

Abstract
A new class of clickable and biodegradable polylactide was designed and prepared via bulk polymerization of 3,6-dipropargyloxymethyl-1,4-dioxane-2,5-dione (1) which was synthesized from easily accessible propargyloxylactic acid (5). A homopolymer of 1 and random copolymer of 1 with l-lactide were obtained as amorphous materials and exhibit low Tg of 8.5 and 34 °C, respectively, indicating their promising potentials for biomedical applications. The statistical nature of random copolymers was investigated by DSC analysis and 13C NMR spectroscopy, which implies the random distribution of terminal alkyne groups along the back bone of copolymers. The efficient click post-modification of this new class of polylactide with alkyl and mPEG azides affords novel hydrophilic biomaterials, which exhibit reversible thermo-responsive properties as evidenced by their tunable LCST ranging from 22 to 69 °C depending on the balance of the incorporated hydrophilic/hydrophobic side chains. These results indicate the generality of this new class of clickable polylactide in preparing novel smart biomaterials in a simple and efficient manner via click chemistry.
Introduction
The great versatility and untapped potential of smart polymeric materials render them one of the most exciting interfaces between chemistry and biology. Because of their dramatic physical property changes in response to small external stimuli changes, they can be potentially applied to solve biological problems, such as in controlled drug and gene delivery,1–3 biosensor design4,5 and tissue engineering.6 Among various smart materials, thermo-responsive polymers have been studied intensively because of the easiness to handle their temperature changes,7,8 such as from room temperature to steady human body temperature. Thermo-responsive materials can exhibit a unique lower critical solution temperature (LCST). At temperatures below LCST, polymer is completely miscible with water due to the extensive hydrogen bonding interactions with the surrounding water molecules, whereas at temperatures above LCST a solution–gel phase separation occurs. Hydrogen bonding with water is disrupted above LCST, and intra- or intermolecular hydrogen bonding and hydrophobic interactions between polymer chains dominate, which results in the changes of polymer solubility.9 LCST can be tuned easily over a broad range of temperatures by adjusting the balance of hydrophilic/hydrophobic segments incorporated into polymer structure.7,10–15 Higher LCST can be achieved by increasing the content of hydrophilic segments to increase overall hydrogen bonding; whereas the incorporation of more hydrophobic groups lowers LCST which could be rationalized by easier disruption of hydrogen bonding between polymer chains and water.16
Thermo-responsive and biodegradable hydrogels prepared from di- or tri-block copolymers of polylactide (PLA) and poly- (ethylene glycol) (PEG) exhibit sol–gel phase transition at variable temperatures depending on the block lengths and polymer concentrations.17–19 In vitro release behavior of both hydrophilic and hydrophobic drugs from thermo-responsive hydrogel was also evaluated.20 However, these block PLA copolymers may encounter unpredictable degradation profiles and uncontrolled drug release behavior because of their structural heterogeneity. Thermo-responsive comb-like PLA polymers were synthesized by ring-opening polymerization (ROP) of lactide monomers functionalized with oligo(ethylene glycol) (OEG) of exact chain lengths,10 and their thermo-responsiveness was related to the chain length of OEG. However, the preparation of those comb-like PLA polymers requires complicated synthesis of each monomer. Furthermore, these block and/or comb-like PLA biomaterials provide no opportunity to further tune the polymer architecture or composition due to lack of functional sites, which makes them less interesting to the material science community.
Pendant terminal-alkyne functionalized PLAs have great flexibility in altering polymer composition by covalently grafting various functional molecules onto polymers via efficient click reaction without backbone degradation thanks to the mild reaction conditions and simple work-up procedures.21–24 Several novel biodegradable PLA materials were prepared via ‘click’ post-modification and exhibit different physical properties, such as increased hydrophilicity,25 thermo-responsive property7 and novel drug conjugates.26–28 Terminal-alkyne functionalized PLA has also found promising applications in nanomaterial-mediated drug delivery via thiol–yne click reaction.29,30 It allows for incorporation of two thiol units to one terminal alkyne unit along PLA backbones in one step at room temperature with high efficiency. These results clearly indicate that terminal-alkyne functionalized PLAs are of great importance in the formation of novel biomaterials with unique physical or chemical properties via alkyne–azide or thiol–yne click reactions. In addition, click post-modification of PLAs would allow for facile preparation of a series of smart biodegradable materials from a single clickable PLA substrate. This would enable high efficiency for specific smart property screening by simply tuning the ratio or composition of different functionalities incorporated into the polymers to modulate stimuli-response at the desired level for a specific application. We have continuous interest in the design and preparation of novel clickable PLA for developing new smart PLA biomaterials via click modification and further studying their self-assembly behavior in aqueous solution. Herein, we report a new class of clickable PLA via ROP of easily-accessible terminal alkyne functionalized lactide monomer (1) and its tunable thermo-responsive property after click post-modification. The LCST is tunable over a broad range including physiological temperature. Copolymerization of 1 with lactide at different feed ratios provides statistically distributed copolymers. We anticipate this new family of clickable PLA would be of great importance for developing efficient, targeted and biodegradable delivery vehicles.
Experimental section
Materials
Propargyloxylactic acid (5) was synthesized according to our previous report.31 The tosylate of triethylene glycol monomethyl ether was prepared following a previous report.32 1-(2-azidoethoxy)-2-(methoxyethoxy) ethane (m3PEGN3)33 and 1-azidodecane (C10H21N3)34 were synthesized using literature procedures. Sodium azide, tosyl chloride (TsCl), p-toluenesulfonic acid monohydrate (TsOH), 4-tert-butylbenyl alcohol (TBBA), triethylene glycol monomethyl ether (m3PEG) and 1-bromo-decane were purchased from Sigma-Aldrich. l-Lactide (LA) was purchased from Sigma-Aldrich and recrystallized from ethyl acetate before use. Stannous octanoate (Sn(Oct)2) was freshly distilled under vacuum and kept in dry box before use. THF and toluene were dried over sodium/benzophenone and distilled before use. The silica gel (60 Å porosity) for column chromatography was purchased from SiliCycle Inc. All other reagents and solvents were ACS grade and used as received unless specified.
Characterization
Melting points were measured on Electrothermo® melting point apparatus. 1H NMR and 13C NMR were recorded on a VXR-500 MHz instrument in CDCl3 unless otherwise noted. Residual CDCl3 was used as the internal standard for both 1H NMR (δ = 7.24) and 13C NMR (δ = 77.0). Analytical thin-layer chromatography (TLC) was performed on silica gel plates with F-254 indicator. FT-IR spectra were taken with Mattson Galaxy FT-IR 3000. HRMS were taken on Waters Xevo G2-S TOF UPLC/TOF MS. Thermogravimetric analysis (TGA) was recorded on Perkin-Elmer TGA 7 from 25 to 850 °C with a heating/cooling rate of 10 °C under an air atmosphere. Differential scanning calorimetry (DSC) measurements were recorded on DSC-Q100 from −40 to 180 °C with a heating/cooling rate of 10 °C under a nitrogen atmosphere and the second heating scans were used for DSC analyses. Dynamic light scattering (DLS) was carried out with a Malvern NanoZS ZetaSizer with a 178 degree backscattering detection to provide size profiles of samples at different temperatures. Polymer molecular weights were determined by gel permeation chromatography (GPC) at 35 °C using two PLgel 10µ mixed-B columns in series. A Waters 2410 differential refractometer detector and monodisperse polystyrene standards were used to calculate the molecular weights. Eluting solvent was THF at a flow rate of 1 mL min−1. Uv-vis spectra were recorded with Evolution 600 BB UV-visible spectrophotometer with a polymer concentration at ~3 mg mL−1.
