

Abstract
AIM: To investigate the role of IKBKAP (inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase complex-associated protein) in the development of enteric nervous system (ENS) and Hirschsprung disease (HSCR).
METHODS: In this study, we injected a morpholino that blocked the translation of ikbkap protein to 1-cell stage zebrafish embryos. The phenotype in the ENS was analysed by antibody staining of the pan-neuronal marker HuC/D followed by enteric neuron counting. The mean numbers of enteric neurons were compared between the morphant and the control. We also studied the expressions of ret and phox2bb, which are involved in ENS development, in the ikbkap morpholino injected embryos by quantitative reverse transcriptase polymerase chain reaction and compared them with the control.
RESULTS: We observed aganglionosis (χ2, P < 0.01) and a reduced number of enteric neurons (38.8 ± 9.9 vs 50.2 ± 17.3, P < 0.05) in the zebrafish embryos injected with ikbkap translation-blocking morpholino (morphant) when compared with the control embryos. Specificity of the morpholino was confirmed by similar results obtained using a second non-overlapping morpholino that blocked the translation of ikbkap. We further studied the morphant by analysing the expression levels of genes involved in ENS development such as ret, phox2bb and sox10, and found that phox2bb, the ortholog of human PHOX2B, was significantly down-regulated (0.51 ± 0.15 vs 1.00 ± 0, P < 0.05). Although we also observed a reduction in the expression of ret, the difference was not significant.
CONCLUSION: Loss of IKBKAP contributed to HSCR as demonstrated by functional analysis in zebrafish embryos.
Keywords: Hirschsprung disease, Enteric nervous system, IKBKAP, Zebrafish, Morpholinos
Core tip: To investigate the functional role of IKBKAP in enteric nervous system (ENS) development, we knocked down the zebrafish ortholog ikbkap using a translation blocking antisense morpholino. Loss of ikbkap caused aganglionosis and a reduced number of enteric neurons, indicating that IKBKAP is important for proper ENS development.
INTRODUCTION
The enteric nervous system (ENS) is an intrinsic, autonomic nervous system in the intestine of vertebrates and is responsible for regulating peristalsis, transmucosal movement of fluids and local blood flow. Neurons and glia in the ENS are organised into ganglions, which form the myenteric (between the intestinal longitudinal and circular muscles) and the submucosal (underneath the submucosal) plexuses. Enteric neurons and glia are originated from vagal neural crest cells (NCCs) during embryonic development. Vagal NCCs migrate from the dorsal neural tube and colonise the intestine, and differentiate into mature enteric neurons and glia[1]. In humans, defective ENS development results in hirschsprung disease (HSCR, MIM142623).
HSCR is a congenital neuropathy, characterised by the absence of enteric ganglion (aganglionosis) and impaired peristaltic movement along variable lengths of distal intestine. HSCR patients develop constipation, diarrhoea, vomiting and sometimes life-threatening colon complications such as enterocolitis. Currently the treatment of HSCR is by surgery in which the aganglionic, non-functioning distal intestine is resected and the normal proximal intestine is reconnected to the anus. HSCR is a complex multifactorial disease, which most commonly presents sporadically (80%-95%) although it can be familial (5%-20%). It displays high heritability, large sex bias (male to female 4:1), high sibling recurrence risk, non-Mendelian inheritance in families and higher frequency in Asian populations (28 per 100000 live births). Coding sequence mutations of RET, which encodes a receptor tyrosine kinase, account for up to 50% of the familial and between 7% and 35% of the sporadic HSCR cases. RET is important for ENS development, as mice without Ret exhibit an HSCR-like phenotype. Mutations in other genes that are important for ENS development, such as endothelin receptor B, transcription factors SRY-box 10 (SOX10) and paired-like homeobox 2b (PHOX2B), are subsequently found to cause HSCR. However, all these known HSCR-causing mutations together only explain a fraction of all HSCR cases, suggesting unknown mutations are yet to be discovered[2]. To uncover new HSCR-susceptibility loci, we fine mapped the chromosomal region 9q31[3], which was previously shown to segregate in families bearing either hypomorphic or non-coding RET mutations[4]. We narrowed this region down to the gene IKBKAP (inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase complex-associated protein) and confirmed the association of this gene to Chinese HSCR patients[3].
