

Abstract
Integration in a soft material of all molecular components necessary to generate storable fuels is an interesting target in supramolecular chemistry. The concept is inspired by the internal structure of photosynthetic organelles such as plant chloroplasts which co-localize molecules involved in light absorption, charge transport, and catalysis to create chemical bonds with light energy. We report here on the light-driven production of hydrogen inside a hydrogel scaffold built by the supramolecular self-assembly of a perylene monoimide amphiphile. The charged ribbons formed can electrostatically attract a nickel-based catalyst, and electrolyte screening promotes gelation. We found the emergent phenomenon that screening by the catalyst or the electrolytes led to two-dimensional crystallization of the chromophore assemblies and enhanced the electronic coupling among the molecules. Photocatalytic production of hydrogen is observed in the three-dimensional environment of the hydrogel scaffold and the material is easily placed on surfaces or in the pores of solid supports.
The development of soft materials that integrate all necessary molecular components to generate storable fuels in the presence of sunlight is an unexplored area of chemistry with potential impact in renewable energy. Such systems could have advantages over the use of large volumes of liquids, dispersions of expensive or toxic inorganic particles, or complex devices. The use of such soft materials with integrated functions and high water content is bioinspired by the internal structure of chloroplasts in plants. These photosynthetic organelles have evolved to co-localize within stacked lipid bilayers in their stroma the protein machinery which integrates light-absorption, charge transport, and the catalytic functions necessary to convert light energy into chemical bonds1,2.
Efforts to emulate natural photosynthetic systems over the past several decades have concentrated on the development of efficient catalysts for water oxidation and proton reduction3-7. In other recent work, catalysts have been coupled to light absorbing CdSe quantum dots8, Si microrods9, and organic dyes10,11 to create artificial photosynthetic systems. Also functional devices capable of performing water-splitting and fuel-generating reactions using earth-abundant resources have been demonstrated12. The development of bionspired soft materials that can be shaped into forms and integrate light-harvesting, charge transport, and catalytic functions to produce solar fuels is an obvious gap. This gap can be addressed through self-assembly strategies for materials in which a bottom-up approach fine tunes all functional aspects of a catalytic system13. Organic systems may have shorter lifetimes than their inorganic counterparts, but could have their own niche in sustainable energy given their soft matter nature and low energy requirements for production.
We report here on a strategy to create supramolecular hydrogels that integrate both light-absorbing chromophores and catalysts into a material for light-driven hydrogen (H2) production. Our work here is focused only on the supramolecular chemistry necessary to create a formable catalytic material and therefore does not explore its possible integration into a photocathode that would not require a sacrificial electron donor. We designed a charged amphiphilic chromophore with the capacity to self-assemble into supramolecular polymers via hydrophobic collapse. At sufficiently high concentrations and under electrostatic screening conditions, charged supramolecular polymers can easily produce a three-dimensional network that takes the form of a gel. These networks could be highly hydrated and host the soluble components necessary to produce the solar fuel. At the same time, much like natural photosynthetic antennae, supramolecular structures of conjugated molecules formed through π orbital overlap should have the capacity to absorb light, split excitons, and transport the charges to catalytic reaction centers. Despite the large body of work on the gelation and light harvesting abilities of conjugated molecules14, the use of π-conjugated gels for artificial photosynthetic applications has not been reported. Through molecular design, we demonstrate here a supramolecular catalytic system that localizes catalysts within chromophore gels using electrostatic attraction.
Results and Discussion
We synthesized a perylene monoimide (PMI) chromophore amphiphile (CA) by covalently functionalizing perylene monoanhydride with a five-carbon linker to a carboxylic acid (Fig. 1a). PMIs are useful visible-light absorbing chromophores because they are highly stable and easy to synthesize, although little is known about their self-assembly properties15-18. We then investigated the structures formed by this CA in water, where the hydrophobic collapse of its aromatic groups should drive self-assembly. Indeed cryogenic transmission electron microscopy (cryo-TEM) of the CA in water revealed high-aspect-ratio ribbons approximately 40 ± 7 nm wide and several microns long (Fig. 1b). Atomic force microscopy (AFM) also showed flat ribbons with an average height of 2.3 ± 0.4 nm (see Fig. 1c). The small-angle X-ray scattering curve of the supramolecular assembly in water has a slope of −2 in the low q region, indicating the existence of flat, ribbon-like nanostructures (Fig. 1d). We observed a scattering minimum that corresponds to a flat structure measuring 1 nm in thickness (Supplementary Fig. 1). This is the expected dimension of a ribbon core composed of interdigitated electron-rich perylene moieties (we expect the hydrated head groups on both faces of the nanostructure to not be visible in this experiment due to their similar scattering to bulk water). Based on this data, we conclude the ribbons consist of interdigitated bilayers of CA molecules (Fig. 1e). Interestingly, the peaks observed in the low q region indicate a lamellar spacing of 27 nm, which is likely due to electrostatic repulsion between these charged ribbons in solution (Supplementary Fig. 2)19.
Figure 1. Self-assembly of PMI CAs into supramolecular ribbons.