Synthesis of 3,6-dipropargyloxymethyl-1,4-dioxane-2,5-dione (1)
Racemic propargyloxylactic acid (5, 10.5 g, 72.9 mmol), TsOH (1.0 g, 5.3 mmol) and 1.5 L of toluene were added into a 2 L round bottom flask sequentially. The resulting mixture was heated gently overnight until all acids were dissolved in toluene to give a slightly yellow solution (Note: Fast heating the reaction mixture to reflux could result in unexpected decomposition of 5). The resulting solution was refluxed for 11 days and water was removed from reaction mixture azeotropically with a Dean–Stark trap. The solution was cooled to room temperature and toluene was removed under vacuum. The residue was re-dissolved in 500 mL of CH2Cl2, and the resulting organic layer was washed with saturated aqueous NaHCO3 (3 × 150 mL), dried over anhydrous MgSO4 and filtered. CH2Cl2 was removed and the residue was dried in vacuo to afford crude monomer 1 as a light brown sticky liquid which was a mixture of trans-diastereomer (RS) and cis-diastereomers (RR, SS) (5.5 g, ~60%). This brown liquid was purified by flash chromatography with CH2Cl2–EtOAc (19 : 1) as eluent to give desired product 1 as a pure trans-diastereomer which was dried in vacuo and kept in dry box; white solid, 2.90 g, yield 31.5%. Rf = 0.60. m.p. 125–128 °C. 1H NMR (500 MHz, CDCl3) δ 5.20 (t, 2H, J = 2.5 Hz), 4.23 (d, 4H, J = 3 Hz), 4.14 (dd, 2H, J = 10 and 2.5 Hz), 4.03 (dd, 2H, J = 10 and 2.5 Hz), 2.50 (t, 2H, J = 3 Hz); 13C NMR (125 MHz, CDCl3) δ 164.09, 77.94, 76.43, 75.94, 69.52, 59.01; IR 3291, 2933, 2878, 2122, 1760, 1463, 1446, 1361, 1311, 1278, 1231, 1198, 1123, 1057 cm−1; HRMS (m/z) [M + H]+ calcd for C12H13O6 253.0712; found 253.0711.
General procedure for bulk polymerizations
Bulk polymerization was carried out in a sealed glass bulb prepared from a 3/8 in. diameter glass tubing with a bulb (1 in. diameter) blown on one end. In a dry box, monomer was added into a 5 mL glass vial and dissolved in minimum amount of THF. Predetermined amounts of catalyst (Sn(Oct)2) and initiator (TBBA) in toluene (0.04 M) were added to the vial separately via syringes. The resulting solution was transferred to the glass bulb via syringe and a small magnetic stir bar was added. The glass bulb was connected to a T-shape vacuum adapter fitted with a stopcock and an air-free Teflon valve via cajon fitting. The outlet of stopcock was sealed with a septum. The polymerization setup was taken out of dry box and connected to a vacuum line to remove all solvents carefully via Teflon valve. The resulting solid mixture was dried under high vacuum for 10 h at room temperature. The bulb was filled back with nitrogen and immersed in oil bath (130 °C) for polymerization. At the desired period of time, the bulb was removed from oil bath, quenched in ice and opened, and polymer was dissolved in CH2Cl2. A small portion of polymer solution was concentrated under vacuum and analyzed by 1H NMR for reaction conversion. The remaining polymer solution was precipitated from cold methanol–hexane twice and dried for 24 h in vacuo at room temperature to afford the desired polymer.
Polymerization of monomer 1
Monomer (1, 126 mg, 0.500 mmol) was polymerized for 40 min with the ratio of monomer-to-initiator ([M]/[I]) at 50. The conversion calculated from 1H NMR was 87%. Precipitation and drying under vacuum gave homo-polymer P1 as a light brownish wax-like sticky solid, 95.0 mg (75.4%). 1H NMR (500 MHz, CDCl3) δ 5.37–5.60 (br, 1H), 4.17–4.27 (br, 2H), 3.90–4.12 (br, 2H), 2.45–2.58 (br, 1H); Mn, NMR = 12 100 g mol−1; GPC (THF): Mn = 10 100 g mol−1, PDI = 1.46. Other polymers with [M]/[I] at 25, 100 and 200 were also prepared.
Copolymerization of 1 and LA
A mixture of 1 (31.5 mg, 0.125 mmol) and LA (18.0 mg, 0.125 mmol) was polymerized for 40 min with [M]/[I] = 100. Precipitation and drying under vacuum gave 30.0 mg of copolymer P(1-co-LA) as a light yellow solid (77.6%). 1H NMR (500 MHz, CDCl3) δ 5.40–5.56 (br, 1H), 5.17–5.33 (br, 1H), 4.18–4.28 (br, 2H), 3.89–4.12 (br, 2H), 2.45–2.56 (br, 1H), 1.45–1.62 (br, 3H); Mn, NMR = 17 100 g mol−1; GPC (THF): Mn = 10 200 g mol−1, PDI = 1.45. Polymerization with feed ratios of 1 to LA at 3 : 1 and 1 : 3 were also conducted.
General procedure for “click” post-modification
The desired amount of terminal-alkyne functionalized polymer, three equivalents of azide and 10 mol% of sodium ascorbate (10 mg mL−1 in water) were mixed and dissolved in DMF in a 20 mL glass vial. The resulting solution was deoxygenated by two freeze–pump–thaw degassing cycles. CuSO4 (5 mol%, 10 mg mL−1 in water) was added to the frozen mixture under a nitrogen purge followed by another freeze–pump–thaw degassing cycle. The glass vial was backfilled with nitrogen, and the reaction mixture was gradually warmed to room temperature and stirred for 12 h. At the end of click reaction, DMF was blown away by gentle nitrogen flow and the resulting viscous liquid was then washed with hexane and ether to afford a sticky wax-like solid after drying under vacuum.
1-Azidodecane (C10H21N3)-grafted P1-g-C10H21
P1 ([M]/[I] = 50, 9.2 mg) and C10H21N3 (40 mg, 0.22 mmol) were dissolved in 6 mL of DMF for click reaction. The decane-grafted P1 was isolated as a slightly brown and sticky solid, conversion: 100%; 19.0 mg, 81.2% yield. 1H NMR (500 MHz, CDCl3) δ 7.5–7.9 (br, 1H), 5.2–5.6 (br, 1H), 4.5–4.8 (br, 2H), 4.2–4.4 (br, 2H), 3.7–4.15 (br, 2H), 1.8–2.0 (br, 2H), 1.1–1.4 (br, 14H), 0.8–0.9 (br, 3H); Mn, NMR = 29 700 g mol−1.
m3PEGN3-grafted P1-g-m3PEG
P1 ([M]/[I] = 50, 7.5 mg) and m3PEGN3 (34 mg, 0.18 mmol) were dissolved in 5 mL of DMF for click reaction. The m3PEG-grafted P1 was isolated as a brownish sticky solid, conversion: 57%; 11 mg, 42% yield. 1H NMR (500 MHz, CDCl3) δ 7.5–7.9 (br, 1H), 5.2–5.6 (br, 1H), 4.5–4.8 (br, 2H), 4.2–4.4 (br, 2H), 3.7–4.15 (br, 4H), 3.4–3.68 (br, 8H), 3.2–3.4 (br, 3H), 2.5 (br, 0.4H, leftover terminal alkyne), 1.1–1.4 (br, 14H), 0.8–0.9 (br, 3H); Mn, NMR = 22 400 g mol−1.
m3PEGN3–C10H21N3 (2 : 1, 7 : 2)-grafted P1-g-C10H21/m3PEG
Two mixtures of m3PEGN3 and C10H21N3 (ratios at 2 : 1 and 7 : 2) were used to graft both PEG and alkyl chains on to P1. Conversions for both reactions were quantitative as evidenced by 1H NMR. Mn, NMR = 29 900 g mol−1 for polymer from 2 : 1 ratio and Mn, NMR = 30 000 g mol−1 for polymer from 7 : 2 ratio.