IKBKAP encodes the protein IkappaB kinase complex associated protein, also known as elongator complex protein 1 (ELP1). It is a scaffold protein that forms the elongator complex with ELP2, 3, 4, 5 and 6. Previous studies on IKBKAP and the elongator complex suggested they performed diverse functions. The elongator complex was found to be associated with RNA polymerase II to regulate transcription[5]. In addition, IKBKAP has also been suggested to be responsible for DNA demethylation[6], JNK activation[7], exocytosis[8], tRNA modification[9], α-tubulin acetylation[10,11], actin organisation[12,13], and cell migration[14] and survival[15]. Knocking down IKBKAP in vitro in human neuroblastoma cells resulted in the down regulation of RET[16], suggesting IKBKAP might regulate RET expression and ENS development. Mutations in human IKBKAP lead to familial dysautonomia (FD, MIM223900), a neuropathy of the autonomic nervous system characterised by abnormal or incomplete neuronal development and progressive neuronal degeneration[17], with more than 60% of patients also being affected with gastrointestinal dysfunction[18]. Interestingly, FD patients were shown to have reduced ganglia and neurons density in the ENS[19] and a co-occurrence of HSCR and FD has been reported[20]. These findings suggested that HSCR and FD might share a common etiology. A number of murine models were established to study the function of IKBKAP in development[21-26]. However, these studies primarily focused on analysing FD-related phenotypes in the central nervous system, dorsal root ganglia, behavioural changes, etc., without thoroughly investigating the ENS. No prior study exists on the role of IKBKAP in the development of ENS and the pathogenesis of HSCR.
Prompted by the above information, we set out to investigate whether IKBKAP plays a role in ENS development. Thus in this study, we knocked down ikbkap in zebrafish embryos using antisense morpholino. We found that ikbkap knock down resulted in aganglionosis and a reduced neuron number in the ENS of zebrafish embryos, and reduced expression of phox2bb (ortholog of human PHOX2B).
MATERIALS AND METHODS
Animals
Wild-type adult zebrafish (Danio rerio) were purchased from local aquariums and embryos were obtained from natural spawning. The maintenance of the zebrafish and the culture of the embryos were carried out as described previously[27]. Embryos were staged by hours post-fertilisation or days post-fertilisation (dpf) at 28.5 °C[28].
Morpholino microinjection
Two antisense morpholinos (Gene Tools LLC) specifically blocking ikbkap translation (5’- TCAGCAGACTGAGGTTTCTCATTGT-3’, 5’- GGTTTATGTTTTCCT CAAGATGAGA-3’) and the standard control morpholino (5’-CCTCTTACCTCAG TTACAATTTATA -3’) were used in the study. The preparation of the morpholinos and the procedure for zebrafish microinjection were carried out as described previously[29]. Morpholino injected embryos were cultured at 28.5 °C to later stages for analysis.
Immunohistochemistry and neuron counting
Morpholino injected embryos were cultured to 5 dpf and immunohistochemistry of the neuronal marker HuC/D was conducted as described[30]. The following antibodies and dilutions were used: anti-HuC/D (16A11, Molecular Probes A-21271) 1 in 1000, anti-IKBKAP (Santa Cruz sc-8336) 1 in 200, and goat-anti-mouse HRP-conjugated secondary antibody (Abcam ab6789) 1 in 1000. DAB+ (Dako K346811) was used for signal development. Images were taken by a standard upright microscope. For neuron counting, HuC/D positive cells in the 30 μm length of distal-most intestine were counted. Results were analysed by the χ2 test or the Student’s t test.
