

Abstract
Superoxide () has been implicated in the pathogenesis of many human diseases including hypertension. Mitochondria-targeted superoxide scavenger mitoTEMPO reduces blood pressure; however, the structure–functional relationships in antihypertensive effect of mitochondria-targeted nitroxides remain unclear. The nitroxides are known to undergo bioreduction into hydroxylamine derivatives which reacts with with much lower rate. The nitroxides of pyrrolidine series (proxyls) are much more resistant to bioreduction compared to TEMPOL derivatives suggesting that mitochondria-targeted proxyls can be effective antioxidants with antihypertensive activity. In this work we have designed and studied two new pyrrolidine mitochondria targeted nitroxides: 3-[2-(triphenyphosphonio)acetamido]- and 3-[2-(triphenyphosphonio) acetamidomethyl]-2,2,5,5-tetramethylpyrrolidine-1-oxyl (mCP2) and (mCP1). These new mitochondria targeted nitroxides have 3- to 7-fold lower rate constants of the reaction with compared with mitoTEMPO; however, the cellular bioreduction of mCP1 and mCP2 was 3- and 2-fold slower. As a consequence incubation with cells afforded much higher intracellular concentration of mCP1 and mCP2 nitroxides compared to mitoTEMPO nitroxide. This has compensated for the difference in the rate of scavenging and all nitroxides similarly protected mitochondrial respiration in H2O2 treated endothelial cells. Treatment of hypertensive mice with mCP1 and mCP2 (1.4 mg/kg/day) after onset of angiotensin II-induced hypertension significantly reduced blood pressure to 133±5 mmHg and 129±6 mmHg compared to 163±5 mmHg in mice infused with angiotensin II alone. mCP1 and mCP2 reduced vascular and prevented decrease of endothelial nitric oxide production. These data indicate that resistance to bioreduction play significant role in antioxidant activity of nitroxides. Studies of nitroxide analogs such as mCP1 and mCP2 may help in optimization of chemical structure of mitochondria-targeted nitroxides for improved efficacy and pharmacokinetics of these drugs in treatment of hypertension and many other conditions including atherosclerosis, diabetes and degenerative neurological disorders in which mitochondrial oxidative stress seems to play a role.
Keywords: Mitochondria, Antioxidant, Superoxide, Endothelial cells, Nitroxide, Hypertension
Graphical abstract
Highlights
-
•
Nitroxides bioreduction into hydroxylamine can reduce their antioxidant properties.
-
•
Mitochondria-targeted proxyls are more resistant to bioreduction vs mitoTEMPO.
-
•
Intracellular concentration of mCP1 and mCP2 nitroxides is higher vs mitoTEMPO.
-
•
mCP1 and mCP2 diminished vascular superoxide and reduced hypertension.
Introduction
Clinical data show that 26% of adult population has hypertension [1]. This disease represents a major risk factor for stroke, myocardial infarction, and heart failure [2]. Hypertension is a multifactorial disorder involving perturbations of the vasculature, the kidney and the central nervous system [3]. Despite treatment with multiple drugs, 37% of hypertensive patients remain hypertensive [4], likely due to the mechanisms contributing to blood pressure elevation that are not affected by current treatments. New classes of antihypertensive agents could therefore add to the currently available therapeutic armamentarium to improve treatment of hypertension.
In the past decade it has become clear that vascular superoxide () production contributes to hypertension [5]. In almost all experimental models of hypertension production is increased in multiple organs, including vasculature where promote vasoconstriction and remodeling, increasing systemic vascular resistance. In the past several years, we have shown that the mitochondria become dysfunctional in hypertension and have defined novel role of mitochondrial in this disease [6,7]. The mitochondria are an important source of and we have shown that scavenging mitochondrial improves endothelial function and attenuates hypertension [6].
Common antioxidants like ascorbate and vitamin E have proven ineffective in preventing cardiovascular diseases and hypertension [8]. These agents unlikely reach important sites of ROS production such as the mitochondria. Experimental studies have shown an important role of mitochondrial reactive oxygen species in the development of endothelial dysfunction, hypertension and atherosclerosis [9,10]. Indeed, we have shown that SOD2 overexpression attenuates hypertension and treatment of hypertensive mice with mitochondria-targeted antioxidants reduces blood pressure [6,7].
The membrane potential of mitochondria within living cells is negative inside (−140 mV). As this membrane potential is far larger than in other organelles within cells, lipophilic cations such as triphenylphosphonium (TPP) selectively accumulate within mitochondria [11]. Antioxidants conjugated to TPP, therefore can be targeted to mitochondria and may be concentrated in the mitochondrial matrix by 1000-fold [12]. The pharmacology of mitochondria-targeted antioxidants is not well understood. For example, previously described mitoquinone (MitoQ10) [12] may have prooxidant and proapoptotic properties due to redox cycling and generation of by quinone [13,14]. Nitroxides are well-known SOD mimetics [15,16]. They are not consumed in the reaction of superoxide dismutation, and this makes their use advantageous over many other antioxidants. Nitroxides have very low toxicity and this makes them perfect candidates for conjugation with lipophilic cations (e.g., triphenylmethylphosphonium) for in vivo use [17]. Furthermore, some phosphonium ions may inhibit oxidation of pyruvate, malate, 2-oxoglutarate and glutamate in heart mitochondria at micromolar concentrations [18] suggesting that triphenylmethylphosphonium conjugates should be used at submicromolar concentrations and tested for side effect on respiration. Indeed, low doses of mitoTEMPO did not affect mitochondrial respiration in control cells but improved mitochondrial function in the presence of angiotensin II [6].