Measurement of LCST
LCST of the grafted polymer samples were tested by measuring absorbance of their aqueous solutions at 450 nm under various temperatures. The aqueous polymer solution (1 mL, 3 mg mL−1) was placed in quartz cuvette and the cuvette was placed into a UV-vis spectrophotometer with a programmed temperature control function (heating only). The absorbance of polymer solutions at 450 nm was taken at desired temperatures after equilibration and plotted as a function of temperature. The midpoint of solution–gel transition was taken as the LCST.
Results and discussion
Synthesis of 3,6-dipropargyloxymethyl-1,4-dioxane-2,5-dione (1)
As shown in Scheme 1, 3,6-dipropargyloxymethyl-1,4-dioxane-2,5-dione (1) was prepared from propargyloxylactic acid (5) which was synthesized via a practical and scalable synthetic pathway developed in our group.31 Briefly, the protected aldehyde 2 was coupled with propargyl alcohol to afford intermediate 3 which was converted to cyanohydrin 4 via acidic hydrolysis and the subsequent cyanation. Upon hydrolysis under acidic condition, acid (5) was obtained in 71% yield over 4 steps. The process is very practical and does not involve column purification of any intermediates. Finally, acid 5 was cyclized by azeotropic reflux of 5 with TsOH in toluene to furnish the desired monomer 1 as a meso-stereoisomer in 32% yield. An X-ray study further verified the stereochemistry of 1 as a meso-(R,S) stereoisomer (Fig. S6†). Methine protons of monomer 1 appear at 5.20 ppm as a triplet (shown in Fig. 1) and show a significant change in chemical shift from that of acid 5 (4.40 ppm, Fig. S3†), indicating the formation of a lactide ring. Both HRMS and FT-IR further confirmed the structure of monomer 1.
Scheme 1.
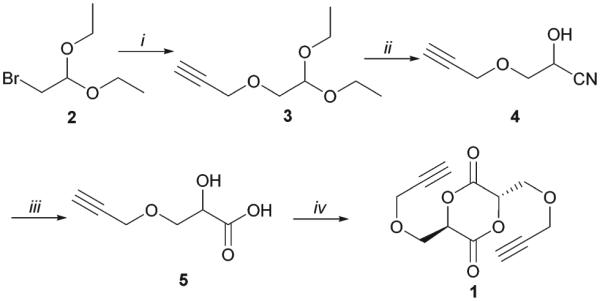
Synthesis of 3,6-dipropargyloxymethyl-1,4-dioxane-2,5-dione (1). Conditions: (i) propargyl alcohol, NaH, THF, 92%; (ii) (a) TFA–H2O–CH2Cl2 (1 : 1 : 4), (b) KCN, AcOH–H2O (2 : 1), 91%; (iii) (a) 6 M H2SO4, MeOH–H2O 1 : 1, (b) 2 M NaOH, CH2Cl2 extraction, (c) 4 M H2SO4, 85%; (iv) TsOH, toluene reflux, 32%.
Fig. 1.

1H NMR spectra (500 MHz) of 1, P1 and P(1-co-LA) in CDCl3.
Synthesis of clickable polylactide P1
Kinetics of bulk polymerization
Due to the poor solubility of monomer 1 in organic solvents, polymerization of 1 in toluene, THF or CH2Cl2 proceeded very slowly, which led to poor molecular weight control. This is also true when copolymerization of 1 with LA was attempted. Therefore, solvent-free bulk polymerization process was adopted to polymerization of 1.
We examined the kinetic behavior of bulk polymerization of 1 for a better control of the resulting polymers. The bulk polymerization of 1 to yield polymer P1 was performed with a ratio of monomer/catalyst/initiator at 50 : 1 : 1 in sealed polymerization bulbs (Scheme 2). The polymerization bulbs were immersed in a 130 °C oil-bath and quenched at the desired polymerization times. The conversion of monomer to polymer was determined by comparing 1H NMR integration of methine protons of monomer 1 (5.2 ppm) with those in polymer P1 (5.5 ppm) (Fig. 1). The ROP of lactide is typically first-order at low conversions35 and can be expressed as eqn (1),
| (1) |
where [M]0 and [M]t are the initial concentration of monomer and its concentration at time t, respectively. [cat]0 is the initial concentration of catalyst and is assumed to be constant for a living polymerization. kp is the rate constant for propagation.
Scheme 2.
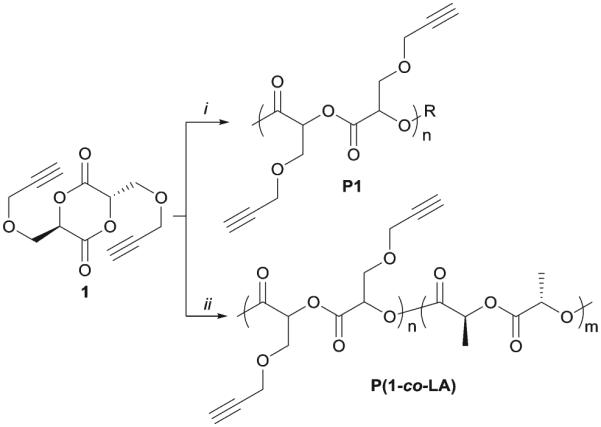
Ring opening polymerization of 1. Conditions: (i) Sn(Oct)2, TBBA, 130 °C; (ii) LA, Sn(Oct)2, TBBA, 130 °C.
Polymerization of lactide often establishes an equilibrium between propagation and depropagation steps with an equilibrium monomer concentration [M]e.36,37 Therefore, eqn (1) is revised to account for [M]e by subtracting [M]e from [M]0 and [M]t, giving eqn (2).
| (2) |
The [M]e was calculated from the remaining content of 1 (6%) in the polymerization mixture at equilibrium, and was used to plot the kinetics of bulk polymerization of 1 (Fig. S7†). As shown in Fig. 2, a nearly linear relationship between −ln{([M]t − [M]e)/([M]0 − [M]e)} and polymerization time t was clearly indicates that the bulk polymerization of 1 exhibits a similar kinetic pattern to that of lactide, and that the ROP of 1 is controlled and facile. Deviation of linear relationship at higher conversions may be due to the undesired transesterification when monomer 1 is nearly consumed.
Fig. 2.
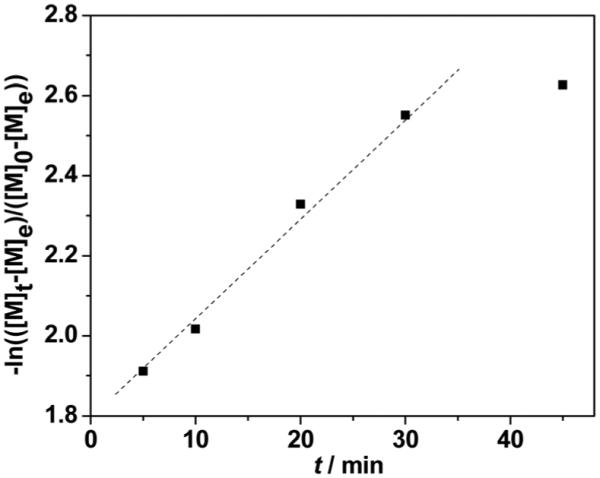
Kinetics of bulk polymerization of 1. Conditions: 130 °C, [1] : [Sn (Oct)2] : [TBBA] = 50 : 1 : 1.
