

Abstract
Vector capsid dose-dependent inflammation of transduced liver has limited the ability of adeno-associated virus (AAV) factor IX (FIX) gene therapy vectors to reliably convert severe to mild hemophilia B in human clinical trials. These trials also identified the need to understand AAV neutralizing antibodies and empty AAV capsids regarding their impact on clinical success. To address these safety concerns, we have used a scalable manufacturing process to produce GMP-grade AAV8 expressing the FIXR338L gain-of-function variant with minimal (<10%) empty capsid and have performed comprehensive dose–response, biodistribution, and safety evaluations in clinically relevant hemophilia models. The scAAV8.FIXR338L vector produced greater than 6-fold increased FIX specific activity compared with wild-type FIX and demonstrated linear dose responses from doses that produced 2–500% FIX activity, associated with dose-dependent hemostasis in a tail transection bleeding challenge. More importantly, using a bleeding model that closely mimics the clinical morbidity of hemophilic arthropathy, mice that received the scAAV8.FIXR338L vector developed minimal histopathological findings of synovitis after hemarthrosis, when compared with mice that received identical doses of wild-type FIX vector. Hemostatically normal mice (n=20) and hemophilic mice (n=88) developed no FIX antibodies after peripheral intravenous vector delivery. No CD8+ T cell liver infiltrates were observed, despite the marked tropism of scAAV8.FIXR338L for the liver in a comprehensive biodistribution evaluation (n=60 animals). With respect to the role of empty capsids, we demonstrated that in vivo FIXR338L expression was not influenced by the presence of empty AAV particles, either in the presence or absence of various titers of AAV8-neutralizing antibodies. Necropsy of FIX–/– mice 8–10 months after vector delivery revealed no microvascular or macrovascular thrombosis in mice expressing FIXR338L (plasma FIX activity, 100–500%). These preclinical studies demonstrate a safety:efficacy profile supporting an ongoing phase 1/2 human clinical trial of the scAAV8.FIXR338L vector (designated BAX335).
Introduction
Hemophilia B is an X-linked congenital bleeding disorder that results from deficient activity of clotting factor IX. In the severe form it may be complicated by recurrent potentially crippling joint and muscle bleeding and, less frequently, by life-threatening hemorrhage including bleeding into the central nervous system. Factor IX protein replacement by regular intravenous infusion is effective; however, treatment is cumbersome, extraordinarily expensive, and only widely available to the estimated 20% of the world's hemophilic individuals who live in more economically resourced countries.1 Gene therapy for hemophilia has been a prized but elusive goal of the biomedical research community.
A human clinical trial conducted between 2001 and 2004 provided proof of concept that viral vectors based on the nonpathogenic dependovirus adeno-associated virus (AAV) can successfully deliver the factor IX gene to the liver.2 This trial established the research pathway for the last decade of effort toward a cure. In that phase 1/2 dose-escalation trial reported by Manno and colleagues, which employed a single-stranded DNA vector based on AAV serotype 2 (for which humans are the natural host), two lower vector doses were shown to be safe but did not result in measurable factor IX expression. Escalation to the planned highest dose (2×1012 vector genomes [VG]/kg body weight) led to transient factor IX expression; however, asymptomatic liver inflammation ensued, with loss of the successfully gene-transduced hepatocytes.2 Subsequent investigation suggested that challenge with recombinant AAV vectors can, in a vector capsid dose-dependent fashion, lead to reactivation of memory T and B cell responses in an individual who has been exposed to wild-type AAV earlier in life. The reactivation of this adaptive immune response appears capable of inciting a cytotoxic T lymphocyte (CTL)-mediated elimination of the hepatocytes that have processed the recombinant virus vector and that present AAV capsid epitopes for immune recognition.3 In light of these findings, our group and other research groups pursued strategies to increase the efficiency of AAV factor IX gene delivery in hopes of achieving clinically meaningful expression while limiting vector doses to levels that are lower than the doses associated with apparent CTL-mediated immune response.4–6
A clinical trial sponsored by St. Jude Children's Research Hospital and conducted at the University College of London (SJCRH/UCL) achieved the first unequivocal clinical success for hemophilia gene therapy. Persistent expression of 1–6% normal factor IX activity was demonstrated in all six individuals receiving the “scAAV2/8-LP1-hFIXco” vector.7 The advances incorporated into the SJCRH/UCL vector included (1) the use of a self-complementary rather than single-stranded AAV genome form; (2) codon optimization of the factor IX sequence; and (3) use of the capsid from AAV8 (a rhesus macaque serotype) rather than AAV2 (for which humans are the natural host), associated with improved liver tropism and allowing (4) peripheral venous rather than direct intraportal venous vector infusion. In this SJCRH/UCL trial, evidence of capsid dose-dependent immune hepatitis was observed once again after escalation to a dose of 2×1012 VG/kg body weight. The persistence of factor IX expression despite immune targeting of the vector-transduced hepatocytes was credited by these investigators to the institution of immune suppression with a course of corticosteroids in most individuals treated at this highest vector dose. Of note is that the production methods used to generate the clinical vector resulted in vector stocks that contained at least 80% empty AAV capsids,8 so that the dose of 2×1012 VG/kg was associated with an exposure to at least 1×1013 potentially immunogenic AAV capsids per kilogram.
Gain-of-function variants of factor IX carrying a single amino acid change at amino acid 338 have been described,9,10 as well as their incorporation into gene therapy vectors for preclinical applications in vivo.11–15 An AAV vector that incorporates the improvements employed in the SJCRH/UCL factor IX vector and in addition directs the expression of a gain-of-function FIXR338L variant is currently undergoing evaluation in a phase 1/2 clinical trial for hemophilia B, and its preclinical evaluation is described herein (www.clinicaltrials.gov/show/NCT01687608). These investigations suggest that scAAV8.FIXR338L vector (now being tested in a human clinical trial by Baxter Healthcare [Deerfield, IL] and labeled BAX335) has the potential to achieve clinically meaningful circulating factor IX activity without relying on exposure to doses of vector capsid that have been associated in previous trials with hepatic inflammation and rescue immunosuppression.
Materials and Methods
AAV vector production
The construct pTTRhFIXopt, which directs the expression of a codon-optimized wild-type human factor IX (FIX) sequence from a liver-specific transthyretin (TTR) promoter, has been described previously.6 A QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) was used to generate single nucleotide changes in this expression cassette, resulting in the cassettes encoding alanine (FIXR338A), leucine (FIXR338L), or glutamine (FIXR338Q). The FIX expression cassettes were subsequently tested for expression after packaging in self-complementing AAV vectors.
In separate experiments, the sequence of the LP1 promoter/enhancer/SV40 intron from the scAAV2/8-LP1hFIXco construct was synthesized and substituted in the FIXR338L expression cassette so as to create an expression cassette to compare the effect of transcriptional regulatory elements in the scAAV2/8-LP1-hFIXco vector and the scAAV8.FIXR338L vector16 while maintaining all the FIX transgene and AAV backbone elements identical between the vectors. All recombinant AAV8 vectors were produced at the University of North Carolina at Chapel Hill Vector Core Facility, using a triple transfection protocol as previously described.17 Suspension HEK293 cells were grown in the WAVE bioreactor system (GE Healthcare, Piscataway, NJ) to produce Good Manufacturing Practice (GMP) vector, using a manufacturing process that is both scalable and animal component free.
