

Abstract
Molecular beacons (MBs) are dual-labeled oligonucleotides that fluoresce only in the presence of complementary mRNA. The use of MBs to target specific mRNAs allows sorting of specific cells from a mixed cell population. In contrast to existing approaches that are limited by available surface markers or selectable metabolic characteristics, the MB-based method enables the isolation of a wide variety of cells. For example, the ability to purify specific cell types derived from pluripotent stem cells (PSCs) is important for basic research and therapeutics. In addition to providing a general protocol for MB design, validation and nucleofection into cells, we describe how to isolate a specific cell population from differentiating PSCs. Using this protocol, we have successfully isolated cardiomyocytes differentiated from mouse or human PSCs with ~97% purity as confirmed using electrophysiology and immunocytochemistry. After designing MBs, their ordering and validation requires two weeks, and the isolation process requires three hours.
INTRODUCTION
The ability to separate different cell types is important for a wide range of biological and medical studies, including the quantification of cells with specific phonotypes for disease diagnosis, the isolation of terminally differentiated induced pluripotent stem cells (iPSCs) at different stages of maturation, and the selection of cells from a mixed cell population that possess unique characteristics or functions. In most cases, the selection and separation methods rely on cell physical properties (e.g. size, shape, stiffness, etc.), cell surface protein expression, or genetic modifications. In particular, cells derived from pluripotent stem cells (PSCs), including embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) 1,2, are becoming a powerful tool that dramatically changes how pharmaceuticals are developed and validated for therapies by allowing patient-specific mechanistic studies, and personalized drug testing for efficacy and toxicology. For example, researchers have used cells derived from PSCs to model genetic diseases such as long QT syndrome 1 (LQT1) 3,4. PSC-based disease modeling is challenging, however, since many disorders affect only specific, terminally differentiated, cell populations. Currently available PSC differentiation systems typically generate mixed populations containing undifferentiated cells or undesirable cells which could cause teratoma formation or interfere with high throughput quantification5. Thus, purification of tightly controlled populations of terminally differentiated cells derived from PSCs is desirable to prevent detrimental effects.
Methods developed to isolate specific populations of differentiated cells derived from PSCs
Various techniques have been developed to isolate specific cell types from differentiating PSCs including positive selection6,7, negative selection8, genetic modification9,10, or metabolic negative selection11,12. The most popular method for isolating specific populations of cells is to use antibodies to target surface proteins6,7. However, the lack of specific cell surface proteins that can be targeted by conventional antibody-based fluorescence-activated cell sorting (FACS) remains one of the major challenges commonly encountered when isolating terminally differentiated cells from differentiating PSCs. Several methods that do not require specific antibodies are available, including the classic purification technique that relies on a fluorescent reporter gene driven by a promoter such as NKX2.5, ISL1 or MHC in genetically modified cell lines 6,7. However, such reporter-gene based methods may not be applicable to certain PSCs such as iPSCs where selecting a line with the reporter gene (such as GFP) integrated at a single, correct genomic location is very challenging. Alternatively, non-genetic approaches such as the use of a Percoll gradient13 or the use of cell metabolism12,14 have been developed. While these methods are useful in specific applications, they are limited to targeting specific cellular phenotypes which may be dynamic during the differentiation process6. Together, these methods may lack the required detection specificity due to their not using a specific molecular marker highly expressed in target cell types. To address the limitations of the above approaches, we developed a method to isolate specific cell types by directly targeting intracellular mRNAs using molecular beacons (MBs) and sorting via FACS.
Development of the protocol
MBs are dual-labeled oligonucleotides ~15–30 bases long with a fluorophore on one end and a quencher molecule on the other end (Figure 1A) 15. Since their development in 1996 15, MBs have been used to identify specific mRNA or DNA sequences in solution 16,17, and to visualize the intracellular localization of mRNA transcripts in individual living cells 18,19. MBs excel in both types of applications because they fluoresce only when hybridized to complementary oligonucleotides, a property that confers molecular specificity and target versatility. In the absence of oligonucleotide target, MBs assume a hairpin conformation that brings the fluorophore and quencher moieties into contact, resulting in significant quenching of the fluorophore and very low background fluorescence (Figure 1A). Hybridization of the MB with target oligonucleotide sequence opens the hairpin, thus physically separating the fluorophore from the quencher, restoring fluorescence upon excitation (Figure 1B).
Figure 1. Molecular beacon structure and control molecular beacons.

(a) A schematic of a molecular beacon in a stem-loop hairpin conformation. The stem brings the 5′ dye and 3′ quencher together to quench the fluorescence signal. The loop region with 15–30 base pairs is complementary to the target sequence of specific mRNA thus providing specificity. (b) A schematic depicting a molecular beacon in an open conformation after hybridizing to its complementary target mRNA sequence. Hybridization induces a conformational change that separates the fluorophore from the quencher, resulting in a >10-fold increase in fluorescence signal. (c) A schematic of a positive control molecular beacon with a fluorescent dye attached to both the 5′ and 3′ ends, thus is constitutively fluorescent. This probe is used to determine if MBs can be delivered into different cell types efficiently and uniformly. (d) A schematic depicting a specific negative control beacon. The four mutated bases should prohibit hybridization of the probe to the target region of specific mRNA. When delivered into 3T3 cells as negative control cells, this control beacon should yield very low background signal, confirming the MB specificity. (d) A predicted secondary structure of a cardiac specific gene, with a single stranded region highlighted and expanded.
It was demonstrated in proof of concept studies that MBs could be used in a flow cytometry assay to identify cells expressing a specific mRNA in a mixed cell population without affecting the expression of this mRNA 20. We have recently developed a MB based method for the isolation of cardiomyocytes (CMs) from a differentiating culture of PSCs by targeting highly expressed specific mRNAs21,22. Specifically, we designed and validated MBs targeting cardiac troponin T (cTNT or TNNI3) or myosin heavy chain genes (α/βMHC or MYH6/7) mRNAs respectively that are highly expressed only in CMs. MBs were then delivered into differentiating mouse or human PSCs using nucleofection followed by FACS sorting of CMs based on MB signal. Control studies confirmed that CM specific MBs had low background signal in all non-CM cells in the differentiating culture of PSCs, confirming detection specificity. We found that 97% of mouse or human cells isolated using FACS based on MB signal demonstrated CM-like characteristics in assays including electrophysiology, flow cytometry, and immunocytochemistry. This approach, using a FACS sorter to purify CMs based on MB signal from specific mRNA expression, is very versatile and can be adopted for the isolation of other cell types from differentiating PSCs. Although the later steps of the procedure (step 18 onwards) uses the isolation of CMs from differentiating human PSCs as an example application, this part of the protocol can be readily modified to isolate other specific cell types providing MBs have been successfully designed to target genes highly expressed in the cell type of interest.
Experimental Design
There are five essential elements of the MB-based protocol for the isolation of a specific cell type from differentiating cells (Figure 2): (1) identification of specific mRNAs that are highly expressed only in the desired cell type (Figure 2A); (2) design and validation of MBs targeting specific mRNAs with high signal-to-background ratio (Figure 2B); (3) delivery of MBs into a mixed population of cells, for example from differentiating PSCs, with high efficiency (Figure 2C); (4) isolation of cells with FACS based on MB signal (Figure 2D); and (5) validation of cell characteristics (Figure 2E). Although the experimental design described here uses the isolation of CMs as an example, the MB based method can be readily modified for the isolation and enrichment of other cell types by designing MBs to target the specific gene(s) highly expressed in the particular cell type, as described in the procedure below.
Figure 2. Major elements of procedure.
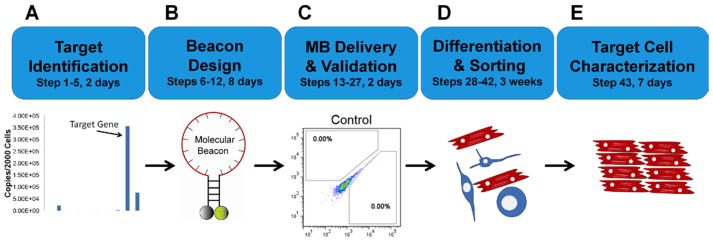
(a) The target gene(s) abundantly expressed in a cell type of interest are identified and confirmed using RT-PCR in Steps 1–5. (b) MBs are designed by identifying unique target sequences that are accessible, and choosing a stem region and an appropriate dye-quencher pair. (c) MBs are tested in solution to check the quality of MB synthesis and in positive/negative control cells to ensure that a high fluoresce signal is generated only in the specific cell type of interest. (d) Stem cells are differentiated into target cells (e.g., CMs), MBs are delivered into cells in the entire culture of mixed cell population, and MB positive cells are isolated using FACS. (e) MB-positive cells are characterized to confirm that they are the right cell type with high purity using RT-PCR, immunocytochemistry, and electrophysiology (for CM).