Fig. 3 shows the evolution of number-average molecular weight (Mn) and polydispersity index (PDI) from GPC with time during the bulk polymerization. As polymerization proceeds, Mn increases, reaches a maximum at high conversion (<90%) and then drops. The PDI is in the range of 1.2–1.5 when conversions are below 90% and then rapidly increases at higher conversions (Fig. 3 and Table S1†). This behavior is a typical characteristic of ROP of lactides.36,37 Mn increases linearly with a narrow PDI at low conversion because of its living nature. A lower Mn with wider PDI at longer polymerization time may relate to the intermolecular or intramolecular transesterification of PLA chains at high conversion (>90%).36 And the unexpected dramatic increases of both Mn and PDI at the extended melt polymerization (t = 180 min) is probably due to the cross-linking of triple bonds.38
Fig. 3.
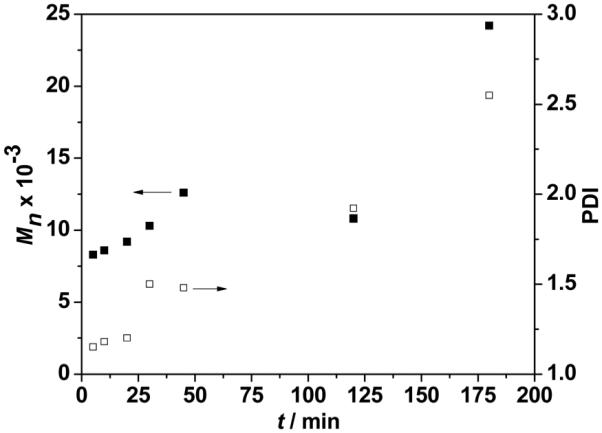
The molecular weight (Mn) (■) from GPC and PDI (□) versus time for bulk polymerization of 1. Conditions: 130 °C, [1] : [Sn(Oct)2] : [TBBA] = 50 :1: 1.
The results of bulk polymerization of 1 at varied [M]/[I] from 25 : 1 to 200 : 1 were collected and are listed in Table 1. It shows that the average degree of polymerization, Xn, determined from 1H NMR spectra are close to their theoretical values after correction for conversions at low [M]/[I] ratios. However, Xn measured from 1H NMR at high [M]/[I] ratio shows a lower value compared to the theoretical value (entry 4, Table 1), and this could be due to a less accurate end-group proton integration in 1H NMR of high molecular weight polymer and increased transesterification at longer polymerization time as evidenced by the slightly higher PDI. Mn increases with increasing [M]/[I] ratios, which is consistent with GPC profiles which shift to shorter elution times with increasing [M]/[I] ratios as shown in Fig. S8,† since larger polymers elute faster in GPC.39 The PDI as determined from GPC is fairly narrow around 1.4–1.6 for all of the polymers.
Table 1.
Bulk polymerizations of 1 at 130 °Ca
| Entry | M | [M]/[I] | t/min | Conv.b (%) | X n c | X n d | PDIe | Tgf (°C) |
|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 25 | 30 | 90 | 23 | 22 | 1.48 | 2.7 |
| 2 | 1 | 50 | 40 | 87 | 44 | 48 | 1.46 | 7.9 |
| 3 | 1 | 100 | 60 | 80 | 80 | 65 | 1.55 | 8.3 |
| 4 | 1 | 200 | 100 | 85 | 170 | 135 | 1.60 | 8.5 |
Using Sn(Oct)2 as a catalyst and TBBA as an initiator.
Measured by 1H NMR.
Calculated from [M]/[I] and corrected for conversion.
Determined from 1H NMR using end group analysis.
Measured by GPC in THF.
Determined by DSC.
Thermal properties of homopolymers P1
Thermal properties of polymeric materials define their process and application temperature range. Glass transition temperatures (Tg) of homopolymers were measured by DSC and are listed in Table 1. The DSC traces of homopolymers are shown in Fig. S9.† P1 from a low [M]/[I] ratio of 25 : 1 exhibits a Tg around 2.7 °C and Tg increases to a maximum of ~8.5 °C upon increasing the molecular weight of P1. Melting and crystallization transitions were not observed for P1 in either heating or cooling scans, which indicates the amorphous state of polymers and its importance for biomedical applications.40 The Tg of P1 decreases rapidly compared to that of PLA with a Tg of 55–60 °C due to the disruption of backbone–backbone interactions by the longer and more flexible side chain (propargyl-oxy group over methyl group) along backone.36,41
The thermal stability of polymers P1 is shown by their TGA traces in Fig. 4. The onset decomposition temperature (5% weight loss) for P1 with [M]/[I] ratio at 25 : 1 is ~210 °C and gradually increases to ~255 °C upon increasing [M]/[I] to 100 : 1 and 200 : 1. After a rapid weight loss until 330 °C, all polymers display a weight loss plateau between 335–620 °C which might be due to the stabilizing effect of cross-linked pendant terminal alkyne groups upon heating. Indeed, alkyne groups are known to act as cross-linkers and used to make materials flame-retardant.38,42–44 Due to the higher amount of alkyne groups, polymers with higher molecular weights exhibit slightly longer weight loss plateau and higher complete weight loss temperatures after second rapid decomposition (P1, Fig. 4). This may also explain the unexpected high molecular weight of homopolymer P1 by forming cross-linked polymer (Fig. 3) from the extended melt polymerization (180 min).
Fig. 4.
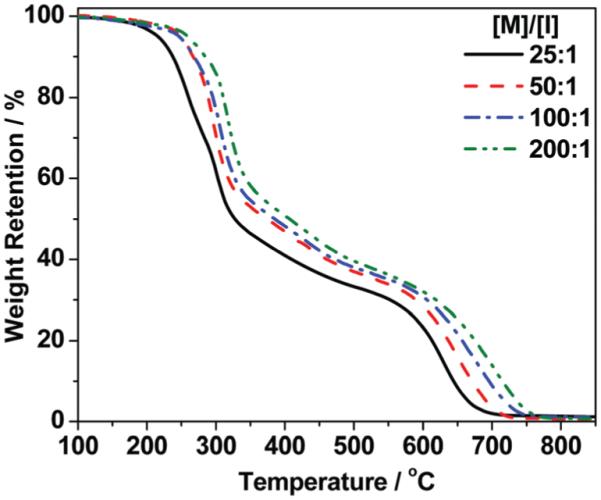
TGA data for homopolymers P1 with different [M]/[I] (130 °C).
Synthesis of statistically random copolymers of P(1-co-LA)
Copolymerization is commonly used to prepare copolymers with properties different from homopolymers by controlling density and/or position of functional groups along backbone. A series of copolymers P(1-co-LA) of 1 and LA were prepared via bulk polymerization and analyzed under the same conditions as for homopolymers (Table 2). The [M]/[I] ratio was kept at 100 while monomer ratios varied with 25%, 50% and 75% LA content (mol%). The percent conversions of LA were high (~96%) and the percent conversions of 1 were estimated to be ~85% from polymerization kinetics due to the peak overlap in 1H NMR spectra. As shown in Table 2, the experimental molecular weights from 1H NMR match well with the calculated Mn values of copolymers with PDI around 1.5 from GPC.
Table 2.