Animal care and use
All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill. All animal experiments were performed in hemophilia B (FIX–/–) mice maintained in a C57BL/6 strain background,18 or hemostatically normal C57BL/6 mice (Jackson Laboratories, Bar Harbor, ME) as noted. Adult male hemophilia B mice were injected with the indicated doses of AAV vector via the tail vein (for peripheral intravenous delivery) or via open abdominal surgery with direct visualization and infusion of the superior mesenteric vein (for portal vein delivery). Mice were 7–10 weeks of age, weighing 20–25 g at the time of experiments, and experimental groups were matched for age and weight. Portal vein and tail vein administration,6 tail transection bleeding challenge,19 and joint capsular puncture bleeding challenge were performed as described previously.20,21 Blood for all studies was collected from the retroorbital venous plexus under anesthesia using isoflurane as previously described,21 with the exception of blood for thrombin–antithrombin (TAT) evaluation, which was obtained under anesthesia from an open puncture of the inferior vena cava, performed as a terminal procedure.
Human factor IX antigen, activity, and antibody assays and thrombin/antithrombin III assay
The human factor IX antigen, one-stage human factor IX activity, human factor IX-directed IgG, and human factor IX Bethesda inhibitor assays were performed as previously described and are specific for human factor IX (although not specifically standardized for factor IX variants, e.g., FIXR338L).21,22 The specific activity of factor IX expressed as units of factor IX activity per milligram of protein (U/mg) was calculated by dividing the factor IX activity (U/ml) by the concentration of the factor IX protein (factor IX antigen) (mg/ml). TAT complexes were measured from platelet-poor citrated plasma collected as a terminal puncture of the inferior vena cava 4 weeks after factor IX vector administration, using an Enzygnost TAT micro ELISA system (Siemens Healthcare Diagnostics, Tarrytown, NY).
Histopathological examination at necropsy of hemophilic mice after AAV8-mediated expression of wild-type human factor IX and FIXR338L
At the time of terminal sacrifice of hemophilic mice after expression of either transgenic human WTFIX or human FIXR338L from AAV vectors, mice were killed by CO2 asphyxiation for histopathological examination. Mice were examined at time of necropsy for gross signs of hemorrhage and all tissues were immersion-fixed in either Bouin's fixative (RICCA Chemical, Arlington, TX) or 10% neutral buffered formaLin, trimmed, processed, sectioned, and stained with hematoxylin and eosin (H&E) by routine methods, and a panel of 18 organs and tissues was evaluated microscopically for histopathological changes (C.J.B., blinded to experimental manipulation). Heart, lung, liver, spleen, kidney, and brain were evaluated for the presence of fibrosis and/or microvascular thrombus formation by immunohistochemistry for fibrinogen, using AB34 from Abcam (Cambridge, MA), and additional evaluation with Masson's trichrome (collagen stains blue, muscle red) and phosphotungstic acid hematoxylin (PTAH), which stains collagen pink but fibrin and muscle blue.
Effect of empty capsids on AAV8 transduction in the presence of AAV8-neutralizing antibodies
To evaluate whether empty AAV8 capsids in the therapeutic vector preparation exert any positive or negative effect on scAAV8.FIXR338L transduction in vivo (especially in the presence of AAV8 neutralizing antibodies [NAbs]), anti-AAV8 NAbs were generated. C57BL/6 mice were vaccinated with an AAV8 vector expressing an irrelevant transgene (α1-antitrypsin, A1AT). The humoral immune response was boosted with an additional AAV8.A1AT intraperitoneal injection. Two weeks later, serum was collected and pooled. The AAV8 neutralizing titer of the serum was determined by examining the ability of serial dilutions of serum to abrogate the transduction of RC32 cells in tissue culture by an AAV8.GFP vector, as previously described.23 Empty rAAV capsids, a gift from M. Agbandje-McKenna (Department of Biochemistry and Molecular Biology, University of Florida, Gainesville, FL) were generated by a triple transfection protocol and CsCl gradient ultracentrifugation collection, as previously described.24 NAb was passively transferred to FIX–/– mice by tail vein injection, achieving a circulating AAV NAb titer of either 1:16 (moderate titer) or 1:2 (near the lower limit of detection). Mice were then treated via the tail vein with either scAAV8.FIXR338L cGMP-grade vector or a combination of scAAV8.FIXR338L and empty AAV8 particles at the ratios detailed in Table 3 and described in Results.
Table 3.
Effect of AAV8 Neutralizing Antibodies on In Vivo Transduction by scAAV8.FIXR338L and Failure of Excess Empty AAV8 Capsids to Affect Transduction
| Factor IX expression (% factor IX activity) | |||||||
|---|---|---|---|---|---|---|---|
| Condition | AAV8 NAb | scAAV8.FIXR338L (VG/kg) | Empty capsid (EC) (VG/kg) | Week 1 | Week 2 | Week 4 | Week 8 |
| Vector alone | None | 1×1011 | None | 5.6±0.8 | 8.2±2.9 | 7.5±2.1 | 7.9±2.0 |
| Moderate-titer NAb | 1:16 | 1×1011 | None | <1 | <1 | <1 | <1 |
| Moderate-titer NAb+50% EC | 1:16 | 1×1011 | 1×1011 | <1 | <1 | <1 | <1 |
| Moderate-titer NAb+80% EC | 1:16 | 1×1011 | 4×1011 | <1 | <1 | <1 | <1 |
| Week 1 | Week 2 | Week 4 | Week 8 | ||||
|---|---|---|---|---|---|---|---|
| Vector alone | None | 1×1011 | None | 5.0±1.0 | 6.6±1.0 | 5.9±0.6 | No sample |
| Low-titer NAb | 1:2 | 1×1011 | None | <1 | <1 | <1 | No sample |
| Low-titer NAb+50% EC | 1:2 | 1×1011 | 1×1011 | <1 | <1 | <1 | No sample |
| Low-titer NAb+80% EC | 1:2 | 1×1011 | 4×1011 | <1 | <1 | <1 | No sample |
| Week 1 | Week 2 | Week 4 | Week 8 | ||||
|---|---|---|---|---|---|---|---|
| Vector alone | None | 1×1011 | None | 7.0±3.1 | 11.5±3.2 | 12.8±5.5 | 12.8±4.7 |
| 95% EC | None | 1×1011 | 1.8×1012 | 7.9±2.7 | 10.5±3.5 | 13.0±4.5 | 12.5±3.5 |
AAV8, adeno-associated virus serotype 8; EC, empty capsids; NAb, neutralizing antibody; VG, vector genomes.
Note: Serum from syngeneic (C57BL/6) mice, previously vaccinated with AAV8.A1AT vector, was used to passively transfer titratable AAV8 neutralizing antibodies (NAbs) into previously untreated C57BL/6 FIX–/– mice. Mice received either no AAV8 NAb, or NAb adequate to achieve low neutralizing titers (1:2), or to achieve moderate NAb titer (1:16). Subsequently, all mice were treated with the same dose of FIXR338L expression vector. Various cohorts of mice received the scAAV8.FIXR338L GLP vector alone (∼5–10% empty capsid contamination) or the GLP vector mixed at a 1:1 ratio with AAV8 empty capsids or the GLP vector.
