

Abstract
Mitochondria contribute to macrophage immune function through the generation of reactive oxygen species, a byproduct of the mitochondrial respiratory chain. MCJ (also known as DnaJC15) is a mitochondrial inner membrane protein identified as an endogenous inhibitor of respiratory chain complex I. Here we show that MCJ is essential for the production of tumor necrosis factor by macrophages in response to a variety of Toll-like receptor ligands and bacteria, without affecting their phagocytic activity. Loss of MCJ in macrophages results in increased mitochondrial respiration and elevated basal levels of reactive oxygen species that cause activation of the JNK/c-Jun pathway, lead to the upregulation of the TACE (also known as ADAM17) inhibitor TIMP-3, and lead to the inhibition of tumor necrosis factor shedding from the plasma membrane. Consequently, MCJ-deficient mice are resistant to the development of fulminant liver injury upon lipopolysaccharide administration. Thus, attenuation of the mitochondrial respiratory chain by MCJ in macrophages exquisitely regulates the response of macrophages to infectious insults.
Keywords: MCJ, reactive oxygen species, TNF shedding, TACE activity, TIMP-3, macrophage
Macrophages play critical roles in the response to infection. They become activated in response to a variety of stimuli and in turn amplify and maintain the inflammatory response [1], and they are critical for the clearance of microorganisms through phagocytosis [2].
Mitochondria are essential engines of the cells, serving as a source of energy through adenosine triphosphate synthesis by oxidative phosphorylation and the generation of reactive oxygen species (ROS), that influence the immune response to pathogens [3–10]. Thus, in response to lipopolysaccharide (LPS), mitochondria are recruited to phagosomes and contribute to antibacterial ROS production [11]. Stimulation with LPS also induces the downregulation of the mitochondrial uncoupling protein, UCP2, in macrophages, increasing ROS production [5, 12]. Consequently, mitochondrial defects result in poor antimicrobial responses. Thus, the partial knockdown of GRIM-19, a component of complex I of mitochondria, results in a higher rate of infection in mice and an imbalanced cytokine response to microorganisms [4].
MCJ (methylation-controlled J protein; also known as DnaJC15 is a mitochondrial protein that belongs to the DnaJ C family of co-chaperones [13–16]. MCJ is a small-sized protein with a transmembrane domain targeted to the inner membrane of mitochondria [13, 15]. We have recently identified the ortholog of human MCJ in mice and shown that is highly expressed in heart, liver and kidney. Within the immune system, MCJ is predominantly expressed in CD8+ cells [17]; however, its expression in the myeloid lineage has not been reported. MCJ associates with and represses the function of complex I of the mitochondrial electron transport chain [13], making it the first defined endogenous inhibitor of complex I.
Herein, we show that MCJ is present in mitochondria in macrophages and is essential for the production of tumor necrosis factor (TNF). The absence of MCJ in macrophages results in increased mitochondrial respiration and, consequently, elevated levels of ROS and ROS-mediated activation of the JNK/c-Jun pathway. This increased activity results in the upregulation of the specific TNF-α–converting enzyme (TACE; also known as ADAM17) inhibitor, TIMP-3, which effectively prevents the shedding of TNF from the plasma membrane. Thus, the absence of MCJ results in decreased production of soluble TNF and, importantly, reduced liver damage upon injection with LPS/D-galactosamine (GalN). MCJ is therefore a critical regulator of mitochondrial homeostasis in macrophages during their response to bacterial infection.
METHODS
Mice
MCJ-deficient mice on a C57Bl/6 (B6) background [13] and wild-type B6 mice were bred at the University of Massachusetts–Amherst and CIC bioGUNE. The institutional animal care and use committees at the University of Massachusetts–Amherst and CIC bioGUNE approved all procedures involving animals.
ShMCJ Plasmid Construction
The pCAGGS vector (Addgene, Cambridge, MA) was used to clone the target sequence against mouse MCJ (Table 1) under the influence of the H1 promoter.
Table 1.
Primers and Oligonucleotides Used in This Study
| Gene | Sequence | Purpose/Sequence Type |
|---|---|---|
| Actb | 5′-GAC GAT GCT CCC CGG GCT GTA TTC-3′ | RT-PCR |
| 5′-TCT CTT GCT CTG GGC CTC GTC ACC-3′ | Real-time qPCR | |
| Dnajc15 | 5′-ACG CCG ACA TCG ACC ACA CAG-3′ | RT-PCR |
| 5′-AAT CTT CCT TGC TGT TGC CGT G-3′ | ||
| Tnf | 5′-AGC CCA CGT CGT AGC AAA CCA C-3′ | Real-time qPCR |
| 5′-ATC GGC TGG CAC CAC TAG TTG GT-3′ | ||
| Adam17 | 5′-TGG GAC ACA ATT TTG GAG CA-3′ | Real-time qPCR |
| 5′-CCT CCT TGG TCC TCA TTT GG-3′ | ||
| Timp-3 | 5′-GGC CTC AAT TAC CGC TAC CA-3′ | Real-time qPCR |
| 5′-CTG ATA GCC AGG GTA CCC AAA A-3′ | ||
| Dnajc15 | 5′-GAT CCC C G CGA GAG GCT AGT CTT ATT T TCA AGA GA AAT AAG ACT AGC CTC TCG C TTT TTG GAA a-3′ | siRNA |
Abbreviations: qPCR, quantitative polymerase chain reaction; RT-PCR, reverse-transcription polymerase chain reaction; siRNA, small interfering RNA.