Mn and Tg of copolymers P(1-co-LA)
| Entry | Ratio LA : 1 | Conv.a (%) | Polymer compositionb LA : 1 | Mnc calc. (×10−3) | Mnd NMR (×10−3) | PDIe | Tgf (°C) | |
|---|---|---|---|---|---|---|---|---|
| 1 | 1 : 3 | 97 | 85g | 38 : 62 | 19.1 | 18.0 | 1.41 | 19.9 |
| 2 | 1 : 1 | 96 | 85g | 53 : 47 | 17.7 | 17.1 | 1.45 | 26.4 |
| 3 | 3 : 1 | 96 | 85g | 77 : 23 | 15.8 | 15.5 | 1.53 | 34.0 |
Determined by 1H NMR.
Calculated from 1H NMR.
g mol−1, calculated from [M]/[I] and corrected for conversion.
g mol−1, calculated from 1H NMR using end group analysis.
Measured by GPC in THF.
Determined by DSC.
Estimated conversions due to peak overlap in 1H NMR.
The onset decomposition temperature of copolymers was ~230–240 °C similar to their homopolymers (Fig. S11†). As discussed previously, pendant terminal alkyne group along polymer backbones act as a stabilizer and slow down thermo-decomposition of copolymers via cross-linking when heating above 300 °C. Higher content of alkyne groups leads to higher level of cross-linking and weight retention above 300 °C, and thus a higher complete thermo-decomposition temperature. DSC analyses show that Tg of copolymers P(1-co-LA) increases from 20 to 34 °C with increasing LA content (Fig. 5a). Melting and crystallization peaks were not observed in any of DSC scans up to 180 °C, indicating that the copolymers are completely amorphous. The absence of any crystallinity, even at the highest level of lactide (77%), reveals that the lactide units/blocks are of insufficient length to crystallize (usually at least 12 lactic acid units are required).45,46 In addition, there was only one glass transition observed for all of the copolymers with different lactide contents, which indicates that the distribution of propargyloxy lactide (1) and LA is statistical in nature (more random than blocks).47 The Tg of the statistical copolymers can be calculated from the Fox eqn (3)48,49 using the weight fractions of monomers in copolymers and Tg of the respective homopolymers.50,51
Fig. 5.
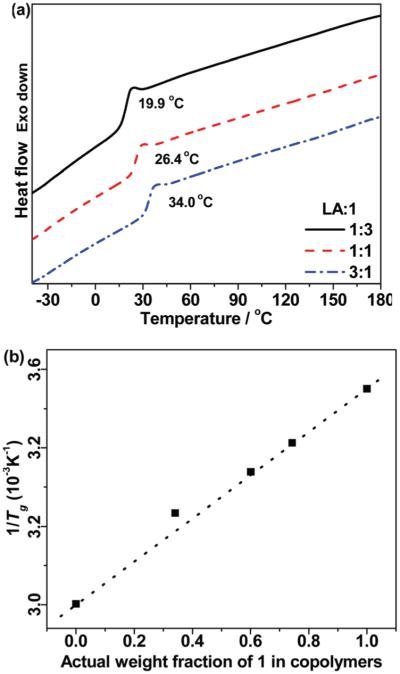
(a) DSC curves (2nd scans) of P(1-co-LA) with different feed ratios of LA to 1. (b) Plot of 1/Tg versus actual weight fraction of 1 in copolymers.
| (3) |
WLA and W1 denote the actual weight fractions of LA and monomer 1. The inverse Tg values of copolymers are plotted as a function of the actual weight fractions of 1 as shown in Fig. 5b. The theoretical value (dashed line) was generated using Fox eqn (3) from the experimental Tg values of homopolymer P1 (8.5 °C) and PLLA (59 °C). The experimental values fit well with the theoretical calculations, which confirms the statistical nature of copolymers and suggests that Tg of these copolymers can be simply predicted and tuned to the desired temperature by changing feed ratios of LA and 1 during polymerization. Defining the Tg of a polymer to a given temperature is of great importance in designing new polymer materials with desired physical and thermal properties,52 for example designing rubbery materials at lower temperatures.
This statistical nature of copolymers was also confirmed by comparing the chemical shift changes of carbonyl region in 13C NMR spectra of homopolymers PLLA, P1 and their copolymer P(1-co-LA), as shown in Fig. 6.7,53,54 The carbonyl signals from P1 and PLLA are well-separated, with resonance of P1 shifted ~3.0 ppm upfield relative to those of PLLA. For random copolymer P(1-co-LA), the “LA” carbonyl carbon shows new broad resonances shifted upfield ~0.3 ppm (marked with “*”). The carbonyl carbon of 1 in copolymer P(1-co-LA) exhibits new broad resonances shifted downfield ~0.2 ppm and a more splitted resonance pattern (marked with “+”). These results indicate a random distribution of monomer 1 and thereof the incorporated terminal alkyne groups along copolymer backbones. Although there are remaining carbonyl resonances in copolymers which are from block PLLA segment and P1 segment, those block segments should be short enough to be omitted to account for the statistical nature of copolymers because of the single glass transition observed in DSC analyses.
Fig. 6.
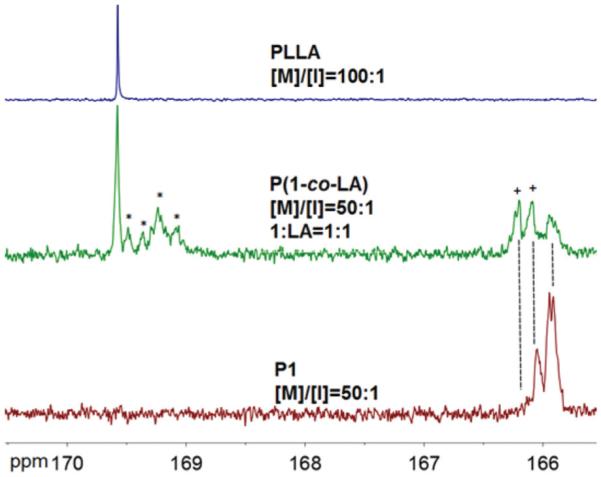
125 MHz 13C NMR carbonyl regions of homopolymers PLLA, P1 and copolymer P(1-co-LA). Solvent: CDCl3.
Click chemistry of clickable PLA (P1) to mPEG/Alkyl-grafted polymers
Click chemistry has been used in post-modification of PLA for generating biomaterials with promising biomedical applications. To prepare mPEG/Alkyl-grafted PLA materials, the mild ‘click’ condition without causing polymer degradation with CuSO4-sodium ascorbate catalytic system in DMF was applied to clickable polymer P1 and azide functionalized PEG or alkane.7,25 1-(2-azidoethoxy)-2-(methoxyethoxy) ethane (m3PEGN3)55 and 1-azidodecane (C10H21N3)56 were prepared following the reported methods and characterized by NMR and FT-IR. The azido groups of both m3PEGN3 and C10H21N3 were readily identified by IR absorbance peak at ~2100 cm−1 in FT-IR spectra and triplet resonance of adjacent methylene protons at ~3.3 ppm in 1H NMR spectra. The clickable PLA P1 prepared from [M]/[I] ratio at 50 : 1 was clicked with m3PEGN3, C10H21N3 or mixtures thereof with different molar ratios (2 : 1 and 7 : 2) for 12 h at room temperature after removal of oxygen as shown in Scheme 3. The obtained click-grafted polymers were isolated as slightly brown and sticky wax-like solids.
Scheme 3.

Click reactions of P1 with alkyl and PEG azides. Conditions: (i) 5% CuSO4, 10% sodium ascorbate, DMF, 12 h, rt under N2.
