

Abstract
Mycobacterium abscessus (M.abs) is a rapidly growing mycobacterial species that infects macrophages, and is an important pathogen in patients with cystic fibrosis. We studied the early stages of M.abs infection of macrophages, with emphasis on the role of heme-oxygenase-1 (HO-1) in this infection. THP-1 cells were activated using TPA into macrophage-like cells and infected with M.abs for different time points. M.abs infection robustly induced HO-1 expression in the THP-1 cells. Production of HO-1 was p38 MAPK-dependent, as p38 inhibitors suppressed HO-1 induction. Pretreatment with HO-1 inhibitors tin-protoporphyrin (SnPP) significantly inhibited M.abs growth inside macrophages. Furthermore, inhibiting HO-1 using HO-1 siRNA or the HO-1 upstream signaling molecule; Nrf2 using Nrf2 siRNA resulted in similar inhibition of M.abs. In contrast, inducing HO-1 did not increase M.abs intracellular growth above control. Products of HO-1 metabolism of heme are bilirubin, biliverdin, carbon monoxide (CO) and iron. The addition of either bilirubin or biliverdin, but not CO, completely restored the SnPP inhibitory effect and partially that with HO-1 siRNA. To understand the mechanisms, we used Syto-62 labeled M.abs to infect macrophages. Interestingly, HO-1 inhibition promoted M.abs-containing phagosome fusion with lysosomes, which should enhance M.abs killing. M.abs infection enhanced THP-1 ROS production as demonstrated by increased DHE, DCF fluorescence, and EPR signal. HO-1 inhibition further increased ROS production in infected macrophages. Our results indicate that HO-1 induction is important for M.abs growth during the early stages of infection, and that the HO-1 products bilirubin and biliverdin, perhaps through modulation of intracellular ROS levels, may be involved.
Keywords: Mycobacterium abscessus, HO-1, THP-1 cells, Oxidative stress
Graphical abstract
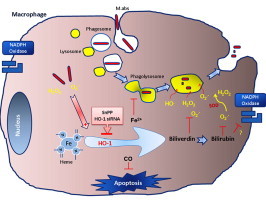
Highlights
-
•
HO-1 induction is important for Mycobacterium abscessus growth inside infected macrophages during the early stages of infection.
-
•
Reducing HO-1 products may enhance the ability of the macrophage to control Mycobacterium abscessus infection.
-
•
HO-1 inhibition increases phagosome–lysosome fusion and thus Mycobacterium abscessus killing.
Introduction
Mycobacterium abscessus (M.abs) is a rapidly growing non-tuberculous mycobacterial (NTM) species that infects macrophages of the lungs and skin and causes a variety of clinical syndromes in humans [1], [2]. It has recently emerged as an important pathogen in patients with cystic fibrosis (CF), causing severe lung disease [3] and multiple complications that prevent lung transplantation [4]. Moreover, despite conventional cross-infection prevention procedures, frequent transmission of multidrug resistant NTM between patients with CF still exists [5].
Heme oxygenase-1 (HO-1) – also known as heat-shock protein 32 – is the rate-controlling enzyme of cellular heme catabolism. This microsomal enzyme acts on heme moieties to produce equimolar amounts of carbon monoxide, iron (Fe), and biliverdin that is in turn converted to bilirubin by biliverdin reductase [6], [7]. The Fe is then stored in ferritin, limiting its ability to participate as a catalyst through Fenton chemistry for production of cytotoxic free radicals [8]. Both biliverdin and bilirubin are thought to play an antioxidant role [9]. It was shown that HO-1 is induced by a variety of stimuli, such as ROS, viral infection and bacterial endotoxins, and appears to be protective in a variety of inflammatory disease states [10], [11], [12] due to its ability to inhibit inflammation and oxidative stress [13]. Moreover, induction of HO-1 suppresses apoptotic cell death through activation of MAPK and PI3K pathways with possible involvement of CO [14], [15], [16], [17]. In THP-1 cells, HO-1 induction counteracted the effect of TNF-induced cell death via Nrf2 activation [18]. This is potentially of importance to mycobacterial infection as it appears that macrophage apoptosis contributes to host defense [19]. The role of CO in mycobacterial infection has been described previously. It was shown that Mycobacterium tuberculosis (M.tb) senses host-derived CO produced by HO-1 induction during macrophages infection [20], and CO activates the expression of dormancy (Dos) regulon [21], and other CO resistance genes such as cor, that protect bacteria from host-derived CO [22], and thus enhances mycobacterial survival in vivo [20], [21], [22]. We have also shown that acquiring Fe is critical to the metabolism and growth of mycobacteria and that limiting Fe availability could be a strategy for novel antimicrobial agents [23], [24].
Little research has been conducted on M.abs pathogenesis. Like other pathogenic mycobacterial species, M.abs infects and multiplies within human macrophages, reflecting their ability to evade macrophage antimicrobial systems. Therefore, understanding the mechanism whereby M.abs successfully infect macrophages and finding approaches to reduce macrophage susceptibility to infection could lead to the development of novel antimicrobial agents to combat the growing clinical problem of multidrug resistant M.abs. The goal of this study is to assess the link between HO-1 and M.abs infection of human macrophages and understand the mechanisms by which HO-1 controls M.abs proliferation inside the infected macrophages.
Materials and methods
Materials
The p38 inhibitors; SB202190 hydrochloride was obtained from (Santa Cruz Biotechnology, Inc., Dallas, TX) and SB203580 from Cell Signaling Technology, Danvers, MA. The carbon monoxide (CO) releasing molecule (tricarbonylchloro(glycinato)ruthenium (II)) was purchased from Sigma, St. Louis, MO. Heme oxygenase-1 siRNA (h), Nrf2 siRNA (h), control siRNA, tin protoporphyrin IX dichloride, bilirubin, biliverdin hydrochloride, protoporphyrin IX cobalt chloride were all obtained from Santa Cruz Biotechnology.
Cell culture
THP-1 cells, a human monocyte-derived cell line, were obtained from the American Type Culture Collection (ATCC, Manassas, VA). The cells were grown in suspension in RPMI medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Thermo Scientific, Waltham, MA). The cells were cultured in 24-well tissue culture flask at a density of 106 cells/well in the presence of 15 ng/ml of 12-O-tetradecanoylphorbol-13-acetate (TPA; Cell Signaling Technology, Danvers, MA) for 24 h to differentiate the cells into a macrophage-like phenotype that adhere to the tissue culture plate. After infection with M.abs (MOI 5), the cells were placed in RPMI medium with 10% FBS and 30 µg/ml gentamicin to inhibit extracellular replication of M.abs. To confirm that SnPP was not cytotoxic at the concentrations that were used, THP-1 cell viability was tested by flow cytometer in the presence of 7-aminoactinomycin D (7-AAD). No toxicity of SnPP was detected in infected or control cells at 24 h.
Cytotoxicity assay
Cytotoxicity was measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay using CellTiter 96 Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI).
Electron paramagnetic resonance spectroscopy
EPR spectroscopy was used to measure superoxide levels in M.abs-infected THP-1 cells. THP-1 cells were activated to macrophages and infected with M.abs for 1 h. Cells were then washed and incubated with antibiotic-free media for different time points. Treated uninfected cells were used as controls. Cells were incubated (30 min at 37 °C) with CMH (200 µM), a cell permeable, superoxide-sensitive spin probe in EPR buffer (pH 7.4). EPR buffer is a Krebs–HEPES buffer consisting of (in mM): 99 NaCl, 4.69 KCl, 2.5 CaCl2, 1.2 MgSO4, 25 NaHCO3, 1.03 KH2PO4, 5.6 d-glucose, 20 HEPES and supplemented with the metal chelators DETC (5 µM) and DF (25 µM). After CMH incubation, macrophages were collected in a cell suspension, and 50 µl of the cell suspension was placed into the Bruker Escan EPR spectrometer. The following EPR settings were used for all experiments: field sweep width, 60.0 G; microwave frequency, 9.75 GHz; microwave power, 21.90 mW; modulation amplitude, 2.37 G; conversion time, 10.24 ms; time constant, 40.96 ms. Levels of superoxide were normalized to the number of macrophages in each sample.