Recently, it has been shown that pretreatment of endothelial cells with the mitochondria-targeted SOD mimetic Mito-CP significantly reduces H2O2- and lipid peroxide-induced cellular oxidative stress [19]. Mito-CP inhibits peroxide-induced inactivation of complex I and aconitase, while restoring the mitochondrial membrane potential. In contrast, the “untargeted” carboxy proxyl (CP) did not protect the cells from peroxide-induced oxidative stress and apoptosis. However, in Mito-CP nitroxide is connected to triphenylmethylphosphonium cation via ester spacer and ester group can be potentially hydrolyzed to give inactive 3-carboxyproxyl (CP).
The main disadvantage of nitroxides is their rapid reduction with cellular antioxidants and enzymatic systems. For example, mitoTEMPO is readily reduced in mitochondria to corresponding hydroxylamine [20]. Hydroxylamine derivatives react with much slower compared with nitroxides [6] therefore bioreduction of nitroxides can significantly reduce antioxidant potentials of nitroxides.
We have hypothesized that increased resistance to bioreduction will be beneficial to mitochondria-targeted nitroxides. The structure-functional relationships in antioxidant properties of mitochondria-targeted nitroxides however remain unclear. In this work we have designed and studied two new pyrrolidine mitochondria targeted nitroxides. The nitroxides of pyrrolidine series (proxyls) are weaker oxidants as compared to TEMPOL derivatives, and they demonstrate higher stability in biological samples [21]. We hypothesized that mitochondria targeted proxyl derivatives are much more resistant to bioreduction compared to TEMPOL and this may improve antioxidant and antihypertensive properties of new nitroxide derivatives. In this work we have designed two new pyrrolidine mitochondria targeted nitroxides mCP1 and mCP1 (Fig. 1), and studied their antioxidant properties, cellular accumulation, their bioreduction and antihypertensive properties.
Fig. 1.
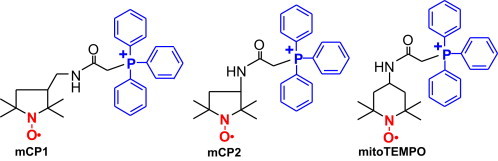
Structures of mitochondria-targeted nitroxides.
Materials and methods
Reagents
Xanthine oxidase was purchased from Roche Molecular Biochemicals (Indianapolis, IN). All other reagents were obtained from Sigma (St Louis, MO).
Synthesis of mCP1 and mCP2
3-Amino-PROXYL (1a) and 3-aminomethyl-PROXYL (1b) were prepared according to the literature methods described by Rozantsev and Hankowsky [22,23]. The nitroxides mCP1 and mCP2 were synthesized according to Fig. 2 similarly to the procedure previously described for mitoTEMPO (mT) [24].
Fig. 2.
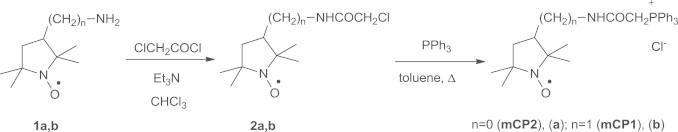
Synthesis of proxyl-based mitochondria-targeted nitroxides mCP1 and mCP2.
3-((2-Chloroacetamido)methyl)-2,2,5,5-tetramethylpyrrolidin-1-oxyl (2b). A solution of 3-aminomethyl-PROXYL (0.9 g or 0.005 mol) and dry triethylamine (1.7 ml or 0.0117 mol) in dry chloroform (20 ml) was placed into 50 ml flat-bottom flask and cooled to − 5 °C. The cold solution was placed into ice bath and chloroacetyl chloride (0.42 ml or 0.005 mol) was added dropwise upon stirring. The resulting dark solution was washed with water (3 × 5 ml), and dried with MgSO4. The chloroform was removed under reduced pressure and the residue was separated by column chromatography (Kieselgel 60, Merck, eluent chloroform) to give 2b. Yellow oil, Found: С, 53.06; Н, 8.08; N, 11.56; Cl 14.10. Calculated for C11H2ON2O2Cl: C, 53.33; H, 8.14; N, 11.31; Cl 14.31 %; νmax (KBr)/см−1 3306 br, 3084 br, 2972, 2934, 2872, 1668, 1543, 1462, 1364, 1313, 1252, 1178, 1159, 1107, 1057, 789, 764, 691 br, 600, 571, 525, 476.
Similarly, 3-(2-chloroacetamido)-2,2,5,5-tetramethylpyrrolidin-1-oxyl (2a) have been prepared from 3-amino-PROXYL (1a), (this compound was first described in [Mao-Man-Jun; Tian, Xuan; Chen, Yao-Zu; Gaodeng Xuexiao Huaxue Xuebao (1998), 19(3), 395–398.], but this publication is not available to authors): yellow crystals, m.p. 130–131°С (hexane-ethyl acetate). Found: С, 51.31; Н, 7.66; N, 11.87; Cl 15.10. Calculated for C10H18N2O2Cl: C, 51.30; H, 7.76; N, 11.99; Cl 15.17 %; νmax (KBr)/см−1 3319, 3076 br, 2982, 2938, 2876, 1676, 1553, 1485, 1408, 1366, 1331, 1299, 1259, 1229, 1196, 1165, 1111, 1045, 806, 772, 687 br, 572, 555, 547, 530, 500.
