

Abstract
Background:
PIM1 kinase is coexpressed with c-MYC in human prostate cancers (PCs) and dramatically enhances c-MYC-induced tumorigenicity. Here we examine the effects of a novel oral PIM inhibitor, AZD1208, on prostate tumorigenesis and recurrence.
Methods:
A mouse c-MYC/Pim1-transduced tissue recombination PC model, Myc-CaP allografts, and human PC xenografts were treated with AZD1208 (n = 5–11 per group). Androgen-sensitive and castrate-resistant prostate cancer (CRPC) models were studied as well as the effects of hypoxia and radiation. RNA sequencing was used to analyze drug-induced gene expression changes. Results were analyzed with χ2 test. Student’s t test and nonparametric Mann-Whitney rank sum U Test. All statistical tests were two-sided.
Results:
AZD1208 inhibited tumorigenesis in tissue recombinants, Myc-CaP, and human PC xenograft models. PIM inhibition decreased c-MYC/Pim1 graft growth by 54.3±39% (P < .001), decreased cellular proliferation by 46±14% (P = .016), and increased apoptosis by 326±170% (P = .039). AZD1208 suppressed multiple protumorigenic pathways, including the MYC gene program. However, it also downregulated the p53 pathway. Hypoxia and radiation induced PIM1 in prostate cancer cells, and AZD1208 functioned as a radiation sensitizer. Recurrent tumors postcastration responded transiently to either AZD1208 or radiation treatment, and combination treatment resulted in more sustained inhibition of tumor growth. Cell lines established from recurrent, AZD1208-resistant tumors again revealed downregulation of the p53 pathway. Irradiated AZD1208-treated tumors robustly upregulated p53, providing a possible mechanistic explanation for the effectiveness of combination therapy. Finally, an AZD1208-resistant gene signature was found to be associated with biochemical recurrence in PC patients.
Conclusions:
PIM inhibition is a potential treatment for MYC-driven prostate cancers including CRPC, and its effectiveness may be enhanced by activators of the p53 pathway, such as radiation.
Prostate cancer afflicts one in six men during their lifetime. Up to 20% of low-risk and 40% of high-risk patients fail primary therapy with surgery and/or radiation, resulting in nearly 30000 deaths annually in the United States (1). Patients who fail primary treatment and develop recurrence often have very limited treatment options. Given the high prevalence of prostate cancer and the high failure rate of current prostate cancer therapies, development of new therapies for aggressive prostate cancer is a high priority. The serine-threonine kinase PIM1 has emerged recently as a possible therapeutic target in prostate and other malignancies (2). PIM1 phosphorylates several target substrates involved in inhibition of apoptosis and promotion of cell cycle progression, eg, BAD, CDC25A, p27KIP1, and the androgen receptor (3,4). Notably, PIM1 dramatically cooperates with c-MYC to promote prostate cancer development in a kinase-dependent manner (5,6) and is required to maintain the tumorigenicity of these tumors (7). These data highlight the potential of Pim1 as a therapeutic target in prostate cancer (8).
AZD1208 is a novel pan-PIM kinase inhibitor that has shown efficacy in models of acute myeloid leukemia (9–11). Here, we examined the efficacy of AZD1208 in treating MYC-driven prostate cancer.
Methods
Mouse Prostate Tissue Recombination
The prostate tissue recombination coupled with lentiviral delivery of MYC/Pim1 have been described (5). SCID mice received AZD1208 by once-daily oral gavage in 0.1% Tween-80 (Sigma-Aldrich), 0.5% Methocel E4M (Gallipot, St. Paul, MN) in water (n = 11) or vehicle (n = 11). Mice were weighed daily and dosed at 10mL/kg-mouse at a drug concentration of 30–45mg/kg-mouse.
In Vitro Clonogenic Assays, Hypoxia Induction, and Radiation
Clonogenic assays and irradiation using Myc-CaP cells (12,13) are described in the Supplementary Methods (available online). 1mL aliquots containing 20×106 Myc-CaP cells were placed into 1mL glass Wheaton vacule vials (Fisher Scientific, Pittsburgh, PA) and sealed by melting the glass. The vials incubated rotating at 37°C to form hypoxic environment. Vials were treated with radiation in the Cs-137 irradiator then incubated for an additional one hour.
Tumor Allografts and Xenografts
Generation of allografts and xenografts in nude mice are detailed in the Supplementary Methods (available online). DU145 cells (ATCC) and CWR22Rv1 (Marja Nevalainen) were authenticated by STR (short tandem repeat) locus profiling at nine loci.
Mouse Allograft Irradiation and Hypoxia
An orthovoltage 300 kVp/10 mA X-ray Pantak irradiator was used to irradiate. Acute hypoxia in hindlimb allografts was induced by temporary rubber-band tourniquet method (14), which causes effective and rapid hypoxia without permanent tissue damage. The X-ray field was shaped by lead blocks to irradiate the hindlimbs containing the allografts, while blocking the majority of the mouse body.
Immunohistochemistry
This was performed as described (5,15) and as detailed in the Supplementary Methods (available online).
RNA-Seq Analysis
RNA was isolated using Trizol (Life Technologies, Grand Island, NY) (Supplementary Table 2, available online) and RNA-seq performed as detailed in the Supplementary Methods (available online). The sequencing files were submitted as project PRJEB5525 to the European Nucleotide Archive (http://www.ebi.ac.uk/ena/).
Affymetrix Gene Profiling
Gene expression was analyzed by Vanderbilt Technologies for Advanced Genomics using Affymetrix Mouse Gene 2.0 ST Arrays. Data normalization, processing and Gene Set Enrichment Analysis (GSEA) analyses were performed as described (7). Data are deposited as GSE59106 in Gene Expression Omnibus (GEO) database.