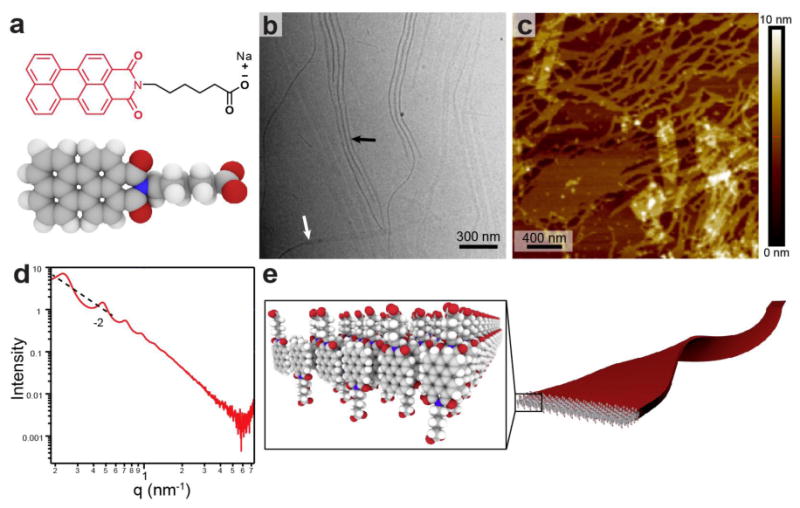
a, Molecular structure and space filling model of PMI-based CA, b, Cryo-TEM micrograph of CA solution (11.5 mM) reveals 40 nm wide supramolecular ribbons. Differences in contrast arise from imaging a ribbon either face-on (white arrow) or edge-on (black arrow). c,AFM image of a dilute CA solution (115 μM) dried on mica shows flat structures with a measured height of 2.3±0.4 nm. d,SAXS profile for aqueous CA solution (11.5 mM) exhibits a -2 slope in the low q region as well as scattering peaks corresponding to a 27.3 nm lamellar spacing between nanostructures. e, Schematic representation of the anti-parallel packing of CA molecules into a highly interdigitated bilayer to form supramolecular ribbons.
When salts are added to aqueous solutions of CA ribbons, we observed the formation of gels. This finding is expected at high enough concentrations when the negatively charged supramolecular polymers are screened by electrolytes. Gels could be irreversibly formed by adding poly(diallyldimethylammonium) chloride (PDDA) (300 mM), CaCl2 (150 mM), or NaCl (300 mM) to a 11.5 mM solution of CA assemblies (Fig. 2a-c and Supplementary Fig. 3). At these concentrations, the gel is comprised of 99% water. Scanning electron microscopy (SEM) of each of the dried gel samples revealed a network of flat, sheet-like structures, consistent with the morphology revealed by other microscopy and X-ray scattering experiments (Fig. 2d-f and Supplementary Fig. 4). Cryo-TEM and SAXS of CA solutions with salt do not reveal a change in morphology with addition of salt (Supplementary Fig. 5 and 1, respectively). Rheological analysis showed that gels formed with PDDA and CaCl2 were two orders of magnitude stiffer than those formed with NaCl (Fig. 2g). Interestingly, we observe a time dependent evolution of the storage modulus in PDDA gels, suggesting a dynamic interaction between the polyelectrolyte and CA ribbons during gel formation.
Figure 2. Gelation of CA ribbons with salts.

Photographs of CA gels prepared by adding aqueous solutions of a, PDDA, b, CaCl2, c, NaCl to aqueous solutions of CA (11.5 mM) and allowing the sample to age overnight. SEM images of CA gelled with d, PDDA, e, CaCl2, f, NaCl. All gels are composed of nanostructures with some variance in their length and width. g, Rheological study of CA gels prepared from each gelling agent showing storage modulus (G', units of Pa) as a function of time. The equilibrium storage modulus increases with higher valency of the electrolytes that promote gelation.
Synchrotron wide-angle X-ray scattering (WAXS) was performed to probe the molecular packing of CA molecules within the self-assembled nanostructures. We observed that all of the CA gels revealed three intense diffraction peaks at 7.3, 17.3 and 18.5 nm−1(Fig. 3a), corresponding to a 2D distorted rectangular unit cell of a crystalline lattice (Fig. 3a inset). The short dimension of the unit cell is consistent with known π-π stacking distances for perylene imides (0.35 nm)20, while the long dimension corresponds to the edge-to-edge distance between adjacent perylene moieties (0.83 nm). CA solutions without added electrolytes showed weaker diffraction peaks than those of the gels (Fig. 3a), suggesting that charge screening allows CA molecules to pack into crystalline assemblies. While perylene-based crystalline nanosheets have been reported previously21, our system represents a unique example of chromophore crystallization within a hydrogel. To corroborate our solution phase X-ray scattering results, we carried out 2D grazing-incidence X-ray scattering (GIXS) on calcium-containing gels dried on silicon substrates (Fig. 3b). The films revealed similar diffraction peaks to those observed in the hydrated gels. The intensity of the meridional peaks relative to the equatorial maxima suggests that the 2D crystal lattice lies parallel to the substrate. The peaks at 3 and 6 nm−1 correspond to the first and second reflection of a 2.1 nm periodicity perpendicular to the substrate surface. This is likely due to lamellar stacking of PMI ribbons on top of one another, since the spacing correlates well with the CA ribbon height obtained by AFM (see Fig. 1c). Taken together, these data indicate that the 2-D crystalline lattice lies within the plane of the ribbon under charge-screening conditions (Fig. 3c).