In situ ROS studies
In situ staining of superoxide (O2•−) and H2O2 levels were determined using the superoxide indicator dihydroethidium dihydroethidium (DHE) and the ROS indicator 5-(6)-chloromethyl-20,70-dichlorodihydrofluorescein diacetate (CM-H2DCFDA). M.abs bacteria were labeled with Syto-62 according to manufacturer's instruction (Invitrogen, Grand Island, NY). TPA-stimulated THP-1 cells were grown on a glass chamber slide and were infected with Syto-62-labeled M.abs for 1 h, and incubated with media for 4 h at CO2 incubator. Thirty minutes before the infection was complete, DHE, and DCF were added to the assigned chambers. After infection was complete, the medium was removed, and chambers were washed, and mounted with Vectasheild mounting medium with DAPI (Vector Laboratories, Burlingame, CA). Images were viewed using Zeiss 510 Meta Confocal Laser Scanning Microscope.
Western immunoblotting
Total protein lysates were prepared in RIPA buffer containing protease inhibitors (Thermo Scientific, Rockford, IL). Lysates were mixed with equal volume of 2× Laemmli loading dye (Bio-Rad, Hercules, CA), boiled for 5 min at 95 °C, and loaded onto SDS-PAGE gels. After running, proteins were transferred to PVDF membranes, blocked with 5% milk in TBST, and probed with primary antibodies (p38 MAPK, Phospho-p38 MAPK, Cell Signaling Technology, Danvers, MA, and Anti-MnSOD, Anti-Catalase, Millipore, Billerica, MA) overnight at 4 °C with constant rocking. Membranes were then washed three times with TBST, incubated with secondary antibodies for 1 h at room temperature, washed three times with TBST, and protein were visualized using Pierce chemiluminescence reagents (Rockford, IL). Densitometry analyses were performed by NIH ImageJ.
Colony forming unit assay
Colony-forming units (CFUs) for M.abs present within THP-1 cells were determined as described previously [23]. In brief, cell culture supernatants were removed, and the plates were washed three times in sterile media. Chilled, chelexed, distilled water was added (0.3 ml/well) and plates were incubated on ice for 10 mins. After that, 1.2 ml lysis buffer (0.05% SDS, 5.0% BSA in Fe-free 7H9) was added to each well, and contents were scraped from plates, transferred to sterile tubes and centrifuged at 14,000g for 15 min at 4 °C. Bacteria were resuspended in 200 µl sterile Fe-free 7H9, and diluted for plating on tryptic soy agar plates (Remel, Lenexa, KS) for 3–5 days at 37 °C. The number of cfu on the plates was counted and the mean CFU was calculated.
Phagosome–lysosomes fusion studies
Macrophage-differentiated THP-1 cell were grown on a glass chamber slide and were infected with Syto-62 labeled M.abs for 1 h in antibiotics-free RPMI medium for 4 h. Two hours before the infection was complete, LysoTracker Red DND-99 (50 nM) was added to the cells to label the lysosomes (Invitrogen, Grand Island, NY). After infection was complete, the medium was removed and cells were washed and mounted with Vectashield mounting medium with DAPI (Vector Laboratories, Burlingame, CA). Co-localization and enumeration studies were performed on Zeiss 510 Meta Confocal Laser Scanning Microscope equipped with 4 lasers: a Blue Diode 405 nm; an Argon Laser 458/477/488,514 nm; a DPSS 561 nm and a HeNe 633 nm. Both, cells containing bacteria only and cells containing bacteria that co-localize with a lysosome were counted in controls and HO-1 inhibited cells. At least 50 cells were counted per condition in each experiment, and at least three independent experiments were performed. The results were expressed as % co-localization (number of cells that contain co-localized bacteria with lysosomes as percent of total cells counted).
SOD, catalase activity and GSH assays
Activity of antioxidant enzymes in macrophages was measured using the OxiSelect Catalase Activity Assay Kit (Cell Biolabs Inc, San Diego, CA) for catalase, GSSG/GSH Quantification kit (DOJINDO Inc. Rockville, MD) for total, oxidized and reduced GSH, and SOD Assay Kit-WST (DOJINDO Inc. Rockville, MD) to measure intracellular SOD activity according to the manufacturers' guidelines.
Analysis of data
Means and standard deviations were calculated from independent experiments. Statistical analysis was done using GraphPad Prism version 5 for Windows (GraphPad Software, San Diego, CA). Differences between three or more means were determined using one-way analysis of variance (ANOVA) with Bonferroni post-hoc tests. Error bars represent mean±SEM. All statistical analyses were considered significant at p<0.05. Each experiment was analyzed relative to its own control group(s).
Results
M.abs infection induces HO-1 expression in macrophages
The role of HO-1 in a variety of infections has been described before [12], [25], [26], [27], [28]. Therefore, we sought to study the level of HO-1 expression in M.abs infected and non-infected macrophages. To do so, we used the human monocytic cell line THP-1. Cells were differentiated into macrophages using TPA and then infected with M.abs. M.abs grew readily inside macrophages as shown by monitoring CFU at day 0 till day 1 (Fig. 1A). As shown in Fig. 1B, M.abs infection significantly increased HO-1 expression in a time-dependent manner, beginning 4 h post-infection (p<0.001) (Fig. 1B, C).
Fig. 1.
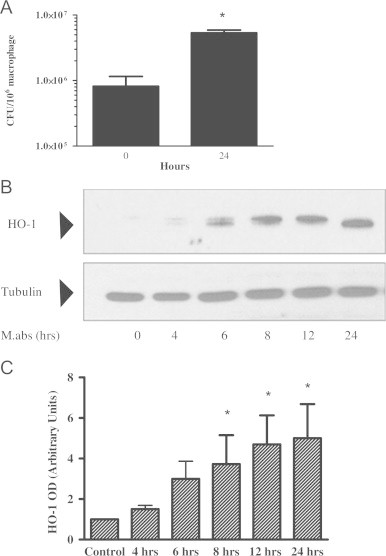
M.abs increased HO-1 expression. TPA-differentiated THP-1 cells were infected with M.abs for 1 h, washed and incubated in media containing gentamicin until harvested at different time points. M.abs cfu were determined and cells were prepared for immunoblotting as described in Materials and methods. (A) M.abs showed robust intracellular growth, as shown by the increase in CFU at days 0, and 1 post-infection. (B) Proteins were run and HO-1 visualized by immunoblotting. Results showed induction of HO-1 after infection. (C) Densitometric analysis of HO-1 bands in (B). M.abs infection resulted in a significant increase in expression of HO-1 (n=4, *indicates p<0.001).
M.abs induces HO-1 expression via p38 MAPK pathway
It has been shown that HO-1 induction is dependent on PI3K/p38MAPK signaling pathway in many cells, including THP-1 cells [29], [30]. When cells were collected at different times after M.abs infection, increased phosphorylated p38MAPK was shown starting at 4 h as shown in Fig. 2A. This is consistent with involvement of the p38MAPK signaling pathway in M.abs HO-1 induction of HO-1. Supporting this possibility, the administration of either of the p38 MAPK inhibitors SB203580 (10 µM), or SB202190 (10 µM) reduced M.abs-induced HO-1 protein expression (Fig. 2B).
Fig. 2.
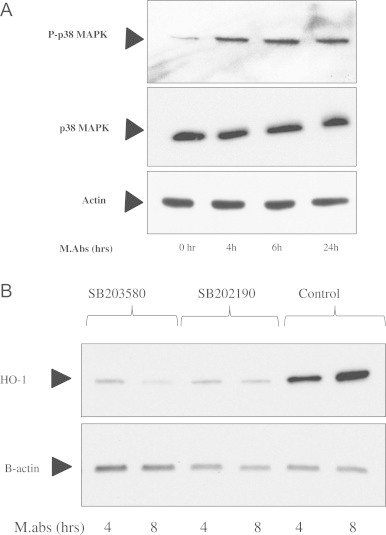
p38 is involved in M.abs-mediated HO-1 induction signal transduction pathway. (A) TPA-differentiated THP-1 cells were infected with M.abs for 1 h, washed and incubated in media as shown in materials and methods. Cells were collected at different time points. Phosphorylated and total forms of p38MAPK were determined by immunoblotting. (B) Addition of the p38 inhibitors; SB203580 and SB202190 reduced HO-1 induction by M.abs.