Statistical Analysis
PIN foci among groups were analyzed using the Chi-squared test. Tumor graft and immunohistochemical quantification was done using a two-tailed, unpaired Student’s t test and by the nonparametric Mann-Whitney rank sum U Test using a type I error (alpha) of .05. All statistical tests were two-sided. Quantitative variables are expressed as mean ± SD. Statistical analyses for genomic data were performed in R with visualizations in R and Spotfire (16,17). The following R packages were used: gplots (18), NMF (19), and reshape2 (20).
Mouse Studies Approvals
All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee at Vanderbilt University and at AstraZeneca.
Results
AZD1208 Inhibition of c-MYC/Pim1-Prostate Tissue Recombinant Grafts
We conducted four independent AZD1208 treatment trials using nude mice carrying prostate tissue recombinant grafts overexpressing c-MYC and Pim1 (Figure 1, A and B). Prostate grafts were generated by coinfecting primary mouse prostate epithelial cells with lentiviruses coexpressing c-MYC or Pim-1 with yellow fluorescent protein (YFP) (5,6), combined with urogenital mesenchyme in collagen and implanted under the renal capsule. AZD1208 (30mg/kg or 45mg/kg) or vehicle was given daily by oral gavage for two or three weeks (Figure 1A). Mice were weighed daily (Supplementary Figure 1A, available online). Two mice in the AZD1208 group receiving 45mg/kg lost between 16% to 19% of body weight, suggesting that dose toxicity level was reached on this schedule. Mice treated with AZD1208 for three weeks at either 30 or 45mg/kg had statistically significantly smaller grafts (P < .003) (Figure 1C). For one set of mice treated with 45mg/kg AZD1208, the grafts were 54.3±39% (P < .001) smaller than controls.
Figure 1.

PIM kinase inhibitor AZD1208 treatment scheme and histological analysis of treated prostate tissue recombinant grafts. A) Scheme for AZD1208 treatment (four trials) of MYC/Pim1-expressing prostate recombination grafts showing the timing and duration of treatment (shaded bars). B) Numbers of mice and individual grafts (number in parentheses) enrolled in the treatment trials. C) Relative change between vehicle-treated and AZD1208-treated tissue recombination graft volumes at harvest. Results are shown as mean ± SD. *Student’s t test P < .05. #Mann-Whitney rank sum test significance at < .05. All statistical tests were two-sided. D) Hematoxylin and eosin (H&E)–stained sections demonstrate more severe lesion (HGPIN) in the vehicle-treated specimens compared with the 30mg/kg AZD1208-treated specimen. Corresponding sections demonstrate yellow fluorescent protein (YFP) immunofluorescence (detected with anti-GFP antibody), indicating the c-MYC/Pim1–transduced cells. E) H&E–stained sections with corresponding YFP by immunohistochemistry (brown) detected with anti-GFP antibody similarly demonstrate worse pathology (HGPIN) in the vehicle-treated specimen compared with the 45mg/kg AZD1208-treated specimen. Scale bars = 0.1mm (short bar), 0.05mm (long bar), respectively, for (D) and (E). H&E = hematoxylin and eosin stain; GFP = green fluorescent protein.
Histological analysis demonstrated more high-grade PIN (HGPIN) lesions in the vehicle-treated graft sections than AZD1208-treated samples (Figure 1, D and E). We could identify prostate glands derived from transduced cells by YFP coexpressed by the bicistronic lentiviral vector along with either c-MYC or Pim1 (5,6). In all vehicle-treated and drug-treated specimens examined, Pim1 and c-MYC colocalized with YFP (Figure 2). No glands could be identified that expressed only c-MYC without Pim1 or vice versa, making YFP a valid surrogate marker for c-MYC and Pim1 expression in this model.
Figure 2.

PIM kinase inhibitor AZD1208 effect on PIN, proliferation, and apoptosis in prostate grafts. A) Incidence of PIN foci is lower in the AZD1208-treated samples compared with vehicle-treated grafts (χ2 P = 1.65x10-7). B) PIN density measured as number of PIN foci per section is decreased in the AZD1208-treated samples compared with vehicle-treated specimens (χ2 P = 3.06x10-7). N = 88 sections were analyzed for (A) and (B). C) Smooth muscle actin (SMA) immunostaining demonstrates substantial thinning of the SMA-containing boundary around abnormal prostate gland lesions in both the vehicle-treated and AZD1208-treated specimens (arrows). D) BrdU-positive nuclei (arrows) are fewer in AZD1208-treated specimens than controls. E) Increased cleaved caspase-3–positive cells (arrows) in AZD1208-treated specimens than the vehicle-treated specimens. Yellow fluorescent protein (YFP) was used to guide analysis to c-MYC/Pim1-expressing areas in “D” and “E”. F) The percentage of BrdU-positive nuclei is lower in the AZD1208-treated (n = 11) specimens than in the vehicle-treated samples (n = 8) (P = .0016). G) The percentage of cleaved caspase-3–positive cells is greater in the AZD1208-treated (n = 11) specimens than in the vehicle-treated specimens (n = 11) (P = .01 for 30mg/kg, P = .039 for 45mg/kg). Results shown as mean ± SD. *Student’s t test P < .05. #Mann-Whitney rank sum test significance at < .05. Scale bars = 0.05mm.
PIN incidence was reduced in a dose-dependent manner in AZD1208-treated samples as determined by a blinded pathologist (Figure 2A). While 91% of 22 analyzed vehicle-treated grafts contained PIN foci, only 50% of 10 grafts and 25% of 12 grafts treated with AZD1208 at 30 and 45mg/kg contained PIN foci, respectively (P < .001) (Figure 2A). AZD1208 treatment also reduced PIN density in a dose-dependent manner (P < .001) (Figure 2B). Both vehicle-treated and drug-treated specimens had lesions with thinning of the smooth muscle actin (SMA) layer (Figure 2C). We next analyzed the YFP-positive gland regions for proliferation and apoptosis by immunostaining for BrdU and cleaved caspase-3, respectively. AZD1208-treated samples at 45mg/kg had a 46±14% (P = .016) lower BrdU index and a 326±170% (P = .039) higher apoptotic index than vehicle-treated controls (Figure 2, D-G). A statistically significant increase in apoptosis of 200±100% (P = .01) was observed in AZD1208-treated specimens at 30mg/kg (Figure 2G). These data are consistent with previous studies showing decreased tumorigenicity of prostate cancer cells after Pim1 knockdown by shRNA (21).