Pivotal Good Laboratory Practice toxicology and biodistribution study in hemostatically normal mice
An 18-week toxicity and biodistribution study of Good Laboratory Practice (GLP) scAAV8.FIXR338L vector was performed in normal male C57BL/6 mice (having normal hemostasis at baseline). The study design is shown in Supplementary Table S1 (supplementary data are available online at www.liebertpub.com/hum). Briefly, scAAV8.FIXR338L vector at a “therapeutic dose” and at a “supraphysiological dose” were compared with vehicle (5% sorbitol in 350 mM NaCl) after tail vein administration to adult mice (n=60 mice per group). The lower dose of 4×1011 VG/kg was based on the cumulative results of the expression studies performed in FIX–/– mice showing that this “therapeutic dose” resulted in >50–100% normal human factor IX activity 40–42 weeks after a single delivery. A 10 times higher dose of 4×1012 VG/kg was studied as a “supraphysiological dose.” At each of four time points after vector delivery (day 3, week 3, week 10, and week 18) 15 mice from each dose group were killed and the following evaluations were completed: blood for complete blood cell counts and morphology, clinical chemistry panel; factor IX antigen, activity and factor IX Bethesda inhibitor; anti-AAV8 IgG, anti-FIX IgG1 (weeks 10 and 18 only); and complete necropsy with gross and microscopic evaluation of 27 tissues along with DNA isolation and PCR analysis for vector biodistribution. After this multitissue examination, the heart, lungs, kidneys, brain, liver, and spleen were independently examined for the presence of fibrosis and/or thrombus formation as described previously (C.J.B., blinded to experimental design). Additional histopathological examination included screening of livers for CD8+ T cells and evaluation of splenocytes for interferon-γ enzyme-linked immunospot (ELISPOT) immune response to AAV8 and FIXR338L protein.25
Statistical methods
Quantitative data are presented as means±SD. The Student t test was used to perform all statistical analyses. P values less than 0.05 were considered a statistically significant difference.
Results
Relative expression of gain-of-function factor IX variants in hemophilic mice using AAV vectors
Single amino acid substitutions for Arg-338 in the factor IX catalytic domain with alanine (FIXR338A), glutamine (FIXR338Q), and leucine (FIXR338L) have each been observed to produce a factor IX variant with increased specific activity.9,10,15 We examined the relative potential advantage of each of these variants for augmenting the clotting activity of factor IX. Each of these substitutions was generated in otherwise identical liver-directed codon-optimized factor IX AAV vectors and injected via the tail vein into hemophilia B mice. The mice were monitored for 29 weeks, and throughout the period of observation each of the variants demonstrated a specific activity that exceeded the expected wild-type factor IX specific activity of ∼200 U/mg protein (Supplementary Fig. S1A and B). On average, FIXR338L gave the greatest advantage in terms of the specific activity of the expressed proteins and resulted in plasma factor IX activity that was approximately two times higher than the FIXR338A and FIXR338Q variants throughout the 29 weeks of observation. Further vector development incorporated the FIXR338L variant as described below.
Expression of WTFIX and FIXR338L from AAV in mice: long-term in vivo expression results
Self-complementary AAV8 vector was generated on the basis of the pTTRhFIXopt expression cassette and expressed the codon-optimized wild-type factor IX cDNA (WTFIX vector). An additional vector, scAAV8.FIXR338L, was generated with identical features except for a single nucleotide change in the factor IX cDNA resulting in the expression of the FIXR338L variant. Each vector was delivered into the portal venous circulation of adult C57BL/6 FIX–/– mice (n=6 mice per group) at a dose of 4×1011 VG/kg and in vivo expression of factor IX antigen (protein) and activity were assayed during 42 weeks of observation. Plasma factor IX protein expression was consistently at least two times higher from the WTFIX vector than the FIXR338L vector (Fig. 1A). The mechanism explaining the mildly lower FIX protein expression from the FIXR338L vector is under investigation. Nevertheless, plasma factor IX activity in the FIXR338L cohort was three to four times higher than in the WTFIX cohort throughout the 42 weeks (Fig. 1B). The improved factor IX activity mediated by scAAV8.FIXR338L reflects the finding that the WTFIX vector led to plasma factor IX specific activity that was close to the expected specific activity of human FIX (200 U/mg), whereas the scAAV8.FIXR338L vector generated more than six times higher specific activity of the expressed FIXR338L protein throughout the period of observation (Fig. 1C). Before sacrifice at 42 weeks postinjection, all mice underwent a tail vein transection bleeding challenge to assess in vivo function of the vector-expressed human factor IX. Untreated hemophilia B mice had prolonged and profuse bleeding with this challenge and the majority died or became moribund and required euthanasia during 8 hr of observation after this bleeding challenge. Mice that received portal vein delivery of either vector survived the tail transection and were active and drinking/feeding normally throughout 8 hr of observation, similar to hemostatically normal control mice that experienced the same bleeding challenge (Fig. 1D). Peak expression from scAAV8.FIXR338L ranged from 300 to 400% normal human factor IX (hFIX) activity, and long-term expression plateaued above ∼125% normal hFIX activity (Fig. 1B). Peak expression from the WTFIX vector was in the range of 60–80% normal, and long-term expression plateaued above ∼30–60% normal human factor IX activity. Mice from both groups were killed at ∼1 year of age and full histopathological examination was performed. No microangiopathic or macroangiopathic thrombi were found on examination of lung, liver, brain, kidney, heart, or spleen of hemophilic mice expressing either physiological levels of wild-type human factor IX or physiological or supraphysiological levels of human FIXR338L.
FIG. 1.

Expression in plasma of wild-type factor IX and FIXR338L from otherwise identical liver-directed expression cassettes in FIX–/– mice. Codon optimization of the factor IX cDNA used in each vector was identical. The Leu-338 variant results from a single nucleotide substitution in the codon-optimized FIX cDNA. The number of C57BL/6 FIX–/– mice that received intraportal venous vector was 13 in the scAAV8.WTFIX treatment group (WTFIX) and 10 in the scAAV8.FIXR338L group. (A) Total human factor IX protein expressed. (B) Specific activity of circulating factor IX. The normal specific activity of recombinant factor IX is ∼200 U/mg. (C) Circulating factor IX activity as assayed in a one-stage clotting assay (factor IX-specific activated partial thromboplastin time) demonstrated sustained FIXR338L expression at supraphysiological levels for most of the 40 weeks of observation. (D) At the end of the follow-up, plasma factor IX activity was measured and then transection of the tail tip was performed at a uniform circumference of 1.5 mm. After this bleeding challenge, hemophilia B mice (FIX–/–) experienced severe bleeding, which resulted in diminished activity or distress requiring euthanasia of 8 of 14 mice. All mice that had received either factor IX gene therapy vector as a single injection 40 weeks earlier survived the tail transection and remained active, similar to the hemostatically normal mice (WT) that underwent the same bleeding challenge.
Liver-directed AAV vector: comparison of transcriptional regulatory elements
We previously reported our effort to identify compact yet strong regulatory elements to direct factor IX gene expression in the liver from self-complementing AAV vectors.6 A similar effort reported by Nathwani and colleagues led to the development of the scAAV2/8-LP1-hFIXco vector used in the SJCRH/UCL human clinical trial.16 Once we established the potential of the FIXR338L transgene to incrementally improve plasma factor IX activity compared with wild-type FIX, we next examined the relative influence of the expression elements (exclusive of the transgene choice) in the scAAV8.FIXR338L vector compared with the clinically tested SJCRH/UCL scAAV2/8-LP1-hFIXco vector. Hemophilic mice received a 4×1011 VG/kg concentration of either scAAV8.FIXR338L or a vector expressing the identical codon-optimized FIXR338L sequence from the LP1 promoter/enhancer/SV40 intron from the SJCRH vector, and the plasma expression of factor IX activity was measured serially over 40 weeks of observation. No consistent advantage in FIXR338L expression from either expression vector was observed (see Supplementary Fig. S2A). This finding also held true regarding correction of the bleeding phenotype. Mice receiving either vector expressed approximately 50% circulating factor IX activity 40 weeks after vector delivery (as recorded in Supplementary Fig. S2A) and demonstrated protection that was comparable to hemostatically normal mice when challenged by tail vein transection at this time point (Supplementary Fig. S2B).