HO-1 inhibition suppresses M.abs growth in macrophages
The induction of HO-1 could be very important for bacterial survival inside the macrophages. HO-1 induces Fe2+ storage as ferritin, thus limiting the generation of free radicals from free Fe2+ by Fenton reaction [8], [31]. Moreover, both biliverdin and bilirubin play an antioxidant role that may contribute to the overall cytoprotective effect of HO-1 [9]. Iron is important for mycobacterial growth and virulence inside infected macrophages. Our laboratory and others have shown that controlling iron availability could inhibit the growth of Mycobacterium tuberculosis and other bacteria growing within human macrophages [23], [32], [33], [34].
Therefore, we examined the effect of altering HO-1 expression and/or HO-1 activity on M.abs growth inside infected macrophages. As shown in Fig. 3, inhibiting HO-1 activity with SnPP significantly (p<0.05) decreased the number of M.abs CFU at 24 h, indicating suppression of bacterial growth in macrophages (Fig. 3A). The CFU determination was made at 24 h rather than 48 h because the infected THP-1 cells began to show evidence of detachment after 24 h, which could confound data interpretation.
Fig. 3.
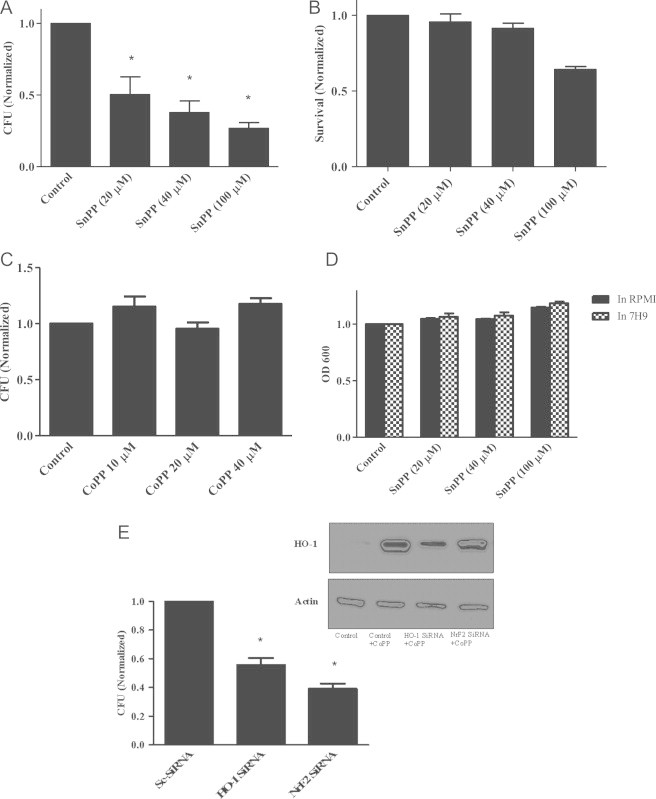
HO-1 inhibition suppresses M.abs growth in macrophages. (A) TPA-differentiated THP-1 cells were cultured in the presence of the HO-1 inhibitor SnPP for 24 h, infected with M.abs for 1 h, and collected 24 h post-infection for CFU determination, as described in materials and methods. SnPP significantly inhibited M.abs growth as detected by CFU (p<0.05, n=5). Results obtained were normalized to vehicle-treated controls. B) TPA-differentiated THP-1 cells were cultured in the presence of different concentrations of SnPP for 48 h and assayed for toxicity by MTT assay. No toxicity was seen using SnPP concentrations up to 40 µM for 48 h. Results were normalized to vehicle-treated controls. (C) No growth inhibition of M.abs was detected when bacteria were cultured in RPMI or 7H9 in the presence of different concentrations of SnPP. (D) TPA-differentiated THP-1 cells were cultured in the presence of different concentrations of CoPP for 24 h, and then infected with M.abs for 1 h. Cells were washed, and cultured in the presence of CoPP for 24 h. M.abs cfu were counted as described in materials and methods. There was no significant difference in M.abs growth in control versus CoPP treated THP-1 cells (n=3, p>0.05). CFU counts were normalized to vehicle-treated controls. (E) TPA-differentiated THP-1 cells were cultured in the presence of siRNA for 24 h and then infected with M.abs 1 hr. Cells were washed and cultured for 24 h before collected for CFU assay as described in materials and methods. Significant inhibition of M.abs growth was observed with treatment of THP-1 cells by HO-1 siRNA (p<0.05, n=3) or Nrf2 siRNA (p<0.05, n=2). CFU counts were normalized to Sc-siRNA-treated controls infected with M.abs. The inset shows decreased HO-1 protein level in the presence of HO-1 siRNA or Nrf2 siRNA. Cells were collected after 24 h of culture in the presence of HO-1 siRNA, Nrf2 siRNA, or CoPP (HO-1 inducer). Control cells treated with HO-1 siRNA shows no detectable protein by immunoblot (not shown). CoPP slightly induced HO-1 in the presence of siRNAs as determined by immunoblot (p<0.05, n=3).
It is important to mention that in these initial experiments, the SnPP was added to THP-1 cell cultures 24 h before infection, and added again after the infection until the time of cell collection. However, when SnPP was added at the time of infection, no inhibition of M.abs growth was observed (not shown). Furthermore, SnPP had no effect on M.abs phagocytosis as evidenced by a similar level of CFU on day 0 post-infection (data not shown). This suggests that the impact of HO-1 activity on M.abs growth is due to a process that occurs early after infection of the host cell.
To rule out any inhibitory effects of SnPP on M.abs or THP-1 cells, cells and bacteria were cultured in the presence of different concentrations of SnPP and monitored for toxicity. As shown in Fig. 3, no toxic effects were observed on either the THP-1 cells (B) or on the bacteria (C) when cultured with SnPP, as determined by the MTT test or OD600 absorbance, respectively. To further confirm our results and ensure that no toxic concentrations of SnPP were used, cells viability was assessed by flow cytometer in the presence of 7-AAD. No significant toxicity of SnPP, was seen in either control or M.abs-infected THP-1 cells at 24 h (not shown). Moreover, when similar experiments were performed in the presence of different concentrations of the HO-1 inducer CoPP, no significant increase in M.abs CFU were detected above untreated control, as shown in Fig. 3D (p>0.05).
To further confirm our results, we inhibited HO-1 by HO-1 siRNA. Inhibiting HO-1 expression by HO-1 siRNA suppressed M.abs growth inside macrophage-differentiated THP-1 cells as demonstrated in Fig. 3E (p<0.05). Moreover, inhibiting NrF2, an upstream signaling factor for HO-1, resulted in a similar effect (Fig. 3E). Collectively, these results show HO-1 suppression at the time of infection inhibits M.abs growth in macrophages.
Impact of the HO-1 products bilirubin, biliverdin, and carbon monoxide on M.abs growth
The mechanism whereby M.abs HO-1 upregulation benefits M.abs intracellular survival was unclear. It has been shown that HO-1 products such as bilirubin, CO, and biliverdin reduce HCV replication in vitro [35], [36]. Therefore, we sought to investigate the effect of HO-1 products on M.abs growth inside infected macrophages. In these set of experiments before infecting them with M.abs we inhibited THP-1 cell HO-1 by SnPP or HO-1 siRNA for 24 h to prevent any increase in HO-1 activity following M.abs infection. The cells were then infected with M.abs, and either bilirubin, biliverdin, or CO was added to the infected cells for another 24 h. Both bilirubin and biliverdin, but not CO, increased the number of CFU recovered from infected macrophages 24 h post-infection (Fig. 4). Bilirubin and biliverdin completely restored the CFU count to control levels with SnPP (Fig. 4A) and partially with HO-1 siRNA (Fig. 4B) treatment. The ability of bilirubin and biliverdin to restore CFU counts could be due to their antioxidant activity.
Fig. 4.