Pim1 can modulate apoptosis by phosphorylating Bad at serine-112 (22). Using YFP as a guide to analyze lentivirus transduced glands, we found that AZD1208 treatment decreased phospho-Bad(S112) immunostaining compared with vehicle (Figure 3, A and B). To test whether Pim inhibition modulated the activity of MYC, we assessed the effect of AZD1208 on the expression of serine-62–phosphorylated MYC and on MYC-regulated targets hexokinase II, Nedd4l, and Prdx6 (7). AZD1208 treatment decreased the expression of phospho-S62-c-MYC and Hk2, with variable effects on Nedd4L and Prdx6 (Supplementary Figure 3, available online).
Figure 3.

AZD1208 treatment effect on phospho-Bad staining in prostate tissue recombinant grafts. A) Vehicle-treated and (B) AZD1208-treated tissue recombinant grafts with adjacent sections stained by hematoxylin and eosin (H&E), yellow fluorescent protein (YFP) (anti-GFP antibody), and phospho-Bad-S112 antibody. Abnormal gland foci are indicated by arrows. Phospho-Bad(S112) expression is reduced within the abnormal gland foci in AZD1208-treated specimens compared with vehicle-treated samples, indicating that PIM kinase function is inhibited in the prostate tissue. Scale bars = 0.1mm (short bar) and 0.05mm (long bar). H&E = hematoxylin and eosin; GFP = green fluorescent protein.
AZD1208 Treatment Inhibition of Established c-MYC-Driven Prostate Cancer
In order to explore the effects of Pim inhibition on established MYC-driven prostate tumors and the effects of combination therapy on recurrence, we turned to Myc-CaP cells, established from the Hi-Myc transgenic model of prostate cancer (13). We first showed that AZD1208 inhibited growth of Myc-CaP colonies in vitro in a concentration-dependent manner (Figure 4, A and B). Next, we established allograft Myc-CaP tumors in nude mice, and after tumors were grown for two weeks (average tumor size = 207.4 ± 34.7mm3) the mice were treated with AZD1208 once daily by oral gavage for three weeks. Drug treatment was well tolerated (Supplementary Figure 1B, available online) and resulted in statistically significant inhibition of tumor growth (Figure 4C). Histologically, AZD1208-treated grafts contained more voids, consistent with the presence of increased apoptotic cells (186±94%, P = .011) (Figure 4E) and reduction in BrdU labeling index (75±73%, P < .001) (Figure 4D). AZD1208 also suppressed the growth of prostate cancer xenografts DU145 and CWR22rv1 (Figure 4, F and G), comparable with the effects of the established cancer drug docetaxel, as well as the growth of bone metastasis–derived PC3 tumor xenografts (discussed below).
Figure 4.

AZD1208 effects on the growth and tumorigenicity of mouse Myc-CaP and human castrate-resistant DU145 and CWR22Rv1 prostate cancer tumors. A) Myc-CaP colonies are decreased by the AZD1208 in a concentration-dependent manner. B) Clonogenic assay with Myc-CaP cells (replicates n = 3 per concentration) demonstrates decreased surviving fraction for colonies reaching 1mm in diameter at AZD1208 concentrations of 300nM and higher (* P < .05). C) Treatment with AZD1208 statistically significantly decreases the growth of Myc-CaP allografts compared with vehicle-treated control (n = 10 per group, *P < .05). D-E) AZD1208-treated tumors show reduced BrdU-positive nuclei (P < .001) and increased cleaved caspase-3–positive cells (P = .011) compared with vehicle-treated control (n = 3 per group). F) Treatment with AZD1208 statistically significantly decreases the growth of human prostate cancer cell line DU145 in xenografts (n = 10 per group, *P < .01). G) AZD1208 treatment impairs tumorigenicity of human prostate cancer cell line CWR22Rv1 xenografts similar to the effects of docetaxel (n = 10 per group, *P < .05). Results shown are mean ± SD for (B-G). *Student’s t test P < .05. #Mann-Whitney rank sum test significance at < .05. All statistical tests were two-sided.
Effect of AZD1208 on Gene Expression in Prostate Tumor Cells
To obtain insights into the mechanisms by which AZD1208 inhibits prostate tumors we performed RNA-seq on Myc-CaP cells treated with vehicle or 1 µM AZD1208 overnight (In Vitro Study–acute treatment) and Myc-CaP tumors in mice treated with vehicle or 45mg/kg AZD1208 for 21 days (In Vivo Study–chronic treatment). Using DAVID functional clustering of statistically significantly altered genes (Supplementary Table 1, available online), we found enrichment of translation processes genes (peptidases, ribosomes), growth arrest genes (cell cycle, chromosomal replication), and perturbed vesicular movement (altered myosin) in AZD1208 treated samples in vivo, suggesting that PIM inhibition impairs tumorigenicity by engaging multiple cancer hallmarks.
Notably, Gene Set Enrichment Analysis revealed striking downregulation of multiple MYC target gene sets, indicating reduced MYC activity in AZD1208-treated cells both in vitro and in vivo (Table 1). PIM1 has been shown to increase MYC protein stability and MYC phosphorylation to enhance activity and to stimulate RNA polymerase II binding to increase the transcription of MYC target genes (23–27). Consistent with these earlier studies, our GSEA analyses found, using a global unbiased approach, strong evidence for downregulation of MYC activity by AZD1208 treatment.
Table 1.