3-[(2-(Triphenyphosphonio)acetamido)methyl]-2,2,5,5-tetramethylpyrrolidin-1-oxyl (mCP1). The 2b (1 g, 0.004 mol) was placed into 50 ml flask containing toluene (15 ml) and triphenylphosphine (2.7 g, 0.01 mol). The mixture was heated to reflux under nitrogen for 15 hours and then cooled in ice bath the precipitate was separated, washed with toluene and purified using column chromatography (silica gel, eluent chloroform-ethanol 50:1). Further purification can be performed by following procedure: the compound was dissolving in chloroform, the solvent was removed under reduced pressure, the resin-like residue was rapidly dissolved in boiling toluene and filtered, the crystalline precipitate formed was filtered off, washed with toluene and dried. The overall yield from 1b was 1.3 g (51 %). Pale yellowish crystals m.p. 236–240°С. Found: С, 68.01; Н, 6.80; N, 5.50; Cl 6.96; P 5.70. Calculated for C29H35N2O2ClP: C, 68.29; H, 6.92; N, 5.49; Cl 6.95; P 6.07 %; νmax (KBr)/см−1 3424 br, 3158 br, 2974, 2928, 2874, 1669, 1560, 1484, 1460, 1438, 1397, 1362, 1324, 1146, 1112, 756, 738, 716.
Similarly, 3-(2-(triphenyphosphonio)acetamido)-2,2,5,5-tetramethylpyrrolidin-1-oxyl (mCP2) was prepared from 2a: the overall yield from 1a was 1.7 g (60 %). Pale yellowish crystals m.p. 211–214 °С. Found: С, 68.05; Н, 6.71; N, 5.62; Cl 7.65; P 6.19. Calculated for C28H33N2O2ClP: C, 67.80; H, 6.71; N, 5.65; Cl 7.15; P 6.24 %; νmax (KBr)/см−1 3425 br, 3182 br, 2974, 2883, 2772, 1670, 1558, 1439, 1365, 1330, 1112, 748, 720, 690, 508, 483.
Cell culture
Human aortic endothelial cells (HAEC) were purchased from Lonza (Chicago, IL) and cultured in EGM-2 medium supplemented with 2% FBS but without antibiotics. On the day before the study, the FBS concentration was reduced to 1%. Cells were washed with Krebs-Hepes buffer prior experiments. Cellular accumulation of mitochondria-targeted nitroxides was determined by EPR analysis of cellular pellet following 70 minutes incubation of HAEC with 0.1 µM nitroxide solution in Krebs-Hepes buffer at 37 °C. Cells were collected by centrifugation at 2500 G for 10 minutes at 4 °C and transferred into 50 µl capillary tube for immediate EPR analysis. Concentration of nitroxide in cell pellet was compared with initial nitroxide level in buffer. Cellular reduction of nitroxides into EPR silent hydroxylamine was measured by flowing time-dependent decay of nitroxide EPR signal in the cellular pellet. In order to test the antioxidant protection of mitochondrial function we pre-incubated HAEC with various concentrations of nitroxides for 15 minutes and then treated with 0.1 mM H2O2 or saline as a vehicle. Mitochondrial respiration was measured after additional 60 minutes incubation in cellular pellet placed in 0.25 ml Krebs-Hepes buffer using Clark electrode (Oxygraph, Hanstech).
Animal experiments
Hypertension was induced by angiotensin II (0.7 mg/kg/day) as described previously [25] using 2–3 month old male C57Bl/6J mice. Seven days after saline or angiotensin II minipump placement, mice received a second minipump for infusion of saline as vehicle, mCP1 or mCP2 as described in the figure legend. Blood pressure was monitored using either the tail cuff method or telemetry as previously described [26,27]. All procedures were performed according to guidelines and approved by IACUC at Vanderbilt University.
ESR experiments
All ESR samples were placed in 50-µl glass capillaries (Corning, New York, NY). ESR spectra were recorded using an EMX ESR spectrometer (Bruker Biospin Corp., Billerica, MA) and a super high Q microwave cavity at room temperature. The ESR settings for field-scan experiments with the spin probe CAT1H were as follows: field sweep, 70 G; microwave frequency, 9.82 GHz; microwave power, 20 mW; modulation amplitude, 0.7 G; conversion time, 41 ms; time constant, 164 ms; and receiver gain, 1 × 105 (n = 4 scans). The rates of H2O2 production were determined by monitoring the amplitude of the low field component of the ESR spectrum of CAT1-nitroxide with the following settings: field sweep, 60 G; microwave frequency, 9.46 GHz; microwave power, 20 mW; modulation amplitude, 2 G; conversion time, 1311 ms; time constant, 5243 ms; and receiver gain, 1 × 105. ESR experiments were repeated at least three times.
Statistics
Experiments were analyzed using the Student Neuman Keuls post-hoc test and analysis of variance (ANOVA). P levels < 0.05 were considered significant.
Results
Synthesis and physical properties of mCP1 and mCP2
In this work we have developed synthesis of two new pyrrolidine mitochondria targeted nitroxides (Fig. 2). The nitroxides mCP1 and mCP2 were synthesized from 3-amino- and 3-aminomethyl-proxyls via chloroacetylation and treatment with triphenylphosphine according to the general Scheme 1 in analogy to the procedure previously described for mitoTEMPO (mT) [24].