Dose–response study of intravenously administered scAAV8.FIXR338L
AAV serotype 8 demonstrates high tropism for liver. The feasibility of liver-directed gene therapy for hemophilia using peripheral intravenous rather than intra-portal venous delivery has been demonstrated in human clinical trials4,7 and is a less invasive approach for individuals with hemophilia, who have an increased risk of bleeding from catheter-directed intrahepatic gene delivery. A range of intravenous doses of scAAV8.FIXR338L from 1.0×1010 VG/kg to 4×1012 VG/kg were tested, resulting in plasma factor IX protein levels that ranged from undetectable up to supraphysiological levels (Fig. 2A). At any given dose across this wide range, the specific activity of the expressed factor IX was five to eight times normal and was sustained for months. As an example, a vector dose of 4×1011 VG/kg yielded plasma factor IX protein levels of 9–10% resulting in 55–65% activity in the human factor IX activity assay (Fig. 2A and B). The increased specific activity of the transgenic FIXR338L protein was also reflected in the observation that for the lowest dose cohort (1.0×1010 VG/kg), by the second week half of the animals had plasma factor IX activity >1%, despite the fact that no circulating factor IX antigen could be detected at the level of sensitivity of the assay (∼10 ng/ml, which is equivalent to ∼0.2% normal protein level) (Fig. 2B). An additional cohort of mice received scAAV8.FIXR338L at 4×1011 VG/kg via the portal vein route to allow direct comparison of R338LFIX expression via portal vein and intravenous delivery. Over the 32 weeks of expression there was no consistent difference in plasma factor IX expression achieved by either the portal vein or intravenous route, confirming previous reports.
FIG. 2.

Dose response after scAAV8.FIXR338L vector was delivered by peripheral (tail) vein. FIX–/– mice (six to eight per group) received the indicated doses of scAAV8.FIXR338L as a single tail vein injection to model the peripheral venous route of dosing planned for human clinical application. So as to allow comparison of tail vein (TV) and portal vein (PV) administration using the same vector dosing examined in Fig. 1, separate groups of age-, weight-, and sex-matched FIX–/– mice received 4.0×1011 VG/kg by PV, with factor IX expression displayed by the green triangles. (A) Circulating factor IX protein. (B) Circulating factor IX activity. At any given dose, the specific activity of plasma factor IX (factor IX activity/factor IX antigen, expressed in units/mg protein) was five to eight times normal, with factor IX expression persisting over months. Plasma was examined at each time point for the development of antibodies directed against factor IX. No factor IX inhibitors (as measured by Bethesda assay) or anti-FIX IgG (as measured by ELISA) was detected in any animal throughout the observation. (C) Dose-dependent protection from hemorrhage in the tail transection bleeding challenge. At 32 weeks after vector delivery, all mice from each dose group had plasma factor IX activity determined (as recorded on the x axis below each column) and then underwent a tail transection bleeding challenge and the amount of blood loss in 10 min after wounding was collected and quantitated by measuring the optical density (OD) of the shed hemoglobin.
After euthanasia after tail transection challenge (see below), pathology of the heart, lungs, kidneys, brain, liver, spleen, representative skeletal muscle, and mesenteric lymph node was examined by microscopy from the two highest dose cohorts (8×1011 and 4×1012 VG/kg). Age-matched FIX–/– mice were included as controls. No micro- or macrovascular thrombosis, fibrosis, fibrin depositions, or organ pathology attributable to scAAV8.FIXR338L treatment was present.
Dose-dependent protection from hemorrhage in tail transection challenge
After 32 weeks of expression, all mice in the tail vein dose–response study underwent tail vein transection to evaluate the hemostatic potential of the expressed FIXR338L (Fig. 2C). At the time of tail vein transection, the cohort of mice that received the lowest dose of scAAV8.FIXR338L did not have measurable plasma factor IX activity and all other doses led to some plasma factor IX expression (the mean factor IX activity for each cohort is recorded in Fig. 2C). The lowest vector dose associated with partial protection from blood loss was 5×1010 VG/kg (at ∼3.6% mean circulating FIX activity); however, three of eight mice at this dose showed hemorrhage similar to or worse than that of saline-treated hemophilic mice. A dose response of increasing protection from blood loss was demonstrated as the dose escalated (p<0.01 for doses 1.5×1011 VG/kg and above when compared with saline-treated FIX–/– mice). At the dose of 1.5×1011 VG/kg every mouse was protected from blood loss.
Dose-dependent protection from the development of hemarthrosis and bleeding-induced joint pathology
The tail transection bleeding challenge measures hemostatic protection from an exsanguinating wound. In the clinical care of hemophilia, the most common location of bleeding is into musculoskeletal sites, with recurrent joint hemorrhage causing the blood-induced joint deterioration (hemophilic arthropathy) that is the most common complication of hemophilia. Our group and others have used a model of induced recurrent joint hemorrhage to study hemarthropathy.20,21,26 The joint bleeding challenge consists of a puncture of the knee joint capsule, using a small-gauge needle. The histopathological features of hemophilic synovitis that ensue in hemophilic mice after this bleeding challenge can be quantified by a murine hemophilic synovitis grading system validated in the Valentino laboratory.27 The system has a 0-to-10 point scale that quantitates increasing evidence of hyperplasia of the synovial lining cells and of increased synovial vasculature replacing the subsynovial space, as well as the presence of hemosiderin iron staining, frank blood and cartilage erosion, or synovial villous formation. This model is useful for examining the potentially protective effects of new therapeutic approaches, because the pathology that develops after this injury clearly distinguishes hemophilic mice from control mice with intact hemostasis. When the joint pathology is graded 2 weeks after injury, hemostatically normal mice exposed to this injury essentially never develop synovitis, with 100% of mice scoring less than 2 on the Valentino synovitis grade (historic mean, 0.14).21 Untreated hemophilic mice essentially never score less than this threshold of 2 points out of 10, whereas the historic mean is ∼4.4 out of 10 for hemophilic mice.
Separate groups of FIX–/– mice were treated intravenously with scAAV8.FIXR338L or the corresponding vector expressing WTFIX at a variety of doses to determine whether the expression of AAV.FIX preserves the health of hemophilic joints. At 4 weeks after vector administration, factor IX activity in the plasma was recorded and animals received a single induced joint hemorrhage; joints were collected 2 weeks later to grade the degree of synovitis that developed, using the Valentino grading system (Fig. 3).
FIG. 3.

Dose-dependent protection from the development of hemarthrosis and bleeding-induced joint pathology is improved by the substitution of FIXR338L. FIX–/– mice were treated via the tail vein with scAAV8.FIXR338L (squares) or the corresponding vector expressing WTFIX (×). Four weeks after vector delivery, the mice received a unilateral right hindlimb knee joint needle puncture to induce hemarthrosis. Two weeks after injury, knee joints were collected and scored for synovitis, using the Valentino mouse synovitis score. (A) At both the lower dose of 2×1010 VG/kg (left) and the higher dose of 2×1011 VG/kg (right), the expression from scAAV8.FIXR338L (squares) results in higher plasma levels of factor IX activity when compared with the WTFIX vector (×). The average synovitis score of 4.4 was previously reported for a large cohort of untreated FIX–/– mice that underwent this bleeding challenge, and this value is represented by the dashed line. A large group of strain-matched hemostatically normal C57BL/6 mice exposed to this bleeding challenge developed a mean synovitis score of 0.14, with a range of 0–2.0 (demarcated by the shaded rectangle). (B) Representative histopathology of knee joints 2 weeks after single knee joint puncture injury. The area bounded by the black rectangle in each top panel (original magnification, ×40) is shown at an original magnification of ×200 in the immediately underlying bottom panel. Normal joint anatomy was preserved in hemostatically normal C57BL/6 wild-type control mice (left) by an open joint space (*), thin flat synovial epithelial cells (arrowhead) supported by an unremarkable fatty and fibrovascular stroma, and subsynovial fat pad (**) composed predominantly of scant small blood vessels (arrows) (bottom left). In contrast, FIX–/– mice given scAAV8.FIXR338L (2×1010 VG/kg) that expressed FIXR338L (middle) demonstrated minimal to mild changes of synovitis (synovitis score, ∼2/10); an increase in the size and number of blood vessels (neovascularization) (arrows) in the subsynovial fat pad (**) is present; however, there is little to no increase in synovial epithelial cell thickness (arrowhead). Mice given WTFIX vector (2×1010 VG/kg) had severe synovitis (synovitis score, >4/10) (right) with a marked increase in synovial thickness in concert with effacement and replacement of the subsynovial fat (**) pad by proliferation of synovial epithelial cells and granulation tissue, admixed with inflammatory cells. As observed in clinically severe hemarthrosis, we see pannus formation (P) when these proliferating cells grow over the articular cartilage, resulting in erosion (*) in combination with marked neovascularization (arrows), and foci of fibrinonecrotic debris (D). Hematoxylin–eosin. Scale bars, 200 μm.