Figure 3. Charge screening induces crystallization within supramolecular polymers.
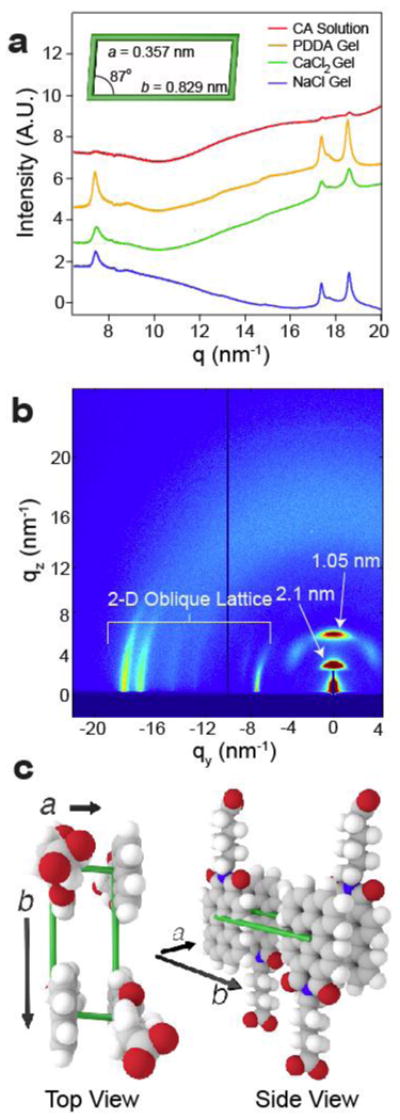
a, WAXS profiles for aqueous CA solution (11.5 mM), gelled with PDDA (5 wt%), CaCl2 (150 mM), or NaCl (300 mM) showing peaks at 7.3, 17.3, and 18.5 nm-1. Gel diffraction peaks correspond to a 2D, distorted rectangular crystalline lattice (inset). b, 2D-GIXS data of CA (11.5 mM) gelled in CaCl2 (20 mM) and dried on a silicon substrate. Three anisotropic peaks appear in the XY plane (parallel to the substrate) that correspond to the same 2D crystalline lattice as observed in solution. Two additional peaks in the Z-direction correspond to a 2.1 nm lamellar spacing. c, Schematic representation of CA unit cell formed under charge screening conditions.
Based on the crystallization of self-assembled perylenes with salt, we can expect the CAs to lose their individual excitonic character and behave as an ensemble. Exciton coupling of perylene-based chromophores results from the interaction of their transition dipole moments in space. For example, when CA was dissolved in water to form ribbons, the absorption maximum blue-shifted to 480 nm (Fig. 4a) relative to monomeric CA in DMSO solutions (λmax = 490 nm, Supplementary Fig. 6). The aggregated chromophores also displayed a fluorescence quantum yield of 1.2% in water relative to 82% when disaggregated in DMSO (Supplementary Fig. 7). These data indicate cofacial π-π stacking of the perylenes (H-aggregated) within the ribbons in water, as observed in other systems22. Upon crystallization with salt, each gel displayed a further blue-shifted absorption maximum (440 nm) and additional features between 500 and 600 nm (Fig. 4a). This further shift in an already H-aggregated system is characteristic of an increase in the intermolecular electronic coupling between the stacked chromophores23. Furthermore, theory predicts that an increase in order enhances electronic coupling23. Therefore, we believe that the observed blue shift is caused by crystallization, likely promoting better alignment of the transition dipoles across many molecules. Taken together, CA crystallization results in stronger excitonic coupling between adjacent PMI molecules, as well as greater π-π overlap leading to increased overall electronic coupling. In addition, we found that by titrating CA solutions with salt, we observed an increase in the population of strongly coupled chromophores (Fig. 4b). Furthermore, absorbance spectroscopy showed a gradual increase in coupling as the solutions of CAs and PDDA were allowed to mix over the course of 21 hours (Fig. 4c). The change in the optical properties of this material can be seen through the change in solution color upon gelation (see Fig. 4c inset). Functionally, the coupling of CAs in water through self-assembly affords the capture of a wider range of photons through spectral broadening. Previous theoretical work has shown that, upon excitation of supramolecular assemblies, strong electronic coupling among chromophores can produce excitons that are decoupled from lattice phonon vibrations23. We therefore hypothesize that the high degree of coupling within the CA ribbon assemblies indicated by our spectroscopic data most likely generates vibrationally decoupled excitons upon light absorption. While this effect is known for other chromophore aggregates24,25, as far as we know our system is the first example of a crystalline, H-aggregated perylene-based assembly that displays the strong coupling regime predicted by theory. Furthermore, this system is novel in that the transition to the high coupling regime can be studied simply by adding salt to CA solutions. Due to this high coupling, we do not expect the exciton to reside on one CA molecule, but rather to spread out over multiple unit cells within the crystalline ribbon26. The exciton produced in these assemblies represents a high potential energy state that can be utilized to drive electron transfer for photocatalytic processes. Further photophysical experiments and theoretical calculations beyond the scope of this work will be required in the future to verify details of our hypothesis.
Figure 4. Optical properties of CA solutions and crystalline gels.