Identification of specific mRNAs highly expressed in target cells
In order to use MBs to isolate specific cells from a mixed cell population, target mRNAs that are highly and uniquely expressed in the cell type of interest must be identified. These mRNAs need to have a relatively high abundance so that, when MBs are hybridized to their mRNA target in the specific cells (such as CMs), the resulting fluorescence signal is at least 5-fold higher than that in other cell types. Genes encoding structural proteins have been used by several groups in the past21,23. However, for isolating novel cell types the gene expression profile must be quantified using qRT-PCR (or DNA microarray) to identify specific mRNAs that have high levels of expression (>10 fold) compared with that in other cell types in the in the PSC differentiation culture (Figure 2a). It may be necessary to choose multiple target genes highly expressed in the cell type of interest so that at least one mRNA will be specific enough and result in high MB signal. This also raises the possibility of using multiple MBs to target a specific subpopulation, such as ventricular CMs rather than all CMs.
MBs can be designed to target human genes, mouse genes, or in some cases both. An MB can be designed for both human and mouse genes only when the selected target sequence is identical for mouse and human genes. Specifically, MBs targeting the MYH6/7 or TNNT2 mRNA described in this protocol were designed for both mouse and human genes, allowing the use of HL-1 CMs (a mouse cell line) for characterization of CM-specific MBs. If an MB is designed for applications with human cells, but the MB targeting sequence does not have a perfect match in the mouse gene, a separate positive control cell line consisting of a standard control cell (e.g., 293 or 3T3 cells) transfected with a plasmid to highly express the target human gene should be used for MB validation. If an MB is designed only for isolating CMs from mouse cells, using HL-1 CMs as a positive control is sufficient.
Design and validation of MBs targeting specific mRNAs
Once specific genes are identified, MBs can be designed by targeting regions of the corresponding mRNA where excellent probe accessibility and signal specificity can be achieved. The target sequences of a specific mRNA may not be accessible due to secondary structure of the mRNA or RNA-binding proteins. To enhance target accessibility, an RNA secondary structure prediction program (e.g., mFOLD) can be used to identify potential single stranded regions of the mRNA (Figure 1D). It is also important to select target sequences with a sufficient (35% – 65%) GC content so that MBs so designed will have a good affinity to their targets. Finally, the NCBI BLAST search is typically performed to identify unique sequences of the mRNA for MBs to target (target sequences), and to minimize the number of sequences that differ from a target sequence by only a few (< 5) bases. Steps 1–10 identify and use three oligonucleotide sequences, the target sequence, a MB with its loop and stem sequences, and a mismatched (mutant) sequence. The target sequence in the specific mRNA needs to be carefully selected to ensure MB signal specificity. The MB loop sequence is complementary to the target sequence, and the MB stem sequence is typically 5 nucleotides long with high GC content. The MB is formed by selecting a dye-quencher pair and conjugating the dye and quencher to the 5′ and 3′ ends of the MB sequence respectively. The most affordable options are the FAM fluorophore with Black Hole Quencher 1 or the Cy3 fluorophore with Black Hole Quencher 2. The use of Cy3 dye is recommended for experiments where another molecule is taking up the FITC/GFP wavelengths, or there is significant autofluorescence. For labs without an oligonucleotide synthesizer facility, it is recommended to order MBs from one of the many high quality oligonucleotide providers such as IDT and MWG Operon, where HPLC purification of the MBs to eliminate the free dye is performed. A mismatched sequence, typically contains 4–6 mutations compared to the target sequence, is used to test the quality of MB synthesis in vitro. Since it is impossible to accurately predict target accessibility in living cells, multiple (three to five) MBs are usually designed and tested against a specific mRNA.
The most important issue with the MB based approach is signal specificity. Ideally, MBs delivered into a mixed cell population exhibit a high level of signal only in target cells but not in other cell types due to the high expression of the gene in target cells. However, in certain cases, a high level of non-specific signal from MBs may occur due to the delivery method (such as microinjection), MB degradation, or MB-protein interactions in cells. To determine MB signal specificity, a negative control random MB was designed whose loop sequence was generated using ‘random walk’ and does not match with any mRNA sequence in the genome of interest. Any signal from the random MBs after delivery into the target cells or positive control cells (cells highly express the target gene, such as the Hl-1 cells for CM isolation) will be non-specific background signal. The ratio of MB signal from specific mRNA to the background signal from random MB gives the signal-to-background ratio, S/N. To have high signal specificity, the MB designed should have a S/N > 5 in target cells.
In addition to using a random MB as a negative control, two assays can be performed to further confirm signal specificity. The first is to use a mutant MB that has 4–5 bases mismatch with the target sequence (Figure 1C). After delivery into the positive control cells (e.g. HL-1 cells), such a mutant MB should only generate a low background signal. The second assay is to down-regulate the expression level of the target mRNA using RNA interference (siRNA) in the positive control cells or to up-regulate the expression level using a plasmid in a cell type unrelated to the target cells, such as mouse embryonic fibroblasts (MEF) for CM isolation. High MB specificity is ensured if the MB signal decreases after down-regulation and increases after up-regulation of the target gene. Plasmid expression in a control cell type (such as MEF) is a reliable method, but is less ideal compared with the siRNA approach, since the MB accessibility might be different from that encountered when the gene is expressed endogenously. Note that the same MB design may have different target accessibilities in human and mouse cells.
Delivery of MBs into a mixed population of cells with high efficiency and throughput
Uniform, rapid delivery of MBs into all cell types in the differentiating culture of PSCs is important. In order for MBs to increase their fluorescence intensity by 5–10 fold upon hybridizing to their complementary mRNA targets, cells need to take up a sufficient number (usually > 2,000) of MBs per transfection. This is based on the estimate that the ideal number of mRNA transcripts per cell is ≥200 for effective isolation using FACS. The large MB/mRNA ratio is to ensure that most (if not all) mRNA targets in a cell are hybridized by MBs.
There are several MB delivery methods, including the use of microinjection, cell penetrating peptides (CPPs), streptolysin O (SLO), and electroporation 24. Microinjection is low-throughput, and often interferes with normal cell function. Although the CPP- or SLO-based delivery of MBs could have high efficiency and throughput, the delivery efficiency is typically cell type dependent and may not work for the mixed cell population resulting from PSC differentiation. In addition CPP or SLO based delivery of MBs might lead to endosomal entrapment and degradation, causing a high level of background signal. Electroporation has been used to deliver oligonucleotides into living cells, but conventional methods may result in low cell viability. Recent advances in electroporation technology (such as nucleofection) have led to a reduction in the harmful events, including heat generation, metal ion dissolution, pH variation, and oxide formation. The advanced electroporation methods lead to cellular internalization of oligonucleotide probes with high transfection efficiency (>90%) and high cell viability (>80%)22. Using nucleofection, it is possible to uniformly deliver MBs into millions of cells within 20 min. Because MBs must be delivered into cells quickly, due to the inherently short functional lifespan of exogenous nucleic acid sequences in a cellular environment25, we recommend delivery via nucleofection. It is also important to isolate cells based on the MB signal within 4 hours to avoid potential artifact caused by MB degradation. MBs may be re-introduced into cells at later time points if necessary, and several groups have shown that the MB signal from each delivery becomes undetectable after 24 hours26. Small numbers of MBs may enter the cell nucleus; however the resulting fluorescence signal should be low since no target mRNA is present in the nucleus and the probability of random genomic integration of MBs is negligible 19,20,24,27,28.
It is also important to ensure that the difference in MB signal between target cells and other cell types is not an artifact due to preferential uptake of MBs by a specific cell type. It is therefore important to determine whether or not all cells in the mixed population take up MBs uniformly (Figure 2C). To this end, the random sequence MB is modified to have two identical fluorophores instead of a dye-quencher pair (Figure 1C) and used as a delivery control probe for delivery studies. After probe delivery into differentiating cells, fluorescence signals are quantified and a relatively uniform signal distribution is expected. This experiment is designed to confirm that the MB signal specificity is due to MB hybridization to target mRNAs rather than MBs preferentially entering one cell type but not others.
