bourboulis EJ, Mouktaroudi M, Bodar E, van der Ven J, Kullberg BJ, Netea MG, van der Meer JW. Crystals of monosodium urate monohydrate enhance lipopolysaccharide-induced release of interleukin 1 beta by mononuclear cells through a caspase 1-mediated process. Ann Rheum Dis. 2009;68:273–278. doi: 10.1136/ard.2007.082222. [DOI] [PubMed] [Google Scholar]
- 20.Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–735. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241–247. doi: 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Menu P, Vince JE. The NLRP3 inflammasome in health and disease: the good, the bad and the ugly. Clin Exp Immunol. 2011;166:1–15. doi: 10.1111/j.1365-2249.2011.04440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.dos Santos G, Kutuzov MA, Ridge KM. The inflammasome in lung diseases. Am J Physiol Lung Cell Mol Physiol. 2012;303:L627–633. doi: 10.1152/ajplung.00225.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuzelova K, Grebenova D, Hrkal Z. Labeling of apoptotic JURL-MK1 cells by fluorescent caspase-3 inhibitor FAM-DEVD-fmk occurs mainly at site(s) different from caspase-3 active site. Cytometry. Part A: the journal of the International Society for Analytical Cytology. 2007;71:605–611. doi: 10.1002/cyto.a.20415. [DOI] [PubMed] [Google Scholar]
- 25.Feldmeyer L, Keller M, Niklaus G, Hohl D, Werner S, Beer H-D. The Inflammasome Mediates UVB-Induced Activation and Secretion of Interleukin-1β by Keratinocytes. Current Biology. 2007;17:1140–1145. doi: 10.1016/j.cub.2007.05.074. [DOI] [PubMed] [Google Scholar]
- 26.Inoue M, Williams KL, Gunn MD, Shinohara ML. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proceedings of the National Academy of Sciences. 2012;109:10480–10485. doi: 10.1073/pnas.1201836109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Denoble AE, Huffman KM, Stabler TV, Kelly SJ, Hershfield MS, McDaniel GE, Coleman RE, Kraus VB. Uric acid is a danger signal of increasing risk for osteoarthritis through inflammasome activation. Proceedings of the National Academy of Sciences. 2011;108:2088–2093. doi: 10.1073/pnas.1012743108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuipers MT, Aslami H, Janczy JR, van der Sluijs KF, Vlaar APJ, Wolthuis EK, Choi G, Roelofs JJTH, Flavell RA, Sutterwala FS, Bresser P, Leemans JC, van der Poll T, Schultz MJ, Wieland CW. Ventilator-induced Lung Injury Is Mediated by the NLRP3 Inflammasome. Anesthesiology. 2012;116:1104–1115. doi: 10.1097/ALN.0b013e3182518bc0. [DOI] [PubMed] [Google Scholar]
- 29.Afshar G, Jelluma N, Yang X, Basila D, Arvold ND, Karlsson A, Yount GL, Dansen TB, Koller E, Haas-Kogan DA. Radiation-induced caspase-8 mediates p53-independent apoptosis in glioma cells. Cancer Res. 2006;66:4223–4232. doi: 10.1158/0008-5472.CAN-05-1283. [DOI] [PubMed] [Google Scholar]
- 30.Ju GZ, Shen B, Sun SL, Yan FQ, Fu SB. Effect of X-rays on expression of caspase-3 and p53 in EL-4 cells and its biological implications. Biomedical and environmental sciences: BES. 2007;20:456–459. [PubMed] [Google Scholar]
- 31.Sidi S, Sanda T, Kennedy RD, Hagen AT, Jette CA, Hoffmans R, Pascual J, Imamura S, Kishi S, Amatruda JF, Kanki JP, Green DR, D’Andrea AA, Look AT. Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3. Cell. 2008;133:864–877. doi: 10.1016/j.cell.2008.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee CL, Blum JM, Kirsch DG. Role of p53 in regulating tissue response to radiation by mechanisms independent of apoptosis. Transl Cancer Res. 2013;2:412–421. [PMC free article] [PubMed] [Google Scholar]
- 33.Edelstein CL, Hoke TS, Somerset H, Fang W, Klein CL, Dinarello CA, Faubel S. Proximal tubules from caspase-1-deficient mice are protected against hypoxia-induced membrane injury. Nephrology Dialysis Transplantation. 2007;22:1052–1061. doi: 10.1093/ndt/gfl775. [DOI] [PubMed] [Google Scholar]