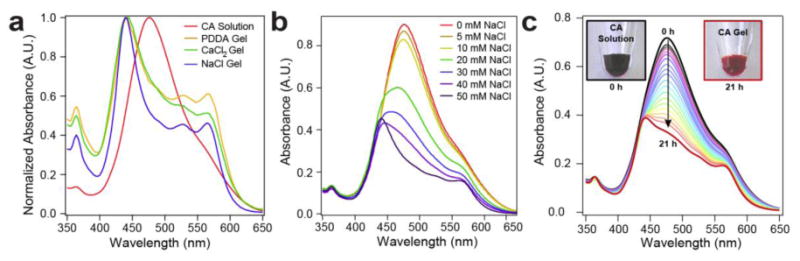
a, CA dissolved in water (red) has an absorbance maximum at 476 nm and a broad shoulder at 530 nm. Normalized absorbance spectra of gels prepared using PDDA, CaCl2, and NaCl all revealed a blue shift in the absorbance maximum to 442 nm and additional features at 529 and 562 nm. Identical line shape for all three gels suggests that the absorbance spectrum of charge-screened CA assemblies does not depend on the composition of the salt. b, Titration of CA solutions with NaCl (up to 50 mM) revealed a dependence of chromophore coupling on salt concentration. c, Absorbance spectroscopy shows changes in light absorption concomitant with gelation by PDDA over the course of 21 hours (data collected every hour, up to 16 hours, and then again at 21 hours). PDDA solution was placed at the top of the cuvette and allowed to diffuse into the CA solution over time. Insets CA solution before and after gelation.
We have therefore explored the use of these CA gels as light absorbing units in a supramolecular catalytic system. Photocatalytic proton reduction to hydrogen gas (H2) requires light absorption by a chromophore and the transfer of two electrons and two protons to a catalyst. Despite the large body of literature on supramolecular materials, there are few examples of their use in light-driven H2 evolution27-30. In designing an artificial photosynthetic system based fully on earth-abundant components, we chose a [Ni(P2PhN2Ph)2](BF4)2 (Ni P2N2) electrocatalyst developed by DuBois and coworkers31, which exhibits high turnover frequencies, low operating overpotentials, and has recently been incorporated into a photodriven system32. As spatial co-localization of chromophore and catalyst is important in natural photosynthesis, we synthesized a cationic analogue of this catalyst by functionalizing its outer ligand sphere with primary amines (Fig. 5a). This high degree of positive charge on the catalyst imparts water solubility and can promote electrostatic complexation with our gel of anionic supramolecular ribbons. Interestingly, we also found the emergent phenomenon that the catalyst itself induces crystallization of the chromophore assemblies and promotes formation of a hydrogel (Fig. 5b). In fact, addition of the catalyst (20 μL at 10 mM) to CA solution (100 μL at 11.5 mM) also leads to the large blue shift in absorbance upon charge screening discussed previously (Fig 5b inset, see Supporting Information for experimental details). This provides evidence for the strong coulombic coupling between the catalyst and the light-harvesting assemblies. Based on these observations, we hypothesize that the carboxylate-terminated ribbon surfaces present electrostatic binding sites for the catalyst. This electrostatic coupling should co-localize catalyst and CA, facilitating electron transfer reactions required for fuel production.
Figure 5. Photocatalytic hydrogen production.
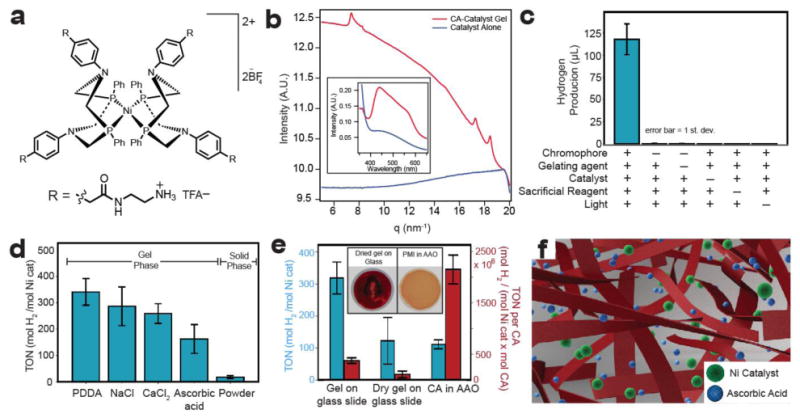
a, Chemical structure of water-soluble proton-reduction catalyst. b, WAXS experiments demonstrate that the addition of catalyst to CA induces crystallization and induces stronger electronic coupling between CA molecules (observed by absorbance spectroscopy, inset). c, H2 evolution experiments show that CA-PDDA gel is capable of photosensitizing nickel catalyst for H2 evolution. No H2 is evolved when chromophore, catalyst, sacrificial reagent, or light is omitted from the system. d, H2 production histogram of CA gels prepared with NaCl, PDDA, CaCl2, and ascorbic acid compared to insoluble, protonated CA. All gel phases produce more H2 than the solid phase CA powder. e, H2 production from CA-PDDA gels prepared on a glass slide, dried on a glass slide (inset, left), and from CA in an AAO (anodized aluminum oxide) filter (inset, right). In one case the data are normalized per mole catalyst (TON) (left) and per mole catalyst and mole chromophore (TON per CA) (right). f, Schematic of gel showing that CA nanoribbons (in red) trap solvent water molecules (not shown for clarity) within a three-dimensional architecture. This open architecture allows integration of ascorbic acid and catalyst with CA ribbons for photoinduced H2 production.