Isolation of target cells with FACS based on MB signal
FACS sorting is currently the only method that can selectively isolate a specific cell population at a rate of millions of cells per hour based on fluorescence. Since MBs can detect cells that express various levels of target mRNAs, the flow cytometry assay may not generate very distinct populations of cells if there is a large variation of target mRNA expression levels in the differentiating PSCs to which MBs are delivered. It is therefore critical to design MBs that target abundantly expressed mRNAs in cells of interest in order to isolate them with high specificity and throughput. It is also important to fine-tune the FACS parameters so that cells with low levels of fluorescence signal will not be isolated. In addition, the FACS output streams should be carefully analyzed for the effect of sorting on both the cell viability and cell yield. Cell viability can be assessed by plating the MB positive population and measuring cell death over a 2-day period after sorting. To assess the yield of the FACS based target cell isolation, cell populations with high MB signal (MB positive) and low MB signal (MB negative) are collected after the sorting experiment, and both populations are compared using qRT-PCR and functional assays. A significant difference in target cell characteristics (such as specific gene expression) between the MB negative and MB positive populations may be used as an indication to confirm that MB based sorting has a high specificity.
Target cell characterization using qRT-PCR and immunocytochemistry
To ensure that a high purity target cell population is isolated from differentiating PSCs, MB positive cells must be tested to ascertain that (i) they highly express the specific gene(s) and (ii) they have low (or zero) expression levels of genes associated with other cell lineages. For example, to isolate CMs, specific markers such as TNNT2, TNNI3, MYH6/7 and MYL2 can be analyzed in the MB positive population using both qRT-PCR and immunocytochemistry to yield population-based results as well as quantitative information on the number of cells expressing specific markers. It is also necessary to demonstrate that the MB positive cells do not express genes that are markers of other cell types, including skeletal muscle, neural progenitor cells, fibroblasts and endothelial cells. To ensure that the MB positive cells represent a high purity population of target cells, a high percentage of cells expressing specific markers and a low percentage of cells expressing markers indicating other cell lineages are required.
Applications
The MB based method presents a novel approach for cell isolation due to its versatility and the ability to target intracellular mRNAs. A major hurdle to the current use of cells differentiated from hPSCs for many applications is the potential tumorigenicity or aberrant tissue formation after cell transplantation. Since many types of cells do not have specific surface protein markers for isolation and it is still impractical to genetically modify hiPSCs to express a selection marker, the MB based method may offer a useful means of purifying specific cells such as CMs generated from PSCs. The MB based method is applicable to the isolation of any cell type that expresses specific gene marker(s) to which corresponding MBs can be designed and validated. No surface protein marker for isolation is required. For example, MBs can be designed to target specific genes that are highly expressed only within neuronal or pancreatic beta cells, so the MB based method can be used to isolate neurons or beta cells generated from PSCs with high purity. MBs could also be used as a tool to evaluate cell differentiation protocols to provide rapid feedback on the yield and efficiency of the process.
In addition to isolating specific cells from the mixed cell population of differentiating PSCs, the MB based approach can also be applied to the isolation and enrichment of pure cell populations after transdifferentiation, in which one mature somatic cell type is transformed into another mature somatic cell type without undergoing a pluripotent state or using a progenitor cell type as an intermediate step. Another potential application of the MB based method described in this protocol is to isolate gene-modified cells after precise genome editing using TALENs and CRISPR/Cas systems. The modification of an endogenous gene is often in the form of single-base mutation (or correction of single-base mutation), deletion or insertion, making selection of gene-modified cells very challenging. With the addition of few wobble mutations in the target gene, MBs could be designed to target the site of modification in the corresponding mRNA, and the gene-modified cells can be isolated using the method described here.
MATERIALS
REAGENTS
CRITICAL: The materials listed here are those needed for the example application of isolating cardiomyocytes from hPSC. For different applications some materials will need to be substituted, for example, antibodies and cell culture media.
Cell line used for target cell purification. We have used human pluripotent stem cell lines, e.g., H1 (WISC Bank), BJ1 (a gift from George Daley at Harvard university) and
Suitable control cell lines. For the example application described here, we use HL-1 CMs (a gift from William Claycomb at Louisiana State University) and mouse embryonic fibroblasts (MEF) (CF-1) cells (ATCC, SCRC-1040)
CRITICAL: Other control cells can also be used. Prepare cells that highly express the MB target gene(s) for validation. For example, HL-1 CMs can be used as a positive control for testing CM-specific MBs. For negative control, various types of non-CM cells such as fibroblasts, or smooth muscle cells or endothelial cells can be used.
DMEM/F-12 (Life Tech, cat. no. 11320-033) - CM Specific
DMEM (Life Technologies, cat. no. 11965-092)
Claycomb medium (Sigma cat. no. 51800C-500ML) - CM Specific
Opti-MEM 1, no phenol red (Life Tech, cat. no. 11058-021)
KnockOut serum replacement (Life Technologies, cat. no. 10828-028) - CM Specific
FBS (ATCC, cat. no. 30-2020)
PBS (Sigma-Aldrich, cat. no. D8537)
Delivery Control MB (MWG, Sequence: ACGACGCGACAAGCGCACCGATACGTCGT, FAM dye added to both 5′ and 3′ end, HPLC purification)
Negative Control MB (MWG, Sequence: ACGACGCGACAAGCGCACCGATACGTCGT, Cy3 dye added to 5′ end and Black Hole Quencher 2 added to 3′ end, HPLC purification)
Activin A (R&D Systems, cat. no. 338-AC-050) (Step #) - CM Specific
BMP4 (R&D Systems, cat. no. 314-BP-010) (Step 12C only) - CM Specific
bFGF (Life Technologies, cat. no. 13256-029) (Step 12C only) - CM Specific
β-Mercaptoethanol (Sigma, cat. no. M7522) - CM Specific
GlutaMAX (Life Technologies, cat. no. 35050-061)
Antibiotic-Antimycotic (Life Technologies, cat. no. 15240-062)
Nonessential amino acid solution (NEAA;, Life Technologies cat. no. 11140-076) - CM Specific
mTeSR1 (STEMCELL Technologies, cat. no. 05857)
Matrigel (BD Biosciences, cat. no. 354277)
Y27632 (Tocris, cat. no. 1254) - CM Specific
Isoproterenol (Sigma, cat. no. I6504) - CM Specific
Cell Line Nucleofector Kit V (Lonza, cat. no. VACA-1003)
Trypsin-EDTA, 0.25% (wt/vol) (Life Tech, cat. no. 25200-056)
Accutase (Life Tech, cat. no. A1110501)
RNAEasy Miniprep Kit (Qiagen, cat. no. 74134)
iScript cDNA Synthesis kit (Biorad, cat. no. 170-8891)
iTaq™ Universal SYBR® Green Supermix (Biorad, cat. no. 172-5122)
Paraformaldehyde 4% (wt/vol) in PBS (Santa Cruz Biotechnology, cat. no. 30525-89-4)
Caution: Paraformaldehyde is harmful; avoid exposure.