Cells
Bone marrow–derived macrophages (BMMs) were generated as described elsewhere [18], using recombinant macrophage colony-stimulating factor. RAW264.7 cells and their derivatives were maintained in Dulbecco's modified Eagle's medium (DMEM; Life Technologies) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin.
Splenic CD8+ T cells, CD4+ T cells, and CD11b+ cells were purified by positive selection, using biotinylated anti-CD8, anti-CD4, and anti-CD11b (BD Biosciences, San Diego, CA); anti-biotin microbeads (Miltenyi Biotec, Auburn, CA); and the MACS system (Miltenyi Biotec). Cells were transfected using X-tremeGENE DNA Transfection Reagent (Roche Diagnostics, Barcelona, Spain) and selected in medium containing 400 µg/mL of G418 (Amresco, Solon, OH) or 3.5–4 µg/mL of puromycin (Sigma Chemical, St. Louis, MO).
Bacteria
Borrelia burgdorferi 297 and 914 [19] were cultured in BSK-H medium (Sigma Chemical). Staphylococcus aureus ATCC13709 (ATCC, Manassas, VA) was cultured in Bacto Heart Infusion broth (BD Biosciences).
In Vitro Stimulation
Cells (106 cells/mL) were stimulated with Porphyromonas gingivalis LPS (1 µg/mL), Escherichia coli LPS (100 ng/mL), S. aureus lipoteichoic acid (LTA; 500 ng/mL), poly I:C (1 μg/mL; Invivogen, San Diego, CA), S. aureus (multiplicity of infection [MOI], 10), and B. burgdorferi strain 297 (MOI, 25). Where noted, cells were pretreated for 1 hour with FCCP, rotenone, N-acetyl cysteine (NAC; Sigma Chemical), or SP600125 (Tocris Bioscience, Bristol, United Kingdom).
Enzyme-Linked Immunosorbent Assay (ELISA)
Serum and cell culture supernatants were collected and assayed for cytokines by ELISA for mouse TNF (BD Bioscience and R&D Systems, Minneapolis, MN).
Reverse-Transcription Polymerase Chain Reaction (PCR) and Real-Time Quantitative PCR (qPCR)
Total RNA was extracted from cells with Trizol (Life Technologies) and was reverse transcribed using Superscript III (Life Technologies). The expression of mouse genes was assessed by real-time qPCR, using SYBR Green PCR Master Mix (Life Technologies) on an ABI Prism 7000 Sequence Detection System thermocycler (Life Technologies). Fold-changes in levels of gene expression were normalized to expression of the gene encoding actin and were quantified by the change-in-threshold method (ΔΔCT). The primers used are listed in Table 1.
Immunoblotting
Cells were lysed in lysis buffer (50 mM Tris, pH 7, 150 mM NaCl, 0.5% sodium deoxycholate, and 1% Triton) containing a protease inhibitor cocktail (Sigma Chemical). For subcellular fractionation experiments, cells (5 × 106) were resuspended in extraction buffer (270 mM sucrose, 26 mM MOPS, pH 6.6, 1 mM EGTA) containing protease inhibitors and disrupted with a Dounce homogenizer. The cleared suspension was subjected to 16 000 × g for 20 minutes at 4°C. The resulting pellet (mitochondrial fraction) and the subsequent 100 000 × g supernatant (cytosolic fraction) were analyzed by immunoblot.
For gradient sucrose fractionation experiments, cells were resuspended in 200 µL of extraction buffer and disrupted by passing through a 26-gauge needle. The precleared supernatant was laid on top of a discontinuous sucrose gradient (120 µL 54% sucrose, 320 µL 40% sucrose, 250 µL 33% sucrose, 250 µL 24% sucrose, and 175 µL 15% sucrose) and centrifuged at 100 000 × g for 3 hours at 4°C, using an SW-41 rotor (Beckman Coulter). One hundred–microliter aliquots from the top were precipitated with 10% trichloroacetic acid and washed with acetone before analyzed by immunoblotting. Inmunoblots were probed with antibodies against MCJ [13], calnexin, Na+/K+-ATPase, TACE, Actin, GAPDH, TIMP-3 (Santa Cruz Biotechnology), COX IV, c-Jun, p-c-Jun, JNK, and p-JNK (Cell Signaling).
Microscopy
Cells were seeded onto 8-chamber slides, washed, fixed in 3.7% paraformaldehyde for 10 minutes and blocked with 5% bovine serum albumin for 60 minutes. Cells were stained with anti-MCJ and mounted with Prolong Gold Anti-fade mounting reagent (Life Technologies). Photomicrographs were taken using a Zeiss Axiovert 200M inverted microscope (Thornwood, NY) equipped with an Apotome and a Hamamatsu Orca camera (Bridgewater, NJ). Some samples were analyzed with a Zeiss LSM 510 Meta Confocal System (Carl Zeiss, Thornwood, NY).
