

Abstract
As many other cancers, pancreatic ductal adenocarcinoma (PDAC) progression is associated with a series of hallmark changes for cancer cells to secure their own growth success. Yet, these very changes render cancer cells highly sensitive to viral infection. A promising strategy may rely on and exploit viral replication for tumor destruction, whereby infection of tumor cells by a replication-conditional virus may lead to cell destruction and simultaneous release of progeny particles that can spread and infect adjacent tumor cells, while sparing healthy tissues. In the present study, we used Myb34.5, a second-generation replication-conditional herpes simplex virus type 1 (HSV-1) mutant in which ICP6 gene expression is defective and expression of the HSV-1 γ134.5 gene is regulated by the cellular B-myb promoter. We found that B-myb is present in experimental PDAC and tumors, and is overexpressed in patients' tumors, as compared with normal adjacent pancreas. Myb34.5 replicates to high level in human PDAC cell lines and is associated with cell death by apoptosis. In experimental models of PDAC, mice receiving intratumoral Myb34.5 injections appeared healthy and tumor progression was inhibited, with evidence of tumor necrosis, hemorrhage, viral replication, and cancer cell death by apoptosis. Combining standard-of-care chemotherapy with Myb34.5 successfully led to a very impressive antitumoral effect that is rarely achieved in this experimental model, and resulted in a greater reduction in tumor growth than chemotherapy alone. These promising results warrant further evaluation in early phase clinical trial for patients diagnosed with PDAC for whom no effective treatment is available.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is highly resistant to standard treatments and still the fourth leading cause of cancer-related death in Western countries.1 Its incidence has increased over the last 50 years. Since 1997, Gemcitabine was the only approved first-line treatment for patients with unresectable locally advanced or metastatic pancreatic cancer.2 However, the 5-year survival rate is only 2%, with 1-year survival rates ranging from 17% to 23%.2 Recently, phase II and III trials exploring gemcitabine-based combinations with erlotinib,3 Folfirinox,4 or nab-Paclitaxel5 were found to improve overall survival of patients with advanced tumors. However, standard of care for management of patients diagnosed with locally advanced tumor is still awaited. An interesting therapeutic approach may rely on viral replication for tumor destruction, by which infection of single-tumor cells may induce cell destruction and release of progeny virions that can further spread to the entire tumor cells population.6 Indeed, PDAC carcinogenesis is associated with a series of hallmark changes for cancer cells to secure their own growth success.7,8 Yet, these very changes (among them lack of interferon response, elevated metabolic activity, and disengagement of cell cycle control) render cancer cells highly sensitive to viral infection.
Several viruses have been explored for their ability to replicate and to inhibit PDAC growth in experimental models.9–11 Genetically engineered conditionally replicating herpes simplex virus type 1 (HSV-1) is promising for PDAC therapy because (1) they infect various tumor cell types, (2) they do not integrate into the genome of infected cells, (3) they have been shown to be safe in clinical trials, (4) there are several available anti-HSV-1-specific drugs, such as acyclovir to control unwanted infection, and (5) total cell killing can be achieved rapidly with a relatively low multiplicity of infection (MOI).11
The most widely used strategy to restrict HSV-1 replication to cancer cells is to delete genes that are important for viral replication and to evade innate immune responses, including the IFN-related protein kinase (PKR) antiviral response.6 ICP6 encodes the large subunit of viral ribonucleotide reductase (RR), an enzyme involved in de novo synthesis of deoxynucleotides.12 In the absence of viral RR, virus replication depends on host cell RR activity, which has been reported to be higher in cancer cells.13 Consequently, HSV-1 mutants with deletions in the ICP6 gene preferentially replicate in actively dividing cells such as malignant cells. Viral double-stranded RNAs activate various cellular antiviral responses, including PKR, which phosphorylates and inactivates the protein initiation factor eIF-2α, thus inhibiting protein synthesis and host cell growth. Among other functions that evolved to overcome the innate cellular responses against infection, viral γ134.5 interacts with cellular protein phosphatase-1α, resulting in dephosphorylation of eIF-2α, thus inhibiting the action of PKR.6,11 This allows the re-initiation of protein translation and robust viral replication. Deleting γ134.5 may restrict viral cytopathic effect and replication to malignant cells in which eIF-2α phosphorylation is deregulated because of the activation of the K-Ras pathway.14
However, first-generation mutant HSV-1 viruses with ICP6, γ134.5, or both deletions have demonstrated modest antitumoral activity in PDAC when injected into tumors. Indeed, while ICP6 deletion can be efficiently complemented by cellular RR, defective γ134.5 mutants display significantly attenuated replication and lysis of cancer cells, when used in vivo.15 Accordingly, we used in this study a second-generation replication-conditional HSV-1 mutant (Myb34.5) in which ICP6 expression is defective and γ134.5 expression is driven by the cellular promoter B-myb.16 We demonstrate that Myb34.5 successfully infects and replicates into PDAC-derived cells, both in vitro and in vivo, to inhibit cell proliferation and tumor growth. In addition, Myb34.5 treatment translates to improved therapeutic efficacy of chemotherapy. This preclinical study stems for the use of Myb34.5 oncolytic virus in patients with locally advanced PDAC for whom standard of care is still awaited.