Triton X-100 (Sigma-Aldrich, cat. no. X100-500ML)
Bovine Serum Albumin (Sigma-Aldrich, cat. no. A9576)
Tween-20 (Sigma-Aldrich, cat. no. P9416)
Anti-ACTN2 (Sigma-Aldrich, cat. no. SAB4503474-100UG) - CM Specific
Anti-TNNT2 (Thermo Scientific, cat. no. MA5-12960) - CM Specific
Mouse anti-cTnT (Thermo, cat. no. MS-295-P0) - CM Specific
Anti-cTnI (Abcam, cat. no. ab10231) - CM Specific
Goat anti-mouse IgG-Alexa Fluor 594 (Life Tech, cat. no. A-11001)
Goat anti-rabbit IgG-Alexa Fluor 488 (Life Tech, cat. no. A-11005)
DAPI (Sigma-Aldrich, cat. no. D8417-5MG)
Cy3 goat anti-mouse (Jackson, cat. no. 115-166-003)
Saponin (Sigma, cat. no. 47036-50G-F)
PBS (Sigma, cat. no. D8537)
Nuclease Free Tris-EDTA buffer, pH 7.0 or 8.0 (Life Tech, cat. no. AM9850G or AM9855G)
T-75 flasks (Fisher Scientific)
Liquid nitrogen
Ethanol, 70% (vol/vol)
Milli-Q water
Borosilicate glass (World Precision Instruments, Inc., cat. no. PG52151-4) - CM Specific
EQUIPMENT
Real-Time PCR Machine (Life Tech, 7500 Fast or equivalent system)
Benchtop centrifuge (e.g., Eppendorf 5415R or equivalent system)
Plate Reader (Tecan, Safire or equivalent system)
Laminar flow hood (Labconco or equivalent system)
Lab-Tek eight-well chambered cover glass (Thermo Scientific, 155411 or similar cultureware)
Cell culture multiwall plates, 6 wells and 12 wells (Greiner Bio-One, 657160 and 665102 or simuilar cultureware)
Centrifuge (e.g., Beckman Coulter Avanti J-30I or equivalent system)
Liquid aspirator setup
Nucleofector™ 2b Device (Lonza)
Flow Cytometer (e.g., Accuri C6, BD or equivalent system)
Flow Activated Cell Sorter (e.g., FACSaria III, BD or equivalent system)
0.22-μm filter (Nalgene or equivalent system)
Inverted Fluorescence Microscope (e.g., Zeiss Axiovision or equivalent system)
P-87 Flaming/Brown puller (Sutter Instrument Company, Novato, CA or equivalent system) - CM Specific
EPC 7 amplifier (List Medical, Darmstadt, Germany or equivalent system) - CM Specific
Origin 6.0 software (Microcal Inc., Northampton, MA or equivalent software) - CM Specific
REAGENT SETUP
Oligonucleotide Solutions
Resuspend oligonucleotides (MBs, target and mutant oligonucleotides designed in steps 1–10) in a nuclease free buffer to 100 μM at the recommended pH (either 7.0 or 8.0 depending on additional molecules). Make several 10 μL aliquots and freeze at −20 °C to maximize their performance and stability (stored for up to 6 months).
Cytokine Culture Media
Add BMP4, Activin A, and FGF2 to DMEM/F12 cell culture media to achieve final concentrations of 10 ng/ml, 3 ng/ml and 5 ng/ml respectively.
END-2 Conditioned Culture Media
Add DMEM/F12 media to confluent mouse endodermal cell line END-2 cells for 1 day. Remove and sterile filter the media.
Nucleofector Solution
The nucleofector solution kit contains two liquids, the solution and the supplement. Store them separately at 4°C where they are stable for 12–18 months according to manufacturer’s specifications. Once the supplement is added to the solution it is stable for 2 weeks in our hands.
Action Potential Measurement Tips- (CM Specific)
Fabricate glass microelectrodes from borosilicate glass and pull using a P-87 Flaming/Brown puller or equivalent system. The tip resistance of the microelectrode should be 40–80 MΩ when filled with a 3 mol/L KCl solution.
EQUIPMENT SETUP
EPC 7 amplifier (or equivalent system, CM Specific)
Adjust the junction potential between the microelectrode solution and the bath solution to zero and compensate for the microelectrodes’ capacitance. Filter and digitize signals at 10 kHz.
PROCEDURE
Cell-specific-MB Design
Timing- 10 days
Step 1: Identify genes highly expressed in target cells based on literature search, experience, and/or prior results. For example, for isolating CMs, cardiac troponin T (cTNT) and α/β myosin heavy chain (MYH6/7) were chosen. Check if the genes identified are also highly expressed in other cell types in the mixed cell population during PSC differentiation; high mRNA expression in other cell types could lead to an inability to separate target cells from mixed cell population.
Step 2: Confirmation of high gene expression levels using RT-PCR. Prepare total RNA from at least 100,000 cells from the specific cell population using the RNeasy mini plus kit according to the manufacturer’s instructions.
Step 3: Convert the RNA into cDNA using the iScript cDNA synthesis kit from BioRad or equivalent system.
Step 4: Perform either Taqman or SYBR Green real-time PCR for each sample in multiplexed reactions performed in triplicate using a 7500 Fast Real-Time PCR system or equivalent. Run all annealing steps at 60°C.
Step 5: Normalize all target genes to GAPDH. Calculate differences in CT values (ΔCT = CT gene of interest − CT GAPDH in experimental samples) for each target mRNA by subtracting the mean value of GAPDH (relative expression = 2−ΔCT). Proceed with the mRNA for which the highest ratios between the target gene and GAPDH were obtained.
Step 6: Prediction of the secondary structure of the target mRNA. Enter the sequence into a secondary structure prediction program (e.g., mFOLD) to predict single stranded regions in the target mRNA.
-
Step 7: Designing the MB loop region. Identify a sequence of 15–30 bases of the target mRNA predicted to be single stranded. Select a region with 35% – 65% GC content, and design a complementary sequence with a predicted melting temperature (Tm) of 60°C in 150 mM NaCl when hybridizes to the target mRNA (the prediction of melting temperature Tm could be performed using the online tool ‘HyTher’, available at http://ozone2.chem.wayne.edu/Hyther/hythermenu.html). This sequence (the reverse complement of the mRNA target sequence) will form the loop of the MB and ensure the MBs will be able to hybridize to the target sequence.
TROUBLESHOOTING
If the positive control cells are from a different species than that of target cells, MBs should be designed to target mRNA sequences that are identical in both species (such as mouse and human). If the target sequence in the gene of interest is not identical for both species, positive control cells should be generated to highly express the target gene by transfecting a cell line unrelated to the target cells with a plasmid encoding the target gene.
-
Step 8: Add a stem to the loop sequence by appending complementary arms of five bases long to the 3′ and 5′ end of the loop sequence respectively. MBs with a total length of more than 40 bases may form a secondary structure therefore should be avoided. It may be desirable to add only one arm with a 5-base sequence complementary to the loop sequence on the opposing end in order to create a shorter MB (shared-stem MB). A 5-base stem with a GC content of ~60% should be strong enough to generate a stem-loop hairpin structure with a Tm of ~50°C, which can be determined using the online tool ‘DNA mfold’ (available at http://www.bioinfo.rpi.edu/applications/mfold/old/dna/).
Critical Step: Do not use Guanine for the first three bases at the 5′ end due to its tendency to quench fluorophores.
TROUBLESHOOTING
Step 9: Use the NCBI BLAST search tool to identify other mRNAs that the MB may hybridize to in the cell type of interest. To do this, use the MB sequence as input. The closest match should be the target mRNA sequence identified in Step 7, and the second closest match should have a mismatch of at least four base pairs compared with the mRNA target sequence.
Step 10: Design a mismatched target sequence. Using the results of the NCBI BLAST search from Step 9, the second closest sequence could be used as the “mismatched sequence” if it contains 4–6 bases not complementary to the beacon loop sequence. If the second closest sequence from the BLAST search has more than 6 bases not complementary to the beacon loop sequence, design a new “mismatched sequence” by mutating 4 randomly selected bases in the target sequence..
-
Step 11: To form the MB, select a dye-quencher pair (as discussed in the Experimental Design section) and conjugate the dye and quencher to the 5′ and 3′ ends of the MB sequence respectively. To reduce background signal, attach the quencher to 3′ end to ensure that all MBs will have a quencher even if the fluorophore is not conjugated to the MB due to a synthesis error. To determine MB performance, the synthetic target oligonucleotide (usually single strand DNA) with the same sequence as target mRNA, and mutant (mismatched) oligonucleotide should also be obtained for MB characterization in solution. Usually MBs with DNA backbones are sufficient for cell isolation. It may be necessary to design and test several (three to five) target-cell specific MBs which can be ordered simultaneously to reduce wait times.
TROUBLESHOOTING
-
Step 12: Re-suspend all oligonucleotides in nuclease free, 0.1x TE, pH 7.0 buffer at 100 uM to make stock solutions.
Critical: Avoid freeze-thaw cycles to preserve stability and function of MBs.
Pause point: MB can be stored at −20°C or −80°C for up to 6 months after synthesis.
MB Validation in Solution
Timing- 2 hours
-
Step 13: Use a buffer mimicking cytosolic conditions (139 mM K, 12 mM Na, 4 mM Cl, etc) and make a 500 μL aliquot of 1 μM cell-specific MBs. Use the same buffer in separate tubes to dilute the target oligonucleotide and mismatched oligonucleotide to 1.5 μM concentration. Make serial dilutions of the target and mismatched oligonucleotides respectively to establish dose dependence.