MYC and p53 gene signatures downregulated by AZD1208 treatment of MycCaP cells/tumors*
| GSEA gene set name | Ref | Size | ES | NES | Nom P | FDR q-val |
|---|---|---|---|---|---|---|
| MYC signatures downregulated in vitro 1 µM AZD1208 overnight | ||||||
| ODONNELL_TARGETS_OF_MYC_AND_TFRC_DN | (35) | 35 | -0.79 | -2.14 | 0 | 0 |
| YU_MYC_TARGETS_UP | (36) | 36 | -0.77 | -2.07 | 0 | 1.17E-04 |
| SCHUHMACHER_MYC_TARGETS_UP | (37) | 70 | -0.63 | -1.94 | 0 | 9.52E-04 |
| SCHLOSSER_MYC_TARGETS_REPRESSED_BY_ SERUM | (38) | 125 | -0.56 | -1.88 | 0 | 0.002 |
| DANG_REGULATED_BY_MYC_UP | (39) | 58 | -0.58 | -1.77 | 0 | 0.011 |
| DANG_MYC_TARGETS_UP | (39) | 123 | -0.5 | -1.69 | 0 | 0.024 |
| SCHLOSSER_MYC_TARGETS_AND_SERUM_ RESPONSE_DN | (38) | 44 | -0.59 | -1.67 | .003 | 0.029 |
| MORI_EMU_MYC_LYMPHOMA_BY_ONSET_ TIME_UP | (40) | 83 | -0.53 | -1.67 | 0 | 0.029 |
| MYC signatures downregulated in vivo 45mg/kg AZD1208 21 d | ||||||
| YU_MYC_TARGETS_UP | (36) | 36 | -0.68 | -1.77 | .001 | 0.01 |
| ODONNELL_TARGETS_OF_MYC_AND_TFRC_DN | (35) | 35 | -0.65 | -1.7 | .001 | 0.024 |
| p53 signatures downregulated in vitro 1 µM AZD1208 overnight | ||||||
| WU_APOPTOSIS_BY_CDKN1A_VIA_TP53 | (41) | 45 | -0.69 | -1.97 | 0 | 5.10E-04 |
| REACTOME_P53_INDEPENDENT_G1_S_DNA_DAMAGE_CHECKPOINT | 48 | -0.63 | -1.81 | 0 | 0.007 | |
| SCIAN_CELL_CYCLE_TARGETS_OF_TP53_AND_TP73_DN | (42) | 22 | -0.73 | -1.78 | 0 | 0.01 |
| REACTOME_P53_DEPENDENT_G1_DNA_ DAMAGE_RESPONSE | 52 | -0.61 | -1.76 | 0 | 0.012 | |
| TANG_SENESCENCE_TP53_TARGETS_DN | (43) | 49 | -0.59 | -1.71 | .004 | 0.02 |
| p53 signatures downregulated in vivo 45mg/kg AZD1208 21 d | ||||||
| TANG_SENESCENCE_TP53_TARGETS_DN | (43) | 49 | -0.74 | -2.06 | 0 | 0 |
| WU_APOPTOSIS_BY_CDKN1A_VIA_TP53 | (41) | 45 | -0.75 | -2.02 | 0 | 0 |
| SCIAN_CELL_CYCLE_TARGETS_OF_TP53_ AND_TP73_DN | (42) | 22 | -0.81 | -1.97 | 0 | 1.05E-04 |
* Shown are gene sets with false discovery rate q-val under 0.05 in Gene Set Enrichment (GSEA) analysis. ES = enrichment score; FDR = false discovery rate; NES = normalized enrichment score; Nom P = nominal P value for enrichment; Ref = reference for gene set; Size = size of the gene set.
We also found evidence of downregulation of p53 transcriptional activity by AZD1208 treatment in vitro and in vivo (Table 1). A link between Pim1 and p53 has been previously suggested by cell culture studies indicating that Pim1 overexpression leads to the stabilization of p53 protein (28). Pim inhibition can therefore be expected to suppress the p53 pathway by reducing p53 stability. Our results indicate that while AZD1208 treatment activates several anticancer mechanisms, such as reduced proliferation, enhanced apoptosis, and reduced MYC activity, it can also suppress the p53 pathway. One implication of this observation is that therapeutic approaches that counteract AZD1208-induced p53 pathway suppression might enhance the drug’s efficacy.
Effect of Radiation and Hypoxia on Pim1 Expression in Prostate Cancer Cells
Pim1 expression can be induced by stresses such as hypoxia and radiation in different cell systems as a cell survival mechanism (29,30). We reasoned that the efficacy of Pim inhibition might be increased in tumor cells exposed to radiation or hypoxia, which induce Pim1 as a prosurvival mechanism. Additionally, activation of the p53 pathway by these stressors may overcome the p53 pathway suppression observed upon AZD1208 treatment. To test these hypotheses, we subjected Myc-CaP cells to radiation and/or hypoxia. Both the 44 and 33kDa isoforms of Pim1 were increased by exposure to radiation+hypoxia (Figure 5A). Clonogenic assays using Myc-CaP cells treated with radiation and AZD1208 at 100nM indicate that AZD1208 functions as a radiation sensitizer (Figure 5B). Myc-CaP tumors subjected to radiation alone or radiation+hypoxia also showed higher Pim1 expression detected by immunohistochemistry, particularly in the nuclei (Figure 5C).
Figure 5.