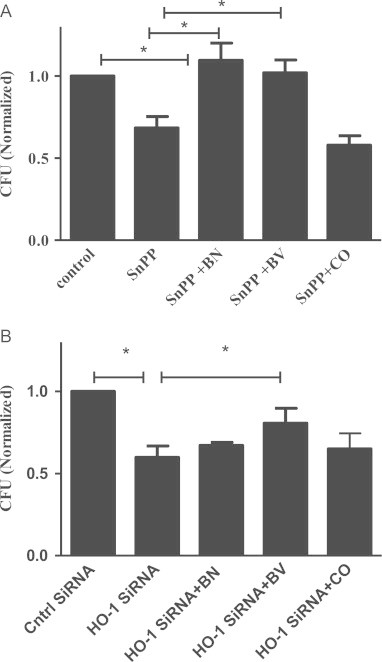
HO-1 enzymatic products restore M.abs growth reduced by HO-1 inhibition. TPA-differentiated THP-1 cells were cultured in the presence of SnPP (30 µM) or HO-1 siRNA for 24 h. Cells were then infected with M.abs and cultured in media with the addition of either bilirubin (10 µM), biliverdin (10 µM), or CO (10 µM) for 24 h. Cells were collected and M.abs CFU assayed. CFU counts were normalized to vehicle-treated controls. (A) Both bilirubin and biliverdin significantly increased CFU in SnPP-treated cells (n=7, p<0.05). (B) Biliverdin significantly increased CFU HO-1 siRNA-treated cells (n=5, p<0.05).
HO-1 inhibition increases phagosome–lysosome fusion
The phagocytosis of mycobacteria by macrophages is followed by phagosome–lysosome fusion [37], [38]. The ability of mycobacteria to inhibit phagosome maturation into a phagolysosome is an important survival mechanisms used by pathogenic mycobacteria [39], [40]. Therefore, we investigated whether HO-1 induction was involved in the ability of M.abs to inhibit phagosome–lysosome fusion. TPA-differentiated THP-1 cells were cultured in the presence of SnPP and HO-1 siRNA for 24 h. Macrophages were then infected for 4 h with Syto-62-labeled M.abs. Two hours before the completion of infection, LysoTracker was added to the cells to label the cells lysosomes. The cells were then viewed under a confocal microscope and the amount of co-localization of bacteria with lysosomes was calculated (Fig. 5). At 4 h after infection there was more co-localization observed in the macrophages with HO-1 inhibition as compared to controls (Fig. 5). This result is consistent with the possibility that HO-1 activity contributes to intracellular M.abs survival by playing a role in limiting phagosome–lysosome fusion.
Fig. 5.
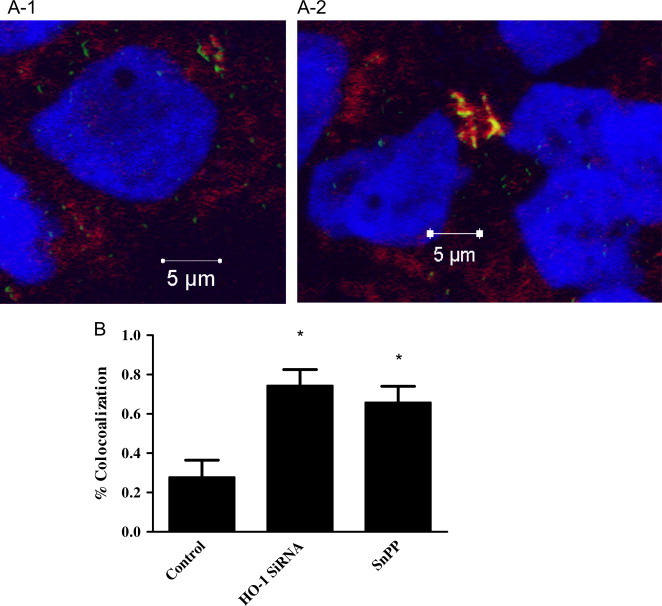
HO-1 inhibition increases M.abs–lysosomal fusion in macrophages. TPA-differentiated THP-1 cells were cultured in the presence of SnPP or HO-1 siRNA for 24 h. Macrophages were infected with Syto-62 labeled M.abs for 1 h, and incubated for 4 h. Lysotracker was added 2 h before preparation for confocal microscopy. (A-1) control cells infected with M.abs (shown in green), lysotracker is shown in red, and nucleus in blue. (A-2) M.abs infection in the presence of HO-1 siRNA. M.abs–lysosomal co-localization is shown in yellow shows (n=5). (B) Percent co-localization in SnPP and HO-1 siRNA treated macrophages as compared to control. HO-1 inhibition resulted in a significant increase in the percentage of M.abs showing co-localization with lysosomes (n=6, p<0.05).
M.abs infection induces oxidative stress and increases ROS production
HO-1 is also known to serve an anti-inflammatory role by decreasing intracellular oxidative stress. To assess the oxidative status of THP-1 cells infected with M.abs, TPA-differentiated THP-1 cells were infected with labeled M.abs for different time points. As shown in Fig. 6, when compared to non-infected control macrophages, M.abs infection increased intracellular superoxide (O2•−) and H2O2 production as determined by DHE and DCFH2, respectively (Fig. 6). To confirm the increased ROS production, M.abs infected macrophages were tested by electron paramagnetic resonance (EPR) using the O2•− sensitive spin probe; CMH. As shown in Fig. 8B, a significant increase in CMH radical was detected after 4 h of M.abs infection, indicating more O2•− production (p<0.05) (Fig. 8B).
Fig. 6.
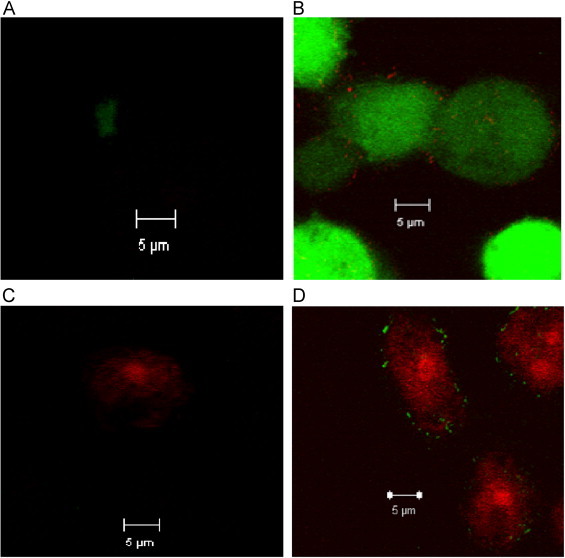
M.abs infection increases THP-1 cell ROS production. TPA-differentiated THP-1 cells were infected with Syto-62 labeled M.abs for 4 h, and loaded with 10 µM DCFH2 or DHE for the 30 min, washed and prepared for confocal microscopy study. Panels A and C are control cells with no infection showing minimal DCF or DHE fluorescence. Panel B shows increased DCF fluorescence and Panel D shows increased DHE fluorescence after 4 h of infection. Syto-62 labeled M.abs bacteria are shown inside the cells in red or green (n=4).
Fig. 8.
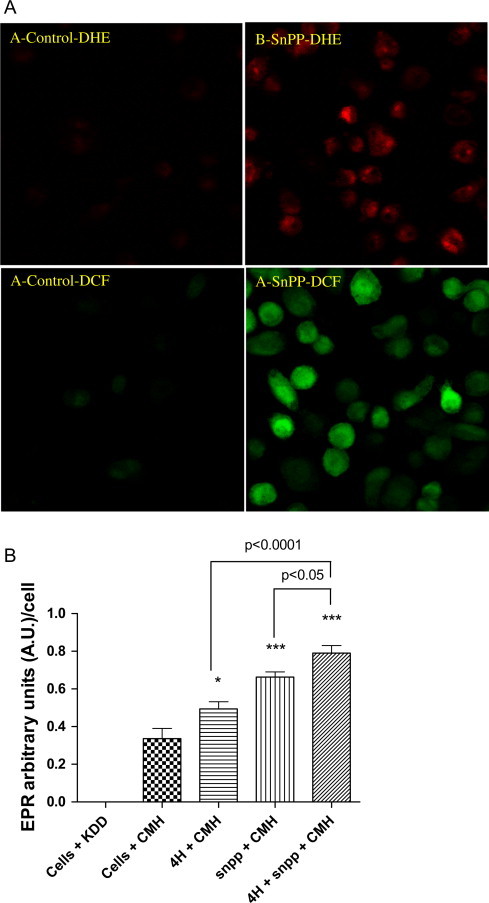
HO-1 inhibition increase ROS production. TPA-differentiated THP-1 cells were infected with M.abs for 1 h, washed and cultured with media for 4 h. Cells were then loaded with DCFH2 or DHE for the 30 min, washed and prepared for confocal microscopy study. (A) HO-1 inhibition using SnPP increased ROS production as shown by DHE (A and B) and DCF fluorescence (C and D). (B) TPA-differentiated THP-1 cells were infected with M.abs for 4 h. Cells were then collected and studied for O2•− production by EPR as described in materials and methods. CMH is the spin probe 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine hydrochloride, KDD is the EPR buffer. A significant increase in O2•− production was observed at 4 h post-M.abs infection (p<0.05), and when infection is combined with HO-1 inhibition (p<0.0001). Inhibiting HO-1 also increased ROS production (p<0.0001) (n=5).