The 1H NMR spectra of these polymers before and after click reactions were recorded and are shown in Fig. 7a. The disappearance of proton resonances at 2.5 ppm (–CH2–CCH) and 4.2 ppm (–CH2–CCH), and the appearance of new broad peaks at 7.6 ppm (H on the triazole ring) and 4.6 ppm indicate the successful click functionalization of P1. This is also confirmed by the appearance of characteristic proton resonances corresponding to m3PEG (3.4–3.7 ppm), methoxy (3.3 ppm) and alkyl groups (0.8–1.5 ppm) in 1H NMR spectra of P1-g-m3PEG, P1-g-C10H21 and P1-g-C10H21/m3PEG (2 : 1 feed ratio of m3PEGN3–C10H21N3). The click reaction between P1 and C10H21N3 proceeded to 100% completion in desired reaction time. However, the click reaction of P1 and m3PEGN3 only run to 57% conversion as shown in 1H NMR (Fig. 7a) and FT-IR spectra (Fig. 7b), and the investigation for this uncompleted click reaction is in progress. Composition of pendant chains of the mix-clicked polymers were determined by 1H NMR spectroscopy to be 58% m3PEG chains and 42% decyl chains from a 2 : 1 feed ratio, and 65% m3PEG chains and 35% decyl chains from a 7 : 2 feed ratio. This clearly indicates that 1-azi-dodecane (C10H21N3) reacts faster than m3PEGN3. These results also imply that the ‘orthogonal’ click functionalization of PLAs can be achieved by simply mixing different azide derivatives to afford grafted PLAs with multiple functionalities. The click functionalization was further substantiated by FT-IR spectra of polymers (Fig. 7b). The absorbance bands of triple bond stretching (2117 cm−1) and terminal alkyne C–H stretching (3282 cm−1) disappeared; new absorbance bands of triazole ring stretching vibration at 3150 and 1460 cm−1 in fingerprint region were observed after click reaction. Furthermore, incorporation of m3PEG and decyl chains also account for the strong absorbance at 2852–2970 and 1200 cm−1 in normalized FT-IR spectra compared to the parent polymer P1.
Fig. 7.

(a) 1H NMR spectra in CDCl3 and (b) FT-IR spectra of P1 and its click-grafted polymers P1-g-m3PEG, P1-g-C10H21 and P1-g-C10H21/m3PEG (2 : 1 feed ratio of m3PEGN3–C10H21N3).
Thermo-responsive property of mPEG/Alkyl-grafted polymers
Various polymer systems have been studied and displayed thermo-responsive behavior.57,58 When heated above LCST, those materials undergo a solution–gel phase separation due to disruption of hydrogen bonding with water. Recent research has shown that the LCST can be easily tuned over a wide range of temperatures by adjusting the balance of hydrophilic/hydrophobic segments.11 Clickable PLA would allow for readily incorporating different organic functionalities via click chemistry and efficiently elaborating new biodegradable thermo-responsive materials. By changing the feed ratios of m3PEGN3 and C10H21N3, four click-grafted polymers, P1-g-C10H21, P1-g-m3PEG, P1-g-C10H21 and P1-g-C10H21/m3PEG with feed ratios of m3PEGN3 and C10H21N3 at 2 : 1 and 7 : 2, were prepared and subjected to thermo-responsive study. P1-g-C10H21 with only decyl side chains grafted is hydrophobic and does not exhibit thermo-responsive property. P1-g-m3PEG and P1-g-C10H21/m3PEG with pendant hydrophilic side chains are hydrophilic biodegradable materials and display thermo-responsive property. The LCST of their aqueous solutions (~3 mg ml−1) was measured using cloud point method by recording their absorbance at 450 nm at different temperatures on a UV-visible spectrophotometer. As shown in Fig. 8a, two mix-grafted polymers P1-g-C10H21/m3PEG with different ratios of hydrophilic and hydrophobic pendant chains (2 : 1 and 7 : 2) exhibit very sharp solution–gel transitions at ~22 and 27 °C which were recorded as a rapid increase of absorbance due to dramatic change of light scattering. P1-g-m3PEG only shows a moderate phase transition with a LCST at ~69 °C. This higher LCST could be attributed to the increased overall hydrogen bonding which requires higher temperature to be disrupted. The dramatic increase in light scattering for all samples was directly visualized and recorded in Fig. 8b as the transition from a transparent solution to a cloudy mixture upon heating above LCST. Notably, it is fully reversible between cloudy mixtures upon heating above LCST and clear solutions upon cooling below LCST for the aqueous solutions of all samples. Moreover, all click-grafted polymers maintain similar thermo-responsive behavior after performing more than 8 heating-cooling cycles, which indicates their relative high structural stability upon heating and cooling of those PLA materials.
Fig. 8.
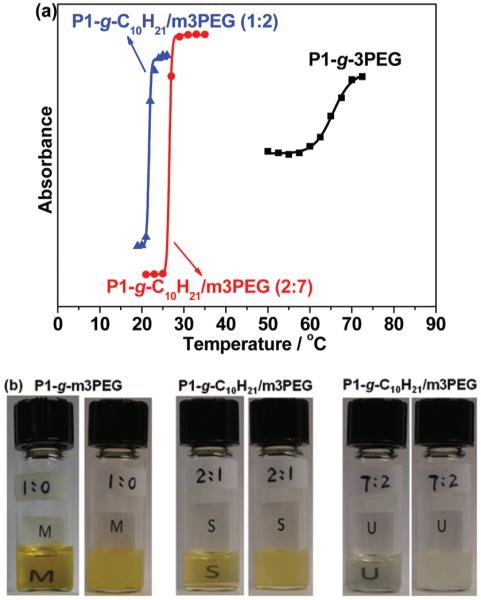
(a) UV turbidity measurements (450 nm) and (b) visualization of sol–gel transition of aqueous solutions of click-grafted PLA polymers at temperatures below (left) and above (right) their LCST.
The thermo-responsiveness of click-grafted PLA polymer solutions was also investigated with DLS as shown in Fig. 9. P1-g-C10H21/m3PEG (7 : 2 for m3PEGN3–C10H21N3) has a hydrodynamic diameter (Dh) of 10 nm below LCST which might correspond to a single hydrated polymer chain.59 Upon heating above its LCST, polymer aggregates and reaches a large Dh value of ~3 μm. P1-g-C10H21/m3PEG (2 : 1 for m3PEGN3–C10H21N3) and P1-g-m3PEG show much larger Dh at 130 and 180 nm blow their LCST, respectively. This might attribute to their less hydrophilic nature and thus formation of bigger aggregates via self-assembly of multiple hydrated polymer chains. These polymer chains dehydrate and form much larger insoluble aggregates (350 nm and 1000 nm) when their aqueous solutions were heated above their LCST. The DLS results further confirm that these click-grafted PLA polymers exhibit thermo-responsive behavior with a sol–gel transition from a hydrated soluble state to an agglomerated insoluble state.
Fig. 9.
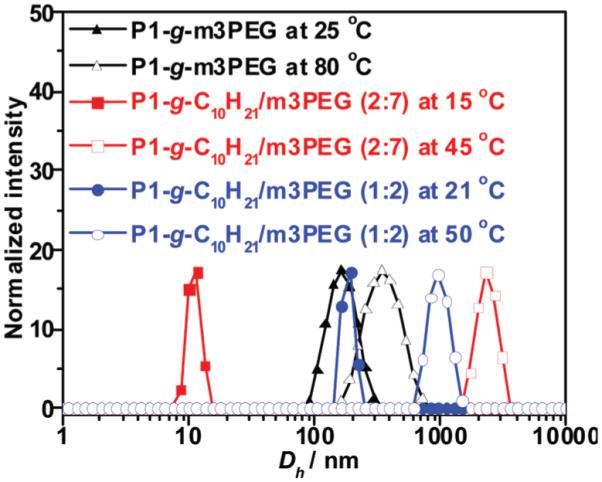
DLS profiles of click-grafted PLA polymers in water at temperatures below (filled symbol) and above (open symbol) their LCST.