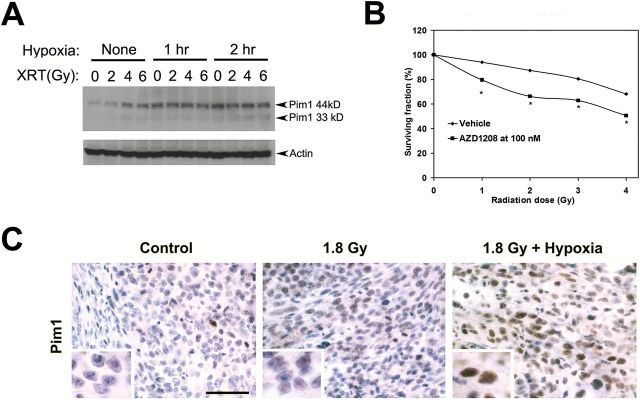
AZD1208 effects on sensitivity of prostate cancer cells to radiation. A) Immunoblots demonstrate induction of Pim1 (44kD and 33 kD isoforms) by radiation and hypoxia in Myc-CaP cells. B) Clonogenic assay demonstrates that AZD1208 acts as a radiation sensitizer by decreasing the surviving fraction when compared with radiation with vehicle (replicates n = 4 per radiation dose). *Student’s t test P < .05. All statistical tests were two-sided. C) Increased Pim1 expression in Myc-CaP tumors treated with radiation and hypoxia. Scale bar = 0.05mm.
Effect of PIM Inhibition and Radiation Treatment on CRPC
Having demonstrated that the PIM inhibitor AZD1208 is effective at sensitizing Myc-CaP cells to radiation, we sought to determine if the sensitizing effects of AZD1208 are also manifested in CRPC in vivo. Myc-CaP tumors were allowed to grow in mice to an average size of 500mm3. Castration of the mice resulted in tumor regression in all cases over six to nine days. Following a recovery period, the grafts regrew in the androgen-deprived mice. At this point, mice were treated with AZD1208 alone, radiation alone, or a combination of AZD1208 and radiation (Figure 6A). AZD1208 at 45mg/kg was given daily for three weeks. After five doses of AZD1208, three once-daily 2 Gy radiation treatments were delivered. As shown in Figure 6B, treatment with either AZD1208 or radiation alone slowed the growth of recurrent tumors, and combination treatment with AZD1208+radiation resulted in more sustained inhibition of tumor growth. The mean graft growth rates for each treatment group were determined for the last eight days of drug treatment, after completing radiation treatment. An exponential fit to each curve revealed the following growth rates: 11% per day for vehicle treatment, 8.4% per day for radiation treatment, 10% per day for AZD1208 treatment, and 5% per day for combined radiation and AZD1208 treatment (Figure 6C).
Figure 6.

AZD1208 and radiation treatment of castrate-resistant recurrent prostate cancer. A) Schematic of Myc-CaP allograft experiment and (B) growth curves of Myc-CaP grafts that were treated with androgen deprivation (surgical castration) and followed over time. Tumors regressed, then started regrowing, after which mice were treated daily with Pim kinase inhibitor AZD1208 at 45mg/kg or vehicle. Additionally, some mice received three once-daily treatments of 2 Gy of radiation (XRT). AZD1208-only and radiation-only treated tumors responded initially, but started growing back. AZD plus radiation-treated tumors show sustained inhibition of growth (n = 6–8 per group). Results shown as mean ± SD. *Student’s t test P < .05. #Mann-Whitney rank sum test significance at < .05. All statistical tests were two-sided. C) Last 8 days of tumor volume measurements with daily growth percentage determined by the exponent coefficient for an exponential curve fit (R2 is the goodness of fit).
MYC and AR Expression in Regressed Myc-CaP Tumors Postcastration
The MYC transgene in Myc-CaP cells is driven by the androgen-regulated PB4 promoter (12). We therefore sought to determine the dynamic pattern of MYC and AR expression in Myc-CaP tumors before castration, in regressed tumors postcastration, and in recurrent tumors postcastration. In control uncastrated mice treated with vehicle or AZD1208, we observed robust nuclear MYC expression (Supplementary Figure 4A, available online). Residual regressed tumors postcastration showed a heterogeneous pattern of MYC, while recurrent tumors regardless of whether they also received AZD1208 or radiation treatment expressed nuclear MYC in a homogenous pattern (Supplementary Figure 4A). AR expression was nuclear and homogenous in intact mice with or without AZD1208 treatment (Supplementary Figure 4B, available online). In regressed tumors, AR expression was heterogeneous and present in both the nuclei and cytoplasm. AR expression becomes more homogeneous in recurrent tumors postcastration with primarily cytoplasmic AR expression in AZD1208-treated tumors postcastration (Supplementary Figure 4B, available online). These results suggest the presence of tumor cells that retain MYC and AR expression in regressed tumors postcastration. These cells may be the origin of recurrent tumors.
Gene Expression Analysis in Cells Derived From AZD1208-Resistant Tumors
The fact that postcastration recurrent Myc-CaP tumors subjected to AZD1208 treatment were able to regrow after a lag period prompted us to examine possible mechanisms of this resistance. To this end, we established a panel of six cell lines, each from independent recurrent tumors, three from vehicle-treated (CxVehicle) and three from AZD1208-treated (CxAZD) surgically castrated animals. Analysis of in vitro growth rates showed no consistent differences between the cells derived from vehicle-treated and AZD1208-treated tumors (data not shown). We next used Affymetrix Mouse Gene 2.0 ST Arrays to profile gene expression changes in the cell lines. Using GSEA, we identified several pathways statistically significantly enriched in CxAZD cell lines compared with CxVehicle cells (Figure 7, A and B). Remarkably, the top enriched gene set is p53_DN.V2_UP, representing genes upregulated in HEK293 cells upon P53 knockdown by RNAi (31). These results suggest that CxAZD cells have lower p53 pathway activity, consistent with our RNA-seq profiling studies of Myc-CaP cells and tumors (Table 1). Another gene set enriched is JAK2_DN.V1_UP, consisting of genes upregulated in human erythroleukemia cells after knockdown of JAK2 by RNAi. PIM kinases have been linked to the Jak/Stat pathway (32). However, Jak/Stat pathway activity was unchanged in CxAZD cells as measured by p-Stat3 levels (Figure 7C). We confirmed reduced p53 protein levels in CxAZD cell lines by immunoblotting (Figure 7C), and intact/postcastration AZD1208-treated tumors expressed reduced levels of p53 protein by immunohistochemistry (Figure 7D). On the other hand, AZD1208-treated tumors exposed to radiation expressed high levels of p53. Thus, reduced p53 expression may represent a mechanism of resistance to AZD1208 by tumors that can be overcome by radiation treatment, which induces p53. This may provide one possible (but not the only) basis for the cooperativity we observed between AZD1208 treatment and radiation in inhibiting Myc-CaP prostate tumor growth. Indeed, treatment of the p53-null human PC3 prostate cancer cell line xenografts with AZD1208 and/or radiation showed that the combination treatment was still more effective in suppressing tumor growth than either single treatment (Supplementary Figure 5, A and B).