For H2 evolution experiments, CA gels were aged overnight and suspended in nickel catalyst-containing solutions (19.5 μM) with ascorbic acid (0.85 M, pH adjusted to 4 with NaOH) as a proton source and sacrificial electron donor. Analysis of sample headspace by gas chromatography after 18 hours of illumination with a white light source (output 400 – 700 nm, ∼250 mW/cm2, Supplementary Fig. 8) showed the production of 118 ± 17 μL H2 (Fig. 5c). Interestingly, H2 was effectively not produced in control experiments in which CA, ascorbic acid, or catalyst, was omitted from the system, indicating that the presence of each of these components within this soft material is necessary to complete the electron transfer pathway for photocatalytic H2 evolution. Furthermore, gels kept in the dark did not produce H2 in the presence of ascorbic acid and catalyst, indicating that the catalyst must be photoactivated by the CA gel (Fig. 5c). We observed a dependence of H2 production in the system on ascorbic acid concentration at pH 4 (Supplementary Fig. 9). The maximum H2 output was observed at pH 4 where both the sacrificial electron donor and proton source were in equal concentrations (pKa = 4 for ascorbic acid) (Supplementary Fig. 9). In order to estimate the proportion of catalyst electrostatically bound to the gel, we added catalyst solution to a PDDA gel, let the system age for two hours, then washed the gels with pure water three times to remove any free catalyst. We observed an 80% loss in H2, compared to unwashed controls, suggesting that some of the catalyst was tightly bound through electrostatic forces to the nanoribbon network that forms the gel (Supplementary Fig. 10). As mentioned previously, the strongly coupled crystalline assemblies should delocalize the exciton across many CA units within the ribbon. The oxidative or reductive quenching of the exciton are both thermodynamically favorable processes in our system, suggesting there are two possible pathways for H2 production (Supplementary Fig. 11). We envision the dominant photoconversion mechanism to involve the reductive quenching of these excitons, which in turn generates free electrons for transfer to the electrostatically bound catalyst. We believe this to be the dominant mechanism given the large concentration of ascorbic acid relative to catalyst (10,000: 1) in the system.
The degree of hydration within the CA gel plays an important role in co-localizing the soluble components within a 3D network. H2 was produced by gels formed with different charge screening conditions with PDDA giving the highest catalytic turnover number (TON) = 340±50 at a turnover frequency (TOF) of 19 h-1. While there are examples of H2 production from gel systems based on Ru(bpy)3 and platinum27, to date examples are not known of photocatalytic H2 evolution from a π-conjugated small molecule hydrogel. The catalyst's activity in our system is similar to a previously reported photocatalytic system utilizing a homogenous solution of an organic chromophore and an unfunctionalized Ni P2N2 catalyst (TOF = 18 h-1, TON = 2700 in 150 hours)32. In attempting to prepare an ungelled control for H2 production, we found that the addition of ascorbic acid to our CA solution induced gelation. The ascorbic acid gels (Supplementary Fig. 12) showed similar H2 production to the other gels (see Fig 5d). All gels showed dramatically more H2 compared to a solid precipitate of protonated (neutral) CA. In contrast to the open network seen in the gels, SEM of this precipitate (Supplementary Fig. 13) showed a compact structure suggesting that photocatalytic H2 production only occurs at the particle's surface. These results demonstrate the importance of a hydrated gel architecture for photocatalytic H2 production using designed soft materials. We envision these gels to consist of porous networks of ribbons that allow diffusion of catalyst and ascorbic acid to enhance photocatalysis (Fig. 5f). While CdSe-based systems report some of the highest activities observed for H2 evolution8,33, it would be environmentally useful to optimize soft organic materials as catalytic systems for solar fuel production.
Performing photocatalysis on a support could provide technical advantages for handling and recycling. Therefore, we prepared the photocatalytic gel formed using PDDA on glass (Fig. 5e). These gels afforded similar H2 production to the free-floating PDDA gels in solution. When the gels were dried on glass prior to immersion in the catalyst/ascorbic acid solution, we observed diminished H2 production suggesting that gel rehydration is not possible, and further supporting the importance of a hydrated network in H2 production. We also filled the 200 nm diameter pores of an anodic aluminum oxide (AAO) membrane with CA and performed photocatalytic H2 measurements by immersing the AAO substrates in the ascorbic acid/catalyst solution described previously (Fig. 5e). We observed TONs from these samples lower than those of gels cast on glass slides. However, we found that the AAO samples contained one-tenth the amount of CA compared to PDDA gels (determined by dissolving the AAO in base and quantifying the amount of CA present using absorbance spectroscopy), (Supplementary Fig. 14). Plotting the data with respect to TON per CA (Fig 5e), we found that the AAO samples gave the largest TON per CA values. We speculate that the local environment and interactions between chromophore molecules and catalyst might be different within the pores of the AAO substrate relative to bulk gels. This difference may account for the enhancement of H2 production in the AAO substrate even though the pores contain less chromophore than bulk gels. While these experiments demonstrate the activity of these gels on scaffolds, the system still requires the use of ascorbic acid as a sacrificial electron donor. Incorporation of these gels with transparent conducting oxides could enable the development of photocathodes that would avoid the use of stoichiometric reagents. We have cast CA-PDDA gels onto ITO-coated glass substrates and did not observe loss in H2 evolution relative to glass substrates (Supplementary Fig. 15). These data demonstrate that hydrogen evolution in the CA scaffold is not affected when the gel interfaces this transparent conducting oxide. A detailed understanding of the ITO-gel interface will be crucial for incorporating CA materials into photocathodes.