Real-time-polymerase chain reaction and quantitative polymerase chain reaction
Morpholino-injected embryos were cultured to 1 dpf. 30 embryos from each treatment were pooled, dechorionated, and homogenised, and RNA was extracted by TRIzol® Reagent (Life Technologies 15596-026) according to the manufacturer’s protocol. 1 μg of RNA was used for cDNA synthesis by the Reverse Transcription System (Promega A3500) using oligo-dT primer. For qualitative real-time-polymerase chain reaction (RT-PCR), standard PCR was performed using AmpliTaq Gold® (Applied Biosystems) and the products were resolved in 0.8% agarose gel. The following primer pairs were used: ret: 5’-GTCCTGTTACTGGGCGGTTA-3’, 5’-GTGGCTCATCCGTTTTCAGT-3’; phox2bb: 5’-AGGAGCTCGCGCTTAAGATT3’, 5’-TTGCCTCTTTGCTGTCCTCT-3’; sox10: 5’-CAGCCAATCGCATTACAAGA-3’, 5’-GGTGGGAGATACTGGTCGAA-3’; elfa1: 5’-CTTCTCAGGCTGACTGTGC-3’, 5’-CCGCTAGCATTACCCTCC-3’. Quantitative polymerase chain reaction (q-PCR) was conducted as described previously[31] using TaqMan® Gene Expression Assays (Applied Biosystems) for ret (Dr03119148_g1), phox2bb (Dr03423610_g1) and beta-actin (Dr03432610_m1). Relative gene expression was determined by the Livak method[32].
RESULTS
Loss of ikbkap resulted in aganglionosis and a reduced number of enteric neurons
We injected an antisense morpholino that blocked the translation of ikbkap protein into 1 to 4-cell stage zebrafish embryos to study the effect of ikbkap loss-of-function in ENS development. The morpholino successfully blocked the translation of ikbkap protein, as shown in the ikbkap antibody staining of 3 dpf embryos (Figure 1). In the control morpholino injected embryo (control), ikbkap was expressed in the intestinal epithelium and surrounding tissues while the expression was absent in the ikbkap morpholino injected embryos (morphants). At 5 dpf, HuC/D positive enteric neurons were found along the entire intestine as shown in the control (Figure 2A). Aganglionosis in the distal intestine (Figure 2B) was observed in 6.4% (7 in 110, Figure 2D) of the morphants that mimicked HSCR in humans. For the rest of the morphants, the number of enteric neurons seemed to be reduced (Figure 2C) and this was confirmed by enteric neuron counting at the 30 μm-length region in distal-most intestine (Figure 2E). On average the morphants had fewer enteric neurons at the distal intestine than the control and the difference was statistically significant (P < 0.05). The experiment was repeated with a second non-overlapping morpholino blocking ikbkap translation and similar results were observed (data not shown).
Figure 1.
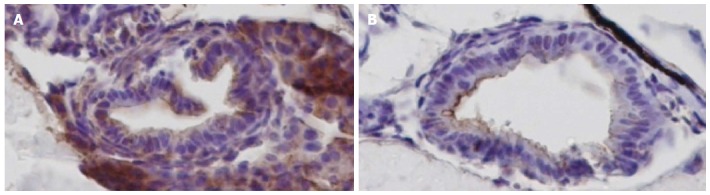
Ikbkap depletion by morpholino. A: In 3 dpf control embryo ikbkap protein was detected by antibody staining (reddish-brown, × 400); B: 3 dpf ikbkap morphant embryo. The level of ikbkap was greatly reduced (magnification × 400).
Figure 2.