Figure 7.

GSEA analysis of gene expression changes in AZD1208-resistant castration-resistant prostate cancer (PC) cells. A) The list of the top 10 gene sets enriched in tumor cell lines established from recurrent, castrated, AZD1208-treated mice (CxAZD) relative to cell lines established from castrated, recurrent, vehicle-treated recurrent tumors (CxVehicle). B) Enrichment analysis plots showing enrichment of genes upregulated in P53 knockdown cells (left panel) and JAK2 knockdown cells (right panel) in CxAZD tumor cell lines. C) Immunoblots showing reduced p53 protein levels in AZD1208-treated castrated Myc-CaP allograft cell lines, but unchanged p-Stat3 and Stat3 levels. D) Reduced p53 expression in AZD1208-treated Myc-CaP tumors by immunohistochemical staining both before and after castration (upper four panels), while tumors exposed to radiation treatment (XRT) or radiation plus AZD1208 (XRT/AZD) expressed high levels of p53 (bottom two panels). Scale bar = 0.05mm E) Gene expression profile of AZD1208-resistant recurrent tumor cells is associated with human prostate cancer recurrence. Cross-species Gene Set Enrichment Analysis analysis shows association of gene expression signature from AZD1208-treated recurrent tumors with biochemical recurrence in prostate cancer patients from (44).
Finally, we sought to determine if genes dysregulated in postcastration AZD1208-resistant recurrent tumors are associated with clinical outcome in human prostate cancer. Using cross-species GSEA analysis, we found that genes upregulated in AZD1208-resistant tumors are statistically significantly enriched in the prostate tumors of patients who experienced biochemical recurrence (Figure 7E). Conversely, genes downregulated in AZD1208-resistant tumors are statistically significantly enriched in patients without recurrence.
Discussion
In this report, we demonstrate the potential for PIM kinase inhibition by the small molecule inhibitor AZD1208 in treating multiple models of mouse and human prostate cancer. AZD1208 appeared to exert its antitumor effects by engaging multiple anticancer pathways. It promoted apoptosis and inhibited proliferation, translation, and vesicular transport. Remarkably, our RNA-seq and microarray studies indicate that AZD1208 treatment leads to suppression of the MYC pathway in both in vitro studies with acutely treated cells as well as in vivo chronically treated tumors.
Pim1 has been intimately linked to MYC in tumorigenesis. Our group has shown dramatic cooperativity between MYC and Pim1, in a kinase-dependent manner, and in mouse (33) and human prostate cancer (23). Several studies suggest that Pim1 may cooperate with MYC by amplifying MYC activity. Pim1 could oppose MYC-induced apoptosis, stabilize MYC protein, and enhance MYC transcriptional activity (21,23,33). Furthermore, Pim1 decreases the activity of protein phosphatase 2A, which may mediate the levels of MYC and the phosphorylation of proteins needed for increased protein synthesis, thereby enhancing the tumor growth of prostate cancer cells (24). Our global gene profiling studies point to PIM kinase promotion of MYC transcriptional activity as a major target of AZD1208.
While Pim inhibition by AZD1208 treatment promoted many antitumor processes, it also suppressed the p53 tumor suppressor pathway. In cell culture models, Pim1 overexpression has been shown to stabilize p53 (28). Suppression of the p53 pathway by AZD1208 and the observation that Pim1 is induced by radiation and hypoxia to promote cancer cell survival provide potent rationales for combining AZD1208 and radiation therapy. Radiation therapy is commonly used in the treatment of both localized and metastatic prostate cancer. However, prostate tumors have hypoxic regions that often make them more resistant to radiation treatment and predict clinical outcome (34). Our results in Myc-CaP castration-resistant tumors and in PC3 xenografts indicate that AZD1208 cooperates with radiation in the treatment of prostate cancer.
Myc-CaP tumors are sensitive to androgen deprivation, but can develop castration resistance in a relatively rapid and reliable manner (13). Since the MYC oncogene is the initiating oncogene in Myc-CaP cells and is under the control of the androgen-sensitive PB4 promoter, we determined whether MYC expression is retained in the regressed tumors in castrated mice. Analyses of regressed tumors indicate that both AR and MYC are expressed in a heterogeneous manner. Thus, some cells in the regressed tumor maintain AR and transgenic MYC expression in the face of castration levels of androgens and may be responsible for driving tumor recurrence. When tumors recurred after castration, MYC and AR were expressed in a homogenous manner in tumor cells. Treatment of recurrent tumor with either radiation or AZD1208 slowed tumor progression but tumors started regrowing after a lag period with decreased growth rates (76% and 90% of the control, respectively). However, combining AZD1208 with radiation treatment statistically significantly impaired tumor recurrence, with a supra-additive effect (growth rate 45% of the control). Our genomic profiling studies of tumor cell lines isolated from postcastration recurrent tumors with and without AZD1208 treatment identified suppression of the p53 pathway in drug-treated tumors. It is likely that our observation of reduced p53 levels in AZD1208-treated cells and tumors is because of the inhibition of p53 stabilization by Pim1. The reduced levels of p53 in AZD1208-treated cells may be involved in the incomplete response to drug treatment we observed. Since radiation therapy was able to strongly induce p53 expression in AZD1208-treated tumors, this may account for the more effective antitumor effect of combining AZD1208 with radiation. Furthermore, the gene signature of AZD1208-resistant tumors is associated with biochemical recurrence in prostate cancer patients. These results are encouraging for the treatment of castrate-resistant prostate cancer with Pim kinase inhibitors and suggest that radiation treatment may be more effective with concurrent Pim kinase inhibition.