Materials and Methods
Cells and viruses
Human PDAC-derived Capan-2 cells and BxPC-3 were grown in RPMI medium supplemented with 10% fetal calf serum, L-glutamine, antibiotic and antimycotic cocktail (Invitrogen), and Plasmocin (InvivoGen). MIA PACA-2 cells were grown in DMEM containing 4.5 g/liter glucose (Invitrogen), 10% fetal calf serum, L-glutamine, antibiotics, Fungizone, and Plasmocin (InvivoGen). Cell lines were grown in a humidified incubator at 37°C in 5% CO2. HSV-1 F strain (wild-type HSV-1) was provided by Dr. A. Epstein. Recombinant HSV-1 virus Myb34.5 (defective for ICP6 expression; γ134.5 expression regulated by the B-myb promoter) and MGH1 (defective for both ICP6 and γ134.5 expression), both derived from F strain, were kindly provided by E.A. Chiocca (chairman, Department of Neurosurgery, Brigham and Women's Hospital, Harvard Medical School, Boston, MA).
Viral replication and cytotoxicity assays
Virus were grown and tittered using Vero cells as described elsewhere.16 For cytotoxicity assays, human pancreatic cancer-derived cells were seeded at 1×104 cells (Mia PACA-2, Capan-2) or 2×104 cells (BxPC-3) per well in a 96-well dish and grown in complete medium. Twenty-four hours later, cells were infected with F-strain, MGH1, and Myb34.5 virus at the indicated multiplicities of infection. Six days later, cell viability was determined by fluorometric method using CellTiter-Fluor Cell Viability Assay (Promega) according to manufacturer's instruction. Cells were co-treated by gemcitabine and Myb34.5 has previously described.17
Ethics statement and experimental protocol
All animal experiments were conducted according to the national ethics guidelines for experimental research, and protocol No. 05/1037/12/13 was approved by the regional Midi-Pyrenees's ethics committee for animal experimentation and were performed in accordance with the Guide for the Care and Use of Laboratory Animals (U.S. National Institutes of Health). Human PDAC-derived Mia PACA-2 or Mia PACA-2 Lucia cells were implanted in the tail of pancreas of athymic mice as previously described.18 Myb34.5 was injected in level 2 animal safety facility in exponentially growing tumors at 15 days following tumor induction. Control animals received PBS. At the time of injection, tumor size was 150±12 mm3.
For noninvasive tracking of tumor growth, blood samples were collected prior to and following Myb34.5 injection. Lucia production was measured in 5 μl of serum using coelenterazine (50 μM) as a substrate, as already described.18,19 Gemcitabine (125 mg/kg) was injected i.p. every 3 days for 2 weeks. Control animals received 0.9% NaCl i.p. At 15 days after Myb34.5 injection, animals were killed and tumors were measured with a caliper and weighted. Tumor volume was calculated using the formula V=(W2×L)/2, where W is width and L is length. Tumors were formalin fixed, paraffin embedded, and stored at 4°C. H&E staining confirmed the presence of tumor(s) in each pancreas. For Western blot studies, tumors were frozen in liquid nitrogen and stored at −80°C until use.
TUNEL assay and immunostaining for B-myb and HSV-1
Mia PaCa-2 tumors were harvested and fixed in formalin. Four-micrometer-thick sections were prepared from paraffin-embedded sections and rehydrated. DNA fragmentation was performed using In situ Apoptosis Detection Kit according to the manufacturer's indications (Takara Bio Inc.). For immunostaining, sections were incubated for 10 min in protein block, serum-free reagent following antigen retrieval to reduce background staining (DakoCytomation). Slides were next incubated overnight at 4°C with anti-HSV-1 (DakoCytomation, clone B0114, dilution: 1:100) or B-myb (Santa Cruz, clone C-20, dilution: 1:250) antibodies. Slides were quickly rinsed in distilled water, washed twice in PBS, and incubated for 30 min at room temperature with Envision+ system-HRP (DakoCytomation). After washing in distilled water, slides were incubated in AEC+ reagent and counterstained with Mayer's hematoxylin. Immunostaining was recorded with an optical microscope using VisioLab2000 image analyzer (Biocom).
Western blotting
Proteins were extracted from infected cells or tumors, resolved on SDS-polyacrylamide gels, and transferred to nitrocellulose membrane. After room-temperature blocking for 1 hr, blots were incubated overnight at 4°C with antibodies against B-myb (Santa Cruz; clone C-20, dilution: 1:500), GAPD (Santa Cruz; clone 6C5, dilution: 1:1000), β −actin (Santa Cruz; clone C4, dilution: 1:1000), HSV-1 (DakoCytomation; clone B0114, dilution: 1:1000), cleaved caspase-3 (Cell Signaling; dilution 1:500), and cleaved PARP (Cell Signaling; dilution 1:1000). Secondary HRP-conjugated antibodies (dilution 1:10,000; Perbio Science) were added, and blots were incubated for 1h at room temperature. Immunoreactive proteins were visualized using Clarity ECL (Biorad) and imaged with ChemiDoc XRS+ (Biorad).