In general, m3PEG segments click-grafted onto polymers form hydrogen bonding with water to facilitate the dissolution of polymers and exhibit reversible dehydration and hydration process with variations in temperatures. This changes the local environment of polymer chains, and results in a shift of their NMR resonances and broader peaks. Indeed, when heating the aqueous solutions of P1-g-C10H21/m3PEG and P1-g-m3PEG above their LCST, proton chemical shifts of the triazole rings (~7.6 ppm), of the mPEG units (3.4–3.7 ppm), of the methoxy groups (3.3 ppm) and of the alkyl groups (0.8–1.5 ppm) all shift downfield in a similar pattern as shown in their 1H NMR spectra (Fig. S13†). Each of these proton resonances is slight broader above their LCST compared to those below LCST due to the phase transition from a soluble to an insoluble state. This clearly further indicates the solution–gel phase transition of click-grafted polymers in water when heated above their LCST. The relationship between LCST and the actual molar fraction of m3PEG in grafted side chains of polymers is plotted in Fig. 10, which shows a nearly linear relationship consistent to literature reports.7,60–62 This suggests that LCST of thermo-responsive PLA biomaterials may be tailored to the desired temperatures for different applications via simply changing the feed ratios of azide side chains during the click functionalization, for example a PLA biomaterial with LCST near physiological temperature.63,64
Fig. 10.
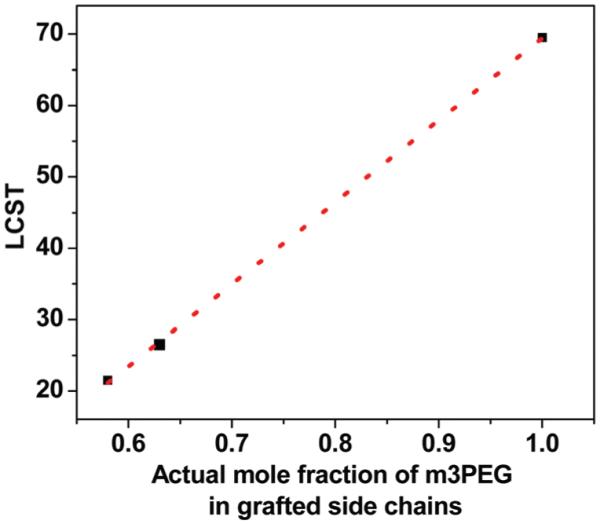
Plot of LCST versus actual mole fraction of m3PEG in click-grafted side chains of PLA polymers. Dashed line is the linear fit.
Conclusions
A new class of clickable and biodegradable PLA was described and investigated. Bulk polymerization of 3,6-dipropargyloxy-methyl-1,4-dioxane-2,5-dione (1) synthesized via dimerization of easily accessible propargyloxylactic acid (5) afforded clickable polylactide. The polymer composition and physical properties can be readily altered via click chemistry of the PLA pendant terminal alkyne groups. The kinetics of bulk polymerization of 1 was studied using Sn(Oct)2 as a catalyst and 4-tert-butyl benzyl alcohol as an initiator. The random copolymerization of monomer 1 with l-lactide provided statistical copolymers evidenced by DSC analysis and 13C NMR spectroscopy. The values of Tg of prepared copolymers fit well with Fox equation which further confirms their statistical nature. The click chemistry of this new class of PLA with 1-azidodecane and m3PEGN3 proceeded efficiently and afforded hydrophilic PLA biomaterials. Turbidity test, DLS and NMR results suggest these click-grafted polymers exhibit reversible thermo-responsive property with LCST ranging from 22 to 69 °C. A tunable LCST by varying the balance of incorporated hydrophilic/hydrophobic side chains was observed, which is of great importance for designing novel thermo-responsive biomaterials.
Supplementary Material
Acknowledgements
This manuscript is in memory of my advisor, Prof. Gregory L. Baker, who passed away unexpectedly while this paper was under preparation. The authors are grateful to Prof. William D. Wulff at Michigan State University for his invaluable guidance, discussions and assistance during preparation of this manuscript. This work was funded by NIH 5RC2ES018756-02.
Footnotes
Electronic supplementary information (ESI) available: X-ray data of 1, GPC, DSC, TGA and NMR data of monomer 1, homopolymers and click-grafted polymers. CCDC 1029799. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4py01425a
References
- 1.Hatefi A, Amsden B. J. Controlled Release. 2002;80:9–28. doi: 10.1016/s0168-3659(02)00008-1. [DOI] [PubMed] [Google Scholar]
- 2.Cunningham A, Oh JK. Macromol. Rapid Commun. 2013;34:163–168. doi: 10.1002/marc.201200532. [DOI] [PubMed] [Google Scholar]
- 3.Yang JH, Zhang P, Tang L, Sun P, Liu WG, Zuo AJ, Liang DC. Biomaterials. 2010;31:144–155. doi: 10.1016/j.biomaterials.2009.09.027. [DOI] [PubMed] [Google Scholar]
- 4.Hu J, Liu S. Macromolecules. 2010;43:8315–8330. [Google Scholar]
- 5.Abraham AA, Fei R, Coté GL, Grunlan MA. ACS Appl. Mater. Interfaces. 2013;5:12832–12838. doi: 10.1021/am4040653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reed JA, Lucero AE, Hu S, Ista LK, Bore MT, Lopez GP, Canavan HE. ACS Appl. Mater. Interfaces. 2010;2:1048–1051. doi: 10.1021/am900821t. [DOI] [PubMed] [Google Scholar]
- 7.Jiang X, Vogel EB, Smith MR, III, Baker GL. Macromolecules. 2008;41:1937–1944. [Google Scholar]
- 8.Dimitrov I, Trzebicka B, Müller AHE, Dworak A, Tsvetanov CB. Prog. Polym. Sci. 2007;32:1275–1343. [Google Scholar]
- 9.Fujishige S, Kubota K, Ando I. J. Phys. Chem. 1989;93:3311–3313. [Google Scholar]
- 10.Jiang X, Smith MR, III, Baker GL. Macromolecules. 2008;41:318–324. [Google Scholar]
- 11.Chen GH, Hoffman AS. Nature. 1995;373:49–52. doi: 10.1038/373049a0. [DOI] [PubMed] [Google Scholar]
- 12.Chen S, Pieper R, Webster DC, Singh J. Int. J. Pharm. 2005;288:207–218. doi: 10.1016/j.ijpharm.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 13.Tachibana Y, Kurisawa M, Uyama H, Kobayashi S. Biomacromolecules. 2003;4:1132–1134. doi: 10.1021/bm034123y. [DOI] [PubMed] [Google Scholar]
- 14.Shimokuri T, Kaneko T, Akashi M. J. Polym. Sci., Part A: Polym. Chem. 2004;42:4492–4501. [Google Scholar]
- 15.Chen C, Wang Z, Li Z. Biomacromolecules. 2011;12:2859–2863. doi: 10.1021/bm200849m. [DOI] [PubMed] [Google Scholar]
- 16.Feil H, Bae YH, Feijen J, Kim SW. Macromolecules. 1993;26:2496–2500. [Google Scholar]
