

Abstract
Rationale
Seizures during status epilepticus (SE) cause neuronal death and induce cyclooxygenase-2 (COX-2). Pilocarpine-induced SE was used to determine if COX-2 inhibition with NS-398, when administered alone or with diazepam, decreases the duration and/or intensity of SE and/or reduces neuronal injury in the rat hippocampus.
Methods
Electroencephalogram (EEG) electrodes were implanted in male Sprague-Dawley rats. SE was induced with lithium-pilocarpine, and continuous EEG and video monitoring were performed for 24 h. Rats were divided into four groups (n = 8-14 rats/group) and received NS-398, diazepam, NS-398 and diazepam, or vehicle 30 min after the first motor seizure. Six hours later, NS-398 injection was repeated in the NS-398 and in the NS-398 + diazepam groups. The duration of SE (continuous spiking) and the EEG power in the γ-band were analyzed. FluoroJade B staining in the dorsal hippocampus at 24 h after SE was analyzed semi-quantitatively in CA1, CA3 and hilus.
Results
The duration and intensity of electrographic SE was not significantly different across the four groups. In rats treated with NS-398 alone, compared to vehicle-treated rats, neuronal damage was significantly lower compared to vehicle-treated rats in CA3 (27%) and hilus (27%), but neuroprotection was not detected in CA1. When NS-398 was administered with diazepam, decreased neuronal damage was further obtained in all areas investigated (CA1: 61%, CA3: 63%, hilus: 60%).
Conclusions
NS-398, when administered 30 min after the onset of SE with a repeat dose at 6 h, decreased neuronal damage in the hippocampus. Administration of diazepam with NS-398 potentiates the neuroprotective effect of the COX-2 inhibitor. These neuroprotective effects occurred with no detectable effect on electrographic SE.
Keywords: cyclooxygenase-2 (COX-2), inflammation, neuroprotection
Status epilepticus (SE) is a neurologic emergency that requires prompt intervention to prevent long-term morbidity and mortality. Both human clinical studies and rodent models of SE indicate that prolonged seizure activity causes neuronal death via several pathways, and rapid treatment of SE reduces this injury and subsequent morbidity (Alldredge et al., 2001; Klitgaard et al., 2002; Lowenstein and Alldredge, 1998; Morimoto et al., 2004). Benzodiazepines, such as diazepam and lorazepam, are widely used as first-line agents to terminate seizure activity, but unfortunately, SE becomes progressively more refractory to these agents after 30-40 minutes (Jones et al., 2002; Naylor et al., 2005; Walton and Treiman, 1988). Thus, developing therapies for treating SE that more effectively terminate later-stage seizure activity, enhance neuroprotection, or suppress the long-term consequences of SE is an important goal.
Inflammatory mediators contribute to the negative consequences of SE (Ravizza et al., 2011; Vezzani et al., 2012). The cyclooxygenase-2 (COX-2) pathway, in particular, has been studied in many animal models of SE and reportedly affects neuronal excitability (Chen and Bazan, 2005; Zhang et al., 2008), susceptibility to neuronal death (Ho et al., 1998; Kawaguchi et al., 2005), and influx of several antiepileptic drugs across the blood brain barrier (Bauer et al., 2008; Schlichtiger et al., 2010; van Vliet et al., 2010), all of which may influence the severity of SE. COX-2 is an inflammatory enzyme involved in prostaglandin synthesis that is expressed under basal conditions in excitatory neurons from cortex, hippocampus, hypothalamus and amygdala (Adams et al., 1996; Kaufmann et al., 1996; Yamagata et al., 1993). Increased synaptic activity associated with seizures markedly amplifies the expression of COX-2 within 30 min from the beginning of seizures, with the peak effect estimated to be about 24 h in different animal models of seizures (Jung et al., 2006; Kawaguchi et al., 2005; Takemiya et al., 2006; Voutsinos-Porche et al., 2004). COX-2 knockout mice showed decreased incidence of after-discharges, reduced after-discharge duration, and delayed induction of convulsive seizures compared to control mice after rapid kindling, suggesting that COX-2 facilitates the recurrence of seizures (Takemiya et al., 2003). The COX-2 pathway has also been implicated in regulating the transport of several antiepileptic drugs across the blood brain barrier (Schlichtiger et al., 2010; van Vliet et al., 2010). Thus hypothetically, COX-2 inhibitors could directly suppress SE, facilitate transport of antiepileptic drugs to brain tissue, and reduce subsequent damage to neurons. However, when attempts to study the effect of COX-2 inhibition on the severity of SE and resultant neuronal damage using primarily behavioral observation of seizure activity, blocking of the COX-2 pathway has given mixed results, with some studies showing neuroprotection (Bauer et al., 2008; Jung et al., 2006; Kunz and Oliw, 2001b; Polascheck et al., 2010; Takemiya et al., 2006), and others showing no neuroprotection (Holtman et al., 2010; Holtman et al., 2009; Pekcec et al., 2009) or even exacerbation of neuronal death (Baik et al., 1999; Kim et al., 2008).
The aim of the present study was to investigate the effect of COX-2 inhibition, both alone and in the presence of diazepam, on the severity of SE and on neuronal damage. Brain electrical activity was monitored continuously for 24 h with subdural EEG electrodes, thus allowing analysis of the effect of COX-2 and diazepam on the electrographic SE, not just behavioral seizure activity. Neuronal damage in the hippocampus was assessed by FluoroJade B staining of brain sections obtained 24 h after induction of SE. We hypothesized that inhibition of COX-2 decreases the duration and/or intensity of electrograpic SE, possibly through decreased pharmacoresistance to benzodiazepines, with resultant neuroprotection.
Experimental Procedures
Experimental animals
Adult male Sprague-Dawley rats (Charles River, USA) weighing 250 - 400 g were kept under 12 h light/dark conditions with free access to food and water. All procedures were approved by the University of Utah Animal Care and Use Committee.
Electrode implantation
The animals were anesthetized with isoflurane (2%) and placed in a stereotaxic device. Bipolar electrodes (MS333-3-B, Plastics One, Roanoke, VA) were used for subdural recordings. Two holes (500 μm) were drilled on the right side of the midline under the bregma, and one lead was placed into each of the craniotomies to provide differential recordings. A third lead was placed in a third craniotomy left of the midline to be used as the ground electrode. Three additional screws were implanted in the skull, then the electrodes were fixed in place with dental cement and the skin was sutured around the skull. After surgery, the animals were put on a 12-h light/dark cycle and fed standard rat chow ad libitum. After recovery from the surgery (≥7 days), the animals were treated with pilocarpine.