One limitation of this study is that the generalizability of PIM inhibitor effects on human prostate cancer is not known. Although five models of prostate cancer were investigated, primary patient-derived prostate cancer tissue was not examined. Nevertheless, this work is promising for the potential application of PIM inhibitors in patients. Pan-PIM kinase inhibitors are currently being tested in Phase 1 clinical trials. The data presented in this article support a therapeutic role for Pim kinase inhibition in the context of MYC overexpression or in combination with radiation treatment.
Funding
Part of this work was supported by an internal grant from the Department of Radiation Oncology at Vanderbilt University Medical Center to ANK. This work was supported by grants R01CA123484 and R01CA167966 from the National Cancer Institute (NCI) to SAA and by AstraZeneca.
Supplementary Material
We thank Dr. Michael Freeman at Vanderbilt University Medical Center for assistance with radiation-related experimental design and use of the irradiation equipment. We thank members of the Abdulkadir lab, especially Meejeon Roh, Irina Doubinskaia, Erin Martinez, and Monica Logan for their helpful discussions and assistance with methods, resources, and technical assistance. We thank Shenghua Wen, Zhong-Ying Liu, and Marat Alimzhanov for technical assistance with the tumor xenograft models. We thank the American Board of Radiology Holman Research Pathway for support of protected research time to ANK. The study sponsor, NCI, had no role in the study design, collection, analysis or interpretation of data, the writing, nor decision to submit the manuscript for publication. Coauthors employed by study sponsor, AstraZeneca, collaborated with the senior and first authors in designing the study, collecting, analyzing, and interpreting data, and in the writing of the manuscript. All authors approved the submission of the manuscript for publication.
EK, MA, SG, and DH are employees of AstraZeneca Pharmaceuticals. The other authors declare no conflicts of interest.
References
- 1. Siegel R, Ma J, Zou Z, et al. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. [DOI] [PubMed] [Google Scholar]
- 2. Nawijn MC, Alendar A, Berns A. For better or for worse: the role of Pim oncogenes in tumorigenesis. Nat Rev Cancer. 2011;11(1):23–34. [DOI] [PubMed] [Google Scholar]
- 3. Aho TL, Sandholm J, Peltola KJ, et al. Pim-1 kinase promotes inactivation of the pro-apoptotic Bad protein by phosphorylating it on the Ser112 gatekeeper site. FEBS Lett. 2004;571(1–3):43–49. [DOI] [PubMed] [Google Scholar]
- 4. Al-Ubaidi FL, Schultz N, Egevad L, et al. Castration therapy of prostate cancer results in downregulation of HIF-1alpha levels. Int J Radiat Oncol Biol Phys. 2012;82(3):1243–1248. [DOI] [PubMed] [Google Scholar]
- 5. Hittelman WN, Liao Y, Wang L, et al. Are cancer stem cells radioresistant? Future Oncol. 2010;6(10):1563–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aggarwal P, Vaites LP, Kim JK, et al. Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase. Cancer Cell. 2010;18(4):329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Anderson PD, McKissic SA, Logan M, et al. Nkx3.1 and Myc crossregulate shared target genes in mouse and human prostate tumorigenesis. J Clin Invest. 2012;122(5):1907–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Magnuson NS, Wang Z, Ding G, et al. Why target PIM1 for cancer diagnosis and treatment? Future Oncol. 2010;6(9):1461–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Keeton E, McEachern K, Alimzhanov M, et al. Efficacy and biomarker modulation by AZD1208, a novel, potent and selective pan-Pim kinase inhibitor, in models of acute myeloid leukemia. In: In: Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research. Chicago, IL, 2012: Abstract 72, p. 2796. Philadelphia (PA): AACR, Cancer Res. 2012;72(8 Suppl):Abstract nr 2796. [Google Scholar]
- 10. Keeton E, Palakurthi S, Alimzhanov M, et al. AZD1208, a Novel, Potent and Selective Pan PIM Kinase Inhibitor, Demonstrates Efficacy in Models of Acute Myeloid Leukemia. ASH Annual Meeting Abstracts. 2011;118(21):1540. [Google Scholar]
- 11. Keeton EK, McEachern K, Dillman KS, et al. AZD1208, a potent and selective pan-Pim kinase inhibitor, demonstrates efficacy in preclinical models of acute myeloid leukemia. Blood. 2014;123(6):905–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ellwood-Yen K, Graeber TG, Wongvipat J, et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell. 2003;4(3):223–238. [DOI] [PubMed] [Google Scholar]
- 13. Watson PA, Ellwood-Yen K, King JC, et al. Context-dependent hormone-refractory progression revealed through characterization of a novel murine prostate cancer cell line. Cancer Res. 2005;65(24):11565–11571. [DOI] [PubMed] [Google Scholar]
- 14. Hagtvet E, Roe K, Olsen DR. Liposomal doxorubicin improves radiotherapy response in hypoxic prostate cancer xenografts. Radiat Oncol. 2011;6:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim J, Eltoum IE, Roh M, et al. Interactions between cells with distinct mutations in c-MYC and Pten in prostate cancer. PLoS Genet. 2009;5(7):e1000542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. R Development Core Team. R: A Language and Environment for Statistical Computing. In. Vienna, Austria: the R Foundation for Statistical Computing; ISBN: 3-900051-07-0. Available at: http://www.R-project.org; 2011. [Google Scholar]
- 17. TIBCO Software. In. Somerville, MA; http://spotfire.tibco.com. [Google Scholar]