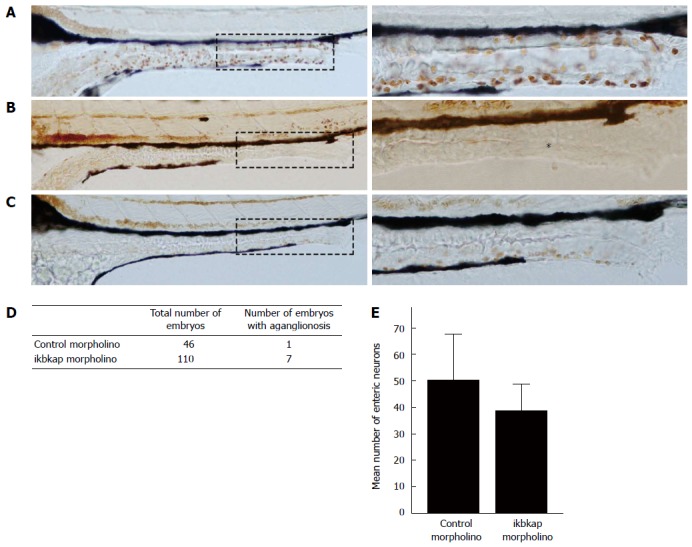
Loss of ikbkap resulted in Hirschsprung disease-like phenotype. A: Control embryo showed normal enteric nervous system (ENS) development with enteric neurons present along the whole intestine (reddish-brown); B: Morphant displayed complete absence of enteric neuron in distal intestine (asterisk); C: Morphant showed milder phenotype of reduced number of enteric neurons. Magnification × 200 and × 400; D: Summary of aganglionosis observed in morphants (n = 110) and control (n = 46). The difference was statistically significant (P < 0.01) by χ2 test; E: Summary on the number of enteric neurons in morphants (n = 17) and control (n = 17). The difference was statistically significant (38.8 ± 9.9 vs 50.2 ± 17.3, P < 0.05) by Student’s t test.
Loss of ikbkap down regulated phox2bb expression
It was reported that knocking down IKBKAP in human neuroblastoma cells resulted in reduced RET expression[16]. We speculated that a similar phenomenon would happen in our embryos injected with ikbkap morpholino, which could account for the reduced enteric neuron number and aganglionosis observed. To test this hypotheses we extracted total RNA from ikbkap morphants and control embryos and checked the expression of ret, phox2bb and sox10 by qualitative RT-PCR. Expression of phox2bb was markedly reduced, while there was a marginal down regulation of ret with sox10 expression unaffected (Figure 3A). We verified the results by q-PCR (Figure 3B). Expression of phox2bb was significantly reduced (P < 0.05). Although an apparent reduction in the level of ret transcript was observed, the difference was not statistically significant.
Figure 3.
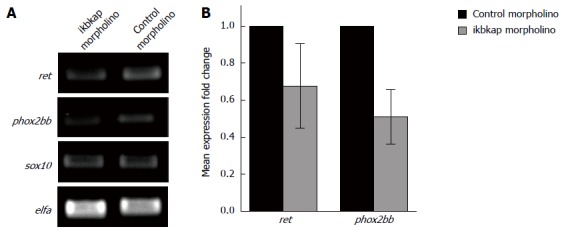
Loss of ikbkap caused phox2bb down-regulation. A: Qualitative real-time polymerase chain reaction for the expressions ret, phox2bb and sox10. elfa served as internal control; B: Quantitative-polymerase chain reaction for ret and phox2bb. Down-regulation of phox2bb was statistically significant (0.51 ± 0.15 vs 1.00 ± 0, P < 0.05) by Student’s t test.
DISCUSSION
The association of IKBKAP to Chinese HSCR patients led to the hypothesis that IKBKAP could contribute to the pathogenesis of HSCR. As shown in the current study, knocking down ikbkap in zebrafish embryos resulted in aganglionosis and a reduced number of enteric neurons, a phenocopy of HSCR in humans. This result indicated that ikbkap is involved in ENS development, and the disruption of ikbkap function results in an HSCR-like phenotype in zebrafish.
The ikbkap morphants showed a down regulation of phox2bb, the ortholog of human PHOX2B. PHOX2B is expressed in NCCs that eventually give rise to mature enteric neurons and glia[33]. It has also been shown that PHOX2B together with SOX10 and NKX2-1 regulate RET expression[34]. Together these pieces of evidence suggest PHOX2B is essential for proper ENS development. Indeed, Phox2b knockout mice or zebrafish display a phenotype reminiscent of that seen in human HSCR patients[33,35]. In humans, PHOX2B mutations are responsible for several neurocristopathies, including neuroblastoma and congenital central hypoventilation syndrome, both of which can have HSCR as part of their phenotypic spectrum[36-38]. The finding of phox2bb down-regulation in our study suggests that ikbkap was acting upstream of phox2bb. What remains unclear is the mechanism by which ikbkap regulates phox2bb expression. ikbkap protein is one of the subunits in the highly-conserved elongator complex that is associated with RNA polymerase type II. The elongator complex is made up of six subunits, and ikbkap acts as a scaffolding protein that holds the complex in its proper configuration[39]. Knocking down ikbkap might disrupt the integrity of the complex and hence the elongation of the target transcripts including phox2bb, thereby reducing their expressions. In addition, increasing evidence shows that the elongator complex is also involved in regulating t-RNA modification and translational control, independently of the proposed regulatory function on transcript elongation. Inactivation of the elongator complex resulted in defective t-RNA modification and reduced expression of target proteins[9,40]. It is therefore plausible that depletion of ikbkap disturbs the translation of target proteins that are involved in regulating phox2bb transcription. Lastly, a report demonstrated that the elongator complex is responsible for genome demethylation during embryogenesis[6]. As DNA demethylation is an important epigenetic control to activate gene transcriptions, failure in the process due to the loss of ikbkap might cause the phox2bb locus to remain methylated and hence reduce its expression. Further research is required to delineate the molecular connection between ikbkap and phox2bb.