Video and EEG recording
Implanted animals were placed into cages with commutators (Plastics One, Roanoke, VA) and connected to cables via their skull caps for EEG recordings. Signals were amplified using EEG100C amplifiers (high-pass filter, 1 Hz; low-pass filter, 35 Hz; 5,000× gain), digitized at 500 Hz using an MP150 digital-analog converter, and acquired with AcqKnowledge acquisition software (BioPac Systems, Santa Barbara, CA). While the rats were tethered in these cages, they were continuously video monitored using eight color cameras linked to a multiplexer to allow for eight animals to be recorded on one DVD. Recordings were obtained for 24 h across three DVD recorders (DMR-ES20, Panasonic), each recording for an 8-h period.
SE induction
Animals were connected to the video-EEG recording system and pretreated with LiCl (127 mg/kg, i.p., 18 h before pilocarpine injection, Fig. 1A). Scopolamine bromide (1 mg/kg, ip) was administered 30 min before the injection of pilocarpine (50 mg/kg, ip). For one set of experiments, animals were grouped in two categories: 1) animals receiving a vehicle injection, 0.5% methylcellulose, (vehicle group, n = 14), and 2) animals receiving COX-2 inhibitor (NS-398, 10 mg/kg, i.p., Cayman Chemical, MI USA, NS-398 group, n = 11) 30 min after the first convulsive seizure. The injections were repeated after 6 h. This concentration of NS-398 has been reported to significantly reduce COX-2 activity in the brain as measured by PGE2 activity in a rat model of kainate-induced SE (Takemiya et al., 2006). Based on our experience with pilocarpine-induced SE model, the mortality rate in the vehicle group is about 35%, so we used a higher number of animals in this group than in the NS-398 group. In a second set of experiments, three different groups of rats received: 1) vehicle (n = 14), 2) diazepam (10 mg/kg, n = 8), and 3) diazepam + NS-398 (10 mg/kg each, n = 9) 30 min after the first convulsive seizure. NS-398 injection was repeated at 6 hours in diazepam + NS-398 group, while the other groups received vehicle. EEG-video recordings were obtained for 24 h after induction of SE (Fig. 2). During this period of time, the mortality rate was: 29% and 36% in vehicle groups, 9% in NS-398 group, 0% in diazepam group, and 11% in diazepam + NS-398 group.
Figure 1.

A: Schematic representation of the experimental design. Subdural EEG electrodes were implanted, and 1 week later LiCl (127 mg/Kg) was injected i.p. The following day, scopolamine bromide (1 mg/kg, ip) was administered 30 min before the injection of pilocarpine (50 mg/kg, ip). The drugs (NS-398 and/or diazepam, 10 mg/kg each) and the vehicle were administered 30 min after the first motor seizure. NS-398/vehicle injections were repeated 6 h later. Rats were sacrificed 24 h after induction of SE. B: Electrographic SE was considered to be the period of time in which the animal displayed continuous spiking without periods of inactivity between spiking.
Figure 2.

Example of electrographic recordings of pilocarpine-induced SE in a vehicle-injected rat (A), a NS-398-treated rat (B), a diazepam-treated rat (C) and a diazepam+NS-398-treated rat. Upper panel: electrographic recordings during the first 10 h after pilocarpine injection. Lower panel: expanded views of electrographic recordings at the following times: 1) baseline, 2) before first injection of vehicle, NS0398, diazepam or diazepam+NS-398, 3) 30 min after first injection of vehicle, NS-398, diazepam or diazepam+NS-398 and 4) 9.5 h after induction of SE.
Analysis of electrographic SE
An algorithm that extracts the energy in the γ-band was used to evaluate the efficacy of NS-398 and/or diazepam on electrographic SE (Lehmkuhle et al., 2009). This program filters the EEG to reduce the movement artifacts (<20 and >70 Hz), and models the energy with an eighth-order polynomial to estimate the effect of the drug over a 10-h period with 10-min resolution. This approach allowed statistical comparisons to be made across treatment groups at selected time points. To normalize the electrographic seizure data across animals, the time of drug administration was considered 100% and data is presented as percent change in power. The duration of electrographic SE was considered to be the period of time in which the animal displayed continuous spiking activity without periods of inactivity between the spiking (Fig. 1B).
Histology
Histology was performed on brains obtained 24 h after induction of SE. Rats were deeply anesthetized with isoflurane and perfused transcardially with ice-cold saline followed by 10% formalin (pH 7.4). Brains were removed immediately after perfusion, post-fixed in the same fixative for one night at 4°C, and impregnated with 30% sucrose diluted in phosphate buffer. Coronal sections (40 μm) were cut on a sliding microtome in serial order starting at the genu of the corpus callosum (anterior) to the brain stem (posterior), and every third section was mounted serially. In adult rats, pilocarpine has been shown to cause intense neuronal damage throughout the entire hippocampus, with the hilar region of the dentate gyrus showing the greatest damage in the ventral (vs. dorsal) hippocampus (Covolan and Mello, 2000; Covolan and Mello, 2006). Fujikawa (1996) reported that CA3 damage is the same in the dorsal and ventral regions, while CA1 and dentate gyrus showed only slightly greater neuronal damage in the ventral vs. dorsal hippocampus. In the present experiments, the number of FluoroJade B-stained neurons was examined in coronal sections of the dorsal hippocampus, where the amount of damage is adequate to detect a possible neuroprotective effect of the COX-2 inhibitor.
COX-2 immunostaining
In a separate set of experiments, brain sections from naïve (no electrode implanted) animals were immunostained with COX-2 antibody and compared to sections from animals given lithium pilocarpine and vehicle or lithium pilocarpine and NS-398. Tissue from 2-3 animals were used for each group. Brain sections obtained 24 h after SE were serially incubated in 5% normal rabbit serum, COX-2 goat polyclonal antibody (1/200 dilution for 48 h, at 4° C, Santa Cruz Biotechnology), and then with the secondary antibody (biotinylated rabbit anti-goat IgG, 1/200 dilution) followed by avidin-biotin complex (diluted 1/200 in 0.3 Triton in PBS). Diaminobenzidine tetrahydrochloride was used as the peroxidase substrate to visualize sites of antibody binding. Negative controls (incubation without first antibody) were performed in order to check the specificity of the antibody (Voutsinos-Porche et al., 2004).