Phagocytosis Assay
BMMs (106/mL) were cultured in serum- and antibiotic-free medium with B. burgdorferi 914 at different MOIs for 6 hours, as described [20]. The cells were analyzed using an LSR II flow cytometer (BD Biosciences) and FlowJo for Mac, version 8.6 (TreeStar, Ashland, OR).
Intracellular Staining
WT and MCJ-deficient BMMs were stimulated with live B. burgdorferi for 12 hours. Brefeldin A was added during the last 5 hours of the experiment. Cells were surface stained with anti-CD11b–fluorescein isothiocyanate (BD Biosciences), followed by intracellular cytokine staining with anti-TNF-Alexa Fluor 647 (BD Biosciences) or an immunoglobulin G isotype control (eBioscience), using the BD Cytofix/Cytoperm Kit (BD Biosciences).
TACE Activity
Cells (1 × 105) were incubated with 10 µM of TACE FRET Substrate I (Anaspec, Fremont, CA) in black NUNC polystyrene 96-well microtiter plates (Fisher Scientific). Nonspecific TACE activity was determined in cells treated with the metalloproteinase inhibitor TAPI-2 (50 µM; Enzo Life Sciences, Farmingdale, NY). Enzyme activity was monitored using a BioTek Synergy HT microplate fluorescence reader (BioTek, Winooski, VT) at a λex of approximately 355 nm and a λem of approximately 500 nm. Results are expressed as specific activity resulting from subtracting nonspecific activity from total activity.
Seahorse Mitochondrial Flux Analyses
Cells were seeded at a density of 100 000 cells/well in 24-well Seahorse assay plates in DMEM without carbonate. After 1 hour at 37°C without CO2, 3 baseline oxidative consumption rate (OCR) and extracellular acidification rate (ECAR) measurements were performed in a Seahorse metabolic flux analyzer (Seahorse Biosciences, North Billerica, MA), followed by injection with rotenone (1 µM) to determine nonmitochondrial respiration. The values obtained were normalized for the amount of cellular protein.
Detection of ROS
Cells (5 × 105) were incubated with 5 µM CellROX Deep Red Reagent (Life Technologies) for 30 minutes at 37°C, washed, and analyzed by flow cytometry.
Infection with S. aureus
Groups of 6–8-week-old MCJ-deficient mice and wild-type littermates were infected by intramuscular injection of 1 × 105 S. aureus cells (suspended in 0.1 mL of Hank's balanced salt solution [HBSS]) in the right posterior thigh [21]. Mice were euthanized 25 hours after infection, and the thigh muscle at the site of infection was aseptically collected and homogenized in 250 µL of HBSS with a Kontes Pellet Pestle (Fisher Scientific). Aliquots of serial dilutions were plated on Difco Heart Infusion Agar plates (BD Biosciences) to calculate colony-forming units per gram of tissue. Sera were collected at euthanized and analyzed for TNF by ELISA.
Treatment with LPS/GalN and Analysis of Liver Damage
Groups of MCJ-deficient and WT mice were injected intraperitoneally with LPS (32 mg/kg) in GalN (800 mg/kg; Sigma Chemical) and analyzed 6 hours after treatment. Levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were determined in serum. Histological examination was performed in formalin-fixed liver sections stained with hematoxylin-eosin. Apoptotic cell death was determined on frozen liver sections by TUNEL assay using the In-Situ-Cell-Death Detection Kit (Roche Diagnostics).
Statistical Analysis
The results are presented as means ± standard errors. Significant differences between means were calculated using GraphPad Prism v 5.0 (San Diego, CA) using the Student t test. P values of ≤.05 were considered statistically significant.
RESULTS
MCJ Is a Mitochondrial Protein in Macrophages
Although macrophages are not highly metabolically active, mitochondria in these cells play an important role in regulating ROS levels [9]. We therefore sought to determine whether MCJ is expressed in macrophages. The analysis of splenic CD11b+ cells showed that MCJ is expressed in these cells (Figure 1A and 1B). Messenger RNA (mRNA) transcripts for Dnajc15 (Figure 1C) and MCJ protein (Figure 1D) were also detected in RAW cells and primary BMMs, as well as in purified human CD14+ monocytes, by real-time qPCR (data not shown).
Figure 1.

MCJ is a mitochondrial protein expressed by murine macrophages. A, Real-time quantitative polymerase chain reaction (PCR) showing the relative expression levels of Dnajc15 in splenic CD4+ T, CD8+ T, and CD11b+ cells. Expression of Dnajc15 is shown as relative to the level detected in CD4+ T cells. B, ApoTome microscopy of splenic cell populations probed with a rabbit anti-murine MCJ antibody. Blue staining indicates the nuclei of the cells. C, Expression of Dnajc15 in RAW cells and murine bone marrow–derived macrophages (BMMs), detected by PCR. The control represents a reaction in the absence of complementary DNA. D, Confocal microscopy showing expression of MCJ in both RAW cells and BMMs. E, Sucrose gradient showing cofractionation of CytC and MCJ. The fractions taken from the top are marked, as are the increasing concentrations of sucrose. The samples were probed for the membrane marker Na+, K+, ATPase and the endoplasmic reticulum marker calnexin. F, Colocalization by confocal microscopy of MCJ (red) and the mitochondrial marker p22-BID in RAW cells transfected with a construct containing p22-BID fused with green fluorescent protein (green). Colocalization of both proteins was determined with J-Image and is marked as white. The cell displayed is shown in the image taken under diffraction light. G, Total (T), mitochondrial (M), and cytosolic (C) fractions of RAW cells were obtained by differential centrifugation and tested by Western blotting for MCJ, the mitochondrial marker COX IV, and actin.