Since, M.abs increased ROS production, we sought to assess the levels of antioxidant enzymes and compounds in M.abs infected macrophages. We first measured the levels of MnSOD and catalase protein levels in M.abs infected macrophages and controls at different time points. As shown in Fig. 7A, there were no differences between the levels of MnSOD and catalase protein over time (A). Similarly, there were no significant differences in either SOD (Fig. 7B) or catalase activity (C) at the different time points post-infection with M.abs (p>0.05).
Fig. 7.
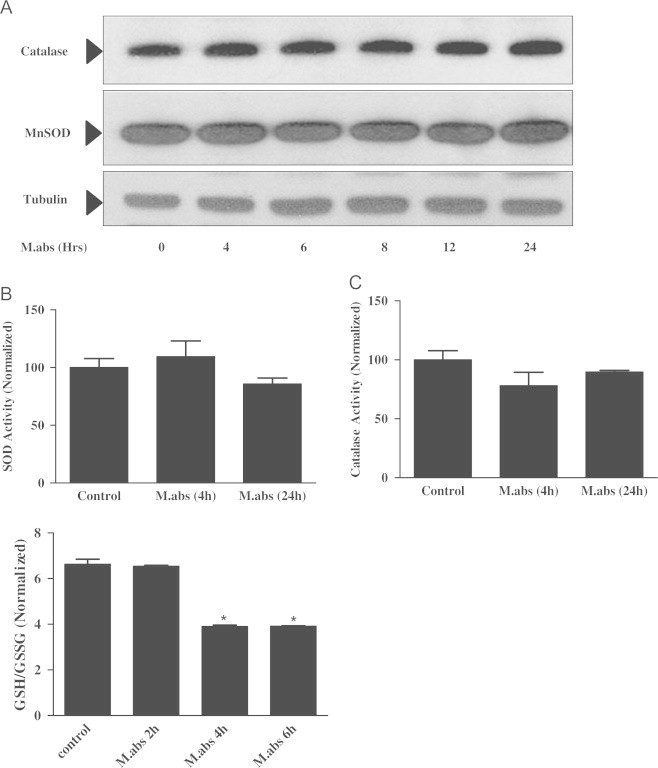
M.abs infection increases THP-1 cell GSH turnover, but not MnSOD or catalase protein expression or activity. TPA-differentiated THP-1 cells were infected with M.abs for 1 h, washed and cultured in media until collected at different time points. Immunoblotting and enzyme activity were determined as described in materials and methods. (A) No difference in catalase or MnSOD protein (Panel A), SOD activity (Panel B, n=3, p>0.05) or catalase activity (Panel C, n=3, p>0.05) was observed between M.abs-infected and non-infected THP-1 cells at different points. (Panel D) Cells were then collected and studied for reduced glutathione (GSH), and oxidized glutathione (GSSG) levels as described in materials and methods. M.abs significantly decreased the ratio of GSH/GSSG, which reflects glutathione turnover (n=3, p<0.05). Data counts in B–D were normalized to uninfected controls.
However, the picture was not the same when we measured the levels of reduced and oxidized GSH in M.abs infected and non-infected THP-1 cells. As observed in Fig. 7D, a decrease in the ratio of reduced/oxidized glutathione (GSH/GSSG) ratio was detected after 4 and 6 h of infection compared to non-infecting controls, consistent with increased GSH turnover in M.abs infected cells as a result of increased oxidative stress (D).
HO-1 inhibition using SnPP increased ROS production as demonstrated by increased DHE and DCF fluorescence (Fig. 8A), and EPR measurements (B). Combining SnPP inhibition with M.abs infection led to more ROS production (Fig. 8B). This suggests that induction of HO-1 following M.abs infection of macrophages may serve to limit to levels of intracellular ROS.
Discussion
Mycobacterium abscessus is a rapidly growing mycobacterial species that can cause severe lung disease. Lung infections caused by M.abs are an emerging problem in the U.S. and many parts of the world, particularly in CF patients. Many M.abs strains are multi-drug resistant, and responsible for difficulties in patients undergoing lung transplantation [41]. M.abs infects and multiplies within human macrophages, indicating an ability of the bacteria to evade macrophage antimicrobial systems. In the current work, we sought to examine the early stages of M.abs infection inside macrophages, with a particular emphasis on the role of HO-1 in this process. We also defined the role of ROS and anti-oxidants inside macrophages infected with M.abs.
Our results show that M.abs infection increased O2•− and H2O2 production as indicated by increased fluorescence of DHE and DCF, respectively. Our EPR results confirmed the increase O2•− during the first hours of infection. Although protein levels and activities of SOD and catalase in the macrophages were not increased upon M.abs infection, reduced GSH was significantly decreased, indicating increased GSH turnover and oxidative stress. In contrast to SOD and catalase, HO-1 was induced during mycobacterial infection. This was not surprising as it was previously reported that HO-1 is induced in both infected macrophages and mice during M. tuberculosis infection [20], [21]. HO-1 is highly inducible in response to a variety of stressful stimuli such as physical stress, reactive oxygen species (ROS), UV irradiation, endotoxins, and hypoxia and provide protection to cells and tissues against oxidative and inflammatory injury [10]. Induction of HO-1 is cytoprotective to host cells. Both biliverdin and bilirubin have antioxidant properties and can protect cells from a variety of ROS insults [9]. In addition, HO-1 prevents free Fe2+ accumulation within cell through sequestrating it by ferritin and thus, limiting the generation of free radicals via the Fenton reaction [8], [31]. CO produced by HO-1 has been reported to suppress apoptosis and modulate levels of pro-inflammatory cytokines, NO, and prostaglandins [42], [43], [44].
Since HO-1 is directly linked to macrophage Fe metabolism, HO-1 induction could contribute to pathogenesis by providing a new source of Fe for bacterial growth and replication. Alternatively, biliverdin/bilirubin production could improve the environment for M.abs growth. Finally, inhibition of macrophage apoptosis resulting from HO-1 induction could enhance M.abs survival in macrophages. In fact, it was found that HO-1 favors pathogen dormancy in M. tuberculosis infections [21], and promotes the establishment of the malaria liver stage of infection [45]. Therefore, we assessed the importance of inhibiting HO-1 on mycobacterial growth inside infected macrophages.
We demonstrated that M.abs infection robustly induces HO-1 expression in the human macrophage-like THP-1 cells. Production of HO-1 was p38 MAPK-dependent, and p38 inhibitors suppressed HO-1 induction. We also demonstrated that pretreatment with HO-1 chemical inhibitors SnPP significantly inhibited M.abs growth inside macrophages, indicating the need of HO-1 for M.abs inside infected macrophages. It is worth mentioning that SnPP was added 24 h before infection to inhibit HO-1, and adding SnPP at the time of infection did not show the same inhibitory effect. Furthermore, we showed that inhibiting HO-1 using HO-1 siRNA or the HO-1 upstream signaling molecule; Nrf2 using Nrf2 siRNA resulted in similar inhibitory effects. In contrast, inducing HO-1 using CoPP did not increase M.abs intracellular growth.
In seeming contrast to our findings, it has been shown that HO-1-deficient (Hmox1−/−) mice were more susceptible to mycobacterial infection, and failed to mount a protective granulomatous response [46], [47]. However, it is possible that the HO-1 effect in these studies extends beyond the macrophages and/or the lung alone since in murine models mycobacterial infection leads to systemic disease. Complete inhibition of HO-1 in whole body tissues might affect other functional roles of HO-1 in mycobacterial resistance.