- 18. Warnes G, Bolker B, Lumley T. gplots: Various R programming tools for plotting data. R package version 2.6.0. http://CRAN.R-project.org/package=gplots. [Google Scholar]
- 19. Gaujoux R, Seoighe C. A flexible R package for nonnegative matrix factorization. BMC Bioinformatics. 2010;11:367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wickham H. Reshaping data with the reshape package. J Stat Software. 2007;20(12):1–20. [Google Scholar]
- 21. Wang J, Anderson PD, Luo W, et al. Pim1 kinase is required to maintain tumorigenicity in MYC-expressing prostate cancer cells. Oncogene. 2012;31(14):1794–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Macdonald A, Campbell DG, Toth R, et al. Pim kinases phosphorylate multiple sites on Bad and promote 14-3-3 binding and dissociation from Bcl-XL. BMC Cell Biol. 2006;7:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim J, Roh M, Abdulkadir SA. Pim1 promotes human prostate cancer cell tumorigenicity and c-MYC transcriptional activity. BMC Cancer. 2010;10:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen WW, Chan DC, Donald C, et al. Pim family kinases enhance tumor growth of prostate cancer cells. Mol Cancer Res. 2005;3(8):443–451. [DOI] [PubMed] [Google Scholar]
- 25. Zhang Y, Wang Z, Li X, et al. Pim kinase-dependent inhibition of c-Myc degradation. Oncogene. 2008;27(35):4809–4819. [DOI] [PubMed] [Google Scholar]
- 26. Zippo A, De Robertis A, Serafini R, et al. PIM1-dependent phosphorylation of histone H3 at serine 10 is required for MYC-dependent transcriptional activation and oncogenic transformation. Nat Cell Biol. 2007;9(8):932–944. [DOI] [PubMed] [Google Scholar]
- 27. Holder SL, Abdulkadir SA. PIM1 Kinase as a Target in Prostate Cancer: Roles in Tumorigenesis, Castration Resistance, and Docetaxel Resistance. Curr Cancer Drug Targets. 2013;14(2):105–114. [DOI] [PubMed] [Google Scholar]
- 28. Hogan C, Hutchison C, Marcar L, et al. Elevated levels of oncogenic protein kinase Pim-1 induce the p53 pathway in cultured cells and correlate with increased Mdm2 in mantle cell lymphoma. J Biol Chem. 2008;283(26):18012–18023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen J, Kobayashi M, Darmanin S, et al. Hypoxia-mediated up-regulation of Pim-1 contributes to solid tumor formation. Am J Pathol. 2009;175(1):400–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim W, Youn H, Seong KM, et al. PIM1-activated PRAS40 regulates radioresistance in non-small cell lung cancer cells through interplay with FOXO3a, 14-3-3 and protein phosphatases. Radiat Res. 2011;176(5):539–552. [DOI] [PubMed] [Google Scholar]
- 31. Elkon R, Rashi-Elkeles S, Lerenthal Y, et al. Dissection of a DNA-damage-induced transcriptional network using a combination of microarrays, RNA interference and computational promoter analysis. Genome Biol. 2005;6(5):R43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen XP, Losman JA, Cowan S, et al. Pim serine/threonine kinases regulate the stability of Socs-1 protein. Proc Natl Acad Sci U S A. 2002;99(4):2175–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang J, Kim J, Roh M, et al. Pim1 kinase synergizes with c-MYC to induce advanced prostate carcinoma. Oncogene. 2010;29(17):2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Turaka A, Buyyounouski MK, Hanlon AL, et al. Hypoxic prostate/muscle PO2 ratio predicts for outcome in patients with localized prostate cancer: long-term results. Int J Radiat Oncol Biol Phys. 2012;82(3):e433–e439. [DOI] [PubMed] [Google Scholar]
- 35. O’Donnell KA, Yu D, Zeller KI, et al. Activation of transferrin receptor 1 by c-Myc enhances cellular proliferation and tumorigenesis. Mol Cell Biol. 2006;26(6):2373–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yu D, Cozma D, Park A, et al. Functional validation of genes implicated in lymphomagenesis: an in vivo selection assay using a Myc-induced B-cell tumor. Ann N Y Acad Sci. 2005;1059:145–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schuhmacher M, Kohlhuber F, Holzel M, et al. The transcriptional program of a human B cell line in response to Myc. Nucleic Acids Res. 2001;29(2):397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schlosser I, Holzel M, Hoffmann R, et al. Dissection of transcriptional programmes in response to serum and c-Myc in a human B-cell line. Oncogene. 2005;24(3):520–524. [DOI] [PubMed] [Google Scholar]
- 39. Zeller KI, Jegga AG, Aronow BJ, et al. An integrated database of genes responsive to the Myc oncogenic transcription factor: identification of direct genomic targets. Genome Biol. 2003;4(10):R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mori S, Rempel RE, Chang JT, et al. Utilization of pathway signatures to reveal distinct types of B lymphoma in the Emicro-myc model and human diffuse large B-cell lymphoma. Cancer Res. 2008;68(20):8525–8534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wu Q, Kirschmeier P, Hockenberry T, et al. Transcriptional regulation during p21WAF1/CIP1-induced apoptosis in human ovarian cancer cells. J Biol Chem. 2002;277(39):36329–36337. [DOI] [PubMed] [Google Scholar]
- 42. Scian MJ, Carchman EH, Mohanraj L, et al. Wild-type p53 and p73 negatively regulate expression of proliferation related genes. Oncogene. 2008;27(18):2583–2593. [DOI] [PubMed] [Google Scholar]
- 43. Tang X, Milyavsky M, Goldfinger N, et al. Amyloid-beta precursor-like protein APLP1 is a novel p53 transcriptional target gene that augments neuroblastoma cell death upon genotoxic stress. Oncogene. 2007;26(52):7302–7312. [DOI] [PubMed] [Google Scholar]
- 44. Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1): 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.