Sucrose gradient analysis of cell extracts from RAW cells revealed that MCJ colocalized in the same fraction as the mitochondrial marker CytC (Figure 1E). Confocal analysis confirmed the colocalization of MCJ with the mitochondrial marker p22 BID-GFP (Figure 1F). Furthermore, differential centrifugation of cell extracts showed the localization of MCJ within the mitochondrial fraction (Figure 1G). These data indicate that MCJ is a mitochondrial protein in macrophages.
MCJ Regulates Macrophage Responses to Infectious Agents and Microbial Components
We generated a stably transfected line of RAW cells expressing small interfering RNA (siRNA) targeting Dnajc15 (shMCJ cells) that efficiently repressed the expression of the protein (Figure 2A). We stimulated shMCJ cells and control cells with several bacterial components and analyzed TNF production by ELISA. Regardless of the stimulus, shMCJ cells produced lower levels of TNF, compared with control cells (Figure 2B).
Figure 2.
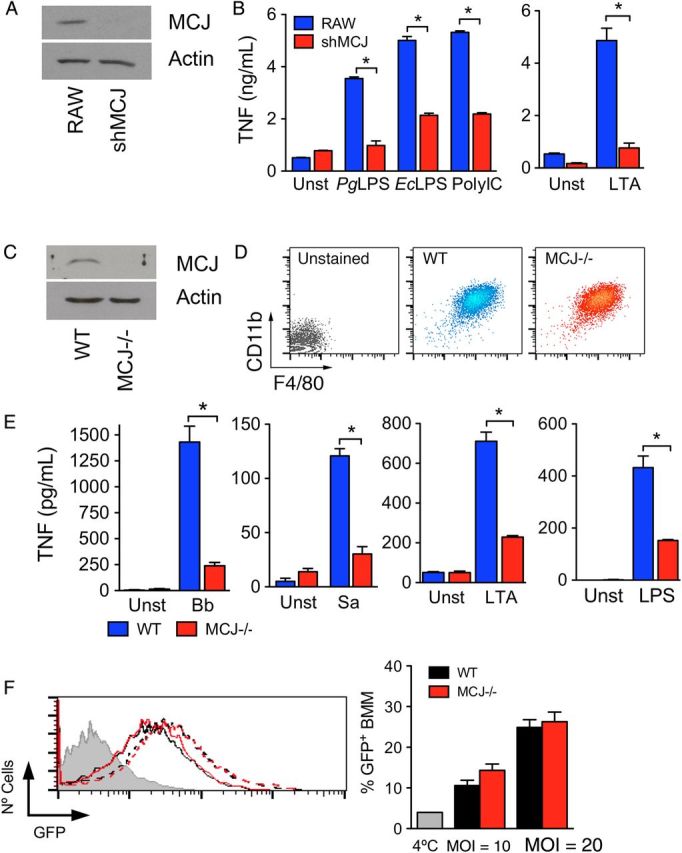
MCJ regulates tumor necrosis factor (TNF) production in response to proinflammatory stimuli. A, shMCJ cells, generated by stable transfection with a plasmid containing small interfering RNA sequences targeting MCJ, show decreased levels of the protein by Western blotting. B, TNF production of RAW (blue bars) and shMCJ (red bars) cells in response to stimulation with several Toll-like receptor (TLR) ligands: PgLPS, lipopolysaccharide (LPS) from Porphyromonas gingivalis; EcLPS, LPS from Escherichia coli; PolyIC, polyinosinic-polycytidylic acid; and LTA, lipoteichoic acid from Staphylococcus aureus. C, Analysis by immunoblotting of MCJ protein levels in bone marrow–derived macrophages (BMMs) from wild-type (WT) and MCJ knockout (KO; MCJ−/−) mice. D, Levels of expression of CD11b and F4/80 in BMMs differentiated from WT and MCJ KO mice. E, TNF production by WT (blue bars) and MCJ KO (red bars) BMMs in response to live Borrelia burgdorferi (Bb; multiplicity of infection [MOI], 25), S. aureus (Sa; MOI, 10), and the TLR ligands LTA and LPS. *P < .05. F, Phagocytosis of B. burgdorferi by WT (black lines) and MCJ-deficient (red lines) BMMs at 2 different MOIs: 10 (continuous lines) and 20 (broken lines). The gray histogram represents a 4°C control to determine binding but not internalization, as well as cell washing efficiency. The histogram on the left shows a representative experiment. The graph on the right corresponds to the percentage of green fluorescent protein (GFP)–expressing cells in triplicates. The experiments are representative of at least 3 performed in each case. Abbreviation: Unst, unstimulated.