Mice given the lowest dose (2×1010 VG/kg) of WTFIX vector (n=6) expressed factor IX activity between 0.8 and 1.4% of normal human factor IX activity in plasma (see scatterplot in Fig. 3A). In contrast, mice given the same dose of scAAV8.FIXR338L (n=6) expressed 1.7–6.2% activity. Most importantly, this expression correlated with relative protection from synovitis. Five of six mice receiving the scAAV8.FIXR338L vector at the low dose displayed significant protection, with scores approaching the threshold score of 2 that separates hemophilic mice from nonhemophilic mice. At the highest dose of 2×1011 VG/kg, the clinically relevant joint protection was more obvious. Mice that received scAAV8.FIXR338L (n=7), but not mice receiving WTFIX vector (n=5), expressed 57–120% factor IX and six of seven mice demonstrated joint protection with histopathological features that did not differ from those of injured hemostatically normal wild-type mice.
Detection of factor IX antibodies in hemophilia B mice after scAAV8.FIXR338L administration
The development of an antibody response directed against replacement factor IX that neutralizes its procoagulant action (an “inhibitor”) is an unusual but potentially morbid complication of the treatment of hemophilia B. Development of inhibitors occurs in about 2–4% of patients with severe hemophilia B. Mice that received gene therapy vector in each of the studies described here were screened at multiple time points for the development of antibodies that interfere with the activity of normal factor IX in an assay of coagulation function (Bethesda inhibitor assay); in addition, at the time of euthanasia the mice were tested for IgG that bound factor IX specifically by ELISA. The results of all antibody studies in the preclinical program are included in Table 1. Also included in Table 1 (assessed by FIX Bethesda assay only) are mice that received AAV vectors and expressed WTFIX or FIXR338L after vector delivery directly into the intraarticular space of the knee joint, following a protocol we have reported previously.21 The intraarticularly treated mice add to our experience with expression of the scAAV8.FIXR338L vector in immunocompetent mice (C57BL/6 strain, expressing HLA mouse H2b haplotype). In this total experience in over 200 mice, including 168 with a factor IX null phenotype, a single factor IX inhibitor antibody has been observed. This arose in a factor IX null mouse whose first exposure to human factor IX was in the context of portal venous vector delivery, accomplished via open abdominal surgery that involves reflection of the intestines out of the abdominal cavity. This factor IX inhibitor was present at a clinically low titer of <1 unit in the factor IX Bethesda assay and a concentration of 0.1 μg/ml IgG; each of these values is at the lower level of sensitivity of the assay.
Table 1.
Factor IX Antibody Formation After scAAV8 Treatment Expressing Either Wild-Type FIX or FIXR338L
| Bethesda inhibitor | Anti-FIX binding IgG | |||||
|---|---|---|---|---|---|---|
| Mouse strain | Route of delivery | Human FIX transgene | Inhibitors/total no. of animals | Titer (BIU) | FIX antibody/total no. of animals | Range (μg/ml) |
| WT C57BL/6 | Peripheral vein | FIXR338L | 0/40 | — | 0/20 | ND |
| FIX–/– C57BL/6 | Peripheral vein | WTFIX | 0/15 | — | 0/15 | ND |
| FIXR338L | 0/88 | — | 0/88 | ND | ||
| FIX–/– C57BL/6 | Intraportal vein | WTFIX | 0/49 | — | 0/49 | ND |
| FIXR338L | 1/40 | <1 | 1/40 | 0.04–0.12 | ||
| FIX–/– C57BL/6 | Intraarticular | WTFIX | 0/72 | — | 0/72 | NP |
| FIXR338L | 0/40 | — | 0/40 | NP | ||
BIU, Bethesda inhibitor units; FIX–/–, factor IX knockout mice; ND, none detected; NP, not performed; WT, wild type.
Thrombin–antithrombin complex formation
In addition to the necropsy evaluations for evidence of thrombosis, the potential for FIXR338L expression to lead to inappropriate activation of coagulation was assessed by measuring plasma TAT complexes. Adult FIX–/– mice received a 2×1012 VG/kg concentration of vector expressing either WTFIX or FIXR338L, whereas control mice were injected with normal saline. Four weeks later, plasma was collected via a single, open terminal puncture of the inferior vena cava. Plasma factor IX activity was determined by a one-stage factor IX activity assay and TAT complexes were measured in an ELISA that has been previously shown to detect mouse TATs (Enzygnost; Siemens Healthcare Diagnostics). As shown in Table 2, scAAV8.FIXR338L expression of FIXR338L at physiological or supraphysiological levels was not associated with elevations in TAT levels when compared with expression of WTFIX, and was not associated with absolute elevations of TAT.
Table 2.
Plasma Thrombin–Antithrombin Complexes: A Marker of Activation of Coagulation After scAAV8FIX Gene Therapy in Hemophilia B Mice
| Transgene | Dose (VG/kg) | n | Human FIX antigen (μg/ml) | Human FIX activity (%) | TAT (ng/ml) |
|---|---|---|---|---|---|
| Untreated | Normal saline | 6 | — | <1 | 31.7±18.3 |
| WTFIX | 1.2×1012 | 6 | 4.17±1.96 | 85.6±26.6 | 27.7±18.8 |
| FIXR338L | 6×1011 | 7 | 0.83±0.34 | 90.7±35.4 | 23.0±12.5 |
| FIXR338L | 2×1012 | 6 | 3.42±1.07 | 340.0±122.6 | 25.9±13.1 |
FIX, factor IX knockout; TAT, thrombin–antithrombin; VG, vector genomes.