- 34.Friedlander RM, Gagliardini V, Hara H, Fink KB, Li W, MacDonald G, Fishman MC, Greenberg AH, Moskowitz MA, Yuan J. Expression of a Dominant Negative Mutant of Interleukin-1β Converting Enzyme in Transgenic Mice Prevents Neuronal Cell Death Induced by Trophic Factor Withdrawal and Ischemic Brain Injury. The Journal of Experimental Medicine. 1997;185:933–940. doi: 10.1084/jem.185.5.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Denes A, G. Lopez-Castejon, D. Brough. Caspase-1: is IL-1 just the tip of the ICEberg? Cell Death & Disease. 2012;3:e338. doi: 10.1038/cddis.2012.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bracey NA, Gershkovich B, Chun J, Vilaysane A, Meijndert HC, Wright JR, Jr., Fedak PW, Beck PL, Muruve DA, Duff HJ. Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. The Journal of biological chemistry. 2014;289:19571–19584. doi: 10.1074/jbc.M114.550624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taxman DJ, Holley-Guthrie EA, Huang MT, Moore CB, Bergstralh DT, Allen IC, Lei Y, Gris D, Ting JP. The NLR adaptor ASC/PYCARD regulates DUSP10, mitogen-activated protein kinase (MAPK), and chemokine induction independent of the inflammasome. The Journal of biological chemistry. 2011;286:19605–19616. doi: 10.1074/jbc.M111.221077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng C, Omura-Minamisawa M, Kang Y, Hara T, Koike I, Inoue T. Quantification of circulating cell-free DNA in the plasma of cancer patients during radiation therapy. Cancer Science. 2009;100:303–309. doi: 10.1111/j.1349-7006.2008.01021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kono H, Chen CJ, Ontiveros F, Rock KL. Uric acid promotes an acute inflammatory response to sterile cell death in mice. The Journal of Clinical Investigation. 2010;120:1939–1949. doi: 10.1172/JCI40124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 41.Zimmet JM, Hare JM. Nitroso-redox interactions in the cardiovascular system. Circulation. 2006;114:1531–1544. doi: 10.1161/CIRCULATIONAHA.105.605519. [DOI] [PubMed] [Google Scholar]
- 42.Pacher P, Nivorozhkin A, Szabo C. Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol Rev. 2006;58:87–114. doi: 10.1124/pr.58.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Derbre F, Ferrando B, Gomez-Cabrera MC, Sanchis-Gomar F, Martinez-Bello VE, Olaso-Gonzalez G, Diaz A, Gratas-Delamarche A, Cerda M, Vina J. Inhibition of xanthine oxidase by allopurinol prevents skeletal muscle atrophy: role of p38 MAPKinase and E3 ubiquitin ligases. PLoS One. 2012;7:e46668. doi: 10.1371/journal.pone.0046668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuraishy A, Karin M, Grivennikov SI. Tumor promotion via injury- and death-induced inflammation. Immunity. 2011;35:467–477. doi: 10.1016/j.immuni.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol. 2013;25:214–221. doi: 10.1016/j.coi.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim PS, Ahmed R. Features of responding T cells in cancer and chronic infection. Curr Opin Immunol. 2010;22:223–230. doi: 10.1016/j.coi.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leighton S, Kok L-F, Halliday GM, Byrne SN. Inhibition of UV-induced uric acid production using Allopurinol prevents suppression of the contact hypersensitivity response. Experimental Dermatology. 2013;22:189–194. doi: 10.1111/exd.12096. [DOI] [PubMed] [Google Scholar]
- 48.Soucy KG, Lim HK, Kim JH, Oh Y, Attarzadeh DO, Sevinc B, Kuo MM, Shoukas AA, Vazquez ME, Berkowitz DE. HZE 56Fe-Ion Irradiation Induces Endothelial Dysfunction in Rat Aorta: Role of Xanthine Oxidase. Radiation Research. 2011;176:474–485. doi: 10.1667/rr2598.1. [DOI] [PubMed] [Google Scholar]