We also generated BMMs from wild-type and MCJ knockout (KO) mice [13]. BMMs from MCJ KO mice did not express detectable levels of MCJ protein (Figure 2C). The absence of MCJ did not affect the generation of BMMs, as assessed by the number of cells differentiated (data not shown) or the expression levels of the surface markers CD11b and F4/80 (Figure 2D). We then measured TNF production in BMMs upon stimulation with bacteria and bacterial components. The levels of TNF in the culture supernatant of MCJ KO BMMs were significantly lower upon stimulation with B. burgdorferi or S. aureus, compared with wild-type macrophages (Figure 2E). Similarly, TNF production was significantly reduced in MCJ KO BMMs stimulated with LTA or LPS, compared with controls (Figure 2E). Since macrophage proinflammatory responses to B. burgdorferi depend on their ability to phagocytose the spirochete [20, 22–24], we analyzed the capacity of MCJ KO BMMs to internalize the bacterium. Phagocytosis of B. burgdorferi was comparable in MCJ KO and wild-type BMMs at 2 different MOIs (10 and 20; Figure 2F). Overall, these data indicate that MCJ regulates the response of macrophages to proinflammatory stimuli.
MCJ Regulates TACE-Mediated Secretion of TNF
To analyze whether the regulation of TNF occurs at the gene expression level, we analyzed by real-time qPCR the levels of Tnf mRNA upon stimulation with LPS or B. burgdorferi. Interestingly, the level of Tnf mRNA was similar in MCJ KO and wild-type BMMs (Figure 3A). Intracellular staining of TNF also showed similar levels in MCJ KO and control BMMs in response to B. burgdorferi stimulation (Figure 3B). Newly formed TNF is translocated to the plasma membrane, where it is released because of the action of the metalloproteinase TACE [25, 26]. The absence of MCJ did not affect the levels of Adam17 mRNA (Figure 3C) or TACE protein (Figure 3D) in either BMMs or RAW cells. However, TACE activity was significantly reduced in shMCJ cells and MCJ KO BMMs, compared with controls (Figure 3E), as well as the human monocytic cell line THP-1, transfected with a plasmid containing shRNA for MCJ (not shown), indicating a functional control of the enzyme by MCJ.
Figure 3.

MCJ regulates tumor necrosis factor (TNF) secretion through the modulation of TACE activity. A, Real-time quantitative polymerase chain reaction (qPCR) of wild-type (WT; blue bars) and MCJ knockout (KO; red bars) bone marrow–derived macrophages (BMMs) either left unstimulated (Unst) or stimulated with lipopolysaccharide (LPS) and live Borrelia burgdorferi (Bb; multiplicity of infection [MOI], 25). Relative expression levels were calculated relative to expression among unstimulated WT BMMs. B, Intracellular staining of TNF in WT and MCJ KO BMMs stimulated with live B. burgdorferi (MOI, 25) for 16 hours. The cells were stimulated in the presence of brefeldin A for the last 5 hours of the assay, prior to being stained for surface CD11b, fixed, permeabilized, and stained for intracellular TNF. C, Relative Adam17 gene expression levels in RAW, shMCJ, WT, and MCJ KO BMMs, measured by real-time qPCR. D, Immunoblots of RAW, shMCJ, WT, and MCJ KO BMMs cell extracts for detection of TACE. Equal loading was determined using an anti-GAPDH antibody. E, TACE activity in RAW and WT BMMs (blue bars) or shMCJ cells and MCJ KO BMMs (red bars). The cells were incubated with a FRET-TACE substrate and analyzed for their activity by fluorescence. The specific activity was determined in relation to nonspecific activity determined with the use of the TACE inhibitor TAPI. Abbreviation: Unst, unstimulated.
The Absence of MCJ Results in Augmented TIMP-3 Expression
TACE activity is inhibited by TIMP-3, a member of the tissue inhibitor of metalloproteinase family [27]. We measured Timp-3 mRNA levels by real-time qPCR in wild-type and MCJ KO BMMs. Cells with repressed expression of MCJ contained increased levels of Timp-3 mRNA (Figure 4A) and protein (Figure 4B), compared with controls. Since TIMP-3 expression is regulated by c-Jun [28], a member of the AP-1 transcription factor, we examined the levels of c-Jun. shMCJ cells and MCJ KO BMMs expressed increased levels of c-Jun, compared with control cells (Figure 4C). Furthermore, luciferase reporter assays for AP1-mediated transcription showed increased AP1-transcriptional activity in shMCJ cells, compared with controls (Figure 4D). Thus, loss of MCJ resulted in elevated c-Jun activity in macrophages.
Figure 4.