FluoroJade B assessment of neuronal damage
Every third brain section of the dorsal hippocampus was immersed in ethanol (100 and 70%), distilled water, and 0.06% potassium permanganate, and then placed in 0.001% FluoroJade B staining solution for 30 min at room temperature (Voutsinos-Porche et al., 2004). After being rinsed in distilled water, dried, immersed in 100% ethanol followed by toluene, and then mounted, sections were examined with a fluorescence microscope (Zeiss Imager Z1) and high-resolution digital images of the hippocampus in each hemisphere were obtained. Digital image of each section was used for the automated counting of the FluoroJade B-stained cells using the AutoMeasure function of the AxioVision 4.3 software from Carl Zeiss. The software could automatically identify the objects of interest based on their brightness, color and descriptive parameters (shape, size). Stained neurons in the area of CA1, CA3 and hilus of the dentate gyrus were counted and the values from left and right hemispheres were added together. The investigator performing the quantification was blinded to the treatment of the animals. The three values were added for each area of the hippocampus and then for the two hippocampi (right and left), in order to have one value for each section (our data showed no difference in neuronal damage in the left vs. right hippocampus). The results obtained from all sections were averaged to have one value for CA1, CA3 and hilus, respectively, for each brain. Those values obtained for each brain (n = 8-10/group) were used in the statistical analysis.
Statistical comparisons
The electrographic seizure activity was modeled with an eighth-order polynomial and statistical analysis was performed using Mann-Whitney U-test. For the assessment of neuronal damage, values are given as mean value ± SEM. Statistical comparisons were performed on the numbers of FluoroJade B positive cells using one-way ANOVA followed by a Tukey test. P < 0.05 was assumed to indicate significant differences.
Results
Lithium Pilocarpine with and without COX-2 inhibition and/or diazepam evokes prolonged SE
Treatment with lithium-pilocarpine evoked prolonged and sustained SE lasting several hours (Fig. 2). The first convulsive seizure, chosen as time point zero for further treatments, was observed clinically as either Racine stage 4 (rearing) or 5 (rearing with falling). Within a few minutes, all animals progressed to Racine stage 5 behavior, and had been in sustained SE when treatment was given at 30 min. During the 24 h video EEG monitoring period, the mortality rate was: 29% and 36% in vehicle groups, 9% in NS-398 group, 0% in diazepam group, and 11% in diazepam + NS-398 group. This trend of reduced mortality with COX-2 inhibition and/or diazepam did not reach statistical significance, but is consistent with previous reports describing this effect (Jiang et al., 2013; Levin et al., 2012; Pitkanen et al., 2005).
Effect of COX-2 inhibition and/or diazepam on duration and intensity of pilocarpine-induced SE
Duration of the electrographic SE did not significantly differ between the groups (Fig. 2 and 3). The period of continuous spiking activity without return to baseline recordings was different from animal to animal and varied from 1.4 to 3.9 h. After continuous spiking ceased, the animals displayed for many hours regular electrographic bursting separated by short periods of inactivity (Fig. 1B). Analysis of the electrographic discharges (algorithm described in the Experimental Procedures section) showed no difference in the intensity of SE in γ-band after treatment with COX-2 inhibitor and/or diazepam when compared to the control conditions (first 10 h from the beginning of SE). Injection of NS-398 and/or diazepam (both at 10 mg/kg) did not cause any detectable difference in the severity of electrographic discharges (Fig.3).
Figure 3.
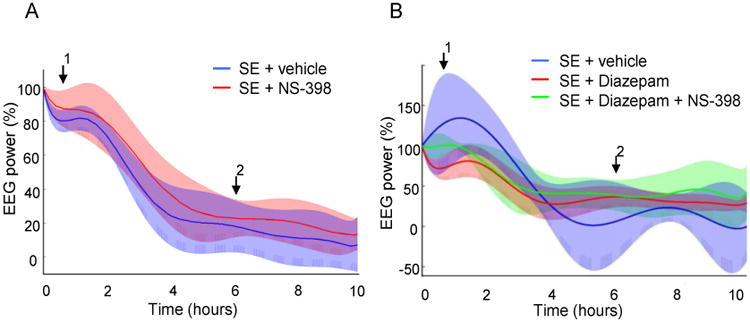
Analysis of subdural EEG recordings obtained for 10 h after the onset of the pilocarpine-induced SE from groups of A: vehicle (n = 12) and NS-398 (n = 10)-treated animals, and B: vehicle (n = 8), Diazepam (n = 8) and Diazepam + NS-398 (n = 7) animals. Percentage change in power in the γ-band showed no difference between the groups. The mean value (colored line) and 95% confidence interval (shaded area) is shown for each group. Arrow 1 indicates the time of the injection of vehicle, diazepam and/or NS-398 at 30 min after the first motor seizure. Arrow 2 indicates the time of the second injection of vehicle or NS-398.
COX-2 expression at 24 h after pilocarpine-induced SE
Based on previous work using a similar dose of NS-398 that showed decreased PGE2 production 24 h after kainate-induced SE (Takemiya et al., 2006), we qualitatively assessed for changes in COX-2 immunostaining 24 h after pilocarpine-induced SE with and without NS-398. Staining with COX-2 antibody of control sections (no SE) showed minimal baseline expression of COX-2 in CA1, CA3 and hilus of the dentate gyrus, confirming the presence of COX-2 enzyme in the hippocampus under normal physiological conditions. For the pilocarpine-treated rats, immunostaining was increased in the vehicle group (SE + vehicle) and decreased in sections from rats treated with COX-2 inhibitor (SE + NS-398). Seizure-induced upregulation of COX-2 was observed in the neurons of the CA1, CA3 and dentate gyrus, as well as in the molecular dendritic layer and hilus of the dentate gyrus, (Fig. 4). The observed reduction in COX-2 immunostaining with NS-398 24 h after SE is consistent with other reports of reduced COX-2 activity or COX-2 products in rat brain after administration of NS-398 (Fathali et al., 2010; Takemiya et al., 2006).
Figure 4.