Gene expression analysis
Total RNA was isolated from patient tumors and normal adjacent tissue with TRIzol Reagent (Life Tech) according to supplier's instructions, and RNA concentration was measured with the ND-1000 NanoDrop spectrophotometer. B-myb mRNA was quantified from 1 μg total RNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific). 18S RNA expression was used as calibrator, as we previously described.20 Relative amounts were calculated by the comparative cycle threshold (CT) method and expressed as 2−ΔCT, where ΔCT=CT(B-myb) – CT(18S). Primers used for B-myb detection were B-myb forward 5′-TCTGGCTCTTGACATTGTGG-3′ and B-myb reverse CGGCAAGGATAGAGACTTGG-3′. Primers for RRM1 were RRM1 forward 5′-CAAGGTCGTGTCCGCAAG-3′ and RRM1 reverse 5′-GGTGCCTGTTTCCGTCTGA-3′. Primers for RRM2 were RRM2 forward 5′-GTGGAGCGATTTAGCCAAGAA-3′ and RRM2 reverse 5′-CATGGCAATTTGGAAGCCATA-3′. Primers for ENT1 were ENT1 forward 5′-GCAAAGGAGAGGAGCCAAGA-3′ and ENT1 reverse 5′-TTCATTGGTGGGCTGAGAGTT-3′. All primers were designed with Primer3. Duplicate qRT-PCR assays were carried out in a StepOnePlus II Real-Time PCR System (Life Tech) with SsoFast EvaGreen supermix (Biorad).
Statistical analysis
Results are expressed as mean±standard error (SE). Data were compared by nonparametric Wilcoxon signed-rank test (*p<0.05, **p<0.01; ***p<0.005) using Graphpad Prism software (Graphpad Software). p<0.05 was considered significant. No statistical method was used to predetermine sample size. The experiments were randomized. The investigators were not blinded to allocation during experiments except for tumor growth experiments in preclinical models, RNA and protein quantification, and histological examinations. No animals were excluded from the study.
Results
B-myb expression in pancreatic cell lines and patients' tumors
Myb34.5 is a genetically engineered HSV-1 mutant derived from F strain and defective in the ICP6 gene, and contains the HSV-1 γ134.5 gene under the control of the E2F-responsive cellular B-myb promoter.16 Host B-myb promoter activity appears essential for optimal Myb34.5 replication and oncolytic activity. In addition, RR-deleted HSV-1 virus such as Myb34.5 rely on endogenous RR expression,6 while Passer et al. described that inhibitors of equilibrative nucleoside transporter 1 (ENT1), a glycoprotein that mediates cellular uptake of nucleosides, are strongly favoring replication, spread, and oncolytic activity of HSV mutants.21
As RNA expression better reflects promoter activity than cellular protein content, we characterized B-myb, together with RRM1, RRM2, and ENT1 RNA levels in tumoral pancreatic cell lines. As shown in Fig. 1A, B-myb RNA is detectable in all cell lines tested with Mia PACA-2>BxPC-3>Capan-2. In addition, RRM1 and RRM2, which favor RR-deleted HSV-1 virus replication, are overexpressed in human pancreatic cancer cell lines with Mia PACA-2 ≫Capan-2>BxPC-3 (Fig. 1A). Last, ENT1, which negatively impacts on HSV-1 replication, is poorly expressed in human-derived human pancreatic cancer cell lines with BxPC-3>Mia PACA-2=Capan-2. In addition, we found that B-myb RNA is overexpressed in tumor samples from patients diagnosed with pancreatic cancer as compared with normal adjacent pancreas (Fig. 1B, fold increase 6.1±2.3, p<0.05) (Fig. 1B), while previous studies have demonstrated RRM1/2 overexpression and ENT-1 down expression in pancreatic cancer.22–24 We conclude that B-myb and additional cellular factors favoring HSV-1 replication are overexpressed in pancreatic cancer experimental models and primary tumors and may be helpful for Myb34.5 virus targeting to pancreatic cancer-derived cells.
FIG. 1.

B-myb expression in pancreatic-derived cell lines and tumors. (A) B-myb, RRM-1, RRM-2, and ENT1 quantification of expression by real-time PCR in pancreatic cancer-derived cell lines. (B) B-myb quantification of expression by real-time PCR in patients' tumors vs. normal adjacent tissue (n=12). *p<0.05.
Myb34.5 efficiently replicates and kills PDAC-derived cell lines in vitro
The in vitro replication rate of Myb34.5 was evaluated in PDAC-derived cell lines using multistep viral recovery experiments. Mia PACA-2, BxPC-3, and Capan-2 cell lines were infected with 0.1 PFU/cell (MOI=0.1) of Myb34.5. Viral replication was determined at 36, 72, and 144 hr postinfection. We found that Myb34.5 virus replicates to high level in the three cell lines tested (Fig. 2A), while F-strain and MGH1 virus replicated to very high and low levels in PDAC-derived cell lines, respectively (Fig. 2B and C). We next investigated cell viability following Myb34.5 infection. Mia PACA-2, BxPC-3, and Capan-2 cell lines were infected with increasing MOIs of Myb34.5 and cell viability was measured by cell viability assay 6 days following infection (Fig. 3A). Viability was decreased by Myb34.5 in all cell lines tested; however, Mia PACA-2 cells were more resistant to the cytopathic effect of Myb34.5, as compared with Capan-2 and BxPC-3 cells. Interestingly, the cytopathic effect of Myb34.5 was consistently greater than that of MGH1 and comparable to HSV-F strain (Fig. 3B and C).