It was reported that knocking down IKBKAP in human neuroblastoma cell line SHSY5Y by shRNA caused a significant reduction in the expression of RET[16]. In our study we have partially replicated that result. The level of ret transcript is apparently reduced in the ikbkap morphant, by both qualitative RT-PCR and q-PCR. However, the reduction is not statistically significant. A hypothesis to explain this difference would be that in the in vivo zebrafish model, a feedback mechanism attenuated the effect of ikbkap knock down on ret expression, while such a mechanism might not exist in the in vitro cell line model. We also noted that in the same study, the authors did not report any change in the expression of PHOX2B. This discrepancy has two possible explanations. First, unlike wild type zebrafish, SHSY5Y bore a 20-nt deletion mutation in the exon3 of PHOX2B[41]. This mutated PHOX2B might be more resistant to the effect induced by the loss of IKBKAP and hence its expression would not be altered. Second, as a cell line derived from human neuroblastoma, there might be other genetic changes such as mutations and chromosome rearrangements in the genome that could counteract the knock down of IKBKAP.
In conclusion, our functional analysis in zebrafish embryos confirmed the role of IKBKAP in ENS development.
ACKNOWLEDGMENTS
The authors thank Professor Anskar Leung and Dr. Alvin Ma of the Zebrafish Core Facility, LKS Faculty of Medicine, HKU for their assistance and advice.
COMMENTS
Background
Hirschsprung disease (HSCR), caused by genetic mutations, is a congenital neuropathy characterized by the absence of enteric ganglion along variable length of distal large intestine and results in chronic constipation. Over the years a number of disease-causing mutations have been found but they only account for a portion of HSCR cases, suggesting there are more to be identified.
Research frontiers
To uncover the unknown HSCR-causing mutations, their laboratory fine mapped chromosome region 9q31, which has been reported to be associated with HSCR, and narrowed it down to the gene IKBKAP and confirmed its association with Chinese HSCR patients. IKBKAP encodes a protein that acts a scaffold in the elongator complex, but it is unclear whether this gene is important for enteric nervous system (ENS) development and HSCR.
Innovations and breakthroughs
To discover if IKBKAP is essential for ENS development and contributes to HSCR, authors depleted the zebrafish IKBKAP orthologs by morpholino-mediated gene knockdown. They found out that depletion of ikbkap resulted in HSCR-like phenotype, along with down regulation of phox2bb. Their report is the first to demonstrate the importance of IKBKAP in ENS development.
Applications
This report enhances the understanding on the cause of HSCR and puts IKBKAP a disease-causing gene, which will be useful in the future in developing novel therapeutic strategies and genetic counselling.
Terminology
Hischsprung disease: A congenital neuropathy characterized by the absence of enteric ganglion, causing chronic constipation; IKBKAP: A human gene encoding a scaffold protein in the elongator complex; morpholino: An antisense oligomers that can mediate knockdown of target gene by blocking either protein translation or mRNA splicing.
Peer-review
The manuscript is well organized and clear, represented with graphs and microscopic photos. The article in general was well made with a sound methodology and a good discussion.