Our data show that the presence of bilirubin and/or biliverdin reversed the negative impact of HO-1 inhibition on M.abs growth in THP-1 cells. This suggests that these HO-1 products may be critical to the growth enhancing effect of HO-1 for M.abs in macrophages. The role of bilirubin and biliverdin as antioxidants extend beyond simply scavenging free radicals [48] to interfering with ROS production. It has been shown that decreased heme content due to HO-1 activation limits heme availability for the assembly of the functional NADPH oxidase subunits [49], [50], and administration of the HO-1 inhibitor SnPP reversed this effect and increased NADPH [50]. The mechanism of inhibition could be partly mediated by bilirubin [50]. This is consistent with our results, where inhibiting HO-1 with SnPP increased ROS production as indicated by EPR and increased DHE and DCF fluorescence. The ability of bilirubin and biliverdin, but not CO, to reverse the effect of SnPP and increase M.abs growth inside macrophages supports a role for HO-1 activity and/or induction for M.abs survival inside macrophages. Further studies are still needed to define the exact mechanisms involved in enhancing M.abs intracellular growth.
The role of ROS in macrophage killing of intracellular mycobacteria is still debated. The enhanced bactericidal effect of increased ROS production in infected macrophages has been previously shown [51], [52], [53]. It has been suggested that M. tuberculosis may be more sensitive to ROS generation than other bacteria [52], [53], and that minimizing the macrophage oxidative burst is one mechanism of mycobacterial intracellular survival [51]. On the other hand, it was reported that an oxidative environment could promote M.abs growth, and adding antioxidants and reducing intracellular oxidative stress could decrease intracellular growth of mycobacteria [54], [55]. Combining our results with others, it could be concluded that both ROS and antioxidants are important for controlled, balanced intracellular environment that promotes mycobacterial killing and provides optimal conditions for functional macrophages against mycobacteria and other pathogens.
The effect of the HO-1 products could involve interaction with bacterial enzymatic activity and cellular functions. Iron inhibits the nonstructural 5B (NS5B) RNA-dependent RNA polymerase of HCV [56]. Moreover, biliverdin, and bilirubin were shown to inhibit HCV NS3/4A protease in liver cells and in cell-free assays [57], [58].
A component of the intracellular survival mechanism of mycobacteria in human macrophages is the inhibition of macrophage phagosome–lysosome fusion [59], [60]. Our results show that inhibiting HO-1 increased mycobacteria–lysosome co-localization and thus phagosome–lysosome fusion in M.abs infected macrophages. Increased phagosome–lysosome fusion in macrophages is important mechanism for mycobacterial killing. Mycobacteria have evolved multiple ways to evade or escape phagosome–lysosome fusion in macrophages. Tools for inhibiting phagosome–lysosome fusion ranged between producing ammonia in abundance [59], alkalizing the intralysosomal compartment [61], altered calmodulin-dependent signal transduction, and abundant iron availability [62]. In the latter, it was shown using dominant negative Rab5, that fusion of phagosomes with early endosomes and an adequate iron supply are important for mycobacteria to inhibit the phagosome maturation process [62]. This is consistent with our results that inhibiting HO-1 enhanced M.abs lysosomal co-localization, i.e. phagosome–lysosome fusion. This could be partially attributed to the reduced Fe availability that might result from lower HO-1 activity.
In summary, our results suggest that inhibiting HO-1 limits M.abs growth inside macrophages at the early stages of infection, and that reducing HO-1 products may enhance the ability of the macrophage to control M.abs infection. Further investigations are still required to define the detailed mechanisms involved and the potential for this to be translated to new forms of therapy against this emerging infection.
Acknowledgments
This work is based upon work supported in part by the Department of Veterans Affairs through a merit review grant to B.E.B (VA Merit Award #00756). The contents do not necessarily represent the view of the Department of Veterans Affairs or the U.S. Government. The study was also partially supported by Free Radicals in Medicine Program (FRMP) pilot project, UNMC. We thank and acknowledge the assistance of the University of Nebraska EPR Spectroscopy Core, which is supported by the National Institute of General Medical Sciences of the National Institutes of Health (P30GM103335), and UNMC Confocal Laser Scanning Microscope Core Facility. We would like to thank Dr. Rebecca Oberley Deegan, at UNMC for her helpful discussions and advice.
References
- 1.Petrini B. Mycobacterium abscessus: an emerging rapid-growing potential pathogen. Acta Pathologica, Microbiologica et Immunologica Scandinavica. 2006;114(5):319–328. doi: 10.1111/j.1600-0463.2006.apm_390.x. 16725007 [DOI] [PubMed] [Google Scholar]
- 2.Griffith D.E., Girard W.M., Wallace R.J. Clinical features of pulmonary disease caused by rapidly growing mycobacteria. An analysis of 154 patients. American Review of Respiratory Disease. 1993;147(5):1271–1278. doi: 10.1164/ajrccm/147.5.1271. 8484642 [DOI] [PubMed] [Google Scholar]
- 3.Chan E.D., Bai X., Kartalija M., Orme I.M., Ordway D.J. Host immune response to rapidly growing mycobacteria, an emerging cause of chronic lung disease. American Journal of Respiratory Cell and Molecular Biology. 2010;43(4):387–393. doi: 10.1165/rcmb.2009-0276TR. 20081053 [DOI] [PubMed] [Google Scholar]
- 4.Román A., Ussetti P., Solé A., Zurbano F., Borro J.M., Vaquero J.M., de Pablo A., Morales P., Blanco M., Bravo C., Cifrian J., de la Torre M., Gámez P., Laporta R., Monforte V., Mons R., Salvatierra A., Santos F., Solé J., Varela A. Guidelines for the selection of lung transplantation candidates. Archivos de BronconeumologÃa. 2011;47(6):303–309. doi: 10.1016/j.arbres.2011.03.007. English Version. [DOI] [PubMed] [Google Scholar]
- 5.Bryant J.M., Grogono D.M., Greaves D., Foweraker J., Roddick I., Inns T., Reacher M., Haworth C.S., Curran M.D., Harris S.R., Peacock S.J., Parkhill J., Floto R.A. Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: a retrospective cohort study. Lancet. 2013;381(9877):1551–1560. doi: 10.1016/S0140-6736(13)60632-7. 23541540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abdalla M.Y., Mathahs M.M., Ahmad I.M. Protective role of heme oxygenase-1 in liver. Biologia. 2012;67(4):623–628. doi: 10.2478/s11756-012-0058-1. [DOI] [Google Scholar]
- 7.Abraham N.G., Lin J.H., Dunn M.W., Schwartzman M.L. Presence of heme oxygenase and NADPH cytochrome P-450 (c) reductase in human corneal epithelium. Investigative Ophthalmology & Visual Science. 1987;28(9):1464–1472. 3114166 [PubMed] [Google Scholar]
- 8.Vile G.F., Tyrrell R.M. Oxidative stress resulting from ultraviolet A irradiation of human skin fibroblasts leads to a heme oxygenase-dependent increase in ferritin. Journal of Biological Chemistry. 1993;268(20):14678–14681. 8325845 [PubMed] [Google Scholar]
- 9.Stocker R., Yamamoto Y., McDonagh A.F., Glazer A.N., Ames B.N. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235(4792):1043–1046. doi: 10.1126/science.3029864. 3029864 [DOI] [PubMed] [Google Scholar]
- 10.Ryter S.W., Alam J., Choi A.M. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiological Reviews. 2006;86(2):583–650. doi: 10.1152/physrev.00011.2005. 16601269 [DOI] [PubMed] [Google Scholar]
- 11.Shan Y., Zheng J., Lambrecht R.W., Bonkovsky H.L. Reciprocal effects of micro-RNA-122 on expression of heme oxygenase-1 and hepatitis C virus genes in human hepatocytes. Gastroenterology. 2007;133(4):1166–1174. doi: 10.1053/j.gastro.2007.08.002. 17919492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abdalla M.Y., Ahmad I.M., Spitz D.R., Schmidt W.N., Britigan B.E. Hepatitis C virus-core and non structural proteins lead to different effects on cellular antioxidant defenses. Journal of Medical Virology. 2005;76(4):489–497. doi: 10.1002/jmv.20388. 15977232 [DOI] [PubMed] [Google Scholar]