MCJ regulates the expression of TIMP-3. A, Real-time quantitative polymerase chain reaction of wild-type (WT; blue bar) and MCJ knockout (KO; red bar) bone marrow–derived macrophages (BMMs) showing the relative expression levels of Timp3. B, Immunoblot for TIMP-3 protein levels in RAW and shMCJ cells. C, Western blot showing c-Jun levels in RAW and shMCJ cells. Equal loading was determined with an anti GAPDH antibody. D, AP-1–mediated transcriptional activity in RAW (blue bars) and shMCJ (red bars) cells transfected with a construct containing 5 × AP-1 consensus sequences driving the expression of the luciferase gene. The cells were either left unstimulated or stimulated for 16 hours with live Borrelia burgdorferi (Bb; multiplicity of infection, 25). E, Immunoblots showing the levels of TIMP-3 and MCJ proteins in RAW, shMCJ, shTIMP-3, and shMCJ/shTIMP-3 cells. F and G, TACE activity (F) and tumor necrosis factor (TNF) induction by lipopolysaccharide (LPS; G) in RAW cells and stable transfectants with shMCJ and/or shTIMP-3. Abbreviation: Unst, unstimulated.
To determine whether the reduced TACE activity in the absence of MCJ was due to the increased expression of TIMP-3, we used an shRNA plasmid targeting the inhibitor. Transfection with shTIMP-3 resulted in reduced levels of TIMP-3 (Figure 4E). Importantly, TACE activity was significantly increased in shMCJ/shTIMP-3 cells (Figure 4F), accompanied with higher levels of TNF in the cell culture supernatants (Figure 4G). These results demonstrate that the upregulation of TIMP-3 expression is responsible for the decreased secretion of TNF by macrophages in the absence of MCJ.
Increased JNK Activity in the Absence of MCJ Results in the Accumulation of c-Jun
JNK activity regulates c-Jun protein stability and expression levels [29]. We therefore analyzed the phosphorylation status of both JNK and its substrate, c-Jun. Both JNK and c-Jun appeared hyperphosphorylated under basal conditions in shMCJ cells, relative to controls (Figure 5A). Treatment with the JNK inhibitor SP600125 resulted in a reduction in the levels of c-Jun in both RAW and shMCJ cells (Figure 5B), indicating that the increased JNK activity in the absence of MCJ results in augmented levels of c-Jun. The activation of JNK proteins can occur both through external and internal signals, most predominantly under stress conditions [30], including oxidative stress [7, 31]. The bioenergetic analysis of RAW and shMCJ cells showed that the absence of MCJ resulted in higher OCR, indicative of elevated cellular respiration (Figure 5C), while the ECAR, indicative of glycolysis, was not affected (Figure 5C). Since increased respiration could lead to augmented ROS levels, we assessed the levels of ROS in shMCJ cells, compared with controls. shMCJ cells contained significant increased levels of cellular ROS (Figure 5D), which were not reduced in the presence of the NADPH oxidase inhibitor apocynin (Figure 5E), suggesting a mitochondrial origin. The treatment of shMCJ cells with the antioxidant N-acetyl cysteine resulted in reduced levels of c-Jun (Figure 5F) and TIMP-3 (Figure 5G), indicating that the enhanced mitochondria-derived oxidative stress resulting from the repressed expression of MCJ induces increased TIMP-3 expression. Indeed, the use of the mitochondrial inhibitor rotenone resulted in decreased c-Jun levels in both shMCJ cells and MCJ KO BMMs, compared with their respective controls (Figure 5I), indicating that the increased levels could be associated with augmented mitochondrial function. Furthermore, the use of the mitochondrial uncoupler FCCP also resulted in increased production of TNF in response to B. burgdorferi by shMCJ cells to levels comparable to those obtained in RAW cells (Figure 5H). Overall, these data suggest that the control of mitochondrial activity by MCJ regulates the level and transcriptional activity of c-Jun, as well as the production of TNF in response to proinflammatory stimuli.
Figure 5.

Increased JNK activity in the absence of MCJ results in the accumulation of c-Jun. A, Western blot analysis of RAW and shMCJ cells for the levels of pJNK, total JNK, pc-Jun, and MCJ. B, RAW and shMCJ cells were treated with 2.5 µM of SP600125 for 6 hours, and the levels of c-Jun were assessed by immunoblotting. C, Seahorse analysis of RAW (blue bars) and shMCJ (red bars) cells. The cells were analyzed under basal conditions and upon addition of rotenone (Rot) for oxygen consumption rates (OCRs) and extracellular acidification rates (ECARs). D, Total reactive oxygen species (ROS) levels in RAW (black histogram) and shMCJ (red histogram) cells. The bar graph in the inset represents the average + standard error of triplicates and is representative of at least 3 independent experiments. E, ROS levels in shMCJ cells pretreated with the NADPH oxydase inhibitor, apocynin (1 µM; red histogram) compared to control-treated cells (black histogram). F and G, shMCJ cells were treated with the antioxidant N-acetyl cytosine (NAC; 20 µM). The levels of c-Jun (F) and TIMP-3 (G) were then determined by immunoblotting after 5 hours and 24 hours of treatment, respectively. H, shMCJ cells and MCJ knockout BMMs were treated with the specified concentrations of rotenone for 16 hours, and the levels of c-Jun were then determined by immunoblotting. I, RAW (blue bars) and shMCJ (red bars) cells were stimulated with live Borrelia burgdorferi (Bb; multiplicity of infection, 25) in the presence of increasing concentrations of the mitochondrial uncoupler FCCP. After 6 hours, the levels of tumor necrosis factor (TNF) were measured in the cell culture supernatants by enzyme-linked immunosorbent assay. Abbreviations: MFI, mean fluorescence intensity; NS, not significant.