Footnotes
Supported by Small Project Funding, the University of Hong Kong, No. 201209176125 to Cheng WWC; Hong Kong Research Grants Council HKU No. 778610M to Tam PKH; Health and Medical Research Fund No. 01121326 to Lui VCH and The University of Hong Kong Genomics Strategic Research Theme.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: May 2, 2014
First decision: May 29, 2014
Article in press: September 16, 2014
P- Reviewer: Bagyánszki M, De Lusong MAA S- Editor: Gou SX L- Editor: A E- Editor: Zhang DN
References
- 1.Young HM, Cane KN, Anderson CR. Development of the autonomic nervous system: a comparative view. Auton Neurosci. 2011;165:10–27. doi: 10.1016/j.autneu.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 2.Tam PK, Garcia-Barceló M. Genetic basis of Hirschsprung’s disease. Pediatr Surg Int. 2009;25:543–558. doi: 10.1007/s00383-009-2402-2. [DOI] [PubMed] [Google Scholar]
- 3.Tang CS, Sribudiani Y, Miao XP, de Vries AR, Burzynski G, So MT, Leon YY, Yip BH, Osinga J, Hui KJ, et al. Fine mapping of the 9q31 Hirschsprung’s disease locus. Hum Genet. 2010;127:675–683. doi: 10.1007/s00439-010-0813-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolk S, Pelet A, Hofstra RM, Angrist M, Salomon R, Croaker D, Buys CH, Lyonnet S, Chakravarti A. A human model for multigenic inheritance: phenotypic expression in Hirschsprung disease requires both the RET gene and a new 9q31 locus. Proc Natl Acad Sci USA. 2000;97:268–273. doi: 10.1073/pnas.97.1.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Otero G, Fellows J, Li Y, de Bizemont T, Dirac AM, Gustafsson CM, Erdjument-Bromage H, Tempst P, Svejstrup JQ. Elongator, a multisubunit component of a novel RNA polymerase II holoenzyme for transcriptional elongation. Mol Cell. 1999;3:109–118. doi: 10.1016/s1097-2765(00)80179-3. [DOI] [PubMed] [Google Scholar]
- 6.Okada Y, Yamagata K, Hong K, Wakayama T, Zhang Y. A role for the elongator complex in zygotic paternal genome demethylation. Nature. 2010;463:554–558. doi: 10.1038/nature08732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holmberg C, Katz S, Lerdrup M, Herdegen T, Jäättelä M, Aronheim A, Kallunki T. A novel specific role for I kappa B kinase complex-associated protein in cytosolic stress signaling. J Biol Chem. 2002;277:31918–31928. doi: 10.1074/jbc.M200719200. [DOI] [PubMed] [Google Scholar]
- 8.Rahl PB, Chen CZ, Collins RN. Elp1p, the yeast homolog of the FD disease syndrome protein, negatively regulates exocytosis independently of transcriptional elongation. Mol Cell. 2005;17:841–853. doi: 10.1016/j.molcel.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 9.Esberg A, Huang B, Johansson MJ, Byström AS. Elevated levels of two tRNA species bypass the requirement for elongator complex in transcription and exocytosis. Mol Cell. 2006;24:139–148. doi: 10.1016/j.molcel.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 10.Creppe C, Malinouskaya L, Volvert M-L, Gillard M, Close P, Malaise O, Laguesse S, Cornez I, Rahmouni S, Ormenese S, et al. Elongator Controls the Migration and Differentiation of Cortical Neurons through Acetylation of α-Tubulin. Cell. 2009;136:551–564. doi: 10.1016/j.cell.2008.11.043. [DOI] [PubMed] [Google Scholar]
- 11.Solinger JA, Paolinelli R, Klöss H, Scorza FB, Marchesi S, Sauder U, Mitsushima D, Capuani F, Stürzenbaum SR, Cassata G. The Caenorhabditis elegans Elongator complex regulates neuronal alpha-tubulin acetylation. PLoS Genet. 2010;6:e1000820. doi: 10.1371/journal.pgen.1000820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johansen LD, Naumanen T, Knudsen A, Westerlund N, Gromova I, Junttila M, Nielsen C, Bøttzauw T, Tolkovsky A, Westermarck J, et al. IKAP localizes to membrane ruffles with filamin A and regulates actin cytoskeleton organization and cell migration. J Cell Sci. 2008;121:854–864. doi: 10.1242/jcs.013722. [DOI] [PubMed] [Google Scholar]