Reaction of with mCP1 and mCP2
We have investigated reaction of with mitochondria targeted nitroxides using xanthine oxidase generating system and cytochrome c as was previously described by Murphy et al. [28] Cytochrome c is rapidly reduced by into ferrocytochrome c (8 × 105 M−1/s) [29] and this reaction is inhibited in the presence of nitroxide radicals. Indeed, mediated cytochrome c reduction was inhibited in dose-dependent manner in the presence of mCP1, mCP2 and mitoTEMPO (Fig. 3A). The rate constants of reaction with nitroxides were estimated from the linear regression of inhibition of cytochrome c reduction (Fig. 3B) expressed as (Vo/V-1), where V and Vo are the rates of cytochrome c reduction in the presence or absence of nitroxide. The slope of this linear regression is proportional to the rate constants of reaction with nitroxides. The sharpest slope for mitoTEMPO indicates the highest rate of reaction with compared with mCP1 and mCP2. Taking into account the concentration of cytochrome c (50 µM) we have estimated the nitroxide rate constants as 2.5 × 105 M−1/s (mitoTEMPO), 7.6 × 104 M−1/s (mCP1) and 3.2 × 104 M−1/s (mCP2). These data support fast reaction between new mitochondria targeted nitroxides and , however mCP1 and mCP2 react with by 3- and 7-fold slower compared with mitoTEMPO.
Fig. 3.
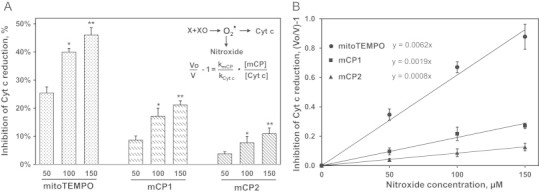
Dose-dependent inhibition of superoxide-mediated cytochrome c reduction by nitroxides (A). Comparison of superoxide reactivity of mCP1, mCP2 and mitoTEMPO based on competitive inhibition of cytochrome c reduction. V and Vo are rates of cytochrome c reduction in the presence or absence of nitroxides. (B) Results represent mean ± SEM for 3–6 repeats per group. ⁎P < 0.001 vs 50 nM nitroxide, ⁎⁎P < 0.05 vs 100 nM nitroxide.
Cellular accumulation and protection of mitochondrial respiration by mCP1 and mCP2
We have previously shown rapid accumulation of mitoTEMPO in intact endothelial cells [6]. In this work we have compared cellular accumulation of mCP1, mCP2, mitoTEMPO and its untargeted analog TEMPOL. EPR analysis of intact HAEC incubated with 5 µM nitroxides showed significant increase in nitroxide accumulation in cellular pellet. It was found that cellular level of mCP1 and mCP2 was increased by 37-fold compared with extracellular nitroxide concentration (Fig. 4A). Cellular accumulation of mitoTEMPO was 25-fold while concentration of untargeted TEMPO was increased only by 4.6-fold vs buffer. These data demonstrate improved cellular accumulation of mCP1 and mCP2 compared with mitoTEMPO.
Fig. 4.
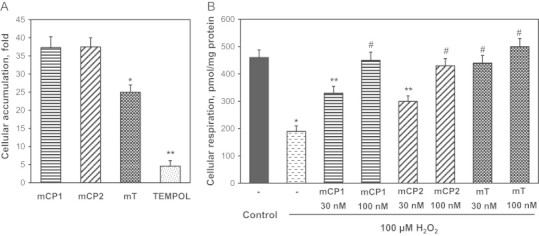
Cellular accumulation of nitroxides (A) and protection of mitochondrial respiration (B) by mCP1, mCP2 and mitoTEMPO (mT). Results represent mean ± SEM for 3–6 repeats per group. ⁎P < 0.001 vs control, ⁎⁎P < 0.01 vs H2O2, #P < 0.05 vs 30 nM mCP1.
To estimate the protective activity of the new mitochondria targeted proxyl nitroxides we studied their effect on HAEC under H2O2-induced mitochondrial oxidative stress recently described by Kalyanaraman group [19]. We have previously shown that H2O2 stimulates production of mitochondrial [7] and therefore scavengers should protect mitochondria. Indeed, it was found that treatment of intact HEAC with low dose of H2O2 reduced mitochondrial respiration by 2.4-fold while supplementation with mCP1, mCP2 and mitoTEMPO protected respiration in a dose-dependent manner (Fig. 4B). Mito-TEMPO was more efficient at low concentration (30 nM) compared with mCP1 and mCP2 which likely due to higher reactivity of mitoTEMPO with . However, higher doses of nitroxides (100 nM) showed similar protection of cellular respiration.
Cellular reduction of mitochondria targeted nitroxides
Nitroxides can be susceptible to bioreduction into EPR silent hydroxylamine form [20]. The increased resistance to bioreduction may contribute to higher cellular level of mCP1 and mCP2 compared with mitoTEMPO (Fig. 5A). We therefore investigated bioreduction of mitochondria targeted nitroxides. It was found that incubation of in intact endothelial cells with mitoTEMPO for 40 minutes reduced intensity of EPR spectra by 41%. Interestingly, incubation mCP1 and mCP2 reduced EPR signals by 13% and 18%, correspondingly (Fig. 5A). Analysis of EPR spectra showed that the reduction rates of mCP1 and mCP2 were 2–3-fold slower compared with mitoTEMPO (Fig. 4B). These data are in line with higher resistance to reduction of proxyl nitroxides compared to TEMPO derivatives.