MCJ KO Mice Are Resistant to Proinflammatory Insults
To assess the role of MCJ in the production of TNF in vivo, we first used an acute infection model with S. aureus [21]. Groups of wild-type and MCJ KO mice were infected with S. aureus ATCC13709 intramuscularly in the left thigh. The mice were analyzed 25 hours later for thigh bacterial burdens and sera TNF levels. The absence of MCJ resulted in significantly decreased sera levels of TNF (Figure 6A) without an appreciable effect on bacterial burdens (Figure 6B). TNF production was dependent on macrophages, since infection of macrophage-depleted mice resulted in highly decreased sera TNF levels (Figure 6C). These data demonstrated that MCJ regulates the activity of macrophages in vivo during bacterial infection.
Figure 6.

MCJ-deficient mice are resistant to bacterial product–induced inflammation. Groups of wild-type (WT; blue bars) and MCJ knockout (KO; red bars) mice were infected intramuscularly in the left thigh with 1 × 105 Staphylococcus aureus ATCC13709. After 25 hours, the sera levels of TNF were determined by enzyme-linked immunosorbent assay (A), and the bacterial burdens in the injected thigh were determined by serial dilutions (B). C, cd11b-Diphtheria toxin (DT) transgenic mice were treated with dyphtheria toxin and infected with S. aureus (Sa), as before, followed by sera TNF determination. D and F, Groups of WT and MCJ KO mice were injected with lipopolysaccharide (LPS)/D-galactosamine (GalN). After 6 hours, the levels of TNF in the sera (D), as well as levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST; F), were determined. E, Mortality rates of WT and MCJ-deficient mice treated with LPS/GalN. G and H, Hematoxylin-eosin–stained sections (G) and results of a TUNEL assay on frozen sections (H) of WT and MCJ KO mouse livers treated with LPS/GalN after 6 hours, compared with untreated controls (0 hours). Abbreviations: CFU, colony-forming units; Sa, staphylococcus aureus; UI, uninfected.
The liver can become sensitive to LPS derived from the gut microbiota. Tissue damage is mediated by LPS-induced TNF production in GalN-sensitized mice and induces fulminant hepatitis [32]. We therefore determined whether the absence of MCJ would result in decreased liver injury in response to the injection with LPS/GalN. Male MCJ KO and wild-type mice were injected with a combination of LPS and GalN and analyzed after 6–8 hours. The levels of TNF were significantly reduced in the sera of mice with absent MCJ expression (Figure 6D). Analysis of survival from LPS-mediated liver injury showed a significantly increased survival of MCJ KO mice, compared with wild-type mice (P < .001, by the log-rank Mantel-Cox test; Figure 6E). The levels of ALT and AST as markers of liver damage were significantly reduced in mice lacking MCJ expression (Figure 6F). Similarly, hematoxylin-eosin staining of liver sections demonstrated lower levels of inflammation in MCJ KO mice, compared with controls (Figure 6G). These results correlated with decreased TUNEL staining in the livers of MCJ KO mice (Figure 6H). Together, these data demonstrate that the absence of MCJ results in decreased liver injury in response to an endotoxin insult.
DISCUSSION
In this report, we uncover a mechanism that regulates proinflammatory cytokine production in macrophages mediated by MCJ, a negative regulator of complex I activity [13]. We show that the loss of MCJ causes a prominent reduction in the production of TNF by macrophages in response to different stimuli. However, MCJ does not affect Tnf gene expression, but affects TACE activity and TNF shedding. While regulation of an enzymatic activity at the cell surface by a mitochondrial protein seems paradoxical, we show that this effect is mediated through upregulation of the expression of the TACE-inhibitor Timp-3 as a result of increased mitochondria respiration, increased ROS production, and activation of the JNK/c-Jun pathway. These data show a novel mechanism by which mitochondrial respiratory chain activity regulation in macrophages can have a major impact in the production of proinflammatory cytokines in response to infectious agents, through the generation of ROS.
An acute increase in ROS is known to have a proinflammatory effect in phagocytic cells and induces the initiation of proinflammatory signaling cascades [33]. However, our results show that the augmented levels of ROS in the absence of MCJ under basal conditions could also have an antiinflammatory effect (ie, reduced production of TNF) through the induction of the JNK/c-Jun pathway and upregulation of molecules such as TIMP-3. These results suggest that sustained increased basal levels of ROS may have a regulatory effect on the overall response to infectious agents and microbial products, resulting in a state of tolerance that prevents the persistent induction of a strong proinflammatory response.
The relevance of the function of MCJ in vivo is substantiated with a model of infection-induced hepatic pathology induced by the injection of LPS/GalN. In this model, hepatic damage and cell death is induced by the stimulation of hepatic macrophages with LPS through the production of TNF [32]. The model mimics endotoxin inclusion into the liver by intestinal bacterial products that reach through the portal vein and that can be exacerbated under increased leakiness conditions. These results underscore the importance of a strict metabolic control of macrophages under normal and pathological conditions.