- 13.Cheishvili D, Maayan C, Cohen-Kupiec R, Lefler S, Weil M, Ast G, Razin A. IKAP/Elp1 involvement in cytoskeleton regulation and implication for familial dysautonomia. Hum Mol Genet. 2011;20:1585–1594. doi: 10.1093/hmg/ddr036. [DOI] [PubMed] [Google Scholar]
- 14.Close P, Hawkes N, Cornez I, Creppe C, Lambert CA, Rogister B, Siebenlist U, Merville M-P, Slaugenhaupt SA, Bours V, et al. Transcription Impairment and Cell Migration Defects in Elongator-Depleted Cells: Implication for Familial Dysautonomia. Mol Cell. 2006;22:521–531. doi: 10.1016/j.molcel.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 15.Hunnicutt BJ, Chaverra M, George L, Lefcort F. IKAP/Elp1 is required in vivo for neurogenesis and neuronal survival, but not for neural crest migration. PLoS One. 2012;7:e32050. doi: 10.1371/journal.pone.0032050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cohen-Kupiec R, Pasmanik-Chor M, Oron-Karni V, Weil M. Effects of IKAP/hELP1 deficiency on gene expression in differentiating neuroblastoma cells: implications for familial dysautonomia. PLoS One. 2011;6:e19147. doi: 10.1371/journal.pone.0019147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naumanen T, Johansen LD, Coffey ET, Kallunki T. Loss-of-function of IKAP/ELP1: could neuronal migration defect underlie familial dysautonomia? Cell Adh Migr. 2008;2:236–239. doi: 10.4161/cam.2.4.6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Axelrod FB. Familial dysautonomia. Muscle Nerve. 2004;29:352–363. doi: 10.1002/mus.10499. [DOI] [PubMed] [Google Scholar]
- 19.Bar-Shai A, Maayan C, Vromen A, Udassin R, Nissan A, Freund HR, Hanani M. Decreased density of ganglia and neurons in the myenteric plexus of familial dysautonomia patients. J Neurol Sci. 2004;220:89–94. doi: 10.1016/j.jns.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 20.Azizi E, Berlowitz I, Vinograd I, Reif R, Mundel G. Congenital megacolon associated with familial dysautonomia. Eur J Pediatr. 1984;142:68–69. doi: 10.1007/BF00442596. [DOI] [PubMed] [Google Scholar]
- 21.Chen YT, Hims MM, Shetty RS, Mull J, Liu L, Leyne M, Slaugenhaupt SA. Loss of Mouse Ikbkap, a Subunit of Elongator, Leads to Transcriptional Deficits and Embryonic Lethality That Can Be Rescued by Human IKBKAP. Mol Cell Biol. 2008;29:736–744. doi: 10.1128/MCB.01313-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dietrich P, Alli S, Shanmugasundaram R, Dragatsis I. IKAP expression levels modulate disease severity in a mouse model of familial dysautonomia. Hum Mol Genet. 2012;21:5078–5090. doi: 10.1093/hmg/dds354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dietrich P, Yue J, E S, Dragatsis I. Deletion of exon 20 of the Familial Dysautonomia gene Ikbkap in mice causes developmental delay, cardiovascular defects, and early embryonic lethality. PLoS One. 2011;6:e27015. doi: 10.1371/journal.pone.0027015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hims MM, Shetty RS, Pickel J, Mull J, Leyne M, Liu L, Gusella JF, Slaugenhaupt SA. A humanized IKBKAP transgenic mouse models a tissue-specific human splicing defect. Genomics. 2007;90:389–396. doi: 10.1016/j.ygeno.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.George L, Chaverra M, Wolfe L, Thorne J, Close-Davis M, Eibs A, Riojas V, Grindeland A, Orr M, Carlson GA, et al. Familial dysautonomia model reveals Ikbkap deletion causes apoptosis of Pax3+ progenitors and peripheral neurons. Proc Natl Acad Sci USA. 2013;110:18698–18703. doi: 10.1073/pnas.1308596110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bochner R, Ziv Y, Zeevi D, Donyo M, Abraham L, Ashery-Padan R, Ast G. Phosphatidylserine increases IKBKAP levels in a humanized knock-in IKBKAP mouse model. Hum Mol Genet. 2013;22:2785–2794. doi: 10.1093/hmg/ddt126. [DOI] [PubMed] [Google Scholar]