Fig. 5.
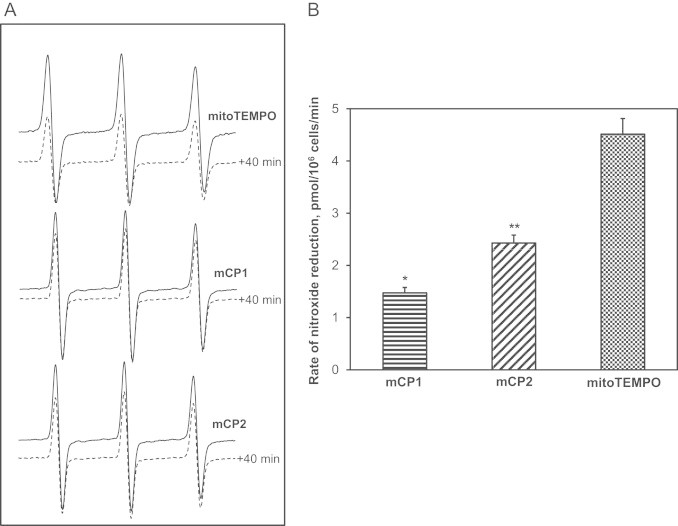
EPR spectra of intact endothelial cells incubated with mCP1, mCP2 and mitoTEMPO (A). Rates of cellular nitroxide reduction during incubation at 37 °C (B). Results represent mean ± SEM for 4–6 repeats per group. ⁎P < 0.01 vs mitoTEMPO, ⁎⁎P < 0.05 vs mitoTEMPO.
In vivo treatment with mCP1 and mCP2 after onset of hypertension
Increased vascular production has been implicated in the pathogenesis of endothelial dysfunction and hypertension [30,31]. We have previously shown that mitoTEMPO supplementation attenuates development of hypertension [6]. Furthermore, treatment of animals after onset of hypertension reduced vascular oxidative stress, improved NO-mediated vasodilatation and reduced blood pressure [6]. Meanwhile, we do not know if other mitochondria targeted nitroxides will reduce blood pressure or antihypertensive effect is specific to mitoTEMPO. We therefore performed additional studies in which mCP1 and mCP2 were administered after the onset of angiotensin II-induced hypertension. Following 9 days of angiotensin II infusion (0.7 mg/kg/day) systolic blood pressure reached 150 mm Hg (Fig. 6A). The subsequent addition of mCP1 or mCP2 (1.4 mg/kg/day) using osmotic minipump resulted in a time-dependent decrease of blood pressure. Infusion of mCP1 and mCP2 significantly reduced blood pressure to 133 ± 5 mm Hg and 129 ± 6 mm Hg while blood pressure in mice infused with saline (vehicle) have continued to rise and reached 163 ± 5 mm Hg at day 14 (Fig. 6A). Of note, treatment of healthy control mice with mitochondria targeted nitroxides did not affect basal blood pressure (101 mm Hg) because does play a role in the regulation of blood pressure in normotensive subjects [6]. These data indicate that mCP1 and mCP2 provide antihypertensive effect similar to mitoTEMPO which we have previously described [6].
Fig. 6.
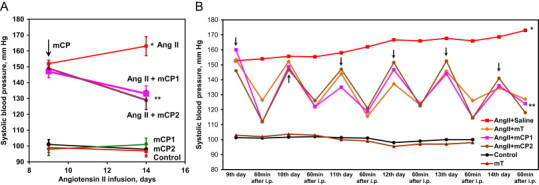
Antihypertensive effect of mitochondria targeted nitroxides in angiotensin II-infused mice. (A) Mice were implanted with osmotic pumps after onset of hypertension containing 1.4 mg/kg/day nitroxide or saline as a vehicle. (B) Mice were treated with the i.p. injection of 1.4 mg/kg/day mCP1, mCP2 or mitoTEMPO (mT). Results represent mean ± SEM for 3–6 animals per group. ⁎P < 0.001 vs control, ⁎⁎P < 0.01 vs Ang II.
In order to compare time-dependent antihypertensive effects of mitochondria-targeted nitroxides we have treated hypertensive mice with daily i.p. injection of saline (vehicle), or 1.4 mg/kg mCP1, mCP2 or mitoTEMPO and measured blood pressure before and after injection. It was found that i.p. treatment with nitroxides acutely reduced blood pressure however this antihypertensive effect was slightly reduced next day after injection (Fig. 6B). Meanwhile the daily treatments increased antihypertensive effect in a time-dependent fission. Interestingly, i.p. treatment with mCP1 and mCP2 provide antihypertensive effect similar to mitoTEMPO (Fig. 6B).
The above studies showing that mCP1 and mCP1 can reduce hypertension do not provide insight into whether antihypertensive effect was associated with reduced level and improved vascular function. Therefore, additional studies were performed of the antioxidant effects of mCP1 and mCP2. Vascular production in isolated aortic vessels from hypertensive mice after onset of angiotensin II induced hypertension was measured using fluorescent probe DHE and HPLC analysis of specific product 2-hydroxyethidium (Fig. 7A) [32]. Both mCP1 and mCP2 reduced vascular and blood pressure similarly to previously report for mitoTEMPO [6] which support pharmacological activity in treatment of vascular oxidative stress by proxyl-based mitochondria-targeted antioxidants.
Fig. 7.