FIG. 2.

Viral infection of pancreatic cancer cells in vitro. BxPC-3, Mia PACA-2, and Capan-2 pancreatic cancer-derived human cell lines were infected with 0.1 multiplicity of infection (MOI) of Myb34.5 (A), HSV-F (B), or MGH (C). Viral titters were determined as described in Materials and Methods at the indicated time.
FIG. 3.

Viral cell killing of pancreatic cancer cells in vitro. BxPC-3, Mia PACA-2, and Capan-2 pancreatic cancer-derived human cell lines were infected with 0.1 MOI of Myb34.5 (A), HSV-F (B), or MGH (C). Cell viability was determined 6 days later as described in Materials and Methods. Data are means±standard error (SE) of three biological replicates per group with three experimental replicates. (D) Mia PACA-2 cells were infected with Myb34.5 at the indicated doses. HSV-1 envelope glycoproteins, caspase-3, and PARP cleavage were detected by Western blotting 72 hr later. Data are representative of three biological replicates per group with three experimental replicates.
We next investigated the molecular mechanisms involved in the antiproliferative effect consecutive to Myb34.5 infection in PDAC-derived cell lines. We performed Western blotting for caspase-3 and PARP at 72 hr postinfection of Mia PACA-2 cells with different doses of Myb34.5. As shown in Fig. 3D, infection with Myb34.5 resulted in detectable expression of glycoproteins of the viral envelope in PDAC cells. In addition, Myb34.5 dose dependently induced caspase-3 and PARP cleavage in PDAC-derived cell lines, two hallmarks of cell death by apoptosis, as compared with control cells. Thus, the B-myb-driven Myb34.5 HSV-1 mutant efficiently replicates in PDAC-derived cell lines, inhibits cell viability, and induces apoptosis, three important properties in view of clinical application of this virus.
Combination of Myb34.5 and chemotherapy for PDAC treatment
Despite its contested efficacy in clinical trials for PDAC patients, gemcitabine became the standard treatment for advanced disease 15 years ago after showing superiority over fluorouracil. Since then, clinical trials of newer cytotoxic or biologic agents for patients with locally advanced tumors free of metastasis (45% at the time of diagnosis) include the “historical” standard-of-care chemotherapy for comparison or combination purposes. Thus, we examined whether Myb34.5 would enhance the efficacy of gemcitabine in PDAC cells. Mia PACA-2 cells were treated for 6 days with increasing amounts of gemcitabine, in combination or not with Myb34.5. Results presented in Fig. 4A demonstrate that gemcitabine and Myb34.5 efficiently kill PDAC-derived cells when administrated at a high dose, while a low dose of virus (0.1 p.f.u.) and gemcitbaine (0.1 μM) failed to significantly impact tumor cell proliferation. However, combining low dose of virus and gemcitabine significantly killed PDAC cells (Fig. 4A). Using Western blot, we found that cancer cell death by apoptosis was augmented by the co-treatment. In addition, treating PDAC-derived cells using gemcitabine had no effect on B-myb expression (Fig. 4B). Thus, we conclude that combining Myb34.5 and standard-of-care chemotherapy results in enhance cell killing and apoptosis induction in human PDAC-derived cells.
FIG. 4.
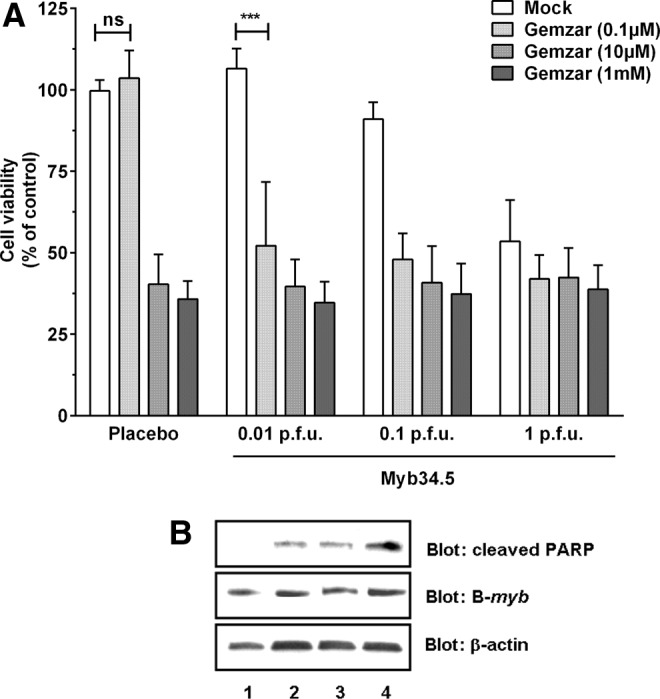
Combination of gemcitabine and Myb34.5 treatments in vitro. Mia PACA-2 pancreatic cancer-derived human cell lines were treated with gemcitabine and/or Myb34.5 at the doses indicated. (A) Cell viability was determined 6 days later as described in Materials and Methods. Data are means±SE of six biological replicates per group with three experimental replicates. (B) Mia PACA-2 cells were treated with 0.1 p.f.u. of Myb34.5 and/or 0.1 μM of gemcitabine. PARP cleavage and B-myb expression were detected by Western blotting 72 hr later. Data are representative of three biological replicates per group with three experimental replicates. 1, mock; 2, gemzar; 3, Myb34.5; 4, gemzar+Myb34.5.