TROUBLESHOOTING
Step 14: Add 25 μL of MB solution to each well of a 384-well black-bottomed plate, then add 25 μL of each concentration of the solutions containing target oligonucleotide or mismatched oligonucleotide to their designated wells. Include one blank well and one well with DNAse 1 as negative and positive controls, respectively. Incubate at 37°C for 5 minutes to allow the solutions to equilibrate.
-
Step 15: Analyzing the fluorescence intensity of MBs using a microplate reader. Use a microplate reader to determine the fluorescence intensity of each well. From this establish a dose dependence curve using the serial dilutions of the target oligonucleotide. The MB signal at the highest target oligonucleotide concentration should generally be between 5 and 30 times higher than the background signal quantified in the negative control experiment in which the signal level of MBs without any target is measured. The MB signal in the well with the highest concentration of mismatched oligonucleotide should be no more than 1.5 times the background signal. Candidate MBs that do not meet these conditions should be disregarded due to lack of specificity and low S/N. Select MBs that satisfy these conditions for further testing.
TROUBLESHOOTING
MB delivery testing with cells
Timing- 1 day
Critical: Nucleofection is an electroporation method for transferring nucleic acids such as DNA and RNA molecules into cells. Under typical nucleofection conditions, most MBs are delivered into cell cytoplasm, with a small amount of MBs in the cell nucleus. Since the target mRNAs are only in the cell cytoplasm, MBs delivered into the cell nucleus should not hybridize to any target nor result in specific fluoresce signal.
Step 16: Plate cells for delivery test (e.g., mouse embryonic fibroblasts) at 50–60% confluency in DMEM medium containing 10% vol/vol FBS and culture them overnight at 37°C.
-
Step 17: Add the delivery control MB (see Reagents section) to nucleofection solution to obtain a MB concentration of 500 nM.
Critical Step: To check the efficiency of MB delivery via nucleofection and select optimal delivery conditions, a delivery control MB (Figure 1C) is designed which has a non-specific, ‘random’ sequence with two FAM dyes conjugated to both 5′ and 3′ ends, allowing the probe to fluoresce without any target when delivered into living cells using nucleofection.
Step 18: Detach and suspend one million cells (from Step 16) per condition, wash with PBS.
-
Step 19: Centrifuge the cells at 90g for 10 minutes. The centrifugation should be gentle to avoid damaging the cells. Remove the supernatant, allowing cell pellet to remain undisturbed.
Critical Step: Ensure that all PBS is removed from the solution to avoid diluting the electroporation solution.
Step 20: Resuspend the cell pellet in the nucleofection solution that contains delivery control MBs (from Step 17).
-
Step 21: After MB delivery, gently centrifuge the cells at 90g for 10 minutes then aspirate the media. Resuspend the cells in PBS and analyze the cells using any standard flow cytometer to determine MB delivery efficiency. For flow cytometry assays, first run an untransfected sample. Use this non-fluorescent sample to set the lower limits of gates that can be used to detect MB positive cells.
TROUBLESHOOTING
Step 22: Optimization of nucleofection parameters. Perform nucleofection using a variety of programs to identify the optimal program with the highest uniform delivery efficiency. Changes in the electroporation program do affect cell viability, so great care must be taken to choose a program that does not kill a large number of cells. Immediately after nucleofection, gently pipette cells from the cuvette into cell culture medium and keep them there for at least 10 minutes.
MB specificity testing in positive and negative control cells respectively
Timing- 1 day
Step 23: Delivery of cell-specific MBs into positive control cells using nucleofection. Use the optimized nucleofection parameters and the procedure described in Steps 16–21 to deliver the cell-specific MBs designed in Steps 1–11 and tested in Steps 13–15 into positive control cells (e.g., HL-1 cells for CM isolation)
Step 24. Delivery of negative control MBs to positive control cells. Repeat Step 23 to deliver the negative control MB (see Reagents section) into a separate aliquot of positive control cells. As a further negative control, deliver the mutant negative control MB (Figure 1D) into a separate aliquot of positive control cells, as described in Step 23.
Step 25: Delivery of cell-specific MBs into other cell types using nucleofection. Deliver the cell-specific MBs into any other cell types you are using as controls (for example, smooth muscle cells, aortic endothelial cells and cardiac fibroblasts typically present in the differentiating PSCs). Use the optimized nucleofection parameters and the procedure described in Steps 16–22
-
Step 26: Analyze all cells after MB delivery using flow cytometry. After MB delivery, incubate cells for 10 min at 37°C, gently centrifuge the cells at 90g for 10 minutes, and then aspirate the media. Resuspend the cells in PBS and analyze the cells using any standard flow cytometer. Use negative control cells to set the gates as in Step 21.
Critical: This step should result in a strong fluorescent signal from cell-specific MBs in positive control cells (Step 23); low (typically ≤3%) fluorescence signal from the negative control (mutant) MBs (Step 24) (Figure 3C, 3D); and low false positive signal resulting from non-specific opening of the random MBs in other cell types (Step 25) (Figure 3B).
Critical Step: The flow cytometry analysis must be performed within 2 hours of MB delivery into the cells. Several groups have shown that non-specific MB signal may increase quickly ~2 hours after MB delivery due to MB degradation, resulting in a high level of false positives in flow cytometry analysis.
TROUBLESHOOTING
Step 27: Selection of the final cell-specific MBs. Repeat Steps 23–26 to select the final MB designs which result in high fluoresce signal in positive control cells (such as HL-1) and very low background fluoresce signal in negative control cells (such as non-CM cells). These MBs will be used for further study for the isolation of target cells. In most of the cases, one cell-specific MB that has satisfactory performance (high positive signal and low background signal) will be sufficient.
Figure 3. Example of control experiments required to demonstrate beacon specificity.

(a)Representative data using a delivery control MB in mouse ESCs (a schematic depicting a delivery control beacon is shown on the right). The random sequence (red nucleotides) delivery beacon with two dyes is used to demonstrate high delivery efficiency and uniformity. All dot plots depict a representative flow cytometry assay where 20,000 events were collected using log scales for both fluorescence and FSC measurements. Each condition was repeated three times to generate the SD shown. (b) Representative flow cytometry data using a negative control MB in mouse HL-1 cells (A schematic depicting a negative control beacon is shown on the right). Random negative control beacons are delivered into target cells to demonstrate that beacon signals do not arise from MB degradation or non-specific intracellular interactions. (c) Representative flow cytometry data using a mutant beacon as a negative control delivered into HL-1 cells to confirm MB signal specificity. A schematic depicting the specific negative control MB with four mutated base pairs in red is shown on the right. (d) Representative flow cytometry data generated using a MB targeting the Myh6 mRNA delivered into negative control mouse ESCs to demonstrate MB specificity. When the MBs are delivered into control cells a low fluorescence signal is generated, demonstrating MB specificity in a cellular environment. A schematic depicting a MB designed to hybridize to a specific target sequence (green nucleotides) is shown.
Stem Cell Differentiation
Critical: Any differentiation protocol may be used for the selection/enrichment of cell type of interest. The following steps represent the method used for the isolation of CMs as an example. Alternatively, prepare CMs differentiated from hPSCs using any of the previously validated protocols 5,29–31
Timing- ~2–3 weeks
Step 28: Culture undifferentiated hPSCs on either mitotically inactivated MEF (STO) cells in DMEM/F12 supplemented with 20% serum replacement, 1% L-glutamine, 1% nonessential amino acids, 100 mM β-mercaptoethanol, and 4ng/ml basic fibroblast growth factor or feeder free culture systems 32. Change the medium every day and transfer hPSCs to new feeder cells every 5 to 7 days. Culture cells for at least one passage after thaw before beginning differentiation.
Step 29: Coating of plates with Matrigel. Add 3 mL Matrigel solution to each 10 cm petri dish. Warm for at least 1 hour at room temperature (21–23 °C), then aspirate liquid just before use.
Step 30: Dissociation of hPSCs from feeder cell culture. When colonies are 80% confluent, remove hPSC culture medium and rinse with DMEM/F-12 medium. Add 3 mL Dispase (1 mg/ml) to hPSC culture and incubate for about 20 min at 37 °C to dissociate them into small clusters (10–20 cells). There may be some batch to batch variation for dissociation time thus frequent microscopic examination ensures successful dissociation especially after 15 min from the addition with Dispase. Once dissociation is completed, add 7ml of DMEM/F12 medium to cell suspension and transfer to a conical tube. Centrifuge the cell suspension at 300 g for 5 minutes to harvest the cells. Repeat this procedure twice to make sure that Dispase is completely removed,
Step 31: Start of 2D hPSC differentiation. Resuspend cells in mTeSR 1 media and transfer cells onto Matrigel-coated plates from Step 29. Culture the cells for 24–48 hrs in mTeSR® media to allow them to expand.