- 27.Westerfield M. The zebrafish book. Eugene, OR: University of Oregon Press; 2000. [Google Scholar]
- 28.Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 29.Rosen JN, Sweeney MF, Mably JD. Microinjection of zebrafish embryos to analyze gene function. J Vis Exp. 2009;(25):1115. doi: 10.3791/1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuhlman J, Eisen JS. Genetic screen for mutations affecting development and function of the enteric nervous system. Dev Dyn. 2007;236:118–127. doi: 10.1002/dvdy.21033. [DOI] [PubMed] [Google Scholar]
- 31.Miao X, Leon TY, Ngan ES, So MT, Yuan ZW, Lui VC, Chen Y, Wong KK, Tam PK, Garcia-Barceló M. Reduced RET expression in gut tissue of individuals carrying risk alleles of Hirschsprung’s disease. Hum Mol Genet. 2010;19:1461–1467. doi: 10.1093/hmg/ddq020. [DOI] [PubMed] [Google Scholar]
- 32.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 33.Pattyn A, Morin X, Cremer H, Goridis C, Brunet JF. The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature. 1999;399:366–370. doi: 10.1038/20700. [DOI] [PubMed] [Google Scholar]
- 34.Leon TY, Ngan ES, Poon HC, So MT, Lui VC, Tam PK, Garcia-Barcelo MM. Transcriptional regulation of RET by Nkx2-1, Phox2b, Sox10, and Pax3. J Pediatr Surg. 2009;44:1904–1912. doi: 10.1016/j.jpedsurg.2008.11.055. [DOI] [PubMed] [Google Scholar]
- 35.Elworthy S, Pinto JP, Pettifer A, Cancela ML, Kelsh RN. Phox2b function in the enteric nervous system is conserved in zebrafish and is sox10-dependent. Mech Dev. 2005;122:659–669. doi: 10.1016/j.mod.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 36.Amiel J, Laudier B, Attié-Bitach T, Trang H, de Pontual L, Gener B, Trochet D, Etchevers H, Ray P, Simonneau M, et al. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat Genet. 2003;33:459–461. doi: 10.1038/ng1130. [DOI] [PubMed] [Google Scholar]
- 37.Trochet D, Bourdeaut F, Janoueix-Lerosey I, Deville A, de Pontual L, Schleiermacher G, Coze C, Philip N, Frébourg T, Munnich A, et al. Germline mutations of the paired-like homeobox 2B (PHOX2B) gene in neuroblastoma. Am J Hum Genet. 2004;74:761–764. doi: 10.1086/383253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berry-Kravis EM, Zhou L, Rand CM, Weese-Mayer DE. Congenital central hypoventilation syndrome: PHOX2B mutations and phenotype. Am J Respir Crit Care Med. 2006;174:1139–1144. doi: 10.1164/rccm.200602-305OC. [DOI] [PubMed] [Google Scholar]
- 39.Glatt S, Séraphin B, Müller CW. Elongator: transcriptional or translational regulator? Transcription. 2012;3:273–276. doi: 10.4161/trns.21525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bauer F, Matsuyama A, Candiracci J, Dieu M, Scheliga J, Wolf DA, Yoshida M, Hermand D. Translational control of cell division by Elongator. Cell Rep. 2012;1:424–433. doi: 10.1016/j.celrep.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Limpt V, Schramm A, van Lakeman A, Sluis P, Chan A, van Noesel M, Baas F, Caron H, Eggert A, Versteeg R. The Phox2B homeobox gene is mutated in sporadic neuroblastomas. Oncogene. 2004;23:9280–9288. doi: 10.1038/sj.onc.1208157. [DOI] [PubMed] [Google Scholar]