Empty vector particles do not circumvent the neutralization of factor IX gene delivery by AAV-neutralizing antibodies
The rAAV8 preparation used in the SJCRH/UCL gene therapy trial contained at least four or five empty capsids, containing no F9 transgene, for each potentially therapeutic F9-containing AAV vector (80–90% empty capsids).8 Our strategy to pursue higher factor IX expression using the FIXR338L and vector stocks with minimal empty capsid is premised on the concept that diminishing the exposure to potentially immunogenic empty capsids may increase rAAV8.F9 safety. Nonetheless, it is conceivable that the presence of empty rAAV capsids might have contributed in some way to the successful gene expression in the SJCRH/UCL trial. For example, some investigators have suggested that empty capsids could serve as a “decoy” for circulating AAV-neutralizing antibodies (AAV NAbs), thereby aiding transduction by the transgene-containing vectors in individuals with Nabs.28 To model the scenario that existed in the SJCRH/UCL trial, we used well-characterized empty AAV8 capsids obtained as a kind gift of M. Agbandje-McKenna. We generated anti-AAV8 NAbs by vaccinating C57BL/6 mice with an AAV8 vector. Serum was collected and pooled, and the AAV8-NAb titer was defined. cGMP-grade scAAV8.FIXR338L, the fully characterized AAV8.F9 vector used in an ongoing human clinical trial, was examined by electron microscopy (EM) and found to contain <10% empty capsids (Fig. 4). NAb was passively transferred to FIX–/– mice by tail vein injection, achieving a circulating AAV NAb titer of either 1:16 (moderate titer) or 1:2 (near the lower limit of detection) (Table 3). Mice were treated next with one of the following: either 1×1011 VG/kg of scAAV8.FIXR338L (<10% empty AAV capsids); or with 1×1011 VG/kg scAAV8.FIXR338L plus 1×1011 VG/kg rAAV8 empty capsids (∼50% empty capsids); or with 1×1011 VG/kg scAAV8.FIXR338L plus 4×1011 VG/kg rAAV8 empty capsids (∼80% empty capsids). A group of mice received no NAb and were treated with scAAV8.FIXR338L at 1×1011 VG/kg to be a positive control for F9 gene delivery. In the absence of NAb, mean plasma factor IX activity after intravenous scAAV8.FIXR338L was 7.4 and 6.7% at 2 and 4 weeks, respectively. Passively transferred NAb at either 1:2 or 1:16 titer prevented transgene expression (FIX activity <1%, lower limit of sensitivity). If the NAb titer was measurable, then AAV8 neutralization was not abrogated by the presence of empty capsids at either the lower dose of empty capsid or the dose that yielded a large excess of empty capsids (expression of FIX activity <1%).
FIG. 4.
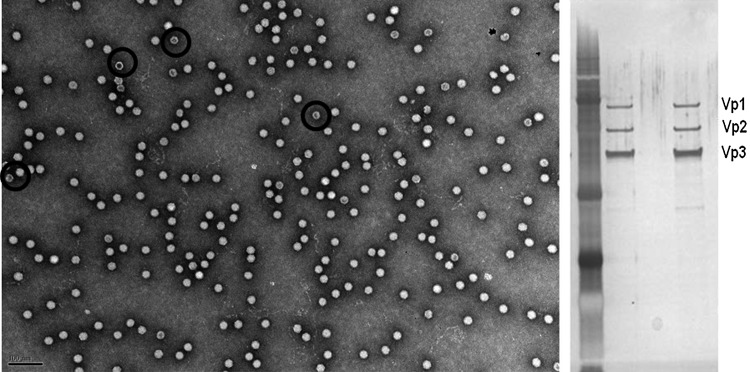
Manufacture of GLP vector produced from scalable suspension cell culture eliminates most contaminating empty AAV capsids. Left: Transmission electron micrograph of the clinical vector used in the dose–response and toxicity/biodistribution studies, demonstrating a favorable ratio of therapeutic particles to defective interfering particles (empty capsids make up less than 10% of vector stock). Black circles are superimposed to show examples of the relatively hollow appearance that is typical of empty vector capsids. Right: Silver stain of clinical vector demonstrates only three proteins, corresponding to the AAV structural proteins Vp1, Vp2, and Vp3, present at the expected ratio of ∼1:1:9. Lanes 2 and 4 contain identical vector.
Pivotal GLP toxicology and biodistribution study in hemostatically normal mice
Plasma FIX activity was measured at all time points, using a one-stage clotting assay, and was expected to measure the animal's baseline normal hemostatic activity (i.e., the baseline activity that results from endogenous mouse factor IX) as well as functional human factor IX expression from the scAAV8.FIXR338L vector. The values reported represent the factor IX activity above mouse factor IX baseline (at each time point correcting for the amount of activity measured in the vehicle-treated group). The mean FIX activity in the 4×1011 VG/kg dose group peaked at ∼200%, subsequently decreasing to a mean of ∼50% above baseline at the 18-week point. The activity in the 4×1012 VG/kg dose group was maintained throughout the study at greatly supraphysiological levels (>1200%) (Supplementary Fig. S3). No factor IX-binding IgG antibodies or factor IX Bethesda inhibitors developed in any animals and there were no differences in blood hematology or clinical chemistry parameters between control and scAAV8.FIXR338L-treated mice. One animal in the lower vector dose group, having no prior signs of toxicity, died on study at day 67 and postmortem examination revealed only chronic changes of mild myocardial fibrosis that were not believed to be large enough to be the direct cause of death. One animal in the high-dose group (supraphysiological FIX expression) died on day 99 with no significant macro- or microscopic abnormalities on postmortem examination. The complete histopathological analysis of all vector-treated mice and saline-treated control mice was unremarkable with the exception of mild focal hepatic necrosis in two of five mice in the high-dose group at the week 10 time point only. The foci were small and serum chemistries for liver enzymes were normal (data not shown). Minimal focal liver necrosis was also noted in control and vector-treated mice at a similar frequency across the groups. Evaluation of livers for CD8+ T cells was negative; thus evidence of cellular immune response to scAAV8.FIXR338L treatment was excluded. Consistent with this finding were results of splenocyte interferon-γ ELISPOT analyses, in which we found only low and transient T cell stimulation to the AAV8 capsid and no T cell response to the factor IX transgene (S.W.J. McPhee and R. Calcedo, data not shown).
Consistent with the known hepatic tropism of AAV8, biodistribution analysis after peripheral intravenous delivery showed the highest concentration was in the liver, the concentration was dose-related, and was highest at the earliest time point (3 days). Full biodistribution data of the detected scAAV8.FIXR338L sequence at each time point in nine organs/tissues (liver, spleen, heart, skeletal muscle, inguinal lymph node, kidney, lung, brain, testis) are included in Supplementary Table S2. On examining the groups exposed to the higher vector dose (see Supplementary Table S2 and Supplementary Fig. S4), on day 3 the organ with the next highest tissue concentration relative to the liver was the spleen (27-fold lower vector copies; i.e., 4.29×106 copies in the liver and 1.58×105 copies in the spleen). The spleen had either the highest or second highest extrahepatic distribution of vector at the day 3 and week 3 time points, using either the lower or higher dose. By week 10, however, vector distribution in the spleen decreased to less than 1% of the vector copy number measured on day 3, and was ∼500- to 600-fold lower than in the liver. At week 3 the extrahepatic concentration of vector in the higher vector dose group was highest in the heart (118-fold lower than in liver). In contrast to the spleen, vector copy numbers in cardiac and skeletal muscle (which are relatively end-differentiated tissues with minimal turnover) remained relatively stable from the 3-week evaluation through the last evaluation at 18 weeks. The heart remained a relatively high site of extrahepatic vector distribution at weeks 10 and 18, although 120- to 150-fold lower than in the liver. At the week 10 and 18 time points, skeletal muscle contained the greatest concentration of vector amongst the extrahepatic tissues.
Discussion
Hemophilia B has joined the list of conditions for which clinical improvement has been achieved by gene therapy.29 The partial correction of the inherited enzyme deficiency in the SJCRH/UCL trial has been persistent and associated with a decrease in clinically apparent bleeding. There is a limitation to the approach used, however, because a dose of 2×1012 VG/kg that can achieve 3–6% plasma factor IX activity (correction to the moderate to mild hemophilia range) is associated with elevations of serum liver transaminases. Elevations in liver enzymes (associated with liver inflammation and injury) were controlled in these subjects with a course of corticosteroid immunosuppression therapy. The investigators have described the observed hepatic inflammation as a manageable toxicity. Nevertheless, the clinical trial populations for the phase 1/2 trials were carefully selected, and approaches that might avoid the need to use vector doses associated with hepatic inflammation are desirable. This is particularly the case because >85% of the U.S. severe hemophilia population over the age of 30 are seropositive due to hepatitis B and C exposure; the majority of those have active disease as evidenced by hepatitis virus RNA on serum PCR testing.