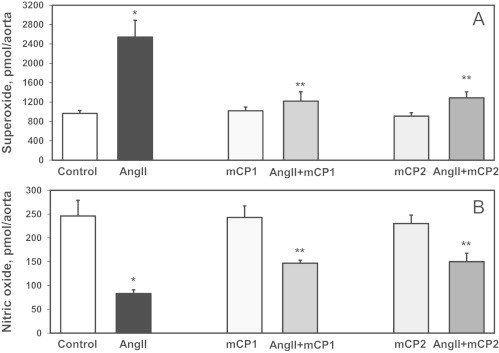
Production of aortic (A) and endothelial nitric oxide (B) in mice treated with mCP1 and mCP2 after onset of angiotensin II-induced hypertension using osmotic pumps. (A) Measurements of vascular (DHE/HPLC) [35] in isolated aortas following 14-days angiotensin II infusion. (B) Detection of endothelial nitric oxide in aortas using electron spin resonance and Fe(DETC)2 as NO spin trap [33]. Results represent mean ± SEM for 5–8 animals per group. ⁎P < 0.01 vs control, ⁎⁎P < 0.05 vs AngII.
Increased production in vessels is leading to inactivation of endothelial nitric oxide and vasoconstriction which contributes to hypertension [30]. We hypothesized that mitochondria-targeted nitroxides will improve vascular nitric oxide level since these treatments reduced vascular . In order test this hypothesis we measured vascular nitric oxide in aortic vessels using electron spin resonance and specific nitric oxide spin trap Fe(DETC)2 as we have previously described [33]. It was found that treatment of hypertensive mice treated with mCP1 or mCP2 prevented oxidation of nitric oxide and significantly improved vascular nitric oxide level (Fig. 7B).
Discussion
In this work we have synthesized novel proxyl-based mitochondria-targeted nitroxides mCP1 and mCP2 and studied their antioxidant properties. The cellular penetration of these nitroxides is driven by the lipophilic triphenyl cation and therefore it is likely to be similar for proxyl and TEMPO based mitochondria targeted nitroxides. Our data however show significant difference in the intracellular reduction of mCP1, mCP2 and mitoTEMPO (Fig. 5). These nitroxides were present in intact cells at higher concentration compared with mitoTEMPO (Fig. 4A) which is in line with greater bioreduction resistance of proxyl based nitroxides (Fig. 5). The advantageous cellular accumulation of mCP1 and mCP2 however was counteracted by reduced reactivity with compared with mitoTEMPO (Fig. 3) which can eliminate the potential advantages of proxyl-based nitroxides. Indeed, both proxyl- and TEMPO-based mitochondria targeted nitroxides showed similar protection of respiration in H2O2-treated endothelial cells (Fig. 4). Furthermore, our in vivo study in angiotensin II-infused mice showed that antihypertensive and antioxidant effects of mCP1 and mCP2 were not significantly different from mito-TEMPO (Figs. 6 and 7). Our data also show that mitoTEMPO is not the only mitochondria-targeted nitroxide exhibiting antihypertensive effect and studies of analogs such as mCP1 and mCP2 may help in optimization of chemical structure of mitochondria-targeted nitroxides for improved, lipophility, antioxidant efficacy and pharmacokinetics.
Antihypertensive effects of TEMPOL and other untargeted nitroxides has been previously shown [34]. Acute treatment of hypertensive animals with TEMPOL caused dose-dependent reductions in blood pressure which was accompanied with vasodilation, increased nitric oxide, reduced sympathetic activity and enhanced potassium channel conductance in blood vessels and neurons [34]. We have proposed that mitochondria-targeted SOD mimetic will specifically reduce mitochondrial and reduce vascular oxidative stress and improve endothelium dependent relaxation. Indeed, we have previously demonstrated that mitoTEMPO attenuated hypertension in dose-dependent manner, reduced production of mitochondrial and H2O2, increased endothelial NO and improved vasodilatation while similar dose of TEMPOL was not effective [6]. Importantly, we found that targeting of SOD mimetic to mitochondria provided beneficial effects at a dose 1000-fold lower than previously reported for TEMPOL [6]. In this work we show similar activity of other mitochondria-targeted mCP1 and mCP2 therefore representing new antihypertensive agents that could add to the currently available therapeutic armamentarium.
These studies confirmed an important role of mitochondrial in the hypertension and demonstrated that mitochondria-targeted nitroxides could have therapeutic benefit in treatment of hypertension.
Acknowledgments
SID and AED were supported by funding from National Institutes of Health grant HL094469 and American Heart Association Innovative Research Grant 15IRG22730049. We thank Alfiya Bikineyeva for technical assistance. IAK was supported by Russian Foundation for Basic Research (12-03-00718-a).