Therapy of PDAC experimental tumors using Myb34.5
We next examined the antitumor efficacy of Myb34.5 against experimental PDAC tumors. In a first set of experiments, Mia PACA-2 cells were engrafted in the pancreas of athymic mice as described before.18,19 Myb34.5 virus was administrated with a single intratumoral injection 2 weeks following tumor induction, a time point at which animals typically have medium-sized tumors (mean 150±12 mm3). We observed no significant variations in body weight or mortality related to Myb34.5 injection (data not shown). These data further underscore the safety profile of Myb34.5 virus in experimental models of cancer.15 Two weeks following treatment, the animals were euthanized and the pancreatic tumors were measured and harvested. Remarkably, Myb34.5 strongly inhibited tumor progression in this very aggressive experimental model of pancreatic cancer (Fig. 5A). Tumor burden was reduced by more than 75% with the highest dose of the virus used. Interestingly, Myb34.5 injection provoked massive tumor necrosis (Fig. 5B), hemorrhage (Fig. 5B, pink area), and inflammatory cell infiltrate (Fig. 5B, arrows), and disorganized tumor cells in comparison to vehicle-treated tumors.
FIG. 5.

Myb34.5 treatment of orthotopic pancreatic xenografts. Mia PACA-2 cells were implanted in the pancreas of athymic mice. Two weeks later, Myb34.5 was injected in exponentially tumors at the indicated doses. In a second set of experiment, tumors were injected with F-strain virus. Control mice were injected by vehicle (PBS). (A) Tumor volume was measured at day 0 and day 15 following intratumoral gene transfer using a caliper. Data are means±SE of eight biological replicates per group. *p<0.05; **p<0.01. (B) Formalin-fixed, paraffin-embedded samples of vehicle or Myb34.5-treated Mia PACA-2 tumor specimens were stained for hematoxylin and eosin. These specimens showed tumor necrosis, hemorrhage (pink area), and inflammatory cell infiltrates (arrows). Representative photomicrographs at 20×. Data are means±SE of three biological replicates per group with three experimental replicates. Pancreatic tissue was harvested for the analysis of HSV-1 (representative photomicrographs at 40×) (C) or DNA fragmentation by TUNEL assay (representative photomicrographs at 20×) (D). (E) Quantification of DNA fragmentation (% of apoptotic cells) in each group. Data are representative of three biological replicates per group with three experimental replicates. *p<0.05; **p<0.01, difference between placebo- and Myb34.5-treated tumors. #p<0.05, difference between tumors treated with 10e8 p.f.u of Myb34.5 and tumors treated with 10e6 or 10e7 p.f.u of Myb34.5. Color images available online at www.liebertpub.com/hum
We next performed immunochemistry in formalin-fixed, paraffin-embedded tumors for HSV-1 envelope proteins to monitor intratumoral viral spread. Two weeks after treatment, HSV-1 proteins were still detected in the Myb34.5-treated tumors, but were absent from normal adjacent pancreas (Fig. 5C). The latter finding strongly suggests that virus successfully replicated while remaining strictly restricted to cancer cells. On the other hand, intratumoral injection of wild-type F-strain virus resulted in massive replication in normal pancreatic tissue (Fig. 5C). We observed strong induction of apoptosis as measured by TUNEL assay in tumors following administration of Myb34.5 (Fig. 5D). Interestingly, increase in tumor cell apoptosis was related to virus doses (Fig. 5E). Cumulatively, the results reported herein provide the first evidence that Myb34.5 exerts antitumor activity against experimental pancreatic cancer, and that is effective following intratumoral route of administration.
Next, we examined whether Myb34.5 would enhance the efficacy of gemcitabine in PDAC xenografts. We recently generated a novel model of PDAC for the noninvasive tracking of tumor growth based on secreted Lucia luciferase.18,19 We engrafted Mia PACA-2 Lucia cells in the pancreas of athymic mice. In vitro, Mia PACA-2 Lucia and parental Mia PACA-2 cells demonstrated similar sensitivity to Myb34.5 virus cytopathic effect (data not shown). Myb34.5 virus was administrated with a single intratumoral injection 2 weeks following tumor induction. Control tumors were injected with vehicle. Tumors were treated twice weekly by i.p. injection of high dosage of gemcitabine (125 mg/kg). Blood samples were collected for orthotopic pancreatic tumors progression monitoring using blood Lucia assay. We found that tumor growth was rapidly and significantly hampered by Myb34.5 injection, and by the combination of gemcitabine treatment and viral infection, while gemcitabine treatment alone resulted in a significant delay in tumor progression inhibition (Fig. 6A).