The current standard of therapy for children with severe hemophilia (which has not been universally adopted for adults with severe hemophilia) is prophylactic replacement with clotting factor protein with a goal to ensure that plasma levels remain in at least the moderate hemophilia range (>1% factor IX activity) at most times. This approach does not completely convert the severe bleeding phenotype to that observed in moderate hemophilia.30 Opinion in the hemophilia community proposes the idea that complete protection from spontaneous joint hemorrhage is the appropriate goal for hemophilia care, rather than a partial conversion to a moderate bleeding tendency.31 An analysis of bleeding in individuals with hemophilia A and clotting factor levels between 1 and 40% demonstrated an 18% reduction in joint bleeding with every percent increase in baseline factor level. The analysis of this population predicted a nearly zero risk for joint bleeding with a baseline factor level of 15% and higher.32 To achieve this degree of disease correction by gene therapy, while avoiding vector doses that approach or exceed those already associated with hepatic inflammation in two human trials, ongoing improvement of AAV.FIX vector efficiency is required. The preclinical program described here demonstrates that the incorporation of gain-of-function factor IX variants at amino acid 338 can improve circulating factor IX activity. The modified FIX proteins demonstrated differences in the level of circulating FIX activity in the animal setting, as well as nuances in protein expression and the ratio of activity/antigen. Our extended studies of transgenic expression of FIXR338L achieved equivalent or improved hemostatic correction with lower exposure to vector than is required when a vector expressing WTFIX is used. Of particular importance is that the protection against bleeding-induced joint disease was improved. The observation is supported by reports in hemophilic animals of FIXR338A (using AAV or adenoviral vectors in mice)11,12,33 or FIXR338L (using lentiviral vectors in mice or AAV vector in dog)13–15,34 demonstrating increased clotting potential from these variants and the absence of thrombosis.
In the current study, survival was normal and evidence of thrombosis was absent on extensive postmortem examination.35,36 This outcome is consistent with the experience in the index case family expressing FIXR338L; clinical thrombosis was experienced by affected members with factor IX activity >700–800% of normal but not in the carrier mother expressing >300% factor IX Padua.10 Of note, supraphysiological expression of transgenic factor VII or factor IX in mouse models has led to thromboembolic premature death in some previous reports.35,36 In our intravenous administration dose–response study of FIXR338L expression, 100% of mice that survived the initial anesthesia and vector administration subsequently lived throughout 8 months of follow-up; this included 12 of 12 mice stably expressing 80–180% FIXR338L activity and 7 of 7 mice stably expressing 300–550% FIXR338L activity.
The substitution of a variant of the factor IX protein introduces the potential risk of immune response to neoepitopes in the factor IX transgene. Clinically, inhibitor antibodies arise in only ∼2–4% of individuals with severe hemophilia B after exogenous factor IX replacement, although the risk is higher in individuals with complete or large gene deletions. Examination of the Factor IX Mutation Database reveals that individuals with missense mutations arising from a single nucleotide variance from wild-type factor IX essentially do not develop inhibitors in the setting of exogenous protein replacement; a single patient with a missense substitution complicated by inhibitor is described among the 1095 unique F9 variants and 3713 individual cases in the database37 (http://www.factorix.org/). All mice used in this report had a factor IX null phenotype. Only individuals who have extensive exposure to factor IX (>150 treatment days) without inhibitor development will participate in phase 1 trials that use this sequence variation.
On the basis of the available preclinical safety and efficacy data reported here, we are currently investigating the liver-directed vector incorporating FIXR338L (scAAV8.FIXR338L, now given the designation BAX335) in a human clinical trial. Manufacturing using scalable suspension cell culture and chromatographic purification resulted in scAAV8 vector clinical stocks with less than 10% empty particle contamination. The dose-escalation design of the ongoing trial has been chosen to target steady state clinical factor IX expression at not greater than 40% of normal factor IX levels for this phase 1/2 safety study. In addition, we demonstrate that the addition of wild-type empty AAV8 capsids to the scAAV8.FIXR338L/BAX335 vector preparation did not provide any evident benefit (e.g., to circumvent AAV8 NAbs) under the experimental conditions used. A report by Mingozzi and colleagues suggested that AAV empty capsids might improve transduction in the presence of AAV NAbs by providing “decoy” particles to remove AAV NAbs, allowing therapeutic particles to infect with less interference from AAV Nabs.28 There are a number of differences in the experimental conditions used in this paper and those of Mingozzi and colleagues. These include differences in vector-manufacturing methods (it is not possible to discern whether their vector stocks contain empty AAV particles in addition to those intentionally supplemented), a higher empty capsid exposure supplemented in their study, and the source of NAb used for the passive transfer of AAV8 NAb (human IVIG in their study compared with specific anti-AAV8 arising after AAV8 infection under our conditions). Given that the only vector-related toxicity that has been observed to date in human trials of AAV gene therapy for hemophilia B is a dose-related immune response to the AAV capsid, our results support our strategy of generating rAAV8 stocks with minimal empty AAV particles as desirable to avoid immune activation.
Supplementary Material
Acknowledgments
This work was supported by grants from NIH NHLBI 1RC3HL103396 (to S.W.J.M. and P.E.M.) and NIH NHLBI P01-HL66973 (to R.J.S., D.W.S., and P.E.M.) and NIH NHLBI P01-HL112761 (to R.J.S. and P.E.M.). T.K. was supported by NIH NIDDK 5R01DK058702-12. J.S. and Z.W. were supported by a Judith Graham Pool Fellowship Grant from the National Hemophilia Foundation. The authors thank Dr. Mavis Agbandje-McKenna for production, characterization, and donation of AAV8 empty capsids and Dr. David Dismuke and Dr. Eric Horowitz for production and characterization of GLP AAV vectors. The authors thank Dr. Roberto Calcedo for performing AAV8 and factor IX ELISPOT assays as a part of the GLP toxicity study. The authors thank Dr. Phillip Owens and Dr. Nigel Mackman for technical assistance with the TAT assay. The authors thank Michael Schadt (Mouse Research Pathology, Section of Comparative Medicine, Yale University School of Medicine) for the routine histology (H&E, MT) and Amos Brooks (Research Histology, Pathology Department, Yale University School of Medicine) for the fibrinogen IHC, and PTAH.
Author Disclosure Statement
Paul E. Monahan receives research support through the University of North Carolina from Asklepios BioPharmaceutical and has received research support in the past from Baxter Healthcare. He holds patents that have been licensed by UNC to Asklepios Biopharmaceutical, for which he receives royalties. He has consulted for Asklepios Biopharmaceutical, for Chatham LLC, and for Baxter Healthcare and has received payment for consultation, services, and speaking. Scott W.J. McPhee and Jade J. Samulski are employees, officers, and shareholders at Asklepios BioPharmaeutical. R. Jude Samulski is the founder and a shareholder at Asklepios BioPharmaceutical. He receives research support through the University of North Carolina from Asklepios BioPharmaceutical. He holds patents that have been licensed by UNC to Asklepios Biopharmaceutical, for which he receives royalties. He has consulted for Baxter Healthcare and has received payment for speaking. Darrel W. Stafford and Joshua C. Grieger hold patents that have been licensed by UNC to Asklepios Biopharmaceutical, for which they receive royalties. Chengwen Li receives research support through the University of North Carolina from Asklepios BioPharmaceutical. No competing financial interests exist for the remainder of the authors.