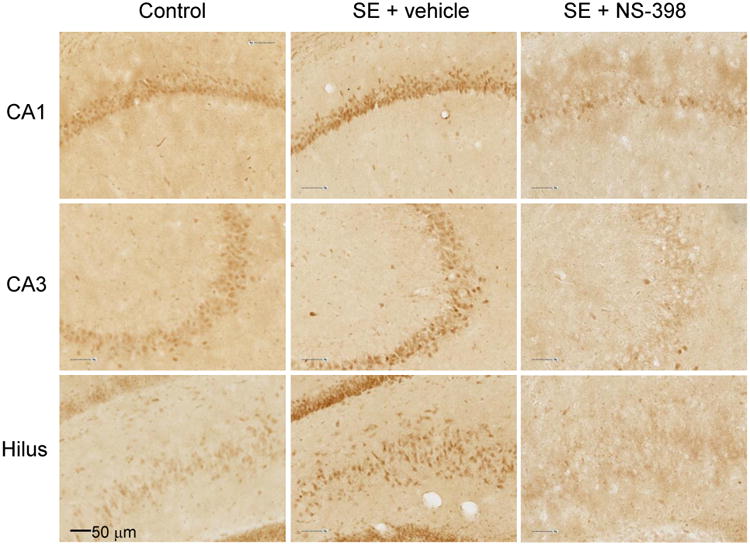
COX-2 immunostaining of sections from the dorsal hippocampus of control rats (no pilocarpine, left column) and rats with pilocarpine-induced status epilepticus (SE) that were treated with vehicle (SE + vehicle) or NS-398 (SE + NS-398). Data show increased expression of COX-2 24 h after induction of SE in vehicle-treated rats. NS-398 decreased COX-2 immunostaining in CA1, CA3 and hilus of the dentate gyrus.
Effect of COX-2 inhibition on neuronal death after pilocarpine-induced SE
Neuronal degeneration was analyzed 1 day after SE in every third section of the dorsal hippocampus. Neuronal damage induced by SE was widespread throughout the left and right hippocampus, and no difference was found between the two hemispheres. For automated counting with the Axiovision program, the entire area of CA1, CA3 and hilus was used for the evaluation of neuronal damage. Treatment with COX-2 inhibitor significantly decreased the number of FluoroJade B-stained neurons in CA3 and hilus of the dentate gyrus (27.1±4% and 26.8±3%, respectively, p<0.05, Fig. 5 and Fig. 6). In the CA1 area, although neuronal damage was decreased by 18±6% in the NS-398-treated group, the results were not statistically significant. When COX-2 inhibitor was associated with diazepam (10 mg/kg), neuronal damage was considerably reduced compared to vehicle rats throughout the entire hippocampus: CA1 (61±3%), CA3 (63±6%) and hilus of the dentate gyrus (60±12%), p < 0.05. Diazepam alone, at the concentration used in this study, did not significantly affect the number of FluoroJade B-stained neurons (Fig. 5 and Fig. 7).
Figure 5.
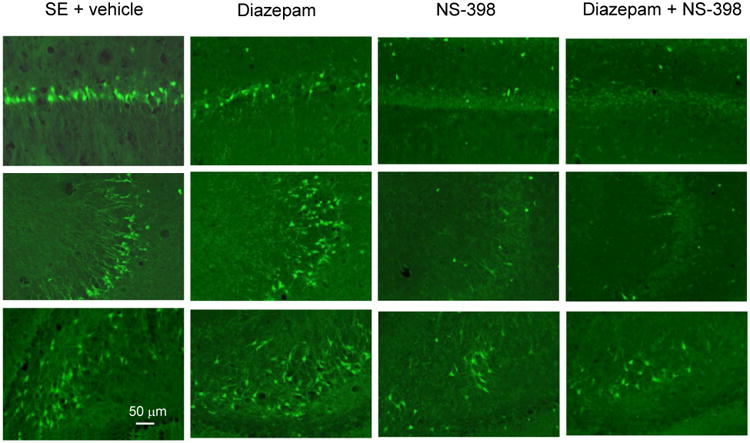
Representative FluoroJade B-staining of sections from the dorsal hippocampus showing widespread neuronal damage in CA1 (top traces), CA3 (middle traces) and hilus (bottom traces) 24 h after pilocarpine-induced SE in vehicle group (SE + vehicle) and in diazepam group (Diazepam, 10 mg/kg). Treatment with COX-2 inhibitor (NS-398) decreased neuronal damage/death most apparent in CA3 and hilus, and to a lesser degree in CA1. Treatment with diazepam plus COX-2 inhibitor (diazepam + NS-398, 10 mg/kg each) decreased cell damage/death throughout the dorsal hippocampus compared to vehicle and diazepam groups.
Figure 6.

The effect of COX-2 inhibitor, NS-398, on neuronal damage 24 h after pilocarpine-induced SE in rats. Counting of FluoroJade B-stained neurons in the CA1, CA3 and hilus of the dentate gyrus show that NS-398 (n=10) significantly decreased the number of degenerating neurons compared to vehicle (n=10) in CA3 and hilus of the dentate gyrus. The neuroprotection induced by COX-2 inhibitor was statistically significant in the CA3 and hilus of the dentate gyrus (* p<0.05), but not CA1.
Figure 7.

The effect of diazepam and COX-2 inhibitor, NS-398, on neuronal damage 24 h after pilocarpine-induced SE. When diazepam was injected alone (SE + Diazepam, 10 mg/kg, n = 8), it did not cause significant neuroprotection compared to the vehicle group (SE + vehicle, n = 9). When diazepam was administered together with NS-398 (10 mg/kg each, n = 8), significant neuroprotection was observed in the CA1, CA3 and hilus of the dentate gyrus (* p<0.05).
Discussion
Three key results were obtained in this study: 1) Treatment with the selective COX-2 inhibitor NS-398--with or without diazepam--did not have a significant effect on the duration or intensity of electrographic SE. 2) COX-2 inhibition significantly decreased neuronal damage in CA3 and hilus in the dorsal hippocampus. 3) This effect was greatly enhanced when NS-398 was associated with diazepam, and neuroprotection was obtained throughout the entire hippocampus.
COX-2 inhibition is neuroprotective within a clinically relevant therapeutic window
Both clinical evidence and animal models of SE have established that neurological outcomes generally depend on the severity and duration of SE. In the LiPC SE model, neuronal death is observed when SE persists beyond 40-60 min (Fujikawa, 1996), and resistance to benzodiazepines occurs within 30-45 min (Jones et al., 2002; Naylor et al., 2005; Pouliot et al., 2013). Access to ambulatory emergency personnel often can take 30-60 min. There is therefore a theoretical window within this first hour where new therapies would be candidates to prevent long-term sequelae if they either 1) reduce the duration of SE, 2) suppress the resistance to benzodiazepines, or 3) prevent neuronal death. Our results suggest NS-398 alone has a modest, but significant, neuroprotective effect alone, but when used in conjunction with a dose of diazepam known to be sub-therapeutic at terminating electrographic SE, the neuroprotective effect is enhanced. This combination initiated at 30 min (with a repeat dose of NS-398 at 6 h) represents a realistic therapeutic intervention to consider for future studies.