We have reported on the supramolecular design of a soft material integrating the necessary chemistry to produce H2 under illumination. The strategy involved the use of chromophore amphiphiles that self-assemble into ribbons with high surface charge density. Charge screening by salts led to the intra-ribbon crystallization of chromophores thus enhancing their electronic coupling. At the same time this screening process converts solutions to soft gels, allowing the physical localization of these materials on surfaces and porous substrates. Using electrostatic coupling of a catalyst to the ribbon network, we observed light-driven H2 production in the presence of a sacrificial electron donor in the gel phase. The catalytic scaffold developed in this work offers a model for integrated chemical systems that may be useful in the production of solar fuels or environmentally interesting chemical transformations.
Methods
Synthesis
The precursor perylene-3,4-dicarboxylic anhydride (PMA) was synthesized using literature procedures.15 (see Supporting information). CA
N-(Hexanoic acid) perylene-3,4-dicarboximide (CA)
To a 100 mL round bottom flask with PTFE stir bar, was added 109.7 mg perylene 3,4-dicarboxylic anhydride (0.341 mmol), 224.3 mg (1.71 mmol) ω-aminohexanoic acid, 43 mg zinc acetate dihydrate (0.339 mmol), and 69.8 mg imidazole (1.03 mmol). The flask was placed under N2 atmosphere and then heated at 130 °C for 50 hours while stirring. The flask was allowed to cool and 50 mL of 2 M HCl was slowly added to both precipitate the product and solubilize the other components. The mixture was diluted to 500 mL with water, filtered, and dried in air to obtain 91 mg of a dark red solid (yield: 61 %). 1H NMR (500 MHz, DMSO) δ 12.02 (s, 1H), 8.57 (t, J = 8.3 Hz, 4H), 8.35 (d, J = 7.9 Hz, 2H), 8.01 (d, J = 8.0 Hz, 2H), 7.66 (t, J = 7.7 Hz, 2H), 4.01 (t, J = 7.6 Hz, 2H), 3.36 (s, 2H), 2.24 (t, J = 7.2 Hz, 2H), 1.64 (t, J = 7.6 Hz, 2H), 1.60 – 1.53 (m, 2H), 1.36 (tt, J = 7.6, 4.8 Hz, 2H). 13C NMR (126 MHz, DMSO) δ 174.46, 162.86, 136.39, 133.69, 130.98, 130.89, 128.85, 128.26, 127.22, 126.85, 125.83, 124.40, 120.75, 120.14, 41.11, 33.45, 27.22, 26.05, 24.20. HR-MS (TOF-ESI, m/z): [M]+ calcd. for C28H21NO4, 435.1464; found, 435.1464; Difference: 1.62 ppm, 0.7 mDa.
The modified DuBois nickel catalyst (Ni-Cat) was prepared using procedures modified from a previously reported nickel electrocatalyst34 (see Supporting Information).
Sample Preparation
CA solutions were prepared by suspending the protonated CA in water, adding one equivalent (per mole) of an aqueous 4 M NaOH solution, and dissolved with the assistance of vortexing and bath sonication. Gels were produced by addition of 20 μL of aqueous electrolyte solution (5 wt% PDDA, 150 mM CaCl2, or 300 mM NaCl) to 100 μL of aqueous CA solution (11.5 mM). For studies requiring different volumes of solution, a 1:5 volume ratio of electrolyte solution to CA solution was used.
Photocatalytic Hydrogen Generation
CA gels with PDDA were prepared in a VWR Microwave Vial 0.5-2 mL (Catalog Number 89079-406), sealed with a rubber septum, and aged overnight (roughly 16 hours). Ascorbic acid solutions (1.7 M) were adjusted to pH 4 using 4 M NaOH (measured using a Fisher Scientific Accumet Research AR50 Dual Channel pH/Ion/Conductivity Meter, calibrated with pH 4.0 and pH 7.0 standard solutions). Ascorbic acid (660 μL, ∼0.85 M ascorbic acid) and catalyst solutions (40 μL, 400 μM) were mixed before addition to CA gels. Vials were sealed with a septum, further tightened with zip-ties, and wrapped in Parafilm to ensure proper sealing. After purging for 10 minutes with N2, the samples were illuminated for 18 hours with a Schott Ace 1 light source equipped with a broadband 21 V, 150 W EKE halogen bulb and fiber optic goosenecks. Samples were placed approximately 1.5 cm from the fiber optic light source (power output 250 W/cm2). For H2 identification and quantification, a 300 μL aliquot was taken from the sample vial (7.7 mL headspace) and injected onto a gas chromatograph (Shimadzu GC-2014) equipped with a 5 Å molecular sieve column, Ar carrier gas, and a thermal conductivity detector. Eight-point calibration curves for H2 and N2 were created using a standard (7% H2 balanced with N2) and integrated peak areas were used to determine the H2 concentration in the sample headspace at STP.