- 17.Jeong B, Bae YH, Lee DS, Kim SW. Nature. 1997;388:860–862. doi: 10.1038/42218. [DOI] [PubMed] [Google Scholar]
- 18.Jeong B, Bae YH, Kim SW. Macromolecules. 1999;32:7064–7069. [Google Scholar]
- 19.Perez C, Sanchez A, Putnam D, Ting D, Langer R, Alonso MJ. J. Controlled Release. 2001;75:211–224. doi: 10.1016/s0168-3659(01)00397-2. [DOI] [PubMed] [Google Scholar]
- 20.Jeong B, Bae YH, Kim SW. J. Controlled Release. 2000;63:155–163. doi: 10.1016/s0168-3659(99)00194-7. [DOI] [PubMed] [Google Scholar]
- 21.Kolb HC, Finn MG, Sharpless KB. Angew. Chem., Int. Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 22.Hein JE, Fokin VV. Chem. Soc. Rev. 2010;39:1302–1315. doi: 10.1039/b904091a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Su H, Zheng J, Wang Z, Lin F, Feng X, Dong X-H, Becker ML, Cheng SZD, Zhang W-B, Li Y. ACS Macro Lett. 2013;2:645–650. doi: 10.1021/mz4002723. [DOI] [PubMed] [Google Scholar]
- 24.Yu Y, Zou J, Cheng C. Polym. Chem. 2014;5:5854–5872. [Google Scholar]
- 25.Coumes F, Darcos V, Domurado D, Li S, Coudane J. Polym. Chem. 2013;4:3705–3713. [Google Scholar]
- 26.Yu Y, Zou J, Yu L, Ji W, Li Y, Law W-C, Cheng C. Macromolecules. 2011;44:4793–4800. [Google Scholar]
- 27.Yu Y, Chen C-K, Law W-C, Weinheimer E, Sengupta S, Prasad PN, Cheng C. Biomacromolecules. 2014;15:524–532. doi: 10.1021/bm401471p. [DOI] [PubMed] [Google Scholar]
- 28.Yu Y, Chen C-K, Law W-C, Sun H, Prasad PN, Cheng C. Polym. Chem. 2015 DOI: 10.1039/C4PY01194E. [Google Scholar]
- 29.Zhang Z, Yin L, Tu C, Song Z, Zhang Y, Xu Y, Tong R, Zhou Q, Ren J, Cheng J. ACS Macro Lett. 2013;2:40–44. doi: 10.1021/mz300522n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Z, Yin L, Xu Y, Tong R, Lu Y, Ren J, Cheng J. Biomacromolecules. 2012;13:3456–3462. doi: 10.1021/bm301333w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Q, Ren H, Baker GL. J. Org. Chem. 2014;79:9546–9555. doi: 10.1021/jo5016135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Q, Ren H, Baker GL. Tetrahedron Lett. 2014;55:3384–3386. [Google Scholar]
- 33.Saha A, Ramakrishnan S. Macromolecules. 2009;42:4956–4959. [Google Scholar]
- 34.Marti MJ, Rico I, Desavignac A, Lattes A. Tetrahedron Lett. 1989;30:1245–1248. [Google Scholar]
- 35.Dubois P, Ropson N, Jerome R, Teyssie P. Macromolecules. 1996;29:1965–1975. [Google Scholar]
- 36.Yin M, Baker GL. Macromolecules. 1999;32:7711–7718. [Google Scholar]
- 37.Jing F, Smith MR, III, Baker GL. Macromolecules. 2007;40:9304–9312. [Google Scholar]
- 38.Unroe MR, Reinhardt BA. J. Polym. Sci., Part A: Polym. Chem. 1990;28:2207–2221. [Google Scholar]
- 39.Settle FA. Handbook of Instrucmental Techniques for Analytical Chemsitry. Prentice Hall PTR; Upper Saddle River, NJ: 1997. p. 856. ch. 4. [Google Scholar]
- 40.Trimaille T, Gurny R, Moller M. J. Biomed. Mater. Res., Phys. 2005;43:1624–1630. [Google Scholar]
- 41.Baker GL, Vogel EB, Smith MR., III Polym. Rev. 2008;48:64–84. [Google Scholar]
- 42.Fournier D, Billiet L, Du Prez F. In: New Smart Materials via Metal Mediated Macromolecular Engineering. Khosravi E, Yagci Y, Savelyev Y, editors. Springer; Netherlands: 2009. p. 157. [Google Scholar]
- 43.Bertini F, Audisio G, Kiji J, Fujita M. J. Anal. Appl. Pyrolysis. 2003;68–69:61–81. [Google Scholar]
- 44.Fournier D, Du Prez F. Macromolecules. 2008;41:4622–4630. [Google Scholar]
- 45.Leemhuis M, Akeroyd N, Kruijtzer JAW, van Nostrum CF, Crommelin DJA, Hennink WE. Eur. Polym. J. 2008;44:308–317. [Google Scholar]
- 46.Leemhuis M, van Nostrum CF, Kruijtzer JAW, Zhong ZY, Feijen J, Hennink WE. Macromolecules. 2006;39:3500–3508. [Google Scholar]
- 47.Fiore GL, Jing F, Young VG, Jr., Cramer CJ, Hillmyer MA. Polym. Chem. 2010;1:870–877. [Google Scholar]
- 48.Tsai Y, Fan C-H, Hung C-Y, Tsai F-J. J. Appl. Polym. Sci. 2008;109:2598–2604. [Google Scholar]
- 49.Kim JH, Jang J, Lee D-Y, Zin W-C. Macromolecules. 2002;35:311–313. [Google Scholar]
- 50.Hiemenz PC, Lodge TP. Polymer Chemistry. CRC Press; Boca Raton, FL: 2007. p. 493. [Google Scholar]
- 51.Sperling LH. Glass-Rubber Transition Behavior, in Introduction to Physical Polymer Science. 4th John Wiley & Sons, Inc.; Hoboken, NJ, USA: 2005. p. 401. [Google Scholar]
- 52.Kraus G, Childers CW, Gruver JT. J. Appl. Polym. Sci. 1967;11:1581–1591. [Google Scholar]
- 53.Guo M, Yu T, Xue Z. Makromol. Chem., Rapid Commun. 1987;8:601–605. [Google Scholar]
- 54.Zhou X-M, Jiang Z-H. J. Polym. Sci., Part B: Polym. Part A. 2007;80:55–65. [Google Scholar]
- 55.Saha A, Ramakrishnan S. Macromolecules. 2009;42:4956–4959. [Google Scholar]
- 56.Marti MJ, Rico I, Desavignac A, Lattes A. Tetrahedron Lett. 1989;30:1245–1248. [Google Scholar]
- 57.Gil ES, Hudson SA. Prog. Polym. Sci. 2004;29:1173–1222. [Google Scholar]
- 58.Aseyev V, Tenhu H, Winnik FM. Adv. Polym. Sci. 2011;242:29–89. [Google Scholar]
- 59.Lavigueur C, Garcia JG, Hendricks L, Hoogenboom R, Cornelissen JJL, Nolte RJM. Polym. Chem. 2011;2:333–340. [Google Scholar]
- 60.Fournier D, Hoogenboom R, Thijs HML, Paulus RM, Schubert US. Macromolecules. 2007;40:915–920. [Google Scholar]
- 61.Kempe K, Neuwirth T, Czaplewska J, Gottschaldt M, Hoogenboom R, Schubert US. Polym. Chem. 2011;2:1737–1743. [Google Scholar]
- 62.Porsch C, Hansson S, Nordgren N, Malmstrom E. Polym. Chem. 2011;2:1114–1123. [Google Scholar]
- 63.Winzenburg G, Schmidt C, Fuchs S, Kissel T. Adv. Drug Delivery Rev. 2004;56:1453–1466. doi: 10.1016/j.addr.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 64.Ward MA, Georgiou TK. Polymers. 2011;3:1215–1242. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.