Previous studies inhibiting COX-2 activity have reported conflicting results regarding neuroprotection with some showing neuroprotection (Jung et al., 2006; Kawaguchi et al., 2005; Kunz and Oliw, 2001b; Polascheck et al., 2010), and others showing either no neuroprotection (Holtman et al., 2010; Pekcec et al., 2009) or aggravation of neuronal damage (Baik et al., 1999; Kim et al., 2008). Several proposed mechanisms have been suggested to explain this discrepancy. Although timing of COX-2 manipulation appeared initially to explain differences in results with earlier pre-SE COX-2 treatment (Baik et al., 1999; Holtman et al., 2010; Takemiya et al., 2006) usually resulting in poorer neuroprotection, subsequent studies by Baik and colleagues comparing pre- and post-SE protocols did not find a significant difference (Kim et al., 2008). While pre-SE COX-2 treatment protocols are important in elucidating COX-2 inhibitor's effects on severity of chronic spontaneous seizure activity, it is unclear how clinically relevant these studies are as a therapeutic intervention for SE. Another interesting hypothesis is that COX-2 is the precursor step to several pathway targets, some that have neuroprotective effects and others that aggravate neuronal damage. It is possible that more selective manipulation at these downstream targets will result in greater neuroprotection without other adverse outcomes. The main product of COX-2 in the brain is prostaglandin E2 (PGE2), which is markedly increased after SE. Low concentrations of PGE2 appear to be neuroprotective (Akaike et al., 1994), while high concentrations may cause neuronal damage (Takemiya et al., 2006; Takemiya et al., 2007). Recent studies investigating neurotoxic mechanisms of COX-2 demonstrate both toxic and paradoxically protective effects of downstream PGE2 receptor signaling pathways (Ahmad et al., 2008; Echeverria et al., 2005; Jiang et al., 2013; Kawano et al., 2006). Therefore, development of more specific inhibitors/receptor antagonists working downstream on the COX-2 inflammatory cascade is necessary and probably the best approach for investigating the role of inflammation in the pathology of seizures. A third explanation is that other method-specific discrepancies (i.e. rat strain, SE severity, protocol differences) account for differences in results. Obviously, any difference in experimental design can be a candidate to explain disparate results between studies. However, given our finding of enhanced neuroprotection after sub-therapeutic diazepam, the use of diazepam or other benzodiazepines in the experimental design warrants closer examination. When previous studies have claimed a detrimental effect of COX-2 inhibition (Baik et al., 1999; Kim et al., 2008), no benzodiazepine was used. In addition, several studies that did not use a benzodiazepine in their protocol concluded that COX-2 inhibition had no effect on neuroprotection (Gobbo and O'Mara, 2004; Holtman et al., 2010; Holtman et al., 2009; Kunz and Oliw, 2001a), although others did show neuroprotection in hippocampus (Kunz and Oliw, 2001b) or, more specifically, CA3 region (Kawaguchi et al., 2005; Takemiya et al., 2006) similar to our present study. Previous studies that did include diazepam (Jung et al., 2006; Polascheck et al., 2010) did show neuroprotection in both CA1 and CA3 regions of hippocampus, again similar to our present study. Thus, even when a variety of different protocols are used, there is a trend for greater neuroprotective effects of COX-2 inhibition when diazepam is included in the experimental design. While multiple experimental factors can influence the results from different studies, the inclusion of diazepam in the experimental design should be considered when attempting to account for differences between results.
In the present study, neuroprotection was assessed in the dorsal hippocampus. Previous work has shown that the area of greatest hippocampal injury after status epilepticus is observed in the ventral hippocampus (Ekstrand et al., 2011; Fujikawa, 1996; Williams et al., 2002), and initiation of spontaneous seizures after status epilepticus is also more often observed in this region compared to dorsal hippocampus (Toyoda et al., 2013). These results suggest that the ventral hippocampus may have particular significance in the later development of epilepsy. Inspection of brain sections containing ventral hippocampus showed that the neuroprotective effect of combined NS-398 and diazepam was observed also in the ventral hippocampus, but quantitative analysis was not performed. For studies assessing for a possible correlation between hippocampal neuronal injury and epileptogenesis, it will be important to include an analysis of ventral hippocampus. However, for assessment of general neuroprotection, analysis of dorsal hippocampus may be sufficient.
COX-2 inhibition and diazepam did not affect the duration or intensity of SE
No effect of NS-398 and/or diazepam on duration of SE was detected. Similarly, using a quantitative algorithm for analyzing the intensity of EEG power in the γ-band, no significant difference between the NS-398 and/or diazepam groups and the vehicle group was observed. This quantitative analysis of the severity of SE has advantages in detecting subtle changes in seizure intensity over many previous assessments of SE, particularly when compared to work where only behavioral observation of SE is used. Even with the known inherent variability observed in SE EEG traces between different animals, this analysis has previously detected effects on SE from other antiepileptic compounds. A positive control for this algorithm was first presented by Lehmkuhle et al. (2009), with propofol significantly decreasing the EEG power compared to control. Subsequently, suppressive effects on the severity of SE were detected for early diazepam treatment (15 min after SE), pentobarbital, and sec-butyl-propylacetamide, but similar to the present study later diazepam treatment (30 min after SE) did not result in a significant difference (Pouliot et al., 2013). Based on in vitro data showing that PGE2, the main product of COX-2 enzyme in the brain, increases membrane excitability, enhances excitatory postsynaptic potentials (EPSPs) in CA1 pyramidal neurons (Chen and Bazan, 2005) and induces glutamate release from astrocytes (Bezzi et al., 1998), we expected COX-2 inhibitor to reduce the aberrant neuronal activity during SE. Our results do not support the hypotheses that COX-2 neuroprotection may be partially mediated by changes in status severity. While, we cannot exclude the possibility that NS-398 may have decreased neuronal excitability, with the effect being too small to be detected with our recording and analysis methods, our results suggest that at least with the lithium pilocarpine model, the evoked SE is not significantly influenced by previously described COX-2 mediated effects on excitability.