FIG. 6.

Combination of Myb34.5 and gemcitabine strongly impacts pancreatic cancer growth. Mia PACA-2 cells were implanted in the pancreas of athymic mice. Two weeks later, 10e8 p.f.u. Myb34.5 was injected in exponentially tumors. Control mice were injected by vehicle (PBS). (A) Non-invasing monitoring of PDAC tumor growth was performed for 28 days before and after intratumoral gene transfer. Results are mean±SD of Lucia levels, expressed as arbitrary units. (B) Tumor volume was measured using a caliper at day 0 and day 15 following intratumoral infection and gemcitabine (Gemzar) treatment. Data are means±SE of six biological replicates per group. **p<0.01; ##p<0.01 difference between Gemzar and Gemzar+Myb34.5 study groups. (C) Tumors were weighted 15 days following intratumoral infection and/or gemcitabine (Gemzar) treatment. Data are means±SE of six biological replicates per group. ***p<0.005; ##p<0.01 difference between Gemzar and Gemzar+Myb34.5 study groups. (D) Western blot for B-myb expression in tumors treated or not by gemcitabine. Data are representative of three biological replicates per group with three experimental replicates.
The animals were euthanized and the pancreatic tumors were measured, harvested, and weighted 2 weeks following infection. We found that gemcitabine treatment or Myb34.5 infection alone statistically inhibited tumor growth and tumor weight to similar extent (Fig. 6B and Fig. 6C). However, combining gemcitabine and Myb34.5 significantly translates to improved therapeutic efficacy of standard-of-care chemotherapy, as compared with gemcitabine alone. Again, treating PDAC tumors using gemcitabine had no effect on B-myb expression (Fig. 6D). Overall, the intratumoral injection of Myb34.5 exerts a strong antitumoral effect against very aggressive experimental PDAC tumors. In addition, combining targeted HSV-1 viruses with standard-of-care chemotherapy resulted in a significant higher inhibition of tumor growth, and warrant further evaluation at the clinical level.
Discussion
PDAC is one of the most lethal human cancers. This is a rare disease but since incidence equals mortality, this diagnosis sounds like a death sentence for most cases because of the lack of curative treatment for advanced disease.1 PDAC has a sophisticated network of biological activities that maintains self-sufficiency in growth signals, is resistant to endogenous antiproliferative signals, evades apoptosis, has limitless replicative potential, and undergoes tissue invasion and metastasis.8,25 This heterogeneity stems for the unchallenged resistance of PDAC to conventional therapeutic approaches (chemotherapy, radiotherapy, etc.) and targeted biotherapies. Consequently, viral therapy has been regarded as a potential new treatment modality because of its specificity and high potency. Indeed, oncolytic virotherapy exploits the ability of virus to kill the target cells and simultaneously to spread to other target cells.6 However, a key requirement is that the virus is specifically targeted to the tumor.
A variety of oncolytic viruses such as adenovirus, herpes simplex virus, influenza virus, Newcastle disease virus, poliovirus, vaccinia virus, and reovirus among others have been developed for cancer therapy, including PDAC.6 HSV-1 is currently actively investigated in preclinical and phase I to III clinical trials, as it is prone to genetic engineering to favor cancer selectivity.11,26
We selected HSV-1-based virus for PDAC therapy because its safety profile is well documented when injected in tumors and total cell killing can be achieved rapidly with a relatively low MOI.11 PKR activation in response to infection is one of the main cellular defenses against viral aggression. Consequently, many viruses have developed sophisticated strategies to overcome the resulting shutoff of protein translation. HSV-1 circumvent the consequences of PKR activation by expressing the γ134.5 protein that interacts with cellular protein phosphatase-1α to dephosphorylate eIF-2α, which prevents from protein translation shutoff.6 However, HSV-1 mutants such as MGH1, which are defective in the γ134.5 protein, demonstrate attenuated replication and virulence also in cancer cells.27 Consequently, in this work, we used a second-generation replication-conditional HSV-1 mutant (Myb34.5) in which ICP6 expression is defective and γ134.5 expression is regulated by the cellular promoter B-myb.16
An important aspect of this study was the investigation of B-myb promoter activity in PDAC samples. We found that B-myb RNA is expressed in PDAC-derived cell lines and experimental tumors, and is much higher in patients' tumors, as compared with normal adjacent tissue. To our knowledge, until this study, there have been no reports in the literature addressing the presence of elevated B-myb in this tumor type. In addition, these findings indirectly demonstrate that B-myb promoter is functional and active in PDAC-derived samples. Last, we found that PDAC-derived cells also expressed very high levels of RRM-1 and RRM-2, while we confirm that ENT1 is poorly expressed. Consequently, HSV-1 may exploit the profound alteration of nucleotide synthesis pathway to replicate at high levels in PDAC cells because of the host cells elevated ribonucleoside activity.6 Taken together, our findings strongly suggest that PDAC is highly permissive to Myb34.5 replication.