Supplementary Material
Acknowledgments
This work was supported as part of the Argonne-Northwestern Solar Energy Research (ANSER) Center, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under Award # DE-SC0001059. Use of the Advanced Photon Source (APS) was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. SAXS experiments were performed at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) located at Sector 5 of APS. DND-CAT is supported by E.I. DuPont de Nemours & Co., The Dow Chemical Company and Northwestern University. WAXS experiments were conducted BioCARS Sector 14 at the APS was supported by grants from the National Center for Research Resources (5P41RR007707) and the National Institute of General Medical Sciences (8P41GM103543) from the National Institutes of Health. GIXS data was collected at Sector 12 of APS. We thank the Biological Imaging Facility (BIF) at Northwestern for the use of TEM equipment, the Electron Probe Instrumentation Center (EPIC) facilities of the Northwestern University Atomic and Nanoscale Characterization Experimental (NUANCE) center for the use of SEM equipment. NMR and MS equipment at the Integrated Molecular Structure Education and Research Center (IMSERC) was supported by the National Science Foundation under CHE-9871268. The authors would like to acknowledge Jessica Lehrman and Yuri Velichko of the Stupp laboratory and Kelly Lefler, Walter Salamant, Michael Vagnini, Brad Veldkamp, and Dan Gardner of the Wasielewski laboratory for helpful discussions.
Footnotes
Author contributions: A.S.W., R.V.K., L.C.P., M.R.W., and S.I.S designed the experiments. A.S.W., R.V.K., M.T.M., A.R.K., A.P.S.S., and D.J.K. performed the experimental work. A.S.W., R.V.K., L.C.P., M.R.W., and S.I.S. analyzed the data and wrote the paper.
Additional Information: The authors declare no competing financial interests.
References
- 1.Umena Y, Kawakami K, Shen JR, Kamiya N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature. 2011;473:55–60. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- 2.Amunts A, Drory O, Nelson N. The structure of a plant photosystem I supercomplex at 3.4 Å resolution. Nature. 2007;447:58–63. doi: 10.1038/nature05687. [DOI] [PubMed] [Google Scholar]
- 3.Hull JF, et al. Highly Active and Robust Cp* Iridium Complexes for Catalytic Water Oxidation. J Am Chem Soc. 2009;131:8730–8731. doi: 10.1021/ja901270f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kanan MW, Nocera DG. In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+ Science. 2008;321:1072–1075. doi: 10.1126/science.1162018. [DOI] [PubMed] [Google Scholar]
- 5.Karunadasa HI, et al. A Molecular MoS2 Edge Site Mimic for Catalytic Hydrogen Generation. Science. 2012;335:698–702. doi: 10.1126/science.1215868. [DOI] [PubMed] [Google Scholar]
- 6.Yin Q, et al. A Fast Soluble Carbon-Free Molecular Water Oxidation Catalyst Based on Abundant Metals. Science. 2010;328:342–345. doi: 10.1126/science.1185372. [DOI] [PubMed] [Google Scholar]
- 7.Helm ML, Stewart MP, Bullock RM, DuBois MR, DuBois DL. A Synthetic Nickel Electrocatalyst with a Turnover Frequency Above 100,000 s-1 for H2 Production. Science. 2011;333:863–866. doi: 10.1126/science.1205864. [DOI] [PubMed] [Google Scholar]
- 8.Han Z, Qiu F, Eisenberg R, Holland PL, Krauss TD. Robust Photogeneration of H2 in Water Using Semiconductor Nanocrystals and a Nickel Catalyst. Science. 2012;338:1321–1324. doi: 10.1126/science.1227775. [DOI] [PubMed] [Google Scholar]
- 9.Boettcher SW, et al. Photoelectrochemical Hydrogen Evolution Using Si Microwire Arrays. J Am Chem Soc. 2011;133:1216–1219. doi: 10.1021/ja108801m. [DOI] [PubMed] [Google Scholar]
- 10.Veldkamp BS, et al. Photoinitiated multi-step charge separation and ultrafast charge transfer induced dissociation in a pyridyl-linked photosensitizer-cobaloxime assembly. Energ Environ Sci. 2013;6:1917–1928. [Google Scholar]
- 11.Poddutoori P, et al. Photoinitiated multistep charge separation in ferrocene-zinc porphyrin-diiron hydrogenase model complex triads. Energ Environ Sci. 2011;4:2441–2450. [Google Scholar]
- 12.Reece SY, et al. Wireless Solar Water Splitting Using Silicon-Based Semiconductors and Earth-Abundant Catalysts. Science. 2011;334:645–648. doi: 10.1126/science.1209816. [DOI] [PubMed] [Google Scholar]