- 13.Lee P.J., Alam J., Wiegand G.W., Choi A.M. Overexpression of heme oxygenase-1 in human pulmonary epithelial cells results in cell growth arrest and increased resistance to hyperoxia. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(19):10393–10398. doi: 10.1073/pnas.93.19.10393. 8816811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kocanova S., Buytaert E., Matroule J.-Y., Piette J., Golab J., de Witte P., Agostinis P. Induction of heme-oxygenase 1 requires the p38MAPK and PI3K pathways and suppresses apoptotic cell death following hypericin-mediated photodynamic therapy. Apoptosis. 2007;12(4):731–741. doi: 10.1007/s10495-006-0016-x. 17219054 [DOI] [PubMed] [Google Scholar]
- 15.Brouard S., Otterbein L.E., Anrather J., Tobiasch E., Bach F.H., Choi A.M., Soares M.P. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. Journal of Experimental Medicine. 2000;192(7):1015–1026. doi: 10.1084/jem.192.7.1015. 11015442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Busserolles J., Megías J., Terencio M.C., Alcaraz M.J. Heme oxygenase-1 inhibits apoptosis in Caco-2 cells via activation of Akt pathway. International Journal of Biochemistry & Cell Biology. 2006;38(9):1510–1517. doi: 10.1016/j.biocel.2006.03.013. 16697692 [DOI] [PubMed] [Google Scholar]
- 17.Lang D., Reuter S., Buzescu T., August C., Heidenreich S. Heme-induced heme oxygenase-1 (HO-1) in human monocytes inhibits apoptosis despite caspase-3 up-regulation. International Immunology. 2005;17(2):155–165. doi: 10.1093/intimm/dxh196. 15611319 [DOI] [PubMed] [Google Scholar]
- 18.Rushworth S.A., MacEwan D.J. HO-1 underlies resistance of AML cells to TNF-induced apoptosis. Blood. 2008;111(7):3793–3801. doi: 10.1182/blood-2007-07-104042. 18202225 [DOI] [PubMed] [Google Scholar]
- 19.Behar S.M., Martin C.J., Booty M.G., Nishimura T., Zhao X., Gan H.X., Divangahi M., Remold H.G. Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal Immunology. 2011;4(3):279–287. doi: 10.1038/mi.2011.3. 21307848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shiloh M.U., Manzanillo P., Cox J.S. Mycobacterium tuberculosis senses host-derived carbon monoxide during macrophage infection. Cell Host Microbe. 2008;3(5):323–330. doi: 10.1016/j.chom.2008.03.007. 18474359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar A., Deshane J.S., Crossman D.K., Bolisetty S., Yan B.S., Kramnik I., Agarwal A., Steyn A.J. Heme oxygenase-1-derived carbon monoxide induces the Mycobacterium tuberculosis dormancy regulon. Journal of Biological Chemistry. 2008;283(26):18032–18039. doi: 10.1074/jbc.M802274200. 18400743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zacharia V.M., Manzanillo P.S., Nair V.R., Marciano D.K., Kinch L.N., Grishin N.V., Cox J.S., Shiloh M.U. Cor, a novel carbon monoxide resistance gene, is essential for Mycobacterium tuberculosis pathogenesis. MBio. 2013;4(6) doi: 10.1128/mBio.00721-13. 24255121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olakanmi O., Britigan B.E., Schlesinger L.S. Gallium disrupts iron metabolism of mycobacteria residing within human macrophages. Infection and Immunity. 2000;68(10):5619–5627. doi: 10.1128/IAI.68.10.5619-5627.2000. 10992462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olakanmi O., Gunn J.S., Su S., Soni S., Hassett D.J., Britigan B.E. Gallium disrupts iron uptake by intracellular and extracellular Francisella Strains and exhibits therapeutic efficacy in a murine pulmonary infection model. Antimicrobial Agents and Chemotherapy. 2010;54(1):244–253. doi: 10.1128/AAC.00655-09. 19917753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shih R.-H., Yang C.-M. Induction of heme oxygenase-1 attenuates lipopolysaccharide-induced cyclooxygenase-2 expression in mouse brain endothelial cells. J. Neuroinflammation. 2010;7:86. doi: 10.1186/1742-2094-7-86. 21118574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cummins N.W., Weaver E.A., May S.M., Croatt A.J., Foreman O., Kennedy R.B., Poland G.A., Barry M.A., Nath K.A., Badley A.D. Heme oxygenase-1 regulates the immune response to influenza virus infection and vaccination in aged mice. FASEB Journal. 2012;26(7):2911–2918. doi: 10.1096/fj.11-190017. 22490782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silva-Gomes S., Appelberg R., Larsen R., Soares M.P., Gomes M.S. Heme catabolism by heme oxygenase-1 confers host resistance to mycobacterium infection. Infection and Immunity. 2013;81(7):2536–2545. doi: 10.1128/IAI.00251-13. 23630967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abdalla M.Y., Britigan B.E., Wen F., Icardi M., McCormick M.L., LaBrecque D.R., Voigt M., Brown K.E., Schmidt W.N. Down-regulation of heme oxygenase-1 by hepatitis C virus infection in vivo and by the in vitro expression of hepatitis C Core protein. Journal of Infectious Diseases. 2004;190(6):1109–1118. doi: 10.1086/423488. 15319861 [DOI] [PubMed] [Google Scholar]
- 29.Yeligar S.M., Machida K., Kalra V.K. Ethanol-induced HO-1 and NQO1 are differentially regulated by HIF-1alpha and Nrf2 to attenuate inflammatory cytokine expression. Journal of Biological Chemistry. 2010;285(46):35359–35373. doi: 10.1074/jbc.M110.138636. 20833713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alam J., Cook J.L. How many transcription factors does it take to turn on the heme oxygenase-1 Gene? American Journal of Respiratory Cell and Molecular Biology. 2007;36(2):166–174. doi: 10.1165/rcmb.2006-0340TR. 16990612 [DOI] [PubMed] [Google Scholar]
- 31.Kehrer J.P. The Haber–Weiss reaction and mechanisms of toxicity. Toxicology. 2000;149(1):43–50. doi: 10.1016/s0300-483x(00)00231-6. 10963860 [DOI] [PubMed] [Google Scholar]
- 32.Gobin J., Moore C.H., Reeve J.R., Wong D.K., Gibson B.W., Horwitz M.A. Iron acquisition by Mycobacterium tuberculosis: isolation and characterization of a family of iron-binding exochelins. Proceedings of the National Academy of Sciences. 1995;92(11):5189–5193. doi: 10.1073/pnas.92.11.5189. 7761471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Antunes L.C., Imperi F., Minandri F., Visca P. In vitro and in vivo antimicrobial activities of gallium nitrate against multidrug-resistant Acinetobacter baumannii. Antimicrobial Agents and Chemotherapy. 2012;56(11):5961–5970. doi: 10.1128/AAC.01519-12. 22964249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olakanmi O., Kesavalu B., Pasula R., Abdalla M.Y., Schlesinger L.S., Britigan B.E. Gallium nitrate is efficacious in murine models of tuberculosis and inhibits key bacterial Fe-dependent enzymes. Antimicrobial Agents and Chemotherapy. 2013;57(12):6074–6080. doi: 10.1128/AAC.01543-13. 24060870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu Z., Wilson A.T., Luxon B.A., Brown K.E., Mathahs M.M., Bandyopadhyay S., McCaffrey A.P., Schmidt W.N. Biliverdin inhibits hepatitis C virus nonstructural 3/4A protease activity: mechanism for the antiviral effects of heme oxygenase? Hepatology. 2010;52(6):1897–1905. doi: 10.1002/hep.23921. 21105106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lehmann E., El-Tantawy W.H., Ocker M., Bartenschlager R., Lohmann V., Hashemolhosseini S., Tiegs G., Sass G. The heme oxygenase 1 product biliverdin interferes with hepatitis C virus replication by increasing antiviral interferon response. Hepatology. 2010;51(2):398–404. doi: 10.1002/hep.23339. 20044809 [DOI] [PubMed] [Google Scholar]