We next infected PDAC-derived cell lines with Myb34.5 virus and report for the first time that Myb34.5 significantly replicates and kills pancreatic cancer cells. Interestingly, we found that Myb34.5 replication correlated well with the expression of B-myb, RRM-1, and RRM-2 in PDAC-derived cell lines. As we previously demonstrated that RNA quantification in fine-needle biopsies of patients' tumors is feasible and indicative of diagnosis,20 B-myb, RRM-1, and RRM-2 RNA levels in PDAC tumors could be used for patients' selection in future clinical trials using Myb34.5. In this study, we could not address the specificity of Myb34.5 for cancer cells as compared with normal pancreatic cells. We found that the viruses tested during this work replicate to high levels to kill normal human pancreatic ductal epithelial cells (HPDE; data not shown). These cells, transformed with E6 and E7 HPV proteins, express high levels of RRM-1, RRM-2, and B-myb and could not be considered as pertinent controls for our experiments. We are currently exploring whether organotypic culture model of thick slices of normal human pancreas may be used to validate Myb34.5 specificity in future studies.28
We next demonstrated that Myb34.5 intratumoral injection resulted in a profound inhibition of the progression of a very aggressive experimental model of PDAC. Myb34.5 injection provoked massive tumor necrosis, hemorrhage, inflammatory cell infiltrate, and scattered tumor cells in comparison to vehicle-treated tumors. Although the molecular basis of this inhibitory effect requires further investigation, we demonstrate herein that Myb34.5 infection induces apoptosis in PDAC cells as determined by PARP, caspase-3 cleavage, and TUNEL assays. Since in our model the mice used were immunocompromised, we can argue that the antineoplastic activity of Myb34.5 is dependent on virus replication and not dependent on host immune responses.
Previous works have reported that the addition of chemotherapeutic agents to the administration of HSV-1 mutants may produce either a synergistic17 or an antagonist29 antiproliferative effect in vitro. During this study, we found that gemcitabine had no effect on B-myb expression, both in vitro and in vivo.
Next, we demonstrated that combining Myb34.5 and gemcitabine co-treatment led to a very impressive in vitro antiproliferative effect with massive induction of cell death by apoptosis. In vivo, standard-of-care chemotherapy and Myb34.5 administration resulted in a rarely achieved antitumoral effect in this experimental model, and resulted in a statistically greater reduction in tumor growth than chemotherapy alone. The later result may have significant clinical implications as we demonstrate herein that Myb34.5 and gemcitabine may be used in combination in PDAC patients, and that Myb34.5 administration may help relieve PDAC resistance to conventional chemotherapy. Taken together, we demonstrate for the first time that HSV-1-based selective Myb.34.5 virus effectively replicates and kills PDAC-derived cell lines, both in vitro and in vivo. In addition, we found that combining oncovirotherapy and chemotherapeutic treatment induces unexpected pancreatic tumor growth inhibition. The present study design involved the treatment of existing tumors with Myb34.5 virus, a paradigm closely related to the clinical scenarios in which this approach may be employed. We have recently conducted the first-in-human phase I gene therapy clinical trial in 24 patients diagnosed with advanced pancreatic cancer (Thergap clinical trial, ClinicalTrials.gov Identifier NCT01274455).
We demonstrated during this trial the feasibility and the safety of transfecting PDAC tumors with nonviral vectors using endoscopic ultrasound (manuscript in review). Interestingly, this route of administration has been used for first-generation, OncoVEXGM-CSF and HF10 mutant HSV-1 virus delivery in patient PDAC tumors in early phase clinical trials.11 The authors reported the absence of adverse side effects and some therapeutic potential in some patients, thus encouraging for phase II clinical evaluation and the development of new protocols based on tumor oncolysis. While there clearly remains significant work to be done, our findings highlight for the first time the therapeutic promise of using Myb34.5 to treat PDAC tumors and warrant further clinical evaluation.