Diazepam enhances neuroprotective effect of COX-2 inhibition
In clinical settings, benzodiazepines represent the first-line drugs for the treatment of SE. When COX-2 inhibitor was injected together with diazepam, the neuronal damage was greatly decreased to less than half compared to the vehicle group. Diazepam is known to be neuroprotective at a higher dose, 20 mg/kg, only when injected within 2 h from induction of SE (Pitkanen et al., 2005). We specifically used a low dose of diazepam that does not cause significant neuroprotection by itself and does not affect the severity or duration of electrographic SE (Pouliot et al., 2013). Diazepam, at 10 mg/kg injected 30 min after the first motor seizure, has a strong sedative effect (Pouliot et al., 2013) and decreases mortality (Pitkanen et al., 2005). However, this concentration of diazepam did not significantly affect the electrographic SE. This finding underscores the importance of confirming by EEG recording the cessation of seizure activity when assessing a therapeutic intervention. Although the precise mechanism of the combined COX-2 and diazepam enhancement of neuroprotection is not known, recent work has shown other examples of combining multiple modulators of inflammation to achieve greater neuroprotection (Kwon et al., 2013). It has been reported that pretreatment with COX-2 inhibitors enhances the delivery of antiepileptic medications to the brain including phenytoin and phenobarbital by reducing the upregulation of the efflux transporter P-glycoprotein (Bauer et al., 2008; Holtman et al., 2010; van Vliet et al., 2010). Although the P-glycoprotein transporter is involved in the brain level regulation of several AEDs including phenobarbital, phenytoin, lamotrigine, and leviteracetam (Luna-Tortos et al., 2008), it is not known to influence the pharmacokinetics and tissue distribution of diazepam (Yamazaki et al., 2001). It is also difficult to explain how COX-2 treatment given after the initiation of SE could act via this mechanism. COX-2 inhibitors enhanced the effect of low dose (sub-protective) diazepam on behavioral seizures and on recovery after SE (Dhir et al., 2006). Our results indicate COX-2 inhibitors may be a potential adjuvant therapy for the acute treatment of SE, but further studies are warranted to determine the specific mechanism of this effect with diazepam.
Conclusions
Inhibition of COX-2 activity 30 min after pilocarpine-induced SE with a repeat dose at 6 h leads to moderate neuroprotection, but does not significantly affect duration or intensity of electrographic SE. The neuroprotective effect is greatly enhanced when COX-2 inhibitor is associated with diazepam, indicating that COX-2 inhibitors may have potential therapeutic applications in association with benzodiazepines, and may improve clinical outcomes following SE.
Highlights.
NS-398 is neuroprotective in hippocampus after pilocarpine-induced status epilepticus.
Co-administration with diazepam enhances neuroprotective effect of NS-398.
NS-398 and diazepam did not affect duration or severity of status epilepticus.
Acknowledgments
This research was supported by the Counter-ACT Program, National Institutes of Health Office of the Director (NIH OD), and the National Institute of Neurological Disorders and Stroke (NINDS), Grants No. N01-NS-4-2359 (CT, WP, and FED) and K08-NS-070957 (JE). We gratefully acknowledge the technical assistance of Katie Ricks and Spencer Clark.
Abbreviations
- COX-2
cyclooxygenase-2
- SE
status epilepticus
- PGE2
prostaglandin E2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Adams J, Collaco-Moraes Y, de BJ. Cyclooxygenase-2 induction in cerebral cortex: an intracellular response to synaptic excitation. J Neurochem. 1996;66:6–13. doi: 10.1046/j.1471-4159.1996.66010006.x. [DOI] [PubMed] [Google Scholar]
- Ahmad AS, Yun YT, Ahmad M, Maruyama T, Dore S. Selective blockade of PGE2 EP1 receptor protects brain against experimental ischemia and excitotoxicity, and hippocampal slice cultures against oxygen-glucose deprivation. Neurotox Res. 2008;14:343–351. doi: 10.1007/BF03033858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaike A, Kaneko S, Tamura Y, Nakata N, Shiomi H, Ushikubi F, Narumiya S. Prostaglandin E2 protects cultured cortical neurons against N-methyl-D-aspartate receptor-mediated glutamate cytotoxicity. Brain Res. 1994;663:237–243. doi: 10.1016/0006-8993(94)91268-8. [DOI] [PubMed] [Google Scholar]
- Alldredge BK, Gelb AM, Isaacs SM, Corry MD, Allen F, Ulrich S, Gottwald MD, O'Neil N, Neuhaus JM, Segal MR, Lowenstein DH. A comparison of lorazepam, diazepam, and placebo for the treatment of out-of-hospital status epilepticus. N Engl J Med. 2001;345:631–637. doi: 10.1056/NEJMoa002141. [DOI] [PubMed] [Google Scholar]
- Baik EJ, Kim EJ, Lee SH, Moon C. Cyclooxygenase-2 selective inhibitors aggravate kainic acid induced seizure and neuronal cell death in the hippocampus. Brain Res. 1999;843:118–129. doi: 10.1016/s0006-8993(99)01797-7. [DOI] [PubMed] [Google Scholar]
- Bauer B, Hartz AM, Pekcec A, Toellner K, Miller DS, Potschka H. Seizure-induced up-regulation of P-glycoprotein at the blood-brain barrier through glutamate and cyclooxygenase-2 signaling. Mol Pharmacol. 2008;73:1444–1453. doi: 10.1124/mol.107.041210. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, Pozzan T, Volterra A. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature. 1998;391:281–285. doi: 10.1038/34651. [DOI] [PubMed] [Google Scholar]
- Chen C, Bazan NG. Endogenous PGE2 regulates membrane excitability and synaptic transmission in hippocampal CA1 pyramidal neurons. J Neurophysiol. 2005;93:929–941. doi: 10.1152/jn.00696.2004. [DOI] [PubMed] [Google Scholar]
- Covolan L, Mello LE. Temporal profile of neuronal injury following pilocarpine or kainic acid-induced status epilepticus. Epilepsy Res. 2000;39:133–152. doi: 10.1016/s0920-1211(99)00119-9. [DOI] [PubMed] [Google Scholar]