- 37.Armstrong J.A., Hart P.D. Phagosome–lysosome interactions in cultured macrophages infected with virulent tubercle bacilli. Reversal of the usual nonfusion pattern and observations on bacterial survival. Journal of Experimental Medicine. 1975;142(1):1–16. doi: 10.1084/jem.142.1.1. 807671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Armstrong J.A., Hart P.D. Response of cultured macrophages to Mycobacterium tuberculosis, with observations on fusion of lysosomes with phagosomes. Journal of Experimental Medicine. 1971;134(3 Pt 1):713–740. doi: 10.1084/jem.134.3.713. 15776571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frehel C., de Chastellier C., Lang T., Rastogi N. Evidence for inhibition of fusion of lysosomal and prelysosomal compartments with phagosomes in macrophages infected with pathogenic Mycobacterium avium. Infection and Immunity. 1986;52(1):252–262. doi: 10.1128/iai.52.1.252-262.1986. 2870027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clemens D.L., Horwitz M.A. Characterization of the Mycobacterium tuberculosis phagosome and evidence that phagosomal maturation is inhibited. Journal of Experimental Medicine. 1995;181(1):257–270. doi: 10.1084/jem.181.1.257. 7807006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sanguinetti M., Ardito F., Fiscarelli E., La Sorda M., D’Argenio P., Ricciotti G., Fadda G. Fatal pulmonary infection due to multidrug-resistant Mycobacterium abscessus in a patient with cystic fibrosis. Journal of Clinical Microbiology. 2001;39(2):816–819. doi: 10.1128/JCM.39.2.816-819.2001. 11158161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brouard S., Otterbein L.E., Anrather J., Tobiasch E., Bach F.H., Choi A.M., Soares M.P. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. Journal of Experimental Medicine. 2000;192(7):1015–1025. doi: 10.1084/jem.192.7.1015. 11015442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Otterbein L.E., Bach F.H., Alam J., Soares M., Tao Lu H., Wysk M., Davis R.J., Flavell R.A., Choi A.M. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nature Medicine. 2000;6(4):422–428. doi: 10.1038/74680. 10742149 [DOI] [PubMed] [Google Scholar]
- 44.Otterbein L.E., Choi A.M. Heme oxygenase: colors of defense against cellular stress. American Journal of Physiology – Lung Cellular and Molecular Physiology. 2000;279(6):L1029–L1037. doi: 10.1152/ajplung.2000.279.6.L1029. 11076792 [DOI] [PubMed] [Google Scholar]
- 45.Epiphanio S., Mikolajczak S.A., Gonçalves L.A., Pamplona A., Portugal S., Albuquerque S., Goldberg M., Rebelo S., Anderson D.G., Akinc A., Vornlocher H.P., Kappe S.H., Soares M.P., Mota M.M. Heme oxygenase-1 is an anti-inflammatory host factor that promotes murine plasmodium liver infection. Cell Host Microbe. 2008;3(5):331–338. doi: 10.1016/j.chom.2008.04.003. 18474360 [DOI] [PubMed] [Google Scholar]
- 46.Silva-Gomes S., Appelberg R., Larsen R., Soares M.P., Gomes M.S. Heme catabolism by heme oxygenase-1 confers host resistance to mycobacterium infection. Infection and Immunity. 2013;81(7):2536–2545. doi: 10.1128/IAI.00251-13. 23630967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Regev D., Surolia R., Karki S., Zolak J., Montes-Worboys A., Oliva O., Guroji P., Saini V., Steyn A.J., Agarwal A., Antony V.B. Heme oxygenase-1 promotes granuloma development and protects against dissemination of mycobacteria. Laboratory Investigation. 2012;92(11):1541–1552. doi: 10.1038/labinvest.2012.125. 22964851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jansen T., Daiber A. Direct antioxidant properties of bilirubin and biliverdin. Is there a role for biliverdin reductase? Frontiers in Pharmacology. 2012;3:30. doi: 10.3389/fphar.2012.00030. 22438843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taillé C., El-Benna J., Lanone S., Dang M.C., Ogier-Denis E., Aubier M., Boczkowski J. Induction of heme oxygenase-1 inhibits NAD(P)H oxidase activity by down-regulating cytochrome b558 expression via the reduction of heme availability. Journal of Biological Chemistry. 2004;279(27):28681–28688. doi: 10.1074/jbc.M310661200. 15123630 [DOI] [PubMed] [Google Scholar]
- 50.Datla S.R., Dusting G.J., Mori T.A., Taylor C.J., Croft K.D., Jiang F. Induction of heme oxygenase-1 in vivo suppresses NADPH oxidase derived oxidative stress. Hypertension. 2007;50(4):636–642. doi: 10.1161/HYPERTENSIONAHA.107.092296. 17679649 [DOI] [PubMed] [Google Scholar]
- 51.Manca C., Paul S., Barry C.E., 3rd, Freedman V.H., Kaplan G. Mycobacterium tuberculosis catalase and peroxidase activities and resistance to oxidative killing in human monocytes in vitro. Infection and Immunity. 1999;67(1):74–79. doi: 10.1128/iai.67.1.74-79.1999. 9864198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taneja N.K., Dhingra S., Mittal A., Naresh M., Tyagi J.S. Mycobacterium tuberculosis transcriptional adaptation, growth arrest and dormancy phenotype development is triggered by vitamin C. PLOS One. 2010;5(5):e10860. doi: 10.1371/journal.pone.0010860. 20523728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vilchèze C., Hartman T., Weinrick B., Jacobs W.R., Jr. Mycobacterium tuberculosis is extraordinarily sensitive to killing by a vitamin C-induced Fenton reaction. Nature Communications. 2013;4:1881. doi: 10.1038/ncomms2898. 23695675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oberley-Deegan R.E., Rebits B.W., Weaver M.R., Tollefson A.K., Bai X., McGibney M., Ovrutsky A.R., Chan E.D., Crapo J.D. An oxidative environment promotes growth of Mycobacterium abscessus. Free Radical Biology and Medicine. 2010;49(11):1666–1673. doi: 10.1016/j.freeradbiomed.2010.08.026. 20807564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oberley-Deegan R.E., Lee Y.M., Morey G.E., Cook D.M., Chan E.D., Crapo J.D. The antioxidant mimetic, MnTE-2-PyP, reduces intracellular growth of Mycobacterium abscessus. American Journal of Respiratory Cell and Molecular Biology. 2009;41(2):170–178. doi: 10.1165/rcmb.2008-0138OC. 19097985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fillebeen C., Rivas-Estilla A.M., Bisaillon M., Ponka P., Muckenthaler M., Hentze M.W., Koromilas A.E., Pantopoulos K. Iron inactivates the RNA polymerase NS5B and suppresses subgenomic replication of hepatitis C virus. Journal of Biological Chemistry. 2005;280(10):9049–9057. doi: 10.1074/jbc.M412687200. 15637067 [DOI] [PubMed] [Google Scholar]
- 57.Lehmann E., El-Tantawy W.H., Ocker M., Bartenschlager R., Lohmann V., Hashemolhosseini S., Tiegs G., Sass G. The heme oxygenase 1 product biliverdin interferes with hepatitis C virus replication by increasing antiviral interferon response. Hepatology. 2010;51(2):398–404. doi: 10.1002/hep.23339. 20044809 [DOI] [PubMed] [Google Scholar]
- 58.Zhu Z., Wilson A.T., Luxon B.A., Brown K.E., Mathahs M.M., Bandyopadhyay S., McCaffrey A.P., Schmidt W.N. Biliverdin inhibits hepatitis C virus nonstructural 3/4A protease activity: mechanism for the antiviral effects of heme oxygenase? Hepatology. 2010;52(6):1897–1905. doi: 10.1002/hep.23921. 21105106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gordon A.H., Hart P.D., Young M.R. Ammonia inhibits phagosome–lysosome fusion in macrophages. Nature. 1980;286(5768):79–80. doi: 10.1038/286079a0. 6993961 [DOI] [PubMed] [Google Scholar]
- 60.Malik Z.A., Iyer S.S., Kusner D.J. Mycobacterium tuberculosis phagosomes exhibit altered calmodulin-dependent signal transduction: contribution to inhibition of phagosome–lysosome fusion and intracellular survival in human macrophages. Journal of immunology. 2001;166(5):3392–3401. doi: 10.4049/jimmunol.166.5.3392. 11207296 [DOI] [PubMed] [Google Scholar]
- 61.Hart P.D., Young M.R., Jordan M.M., Perkins W.J., Geisow M.J. Chemical inhibitors of phagosome–lysosome fusion in cultured macrophages also inhibit saltatory lysosomal movements. A combined microscopic and computer study. Journal of Experimental Medicine. 1983;158(2):477–492. doi: 10.1084/jem.158.2.477. 6193224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kelley V.A., Schorey J.S. Mycobacterium’s arrest of phagosome maturation in macrophages requires Rab5 activity and accessibility to iron. Molecular Biology of the cell. 2003;14(8):3366–3377. doi: 10.1091/mbc.E02-12-0780. 12925769 [DOI] [PMC free article] [PubMed] [Google Scholar]