Step 32: Induction of mesodermal differentiation. Replace media with cytokine media and incubate cells for 2 days.
Step 33: Remove the cytokine media and add END-2 conditioned media, incubate cells for 4 days.
Step 34: Supplementation with Isoproterenol to produce mature CMs. If desired, remove conditioned media and incubate cells with mTeSR® media containing isoproterenol (10 μM), a β–adrenergic receptor agonist, for at least 4 days to generate spontaneously beating CMs.
Deliver MBs into differentiating hPSCs for the isolation of target cells
Timing- 6 hours
Step 35: Gentle dissociation of intact differentiated cells using Accutase. Rinse one million cells from Steps 28–34 with PBS, then incubate them for 20 minutes with 2 ml Accutase at 37°C.
Step 36: Add 6 ml DMEM/F12 containing 10% vol/vol FBS to neutralize the Accutase before centrifuging cells at 90g for 10 minutes and aspirating the supernatant.
Step 37: Resuspend cells in nucleofection solution containing 500 nM of delivery control MBs.
Step 38: Use the optimal nucleofection program identified to deliver MBs into the mixed cell population. Immediately after nucleofection, gently pipette cells from the cuvette into the DMEM/F12 medium containing 10% vol/vol FBS and incubate cells for 30 minutes at 37°C.
-
Step 39: Analysis of signal from delivery control MB in cells using a flow cytometer. Gently centrifuge the cells at 90g for 10 minutes, then aspirate the media. Resuspend the pellet (cells) in PBS and analyze the cells using any standard flow cytometer. Cells in the mixed cell population should show uniform, high fluorescence signal. Use negative control cells to set the gates in flow cytometry assay as in Step 22.
TROUBLESHOOTING
Step 40: Delivery of cell-specific MBs into differentiating hPSCs using nucleofection. Detach one million cells from differentiating hPSCs generated from Steps 28–34 using the procedure described in Steps 35–36 and resuspend the cells in the nucleofection solution containing 500 nM of cell-specific MBs. Use the optimal nucleofection program identified to deliver MBs into the mixed cell population. Use a gentle centrifugation of 90g for 10 minutes to avoid damaging the cells. Immediately after nucleofection, gently pipette cells from the cuvette into at least 1 mL of DMEM/F12 medium containing 10% vol/vol FBS and incubate the cells for 10 minutes at 37°C.
-
Step 41: Analysis of cells using a flow cytometer. Gently centrifuge the cells at 90g for 10 minutes then aspirate the media. Resuspend the cells in PBS and analyze the cells using any standard flow cytometer. Use negative control cells to set the gates in flow cytometry assay as in Step 21.
TROUBLESHOOTING
Step 42: Sorting of cells using FACS. Gently centrifuge the cells at 90g for 10 minutes, then aspirate the media. Resuspend in PBS and sort the MB positive cells using a cell sorter such as the FACS Aria II or a Jazz system. Use slightly more stringent gating conditions than that in the previous analysis (Step 41) to ensure high purity of isolated target cells. Gating strategies can be altered to provide the desired purity level by using more stringent conditions. Plate cells on appropriate dishes (MEA plates, 6 well dishes, etc) in the DMEM/F12 media containing 10% vol/vol FBS and incubate for 24–48 hours for subsequent target cell characterization assays.
Characterize target cells purified with cell-specific MBs
Timing- 2 days
Step 43: Characterize cells as appropriate for your particular experiment. Different assays may be used to confirm the function of the cell type of interest. We use the following to confirm the functionality of MB-sorted CMs and provide them here as an example. Option A describes how to characterize cells via RT-PCR. Option B describes how to characterize cells via immunocytochemistry. Option C describes how to characterize MB-sorted cells by intracellular flow cytometry based on the expression of protein markers. Option D describes how to measure action potential in purified CMs. Other functional assays, such as that for glucose sensitivity in insulin producing cells or P450 activity in hepatocytes may be appropriate in place of option D depending on the type of cells being isolated.
Option A: RT-PCR of MB-sorted cells
Prepare total RNA with the RNeasy mini plus kit according to the manufacturer’s instructions from at least 100,000 cells from the previous step.
Convert the RNA into cDNA using the iScript cDNA synthesis kit from BioRad.
Perform either Taqman or SYBR Green real-time PCR using a 7500 Fast Real-Time PCR system. Run all annealing steps at 60°C.
Normalize all target genes to GAPDH in multiplexed reactions performed in triplicate. Calculate differences in CT values (ΔCT = CT gene of interest − CT GAPDH in experimental samples) for each target mRNA by subtracting the mean value of GAPDH (relative expression = 2−ΔCT). It is recommended to test at least three genes from each potential lineage (e.g., CM, fibroblast, neurons etc. for isolating CMs). The specific group of genes may vary depending on the MB target gene.
Option B: Immunocytochemistry of MB-sorted cells
Rinse the MB positive cells with PBS then fix them using 4% paraformaldehyde for 10 min at room temperature (21–23 °C).
Wash cells twice with PBS and permeabilize them with 0.1 or 0.5% Triton X-100 in PBS for 10 min.
Block with 1% BSA in PBS for 60 min at room temperature and then incubate cells with antibodies specific for the protein marker of the target cell at 4°C overnight for immunostaining. For example, for MB-sorted CMs, one may use anti-ACTN2 (1:100), mouse anti-TNNT2 (1:100), or rabbit anti-cTnI (1:100).
Wash the cells three times with 1% Tween 20 in PBS and incubate with anti-mouse IgG– Alexa Fluor 594 (1:1000) or anti-rabbit IgG–Alexa Fluor 488 (1:1000) in PBS for 1 h at room temperature.
Counterstain the nuclei with DAPI (300 nM).
-
Visualize the samples using a fluorescence microscope or confocal laser-scanning microscope.
Critical Step: MB fluorescence should be reduced to background levels 24 hours after delivery into the cells thus should not interfere with immunocytochemistry.
Option C: Characterization of MB-sorted cells by intracellular flow cytometry based on the expression of protein marker
Rinse the MB positive cells with PBS, incubate them with 2 ml Trypsin at 37°C for 5 minutes.
Wash the MB positive cells three times with PBS and centrifuge at 600g for 5 min at room temperature.
Fix the cells with 2% (wt/vol) PFA for 20 min at room temperature.
Wash the cells two times with PBS and centrifuge at 600g for 5 min at room temperature.
Gently resuspend the cell pellets to the permeabilization buffer containing 5% (vol/vol) FBS and 0.5% (vol/vol) saponin, and incubate for 20 min for permeabilization.
Prepare the primary antibodies at appropriate concentrations in staining buffer with 0.2% saponin, For MB-sorted CMs, the protein marker used is cTnT, but the following steps are valid for many other protein markers also. If using cTnT, use mouse anti-cTnT, in staining buffer with 0.2% saponin, and add them in the tube with a final antibody dilution of 1:100 for cTnT. Incubate overnight at 4°C.
Wash the cells three times with staining buffer and centrifuge at 600g for 5 min at room temperature.
-
Resuspend the cell pellets in staining buffer with saponin with a 1:200 dilution of secondary antibody, Cy3 goat anti-mouse (Jackson). Incubate the suspensions for 1 hr min at room temperature.
Critical Step: Maintain the samples in the dark by covering them with aluminum foil after the addition of secondary antibody.
Wash the cells three times with staining buffer and centrifuge at 600g for 5 min at room temperature.
Resuspend the cells in 100 μl of PBS with 2% (vol/vol) FBS and quantify cTnT positive cells by flow cytometry.
Option D: Measure action potential in purified CMs
Transfer the MB positive cells onto 0.1% fibronectin-coated glass bottom microwell dishes and culture them for 7 days.
Mount the 35-mm dishes on an inverted microscope and heat the stage to 37°C with a heating/cooling bath temperature controller. Perfuse the cells with Tyrode’s solution.
Record intracellular membrane potential using the EPC 7 amplifier in current clamp mode at 37 ± 0.5°C.
Impale individual cells with the sharp microelectrodes and record the action potential signals.