- Covolan L, Mello LE. Assessment of the progressive nature of cell damage in the pilocarpine model of epilepsy. Braz J Med Biol Res. 2006;39:915–924. doi: 10.1590/s0100-879x2006000700010. [DOI] [PubMed] [Google Scholar]
- Dhir A, Naidu PS, Kulkarni SK. Effect of cyclooxygenase inhibitors on pentylenetetrazol (PTZ)-induced convulsions: Possible mechanism of action. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30:1478–1485. doi: 10.1016/j.pnpbp.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Echeverria V, Clerman A, Dore S. Stimulation of PGE receptors EP2 and EP4 protects cultured neurons against oxidative stress and cell death following beta-amyloid exposure. Eur J Neurosci. 2005;22:2199–2206. doi: 10.1111/j.1460-9568.2005.04427.x. [DOI] [PubMed] [Google Scholar]
- Ekstrand JJ, Pouliot W, Scheerlinck P, Dudek FE. Lithium pilocarpine-induced status epilepticus in postnatal day 20 rats results in greater neuronal injury in ventral versus dorsal hippocampus. Neuroscience. 2011;192:699–707. doi: 10.1016/j.neuroscience.2011.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fathali N, Ostrowski RP, Lekic T, Jadhav V, Tong W, Tang J, Zhang JH. Cyclooxygenase-2 inhibition provides lasting protection against neonatal hypoxic-ischemic brain injury. Crit Care Med. 2010;38:572–578. doi: 10.1097/CCM.0b013e3181cb1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikawa DG. The temporal evolution of neuronal damage from pilocarpine-induced status epilepticus. Brain Res. 1996;725:11–22. doi: 10.1016/0006-8993(96)00203-x. [DOI] [PubMed] [Google Scholar]
- Gobbo OL, O'Mara SM. Post-treatment, but not pre-treatment, with the selective cyclooxygenase-2 inhibitor celecoxib markedly enhances functional recovery from kainic acid-induced neurodegeneration. Neuroscience. 2004;125:317–327. doi: 10.1016/j.neuroscience.2004.01.045. [DOI] [PubMed] [Google Scholar]
- Ho L, Osaka H, Aisen PS, Pasinetti GM. Induction of cyclooxygenase (COX)-2 but not COX-1 gene expression in apoptotic cell death. J Neuroimmunol. 1998;89:142–149. doi: 10.1016/s0165-5728(98)00132-5. [DOI] [PubMed] [Google Scholar]
- Holtman L, van Vliet EA, Edelbroek PM, Aronica E, Gorter JA. Cox-2 inhibition can lead to adverse effects in a rat model for temporal lobe epilepsy. Epilepsy Res. 2010;91:49–56. doi: 10.1016/j.eplepsyres.2010.06.011. [DOI] [PubMed] [Google Scholar]
- Holtman L, van Vliet EA, van SR, Queiroz CM, Aronica E, Gorter JA. Effects of SC58236, a selective COX-2 inhibitor, on epileptogenesis and spontaneous seizures in a rat model for temporal lobe epilepsy. Epilepsy Res. 2009;84:56–66. doi: 10.1016/j.eplepsyres.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Jiang J, Quan Y, Ganesh T, Pouliot WA, Dudek FE, Dingledine R. Inhibition of the prostaglandin receptor EP2 following status epilepticus reduces delayed mortality and brain inflammation. Proc Natl Acad Sci U S A. 2013;110:3591–3596. doi: 10.1073/pnas.1218498110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DM, Esmaeil N, Maren S, Macdonald RL. Characterization of pharmacoresistance to benzodiazepines in the rat Li-pilocarpine model of status epilepticus. Epilepsy Res. 2002;50:301–312. doi: 10.1016/s0920-1211(02)00085-2. [DOI] [PubMed] [Google Scholar]
- Jung KH, Chu K, Lee ST, Kim J, Sinn DI, Kim JM, Park DK, Lee JJ, Kim SU, Kim M, Lee SK, Roh JK. Cyclooxygenase-2 inhibitor, celecoxib, inhibits the altered hippocampal neurogenesis with attenuation of spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Neurobiol Dis. 2006;23:237–246. doi: 10.1016/j.nbd.2006.02.016. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Worley PF, Pegg J, Bremer M, Isakson P. COX-2, a synaptically induced enzyme, is expressed by excitatory neurons at postsynaptic sites in rat cerebral cortex. Proc Natl Acad Sci U S A. 1996;93:2317–2321. doi: 10.1073/pnas.93.6.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi K, Hickey RW, Rose ME, Zhu L, Chen J, Graham SH. Cyclooxygenase-2 expression is induced in rat brain after kainate-induced seizures and promotes neuronal death in CA3 hippocampus. Brain Res. 2005;1050:130–137. doi: 10.1016/j.brainres.2005.05.038. [DOI] [PubMed] [Google Scholar]
- Kawano T, Anrather J, Zhou P, Park L, Wang G, Frys KA, Kunz A, Cho S, Orio M, Iadecola C. Prostaglandin E2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat Med. 2006;12:225–229. doi: 10.1038/nm1362. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Chung JI, Lee SH, Jung YS, Moon CH, Baik EJ. Involvement of endogenous prostaglandin F2alpha on kainic acid-induced seizure activity through FP receptor: the mechanism of proconvulsant effects of COX-2 inhibitors. Brain Res. 2008;1193:153–161. doi: 10.1016/j.brainres.2007.12.017. [DOI] [PubMed] [Google Scholar]
- Klitgaard H, Matagne A, Vanneste-Goemaere J, Margineanu DG. Pilocarpine-induced epileptogenesis in the rat: impact of initial duration of status epilepticus on electrophysiological and neuropathological alterations. Epilepsy Res. 2002;51:93–107. doi: 10.1016/s0920-1211(02)00099-2. [DOI] [PubMed] [Google Scholar]
- Kunz T, Oliw EH. Nimesulide aggravates kainic acid-induced seizures in the rat. Pharmacol Toxicol. 2001a;88:271–276. doi: 10.1034/j.1600-0773.2001.d01-116.x. [DOI] [PubMed] [Google Scholar]
- Kunz T, Oliw EH. The selective cyclooxygenase-2 inhibitor rofecoxib reduces kainate-induced cell death in the rat hippocampus. Eur J Neurosci. 2001b;13:569–575. doi: 10.1046/j.1460-9568.2001.01420.x. [DOI] [PubMed] [Google Scholar]