References
- 1.Kearney P.M., Whelton M., Reynolds K., Muntner P., Whelton P.K., He J. Global burden of hypertension: analysis of worldwide data. Lancet. 2005;365(9455):217–223. doi: 10.1016/S0140-6736(05)17741-1. 15652604 [DOI] [PubMed] [Google Scholar]
- 2.Sacco R.L., Benjamin E.J., Broderick J.P., Dyken M., Easton J.D., Feinberg W.M., Goldstein L.B., Gorelick P.B., Howard G., Kittner S.J., Manolio T.A., Whisnant J.P., Wolf P.A. American Heart Association prevention conference. IV. Prevention and rehabilitation of stroke. risk factors. Stroke. 1997;28(7):1507–1517. doi: 10.1161/01.str.28.7.1507. 9227708 [DOI] [PubMed] [Google Scholar]
- 3.Harrison D.G., Marvar P.J., Titze J.M. Vascular inflammatory cells in hypertension. Frontiers in Physiology. 2012;3:128. doi: 10.3389/fphys.2012.00128. 22586409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byrd J.B., Zeng C., Tavel H.M., Magid D.J., O’Connor P.J., Margolis K.L., Selby J.V., Ho P.M. Combination therapy as initial treatment for newly diagnosed hypertension. American Heart Journal. 2011;162(2):340–346. doi: 10.1016/j.ahj.2011.05.010. 21835296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harrison D.G., Gongora M.C., Guzik T.J., Widder J. Oxidative stress and hypertension. Journal of the American Society of Hypertension. 2007;1(1):30–44. doi: 10.1016/j.jash.2006.11.006. 20409831 [DOI] [PubMed] [Google Scholar]
- 6.Dikalova A.E., Bikineyeva A.T., Budzyn K., Nazarewicz R.R., McCann L., Lewis W., Harrison D.G., Dikalov S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circulation Research. 2010;107(1):106–116. doi: 10.1161/CIRCRESAHA.109.214601. 20448215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dikalov S.I., Nazarewicz R.R., Bikineyeva A., Hilenski L., Lassègue B., Griendling K.K., Harrison D.G., Dikalova A.E. Nox2-induced production of mitochondrial superoxide in angiotensin II-mediated endothelial oxidative stress and hypertension. Antioxidants & Redox Signaling. 2014;20(2):281–294. doi: 10.1089/ars.2012.4918. 24053613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griendling K.K., Harrison D.G. Out, damned dot: studies of the nadph oxidase in atherosclerosis. Journal of Clinical Investigation. 2001;108(10):1423–1424. doi: 10.1172/JCI14453. 11714732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodriguez-Iturbe B., Sepassi L., Quiroz Y., Ni Z., Wallace D.C., Vaziri N.D. Association of mitochondrial SOD deficiency with salt-sensitive hypertension and accelerated renal senescence. Journal of Applied Physiology. 2007;102(1):255–260. doi: 10.1152/japplphysiol.00513.2006. 17023572 [DOI] [PubMed] [Google Scholar]
- 10.Ohashi M., Runge M.S., Faraci F.M., Heistad D.D. Mnsod deficiency increases endothelial dysfunction in apoe-deficient mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26(10):2331–2336. doi: 10.1161/01.ATV.0000238347.77590.c9. 16873728 [DOI] [PubMed] [Google Scholar]
- 11.Murphy M.P., Smith R.A. Drug delivery to mitochondria: the key to mitochondrial medicine. Advanced Drug Delivery Reviews. 2000;41(2):235–250. doi: 10.1016/s0169-409x(99)00069-1. 10699318 [DOI] [PubMed] [Google Scholar]
- 12.Murphy M.P., Smith R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annual Review of Pharmacology and Toxicology. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. 17014364 [DOI] [PubMed] [Google Scholar]
- 13.O’Malley Y., Fink B.D., Ross N.C., Prisinzano T.E., Sivitz W.I. Reactive oxygen and targeted antioxidant administration in endothelial cell mitochondria. Journal of Biological Chemistry. 2006;281(52):39766–39775. doi: 10.1074/jbc.M608268200. 17060316 [DOI] [PubMed] [Google Scholar]
- 14.Doughan A.K., Dikalov S.I. Mitochondrial redox cycling of mitoquinone leads to superoxide production and cellular apoptosis. Antioxidants & Redox Signaling. 2007;9(11):1825–1836. doi: 10.1089/ars.2007.1693. 17854275 [DOI] [PubMed] [Google Scholar]
- 15.Krishna M.C., Russo A., Mitchell J.B., Goldstein S., Dafni H., Samuni A. Do nitroxide antioxidants act as scavengers of O2-. or as sod mimics? Journal of Biological Chemistry. 1996;271(42):26026–26031. doi: 10.1074/jbc.271.42.26026. 8824242 [DOI] [PubMed] [Google Scholar]
- 16.Soule B.P., Hyodo F., Matsumoto K., Simone N.L., Cook J.A., Krishna M.C., Mitchell J.B. The chemistry and biology of nitroxide compounds. Free Radical Biology and Medicine. 2007;42(11):1632–1650. doi: 10.1016/j.freeradbiomed.2007.02.030. 17462532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Namiecinski M., Pulaski L., Kochman A., Skolimowski J., Bartosz G., Metodiewa D. Cytotoxicity, cytoprotection and neurotoxicity of novel deprenyl-related propargylamines, stable nitroxide free radicals, in vitro and in vivo. In Vivo. 2004;18(2):171–180. 15113044 [PubMed] [Google Scholar]