Analyze action potentials using the Origin 6.0 software.
TROUBLESHOOTING
Troubleshooting advice can be found in Table 1.
Table 1.
Troubleshooting
| Step | Problem | Possible Reason | Solutions |
|---|---|---|---|
| 7, 8, 13 | MB does not fluoresce in the presence of the target oligonucleotide | The MB stem-loop structure is too stable The MB loop sequence is too short or too AT rich |
The stem should be weakened by decreasing its length and/or GC content The loop length and/or GC content of the MB should be increased |
| 8, 13 | MB fluoresces in the presence of a mismatch oligonucleotide | The hybridization of the MB with the mismatch oligonucleotide is more energetically favorable than the formation of the stem-loop MB structure despite the energy penalties of the mismatches | The stem should be strengthened by increase its GC content and/or length |
| 11 | MB fluorescence cannot be detected in GFP+ cells | The GFP fluorescence is detectable at other wavelengths, and it is obscuring beacon signals because of its intensity | Instead of Cy3 or FAM dyes, switch to a dye with an even more spectrally distinct emission wavelength such as Cy5 Ensure that your analysis method is using an appropriate excitation laser that avoids exciting GFP and maximizes the light absorbed by the MB dye |
| 15 | MB fluorescence cannot initially be detected in positive wells | The MBs have not bound to their targets in the solution | Allow more time for the beacon and target solutions to mix, or mix the solutions using a shake function on the microplate reader, or mix the solutions more vigorously while pipetting |
| 21 | Fewer than 80% of the cells are positive for delivery MB signal | Cell membranes are not porous enough for a long enough period of time for MBs to enter the cell | Continue to test other electroporation parameters to increase the transfection efficiency. If transfection cannot be improved, switch to a more easily manipulated cell type |
| 26 | The negative control cells are generating a false positive signal with MB targeted towards the gene of interest | The control cells actually do express the gene of interest (verify by RT-PCR) The gene is not expressed but a non-specific interaction in the control cells is causing MB to fluoresce (verify by RT-PCR) The gene is not expressed but a non-specific interaction in the control cells is causing MB to fluoresce in the cytoplasm (verify by RT-PCR and microscopy) |
Use a different cell type as a control Check the cells using a microscope to observe subcellular localization. If MBs are apparent in the nucleus, try reducing the time between electroporation and analysis Change the mRNA target region to avoid non-specific interactions. This will require the development of a new MB |
| 26 | The positive control cells do not give a strong MB signal | The MB targeting sequence may be inaccessible | Redesign MB to target a different region of the gene to have better accessibility |
| 39 | Fewer than 80% of the differentiated cells are positive for delivery MB signal | Cell membranes are not porous enough for a long enough period of time for MBs to enter the cell | Continue to test other electroporation parameters to increase the transfection efficiency. If transfection cannot be improved, normalize all values to the transfection percentage. Since the dyes on the delivery control and cell- specific beacon are spectrally distinct they may be delivered concurrently for easier analysis. |
| 41 | The positive control cells do not give a strong cell specific MB signal | The MB targeting sequence may be inaccessible | Redesign MB to target a different region of the gene to have better accessibility |
TIMING
Steps 1–12: 10 days
Steps 13–15: 2 hours
Steps 16–22: 1 day
Steps 23–27: 1 day
Steps 28–34: 2–3 weeks
Steps 35–42: 6 hours
Step 43: 2–7 days
ANTICIPATED RESULTS
We have used the protocol described herein to enrich and isolate homogeneous populations of target cells from PSC differentiation culture with high throughout and high (>95%) purity21. Well-designed MBs targeting mRNAs specific for the cell type of interest should show high signal-to-background ratio (Figure 4A), whereas an MB targeting a poorly selected sequence could result in high non-specific signal (Figure 4A). It is also necessary to perform cellular tests of multiple MBs with positive control cells, even for well designed MBs, to ensure that at least one MB gives high fluorescence signal level, since not all MBs are able to access the target mRNA sequence (Figure 4B). For example, in our published studies21, we delivered four MBs into positive control cells (HL-1 CM), and found that two MBs designed to target the MYH6/7 mRNA displayed high fluorescence signal, whereas two MBs targeting the TNNT2 mRNA showed relatively low signal. We therefore selected the MHC1 MB that gave the highest signal level for subsequent CM isolation studies. Optimally designed MBs will give high fluorescence signal in positive control cells (Figure 4B) and very low signal in all other cell types, for example smooth muscle cells and cardiac fibroblast cells chosen to represent cells that may be present in differentiating PSCs for CM isolation (Figure 4C). It may also be necessary to use negative control MBs to confirm the signal specificity in positive control cells as shown in Figures 3B, 3C. As shown in Figure 4D, when MHC-1 MBs were delivered into cells in the differentiating culture of mouse ESCs, a large amount of cells showed a high level of MB signal, and FACS sorting isolated 49.2% cells as mouse CMs. Since PSC-derived cells may express high, medium, or low levels of the target gene, the FACS results may be less clear-cut than that in the positive control cells, as reflected by the lack of distinct populations in the FACS results shown in Figure 4D. However, the percentage of FACS-isolated cells based on MB signal should correlate well with differentiation efficiency, i.e., the expected proportion of target cells in the differentiating culture. This can be used to fine-tune the FACS protocol and obtain an initial confirmation of appropriate MB function.
Figure 4. Anticipated results of MB validation, optimization and target cell type characterization.

(a) Examples of MB validation by fluorescence measurement in solution where MHC1 beacon displays a normal response to target and mismatch oligonucleotides, and the GAPDH beacon shows a lack of specificity due to high signal level in the presence of mismatched oligonucleotide target. Roughly 10% of MB designs fail to adequately discriminate between target and mismatched oligonucleotides. (b) Delivery of MBs into positive control cells (e.g., HL-1 CM) resulted in different signal levels. For CM isolation, two MBs targeting the MYH6/7 mRNA displayed high fluorescence signal whereas two MBs targeting the TNNT2 mRNA showed relatively low signal. MB design MHC1 gave the highest signal level, therefore should be selected for subsequent use. Roughly 50–80% of MBs fail to adequately discriminate between positive and negative control cells. Data in this histogram represent percentage of MB positive cells based on flow cytometry, error bars represent one SD. (c) The MHC-1 MB was subsequently tested in non-cardiac cell types including cultured smooth muscle cells and cardiac fibroblasts that may be present in a differentiating culture of PSCs. A very low percentage of these cells had high fluorescence intensity, which is expected based on the results shown in Figure 3. All dot plots depict a representative flow cytometry assay where 20,000 events were collected using log scales for both fluorescence and FSC measurements. Each condition was repeated three times to generate the SD shown. (d) The MHC-1 MB was tested in cells derived from mouse ESCs. A high percentage of these cells showed a significantly increased beacon signal, which correlated well with the proportion of TNNT2 positive cells. However, PSC-derived cells may express high, medium, or low levels of the target gene, and the results from FACS assays are likely to be less clear-cut than those seen in the control cells. (e) Results of qRT-PCR of the mixed cell population (PRE) from differentiating mouse ESCs and MB positive cells (POST) after MB based sorting displaying increased expression of CM-specific genes (Tnnt2 and Myh7) and decreased expression of gene markers (Acta2 and Sox17) from alternative lineages. Roughly 75% of MBs validated in previous steps are useful for sorting stem cell derived CMs. Data in these histogram represent comparative RT-PCR with all genes normalized to GAPDH via the ddCT method described in Step 5, error bars represent one SD, t-Tests were carried out to determine significance. (f) Immunocytochemistry results showing >98% of the MB-sorted cells from differentiating mouse ESCs are positive for cardiac marker Actn2. The lower image is an enlargement of the upper image. Scale bars, 20 μm.
After isolating MB-positive cells from the mixed cell population using FACS, it is necessary to confirm the characteristics of the target cells using qRT-PCR, Immunocytochemistry, action potential measurements, or other functional assays. It is expected that the expression of cell-specific genes (e.g., TNNT2 and MYH6/7 for CMs) increase in MB-positive cells compared with that of the mixed cell population (Figure 4E). The expression of gene markers from alternative lineages (e.g., Acta2 and Sox17) should show the reverse trend (Figure 4E). Further, it is anticipated that the vast majority (>98%) of the MB-sorted cells are positive for the specific protein marker, as is the case for MB-sorted CMs that highly expressed the cardiac marker Actn2, as revealed by immunocytochemistry (Figure 4F).