- Kwon YS, Pineda E, Auvin S, Shin D, Mazarati A, Sankar R. Neuroprotective and antiepileptogenic effects of combination of anti-inflammatory drugs in the immature brain. J Neuroinflammation. 2013;10:30. doi: 10.1186/1742-2094-10-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmkuhle MJ, Thomson KE, Scheerlinck P, Pouliot W, Greger B, Dudek FE. A simple quantitative method for analyzing electrographic status epilepticus in rats. J Neurophysiol. 2009;101:1660–1670. doi: 10.1152/jn.91062.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin JR, Serrano G, Dingledine R. Reduction in delayed mortality and subtle improvement in retrograde memory performance in pilocarpine-treated mice with conditional neuronal deletion of cyclooxygenase-2 gene. Epilepsia. 2012;53:1411–1420. doi: 10.1111/j.1528-1167.2012.03584.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenstein DH, Alldredge BK. Status epilepticus. N Engl J Med. 1998;338:970–976. doi: 10.1056/NEJM199804023381407. [DOI] [PubMed] [Google Scholar]
- Luna-Tortos C, Fedrowitz M, Loscher W. Several major antiepileptic drugs are substrates for human P-glycoprotein. Neuropharmacology. 2008;55:1364–1375. doi: 10.1016/j.neuropharm.2008.08.032. [DOI] [PubMed] [Google Scholar]
- Morimoto K, Fahnestock M, Racine RJ. Kindling and status epilepticus models of epilepsy: rewiring the brain. Prog Neurobiol. 2004;73:1–60. doi: 10.1016/j.pneurobio.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Naylor DE, Liu H, Wasterlain CG. Trafficking of GABA(A) receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J Neurosci. 2005;25:7724–7733. doi: 10.1523/JNEUROSCI.4944-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekcec A, Unkruer B, Schlichtiger J, Soerensen J, Hartz AM, Bauer B, van Vliet EA, Gorter JA, Potschka H. Targeting prostaglandin E2 EP1 receptors prevents seizure-associated P-glycoprotein up-regulation. J Pharmacol Exp Ther. 2009;330:939–947. doi: 10.1124/jpet.109.152520. [DOI] [PubMed] [Google Scholar]
- Pitkanen A, Kharatishvili I, Narkilahti S, Lukasiuk K, Nissinen J. Administration of diazepam during status epilepticus reduces development and severity of epilepsy in rat. Epilepsy Res. 2005;63:27–42. doi: 10.1016/j.eplepsyres.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Polascheck N, Bankstahl M, Loscher W. The COX-2 inhibitor parecoxib is neuroprotective but not antiepileptogenic in the pilocarpine model of temporal lobe epilepsy. Exp Neurol. 2010;224:219–233. doi: 10.1016/j.expneurol.2010.03.014. [DOI] [PubMed] [Google Scholar]
- Pouliot W, Bialer M, Hen N, Shekh-Ahmad T, Kaufmann D, Yagen B, Ricks K, Roach B, Nelson C, Dudek FE. A comparative electrographic analysis of the effect of sec-butyl-propylacetamide on pharmacoresistant status epilepticus. Neuroscience. 2013;231:145–156. doi: 10.1016/j.neuroscience.2012.11.005. [DOI] [PubMed] [Google Scholar]
- Ravizza T, Balosso S, Vezzani A. Inflammation and prevention of epileptogenesis. Neurosci Lett. 2011;497:223–230. doi: 10.1016/j.neulet.2011.02.040. [DOI] [PubMed] [Google Scholar]
- Schlichtiger J, Pekcec A, Bartmann H, Winter P, Fuest C, Soerensen J, Potschka H. Celecoxib treatment restores pharmacosensitivity in a rat model of pharmacoresistant epilepsy. Br J Pharmacol. 2010;160:1062–1071. doi: 10.1111/j.1476-5381.2010.00765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemiya T, Maehara M, Matsumura K, Yasuda S, Sugiura H, Yamagata K. Prostaglandin E2 produced by late induced COX-2 stimulates hippocampal neuron loss after seizure in the CA3 region. Neurosci Res. 2006;56:103–110. doi: 10.1016/j.neures.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Takemiya T, Matsumura K, Yamagata K. Roles of prostaglandin synthesis in excitotoxic brain diseases. Neurochem Int. 2007;51:112–120. doi: 10.1016/j.neuint.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Takemiya T, Suzuki K, Sugiura H, Yasuda S, Yamagata K, Kawakami Y, Maru E. Inducible brain COX-2 facilitates the recurrence of hippocampal seizures in mouse rapid kindling. Prostaglandins Other Lipid Mediat. 2003;71:205–216. doi: 10.1016/s1098-8823(03)00040-6. [DOI] [PubMed] [Google Scholar]
- Toyoda I, Bower MR, Leyva F, Buckmaster PS. Early activation of ventral hippocampus and subiculum during spontaneous seizures in a rat model of temporal lobe epilepsy. J Neurosci. 2013;33:11100–11115. doi: 10.1523/JNEUROSCI.0472-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vliet EA, Zibell G, Pekcec A, Schlichtiger J, Edelbroek PM, Holtman L, Aronica E, Gorter JA, Potschka H. COX-2 inhibition controls P-glycoprotein expression and promotes brain delivery of phenytoin in chronic epileptic rats. Neuropharmacology. 2010;58:404–412. doi: 10.1016/j.neuropharm.2009.09.012. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Auvin S, Ravizza T, Aronica E. Glia-neuronal interactions in ictogenesis and epileptogenesis: role of inflammatory mediators. 2012 [PubMed] [Google Scholar]
- Voutsinos-Porche B, Koning E, Kaplan H, Ferrandon A, Guenounou M, Nehlig A, Motte J. Temporal patterns of the cerebral inflammatory response in the rat lithium-pilocarpine model of temporal lobe epilepsy. Neurobiol Dis. 2004;17:385–402. doi: 10.1016/j.nbd.2004.07.023. [DOI] [PubMed] [Google Scholar]
- Walton NY, Treiman DM. Response of status epilepticus induced by lithium and pilocarpine to treatment with diazepam. exp neurol. 1988;101:267–275. doi: 10.1016/0014-4886(88)90010-6. [DOI] [PubMed] [Google Scholar]
- Williams PA, Wuarin JP, Dou P, Ferraro DJ, Dudek FE. Reassessment of the effects of cycloheximide on mossy fiber sprouting and epileptogenesis in the pilocarpine model of temporal lobe epilepsy. J Neurophysiol. 2002;88:2075–2087. doi: 10.1152/jn.2002.88.4.2075. [DOI] [PubMed] [Google Scholar]
- Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF. Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron. 1993;11:371–386. doi: 10.1016/0896-6273(93)90192-t. [DOI] [PubMed] [Google Scholar]
- Zhang HJ, Sun RP, Lei GF, Yang L, Liu CX. Cyclooxygenase-2 inhibitor inhibits hippocampal synaptic reorganization in pilocarpine-induced status epilepticus rats. J Zhejiang Univ Sci B. 2008;9:903–915. doi: 10.1631/jzus.B0820018. [DOI] [PMC free article] [PubMed] [Google Scholar]