- 18.Mildaziene V., Baniene R., Marcinkeviciute A., Nauciene Z., Kalvenas A., Zimkus A. Tetraphenylphosphonium inhibits oxidation of physiological substrates in heart mitochondria. Molecular and Cellular Biochemistry. 1997;174(1–2):67–70. 9309667 [PubMed] [Google Scholar]
- 19.Dhanasekaran A., Kotamraju S., Karunakaran C., Kalivendi S.V., Thomas S., Joseph J., Kalyanaraman B. Mitochondria superoxide dismutase mimetic inhibits peroxide-induced oxidative damage and apoptosis: role of mitochondrial superoxide. Free Radical Biology and Medicine. 2005;39(5):567–583. doi: 10.1016/j.freeradbiomed.2005.04.016. 16085176 [DOI] [PubMed] [Google Scholar]
- 20.Trnka J., Blaikie F.H., Smith R.A., Murphy M.P. A mitochondria-targeted nitroxide is reduced to its hydroxylamine by ubiquinol in mitochondria. Free Radical Biology and Medicine. 2008;44(7):1406–1419. doi: 10.1016/j.freeradbiomed.2007.12.036. 18206669 [DOI] [PubMed] [Google Scholar]
- 21.Jagtap A.P., Krstic I., Kunjir N.C., Hänsel R., Prisner T.F., Sigurdsson S.T. Sterically shielded spin labels for in-cell epr spectroscopy: analysis of stability in reducing environment. Free Radical Research. 2015;49(1):78–85. doi: 10.3109/10715762.2014.979409. 25348344 [DOI] [PubMed] [Google Scholar]
- 22.Rozantsev E.G. Free Nitroxyl Radicals. Plenum Press; New York: 1970. p. 249. [Google Scholar]
- 23.Hankovszky H.O., Hideg K., Lex L. Nitroxyls; VIII. Synthesis of nitroxylphosphinimines; a convenient route to amine, isothiocyanate, aminocarbonylaziridine, and carbodiimide nitroxyls. Synthesis. 1981:147–149. [Google Scholar]
- 24.Kirilyuk I.A., Kaledin V.I., Popova N.A., Nikolin V.P., Vasil’eva E.D., Grigor’ev I.A., Lushnikova E.L., Nepomnyashchikh L.M. Nitroxyl antioxidant tppa-tempo increases the efficacy of antitumor therapy on the model of transplantable mouse tumor. Bulletin of Experimental Biology and Medicine. 2010;150(1):75–78. doi: 10.1007/s10517-010-1073-2. 21161057 [DOI] [PubMed] [Google Scholar]
- 25.Dikalova A., Clempus R., Lassègue B., Cheng G., McCoy J., Dikalov S., San Martin A., Lyle A., Weber D.S., Weiss D., Taylor W.R., Schmidt H.H., Owens G.K., Lambeth J.D., Griendling K.K. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation. 2005;112(17):2668–2676. doi: 10.1161/CIRCULATIONAHA.105.538934. 16230485 [DOI] [PubMed] [Google Scholar]
- 26.Krege J.H., Hodgin J.B., Hagaman J.R., Smithies O. A noninvasive computerized tail-cuff system for measuring blood pressure in mice. Hypertension. 1995;25(5):1111–1115. doi: 10.1161/01.hyp.25.5.1111. 7737724 [DOI] [PubMed] [Google Scholar]
- 27.Widder J.D., Guzik T.J., Mueller C.F., Clempus R.E., Schmidt H.H., Dikalov S.I., Griendling K.K., Jones D.P., Harrison D.G. Role of the multidrug resistance protein-1 in hypertension and vascular dysfunction caused by angiotensin II. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27(4):762–768. doi: 10.1161/01.ATV.0000259298.11129.a2. 17272743 [DOI] [PubMed] [Google Scholar]
- 28.Trnka J., Blaikie F.H., Logan A., Smith R.A., Murphy M.P. Antioxidant properties of mito-TEMPOl and its hydroxylamine. Free Radical Research. 2009;43(1):4–12. doi: 10.1080/10715760802582183. 19058062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ilan Y., Shafferman A., Feinberg B.A., Lau Y.K. Partitioning of electrostatic and conformational contributions in the redox reactions of modified cytochromes c. Biochimica et Biophysica Acta. 1979;548(3):565–578. doi: 10.1016/0005-2728(79)90065-3. 228716 [DOI] [PubMed] [Google Scholar]
- 30.Harrison D.G. Endothelial function and oxidant stress. Clinical Cardiology. 1997;20:II-11–II-17. [PubMed] [Google Scholar]
- 31.Harrison D.G., Cai H., Landmesser U., Griendling K.K. Interactions of angiotensin II with NAD(P)H oxidase, oxidant stress and cardiovascular disease. Journal of the Renin-Angiotensin-Aldosterone System. 2003;4(2):51–61. doi: 10.3317/jraas.2003.014. 12806586 [DOI] [PubMed] [Google Scholar]
- 32.Zhao H., Joseph J., Fales H.M., Sokoloski E.A., Levine R.L., Vasquez-Vivar J., Kalyanaraman B. Detection and characterization of the product of hydroethidine and intracellular superoxide by hplc and limitations of fluorescence. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(16):5727–5732. doi: 10.1073/pnas.0501719102. 15824309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dikalov S., Fink B. ESR techniques for the detection of nitric oxide in vivo and in tissues. Methods in Enzymology. 2005;396:597–610. doi: 10.1016/S0076-6879(05)96052-7. 16291267 [DOI] [PubMed] [Google Scholar]
- 34.Wilcox C.S., Pearlman A. Chemistry and antihypertensive effects of tempol and other nitroxides. Pharmacological Reviews. 2008;60(4):418–469. doi: 10.1124/pr.108.000240. 19112152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dikalov S., Griendling K.K., Harrison D.G. Measurement of reactive oxygen species in cardiovascular studies. Hypertension. 2007;49(4):717–727. doi: 10.1161/01.HYP.0000258594.87211.6b. 17296874 [DOI] [PMC free article] [PubMed] [Google Scholar]