- 13.Aida T, Meijer EW, Stupp SI. Functional Supramolecular Polymers. Science. 2012;335:813–817. doi: 10.1126/science.1205962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Babu SS, Praveen VK, Ajayaghosh A. Functional π-Gelators and Their Applications. Chem Rev. 2014 doi: 10.1021/cr400195e. [DOI] [PubMed] [Google Scholar]
- 15.Feiler L, Langhals H, Polborn K. Synthesis of Perylene-3,4-Dicarboximides - Novel Highly Photostable Fluorescent Dyes. Liebigs Ann. 1995:1229–1244. [Google Scholar]
- 16.Lefler KM, Co DT, Wasielewski MR. Self-Assembly-Induced Ultrafast Photodriven Charge Separation in Perylene-3,4-dicarboximide-Based Hydrogen-Bonded Foldamers. J Phys Chem Lett. 2012;3:3798–3805. doi: 10.1021/jz3018946. [DOI] [PubMed] [Google Scholar]
- 17.Samorì P, et al. Self-Assembly of Perylene Monoimide Substituted Hexa-peri-hexabenzocoronenes: Dyads and Triads at Surfaces. Adv Mat. 2006;18:1317–1321. [Google Scholar]
- 18.Bullock JE, et al. Photophysics and Redox Properties of Rylene Imide and Diimide Dyes Alkylated Ortho to the Imide Groups. J Phys Chem B. 2010;114:1794–1802. doi: 10.1021/jp908679c. [DOI] [PubMed] [Google Scholar]
- 19.Cui H, et al. Spontaneous and X-ray-Triggered Crystallization at Long Range in Self-Assembling Filament Networks. Science. 2010;327:555–559. doi: 10.1126/science.1182340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tasios N, et al. Self-Assembly, Dynamics, and Phase Transformation Kinetics of Donor—Acceptor Substituted Perylene Derivatives. J Am Chem Soc. 2010;132:7478–7487. doi: 10.1021/ja102150g. [DOI] [PubMed] [Google Scholar]
- 21.Shahar C, et al. Self-Assembly of Light-Harvesting Crystalline Nanosheets in Aqueous Media. ACS Nano. 2013;7:3547–3556. doi: 10.1021/nn400484y. [DOI] [PubMed] [Google Scholar]
- 22.Würthner F. Perylene bisimide dyes as versatile building blocks for functional supramolecular architectures. Chem Commun. 2004:1564–1579. doi: 10.1039/b401630k. [DOI] [PubMed] [Google Scholar]
- 23.Spano FC. The Spectral Signatures of Frenkel Polarons in H- and J-Aggregates. Acc Chem Res. 2010;43:429–439. doi: 10.1021/ar900233v. [DOI] [PubMed] [Google Scholar]
- 24.Zsila F, Bikádi Z, Keresztes Z, Deli J, Simonyi M. Investigation of the Self-Organization of Lutein and Lutein Diacetate by Electronic Absorption, Circular Dichroism Spectroscopy, and Atomic Force Microscopy. J Phys Chem B. 2001;105:9413–9421. [Google Scholar]
- 25.Spano FC, Meskers SCJ, Hennebicq E, Beljonne D. Probing Excitation Delocalization in Supramolecular Chiral Stacks by Means of Circularly Polarized Light: Experiment and Modeling. J Am Chem Soc. 2007;129:7044–7054. doi: 10.1021/ja067321g. [DOI] [PubMed] [Google Scholar]
- 26.Agranovich VM. Excitations in Organic Solids. Oxford University Press; 2009. [Google Scholar]
- 27.Okeyoshi K, Yoshida R. Hydrogen generating gel systems induced by visible light. Soft Matter. 2009;5:4118–4123. [Google Scholar]
- 28.Yuhas BD, et al. Biomimetic Multifunctional Porous Chalcogels as Solar Fuel Catalysts. J Am Chem Soc. 2011;133:7252–7255. doi: 10.1021/ja111275t. [DOI] [PubMed] [Google Scholar]
- 29.Jiang DL, et al. Photosensitized Hydrogen Evolution from Water Using Conjugated Polymers Wrapped in Dendrimeric Electrolytes. J Am Chem Soc. 2004;126:12084–12089. doi: 10.1021/ja048912e. [DOI] [PubMed] [Google Scholar]
- 30.Utschig LM, et al. Photocatalytic Hydrogen Production from Noncovalent Biohybrid Photosystem I/Pt Nanoparticle Complexes. J Phys Chem Lett. 2011;2:236–241. [Google Scholar]
- 31.Wilson AD, et al. Nature of hydrogen interactions with Ni(II) complexes containing cyclic phosphine ligands with pendant nitrogen bases. Proc Natl Acad Sci U S A. 2007;104:6951–6956. doi: 10.1073/pnas.0608928104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McLaughlin MP, McCormick TM, Eisenberg R, Holland PL. A stable molecular nickel catalyst for the homogeneous photogeneration of hydrogen in aqueous solution. Chem Commun. 2011;47:7989–7991. doi: 10.1039/c1cc12347e. [DOI] [PubMed] [Google Scholar]
- 33.Das A, Han Z, Haghighi MG, Eisenberg R. Photogeneration of hydrogen from water using CdSe nanocrystals demonstrating the importance of surface exchange. Proc Natl Acad Sci U SA. 2013 doi: 10.1073/pnas.1316755110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jain A, et al. Incorporating Peptides in the Outer-Coordination Sphere of Bioinspired Electrocatalysts for Hydrogen Production. Inorg Chem. 2011;50:4073–4085. doi: 10.1021/ic1025872. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.