References
- 1.Skinner MW. Gene therapy for hemophilia: addressing the coming challenges of affordability and accessibility. Mol Ther 2013;21:1–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manno CS, Pierce GF, Arruda VR, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med 2006;12:342–347 [DOI] [PubMed] [Google Scholar]
- 3.Mingozzi F, and High KA. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood 2013;122:23–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nathwani AC, Gray JT, McIntosh J, et al. Safe and efficient transduction of the liver after peripheral vein infusion of self-complementary AAV vector results in stable therapeutic expression of human FIX in nonhuman primates. Blood 2007;109:1414–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vandendriessche T, Thorrez L, Acosta-Sanchez A, et al. Efficacy and safety of adeno-associated viral vectors based on serotype 8 and 9 vs. lentiviral vectors for hemophilia B gene therapy. J Thromb Haemost 2007;5:16–24 [DOI] [PubMed] [Google Scholar]
- 6.Wu Z, Sun J, Zhang T, et al. Optimization of self-complementary AAV vectors for liver-directed expression results in sustained correction of hemophilia B at low vector dose. Mol Ther 2008;16:280–289 [DOI] [PubMed] [Google Scholar]
- 7.Nathwani AC, Tuddenham EG, Rangarajan S, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med 2011;365:2357–2365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allay JA, Sleep S, Long S, et al. Good manufacturing practice production of self-complementary serotype 8 adeno-associated viral vector for a hemophilia B clinical trial. Hum Gene Ther 2011;22:595–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang J, Jin J, Lollar P, et al. Changing residue 338 in human factor IX from arginine to alanine causes an increase in catalytic activity. J Biol Chem 1998;273:12089–12094 [DOI] [PubMed] [Google Scholar]
- 10.Simioni P, Tormene D, Tognin G, et al. X-linked thrombophilia with a mutant factor IX (factor IX Padua). N Engl J Med 2009;361:1671–1675 [DOI] [PubMed] [Google Scholar]
- 11.Schuettrumpf J, Herzog RW, Schlachterman A, et al. Factor IX variants improve gene therapy efficacy for hemophilia B. Blood 2005;105:2316–2323 [DOI] [PubMed] [Google Scholar]
- 12.Brunetti-Pierri N, Grove NC, Zuo Y, et al. Bioengineered factor IX molecules with increased catalytic activity improve the therapeutic index of gene therapy vectors for hemophilia B. Hum Gene Ther 2009;20:479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cantore A, Nair N, Della Valle P, et al. Hyperfunctional coagulation factor IX improves the efficacy of gene therapy in hemophilic mice. Blood 2012;120:4517–4520 [DOI] [PubMed] [Google Scholar]
- 14.Finn JD, Nichols TC, Svoronos N, et al. The efficacy and the risk of immunogenicity of FIX Padua (R338L) in hemophilia B dogs treated by AAV muscle gene therapy. Blood 2012;120:4521–4523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suwanmanee T, Hu G, Gui T, et al. Integration-deficient lentiviral vectors expressing codon-optimized R338L human FIX restore normal hemostasis in hemophilia B mice. Mol Ther 2014;22:567–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nathwani AC, Gray JT, Ng CY, et al. Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood 2006;107:2653–2661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grieger JC, and Samulski RJ. Adeno-associated virus vectorology, manufacturing, and clinical applications. Methods Enzymol 2012;507:229–254 [DOI] [PubMed] [Google Scholar]
- 18.Lin HF, Maeda N, Smithies O, et al. A coagulation factor IX-deficient mouse model for human hemophilia B. Blood 1997;90:3962–3966 [PubMed] [Google Scholar]
- 19.Gui T, Reheman A, Ni H, et al. Abnormal hemostasis in a knock-in mouse carrying a variant of factor IX with impaired binding to collagen type IV. J Thromb Haemost 2009;7:1843–1851 [DOI] [PubMed] [Google Scholar]
- 20.Hakobyan N, Enockson C, Cole AA, et al. Experimental haemophilic arthropathy in a mouse model of a massive haemarthrosis: gross, radiological and histological changes. Haemophilia 2008;14:804–809 [DOI] [PubMed] [Google Scholar]
- 21.Sun J, Hakobyan N, Valentino LA, et al. Intraarticular factor IX protein or gene replacement protects against development of hemophilic synovitis in the absence of circulating factor IX. Blood 2008;112:4532–4541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monahan PE, Lothrop CD, Sun J, et al. Proteasome inhibitors enhance gene delivery by AAV virus vectors expressing large genomes in hemophilia mouse and dog models: a strategy for broad clinical application. Mol Ther 2010;18:1907–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li C, Narkbunnam N, Samulski RJ, et al. Neutralizing antibodies against adeno-associated virus examined prospectively in pediatric patients with hemophilia. Gene Ther 2012;19:288–294 [DOI] [PubMed] [Google Scholar]
- 24.Halder S, Nam HJ, Govindasamy L, et al. Production, purification, crystallization and structure determination of H-1 parvovirus. Acta Crystallogr Sect F Struct Biol Cryst Commun 2012;68:1571–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brantly ML, Chulay JD, Wang L, et al. Sustained transgene expression despite T lymphocyte responses in a clinical trial of rAAV1-AAT gene therapy. Proc Natl Acad Sci USA 2009;106:16363–16368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sen D, Chapla A, Walter N, et al. Nuclear factor (NF)-κB and its associated pathways are major molecular regulators of blood-induced joint damage in a murine model of hemophilia. J Thromb Haemost 2013;11:293–306 [DOI] [PubMed] [Google Scholar]
- 27.Valentino LA, and Hakobyan N. Histological changes in murine haemophilic synovitis: a quantitative grading system to assess blood-induced synovitis. Haemophilia 2006;12:654–662 [DOI] [PubMed] [Google Scholar]
- 28.Mingozzi F, Anguela XM, Pavani G, et al. Overcoming preexisting humoral immunity to AAV using capsid decoys. Sci Transl Med 2013;5:194ra192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Reilly M, Federoff HJ, Fong Y, et al. Gene therapy: charting a future course—summary of a National Institutes of Health Workshop, April 12, 2013. Hum Gene Ther 2014;25:488–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.den Uijl I, Biesma D, Grobbee D, et al. Turning severe into moderate haemophilia by prophylaxis: are we reaching our goal? Blood Transfus 2013;11:364–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skinner MW. WFH: closing the global gap—achieving optimal care. Haemophilia 2012;18(Suppl. 4):1–12 [DOI] [PubMed] [Google Scholar]
- 32.den Uijl IE, Fischer K, Van Der Bom JG, et al. Analysis of low frequency bleeding data: the association of joint bleeds according to baseline FVIII activity levels. Haemophilia 2011;17:41–44 [DOI] [PubMed] [Google Scholar]
- 33.Lin CN, Kao CY, Miao CH, et al. Generation of a novel factor IX with augmented clotting activities in vitro and in vivo. J Thromb Haemost 2010;8:1773–1783 [DOI] [PubMed] [Google Scholar]
- 34.Kao CY, Yang SJ, Tao MH, et al. Incorporation of the factor IX Padua mutation into FIX-Triple improves clotting activity in vitro and in vivo. Thromb Haemost 2013;110:244–256 [DOI] [PubMed] [Google Scholar]
- 35.Ameri A, Kurachi S, Sueishi K, et al. Myocardial fibrosis in mice with overexpression of human blood coagulation factor IX. Blood 2003;101:1871–1873 [DOI] [PubMed] [Google Scholar]
- 36.Aljamali MN, Margaritis P, Schlachterman A, et al. Long-term expression of murine activated factor VII is safe, but elevated levels cause premature mortality. J Clin Invest 2008;118:1825–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rallapalli PM, Kemball-Cook G, Tuddenham EG, et al. An interactive mutation database for human coagulation factor IX provides novel insights into the phenotypes and genetics of hemophilia B. J Thromb Haemost 2013;11:1329–1340 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.