Table 2.
Validated molecular beacon designs
| Target Gene | Species | Sequence |
|---|---|---|
| Myosin Heavy Chain (cardiac) | Mouse, Human | CCTCCATCTTCTTCTTCACGGAGG |
| Iroquois Homeobox 4 | Mouse | CAGGCAGAGAGTAGAAAGCAGATGCCTG |
| Oct4 | Mouse | CGCAGTCCAGGTTCTCTTGTCTCTGCG |
| Random | No Reactivity | ACGACGCGACAAGCGCACCGATACGTCGT |
Summary.
Molecular beacons are dual-labeled oligonucleotides that fluoresce in the presence of complementary mRNA. This protocol describes how to use them to purify specific cell populations by targeting mRNAs highly expressed only in the desired cell type.
Acknowledgments
This work was supported in part by the National Heart Lung and Blood Institute of NIH as a Program of Excellence in Nanotechnology (HHSN268201000043C), by the NIH grant DP3DK094346, the NSF STC grant CBET-0939511 and ACTSI pilot grant (PHS grant UL1 RR025008 from the CTSA program, NIH, and NCRR). K.B. is a recipient of American Heart Association postdoctoral fellowship.
Footnotes
AUTHOR CONTRIBUTIONS
All authors contributed equally to this work. B.W designed and validated the MBs and K.B differentiated stem cells into cardiac cells and carried out their subsequent characterization. All authors discussed the results and implications and commented on the manuscript at all stages.
COMPETING FINANCIAL INTERESTS
The authors have no competing financial interests.
References
- 1.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 2.Park IH, et al. Disease-Specific Induced Pluripotent Stem Cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sinnecker D, et al. Modeling Long-QT Syndromes with iPS Cells. Journal of cardiovascular translational research. 2013;6:31–36. doi: 10.1007/s12265-012-9416-1. [DOI] [PubMed] [Google Scholar]
- 4.Kim C, et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature. 2013;494:105–110. doi: 10.1038/nature11799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laflamme MA, et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotech. 2007;25:1015–1024. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- 6.Dubois NC, et al. SIRPA is a specific cell-surface marker for isolating cardiomyocytes derived from human pluripotent stem cells. Nat Biotech. 2011;29:1011–1018. doi: 10.1038/nbt.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uosaki H, et al. Efficient and Scalable Purification of Cardiomyocytes from Human Embryonic and Induced Pluripotent Stem Cells by VCAM1 Surface Expression. PLoS ONE. 2011;6:e23657. doi: 10.1371/journal.pone.0023657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halbach M, et al. Electrophysiological integration and action potential properties of transplanted cardiomyocytes derived from induced pluripotent stem cells. Cardiovasc Res. 2013;100:432–440. doi: 10.1093/cvr/cvt213. [DOI] [PubMed] [Google Scholar]
- 9.Huber I, et al. Identification and selection of cardiomyocytes during human embryonic stem cell differentiation. Faseb J. 2007;21:2551–2563. doi: 10.1096/fj.05-5711com. [DOI] [PubMed] [Google Scholar]
- 10.Moore JC, et al. A P19Cl6 GFP reporter line to quantify cardiomyocyte differentiation of stem cells. Int J Dev Biol. 2004;48:47–55. doi: 10.1387/ijdb.15005574. [DOI] [PubMed] [Google Scholar]
- 11.Hattori F, et al. Nongenetic method for purifying stem cell-derived cardiomyocytes. Nat Methods. 2010;7:61–66. doi: 10.1038/nmeth.1403. [DOI] [PubMed] [Google Scholar]
- 12.Tohyama S, et al. Distinct Metabolic Flow Enables Large-Scale Purification of Mouse and Human Pluripotent Stem Cell-Derived Cardiomyocytes. Cell Stem Cell. 2013;12:127–137. doi: 10.1016/j.stem.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 13.Xu C, Police S, Rao N, Carpenter MK. Characterization and enrichment of cardiomyocytes derived from human embryonic stem cells. Circulation research. 2002;91:501–508. doi: 10.1161/01.res.0000035254.80718.91. [DOI] [PubMed] [Google Scholar]
- 14.Hattori F, et al. Nongenetic method for purifying stem cell-derived cardiomyocytes. Nat Meth. 2010;7:61–66. doi: 10.1038/nmeth.1403. [DOI] [PubMed] [Google Scholar]
- 15.Tyagi S, Kramer FR. Molecular beacons: probes that fluoresce upon hybridization. Nature biotechnology. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 16.Tsourkas A, Behlke MA, Bao G. Structure-function relationships of shared-stem and conventional molecular beacons. Nucleic Acids Res. 2002;30:4208–4215. doi: 10.1093/nar/gkf536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsourkas A, Behlke MA, Rose SD, Bao G. Hybridization kinetics and thermodynamics of molecular beacons. Nucleic Acids Res. 2003;31:1319–1330. doi: 10.1093/nar/gkg212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tyagi S. Imaging intracellular RNA distribution and dynamics in living cells. Nature Methods. 2009;6:331–338. doi: 10.1038/nmeth.1321. [DOI] [PubMed] [Google Scholar]
- 19.Rhee WJ, Santangelo PJ, Jo H, Bao G. Target accessibility and signal specificity in live-cell detection of BMP-4 mRNA using molecular beacons. Nucleic Acids Research. 2008;36:e30. doi: 10.1093/nar/gkn039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rhee WJ, Bao G. Simultaneous detection of mRNA and protein stem cell markers in live cells. BMC Biotechnology. 2009 Apr 2;9:30. doi: 10.1186/1472-6750-9-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ban K, et al. Purification of Cardiomyocytes From Differentiating Pluripotent Stem Cells Using Molecular Beacons That Target Cardiomyocyte-Specific mRNA. Circulation. 2013;128:1897–1909. doi: 10.1161/CIRCULATIONAHA.113.004228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen AK, Behlke MA, Tsourkas A. Efficient cytosolic delivery of molecular beacon conjugates and flow cytometric analysis of target RNA. Nucleic Acids Res. 2008;36:e69. doi: 10.1093/nar/gkn331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tyagi S, Alsmadi O. Imaging native beta-actin mRNA in motile fibroblasts. Biophys J. 2004;87:4153–4162. doi: 10.1529/biophysj.104.045153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bao G, Rhee WJ, Tsourkas A. Fluorescent Probes for Live-Cell RNA Detection. Annu Rev Biomed Eng. 2009;11:25–47. doi: 10.1146/annurev-bioeng-061008-124920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen AK, Behlke MA, Tsourkas A. Avoiding false-positive signals with nuclease-vulnerable molecular beacons in single living cells. Nucleic Acids Research. 2007;35:e105. doi: 10.1093/nar/gkm593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen AK, Rhee WJ, Bao G, Tsourkas A. Delivery of molecular beacons for live-cell imaging and analysis of RNA. Methods Mol Biol. 2011;714:159–174. doi: 10.1007/978-1-61779-005-8_10. [DOI] [PubMed] [Google Scholar]
- 27.Heyduk T, Heyduk E. Molecular beacons for detecting DNA binding proteins. Nat Biotech. 2002;20:171–176. doi: 10.1038/nbt0202-171. [DOI] [PubMed] [Google Scholar]
- 28.King FW, Liszewski W, Ritner C, Bernstein HS. High-Throughput Tracking of Pluripotent Human Embryonic Stem Cells with Dual Fluorescence Resonance Energy Transfer Molecular Beacons. Stem Cells Dev. 2011;20:475–484. doi: 10.1089/scd.2010.0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang L, et al. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature. 2008;453:524–528. doi: 10.1038/nature06894. [DOI] [PubMed] [Google Scholar]
- 30.Lian X, et al. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proceedings of the National Academy of Sciences. 2012;109:E1848–1857. doi: 10.1073/pnas.1200250109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boheler KR, et al. Differentiation of Pluripotent Embryonic Stem Cells Into Cardiomyocytes. Circulation research. 2002;91:189–201. doi: 10.1161/01.res.0000027865.61704.32. [DOI] [PubMed] [Google Scholar]
- 32.Xu C, et al. Efficient generation and cryopreservation of cardiomyocytes derived from human embryonic stem cells. Regenerative medicine. 2011;6:53–66. doi: 10.2217/rme.10.91. [DOI] [PMC free article] [PubMed] [Google Scholar]