Acknowledgments
The authors would like to thank Dr. Nicolas Ferry from Agence Nationale de Sécurité du Médicament for his help and advice, and Laetitia Ligat from Toulouse Oncopole Pole of Technology for her help with immunochemistry image acquisition.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Siegel R, Naishadham D, and Jemal A. Cancer statistics, 2013. CA Cancer J Clin 2013;63:11–30 [DOI] [PubMed] [Google Scholar]
- 2.Burris HA, 3rd, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997;15:2403–2413 [DOI] [PubMed] [Google Scholar]
- 3.Moore MJ, Goldstein D, Hamm J, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 2007;25:1960–1966 [DOI] [PubMed] [Google Scholar]
- 4.Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–1825 [DOI] [PubMed] [Google Scholar]
- 5.Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ilkow CS, Swift SL, Bell JC, et al. From scourge to cure: tumour-selective viral pathogenesis as a new strategy against cancer. PLoS Pathog 2014;10:e1003836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones S, Hruban RH, Kamiyama M, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science 2009;324:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yachida S, Jones S, Bozic I, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010;467:1114–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Touchefeu Y, Harrington K, Galmiche J, et al. Review article: gene therapy, recent developments and future prospects in gastrointestinal oncology. Aliment Pharmacol Ther 2010;32:953–968 [DOI] [PubMed] [Google Scholar]
- 10.Wennier S, Li S, and McFadden G. Oncolytic virotherapy for pancreatic cancer. Expert Rev Mol Med 2011;13:e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu C, Li H, Su C, et al. Viral therapy for pancreatic cancer: tackle the bad guys with poison. Cancer Lett 2013;333:1–8 [DOI] [PubMed] [Google Scholar]
- 12.Gammon DB, Gowrishankar B, Duraffour S, et al. Vaccinia virus-encoded ribonucleotide reductase subunits are differentially required for replication and pathogenesis. PLoS Pathog 2010;6:e1000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elford HL, Freese M, Passamani E, et al. Ribonucleotide reductase and cell proliferation. I. Variations of ribonucleotide reductase activity with tumor growth rate in a series of rat hepatomas. J Biol Chem 1970;245:5228–5233 [PubMed] [Google Scholar]
- 14.Mundschau LJ, and Faller DV. Endogenous inhibitors of the dsRNA-dependent eIF-2 alpha protein kinase PKR in normal and ras-transformed cells. Biochimie 1994;76:792–800 [DOI] [PubMed] [Google Scholar]
- 15.Nakamura H, Kasuya H, Mullen JT, et al. Regulation of herpes simplex virus gamma(1)34.5 expression and oncolysis of diffuse liver metastases by Myb34.5. J Clin Invest 2002;109:871–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chung RY, Saeki Y, and Chiocca EA. B-myb promoter retargeting of herpes simplex virus gamma34.5 gene-mediated virulence toward tumor and cycling cells. J Virol 1999;73:7556–7564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eisenberg DP, Adusumilli PS, Hendershott KJ, et al. 5-fluorouracil and gemcitabine potentiate the efficacy of oncolytic herpes viral gene therapy in the treatment of pancreatic cancer. J Gastrointest Surg 2005;9:1068–1077; discussion 1077–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sicard F, Gayral M, Lulka H, et al. Targeting miR-21 for the therapy of pancreatic cancer. Mol Ther 2013;21:986–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delpu Y, Lulka H, Sicard F, et al. The rescue of miR-148a expression in pancreatic cancer: an inappropriate therapeutic tool. PLoS One 2013;8:e55513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bournet B, Pointreau A, Souque A, et al. Gene expression signature of advanced pancreatic ductal adenocarcinoma using low density array on endoscopic ultrasound-guided fine needle aspiration samples. Pancreatology 2012;12:27–34 [DOI] [PubMed] [Google Scholar]
- 21.Passer BJ, Cheema T, Zhou B, et al. Identification of the ENT1 antagonists dipyridamole and dilazep as amplifiers of oncolytic herpes simplex virus-1 replication. Cancer Res 2010;70:3890–3895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maréchal R, Bachet J-B, Mackey JR, et al. Levels of gemcitabine transport and metabolism proteins predict survival times of patients treated with gemcitabine for pancreatic adenocarcinoma. Gastroenterology 2012;143:664–674.e1–6. [DOI] [PubMed] [Google Scholar]
- 23.Nakagawa N, Murakami Y, Uemura K, et al. Combined analysis of intratumoral human equilibrative nucleoside transporter 1 (hENT1) and ribonucleotide reductase regulatory subunit M1 (RRM1) expression is a powerful predictor of survival in patients with pancreatic carcinoma treated with adjuvant gemcitabine-based chemotherapy after operative resection. Surgery 2013;153:565–575 [DOI] [PubMed] [Google Scholar]
- 24.Poplin E, Wasan H, Rolfe L, et al. Randomized, multicenter, phase II study of CO-101 versus gemcitabine in patients with metastatic pancreatic ductal adenocarcinoma: including a prospective evaluation of the role of hENT1 in gemcitabine or CO-101 sensitivity. J Clin Oncol 2013;31:4453–4461 [DOI] [PubMed] [Google Scholar]
- 25.Jones S, Zhang XS, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008;321:1801–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Russell SJ, Peng K-W, and Bell JC. Oncolytic virotherapy. Nat Biotechnol 2012;30:658–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chou J, Kern ER, Whitley RJ, et al. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 1990;250:1262–1266 [DOI] [PubMed] [Google Scholar]
- 28.Rebours V, Albuquerque M, Sauvanet A, et al. Hypoxia pathways and cellular stress activate pancreatic stellate cells: development of an organotypic culture model of thick slices of normal human pancreas. PLoS One 2013;8:e76229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kulu Y, Kawasaki H, Donahue JM, et al. Concurrent chemotherapy inhibits herpes simplex virus-1 replication and oncolysis. Cancer Gene Ther 2013;20:133–140 [DOI] [PMC free article] [PubMed] [Google Scholar]