Overall, these data show that the control of mitochondrial ETC function profoundly affects the proinflammatory response mediated by macrophages. Our data also situate MCJ as a target of potential therapeutic intervention during pathological conditions in which macrophages are involved. The only confirmed mechanism by which the expression of MCJ is regulated involves hypermethylation of CpG islands at the promoter and first exon of the gene in cancer cells, which has been demonstrated in several cancer cell types [14, 16, 17, 34]. It is currently unknown whether MCJ expression is regulated under physiological or infection-induced conditions, as are the mechanisms by which this may occur. Further knowledge and control of MCJ expression could lead to specific therapies against exaggerated proinflammatory production in response to infectious agents, while maintaining the capacity of these cells to eliminate microorganisms by phagocytosis.
Notes
Acknowledgments. We thank Arkaitz Carracedo, for critically reading the manuscript, and Justin Radolf, for providing B. burgdorferi 914.
Financial support. This work was supported by the National Institutes of Health (grants AI-078277 [to J. A.], AT-1576 and CA-172086 [to M. L. M.-C.], and CA-127099 [to M. R]), the Spanish Ministry of Economy Plan Nacional (grant SAF-2012-34610 to JA), the Basque government (grant ETORTEK-2011 to M. L. M.-C.), Educación Gobierno Vasco 2011 (to M. L. M.-C.), and Instituto de salud Carlos III (grant FIS PI11/01588 to M. L. M.-C.).
Potential conflicts of interest. All authors: No potential conflicts of interest.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Ma J, Chen T, Mandelin J, et al. Regulation of macrophage activation. Cell Mol Life Sci. 2003;60:2334–46. doi: 10.1007/s00018-003-3020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- 3.Brune B, Dehne N, Grossmann N, et al. Redox control of inflammation in macrophages. Antioxid Redox Signal. 2013;19:595–637. doi: 10.1089/ars.2012.4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Y, Lu H, Liu Q, et al. Function of GRIM-19, a mitochondrial respiratory chain complex I protein, in innate immunity. J Biol Chem. 2012;287:27227–35. doi: 10.1074/jbc.M112.340315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Emre Y, Hurtaud C, Nubel T, Criscuolo F, Ricquier D, Cassard-Doulcier AM. Mitochondria contribute to LPS-induced MAPK activation via uncoupling protein UCP2 in macrophages. Biochem J. 2007;402:271–8. doi: 10.1042/BJ20061430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jabaut J, Ather JL, Taracanova A, Poynter ME, Ckless K. Mitochondria-targeted drugs enhance Nlrp3 inflammasome-dependent IL-1beta secretion in association with alterations in cellular redox and energy status. Free Radic Biol Med. 2013;60:233–45. doi: 10.1016/j.freeradbiomed.2013.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jang HJ, Kim SJ. Taurine exerts anti-osteoclastogenesis activity via inhibiting ROS generation, JNK phosphorylation and COX-2 expression in RAW264.7 cells. J Recept Signal Transduct Res. 2013;33:387–91. doi: 10.3109/10799893.2013.839999. [DOI] [PubMed] [Google Scholar]
- 8.Spooner R, Yilmaz O. The role of reactive-oxygen-species in microbial persistence and inflammation. Int J Mol Sci. 2011;12:334–52. doi: 10.3390/ijms12010334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. 2011;11:389–402. doi: 10.1038/nri2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seki E, Brenner DA, Karin M. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology. 2012;143:307–20. doi: 10.1053/j.gastro.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.West AP, Brodsky IE, Rahner C, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472:476–80. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kizaki T, Suzuki K, Hitomi Y, et al. Uncoupling protein 2 plays an important role in nitric oxide production of lipopolysaccharide-stimulated macrophages. Proc Natl Acad Sci U S A. 2002;99:9392–7. doi: 10.1073/pnas.142206299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hatle KM, Gummadidala P, Navasa N, et al. MCJ/DnaJC15, an endogenous mitochondrial repressor of the respiratory chain that controls metabolic alterations. Mol Cell Biol. 2013;33:2302–14. doi: 10.1128/MCB.00189-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindsey JC, Lusher ME, Strathdee G, et al. Epigenetic inactivation of MCJ (DNAJD1) in malignant paediatric brain tumours. Int J Cancer. 2006;118:346–52. doi: 10.1002/ijc.21353. [DOI] [PubMed] [Google Scholar]
- 15.Schusdziarra C, Blamowska M, Azem A, Hell K. Methylation-controlled J-protein MCJ acts in the import of proteins into human mitochondria. Hum Mol Genet. 2013;22:1348–57. doi: 10.1093/hmg/dds541. [DOI] [PubMed] [Google Scholar]
- 16.Shridhar V, Bible KC, Staub J, et al. Loss of expression of a new member of the DNAJ protein family confers resistance to chemotherapeutic agents used in the treatment of ovarian cancer. Cancer Res. 2001;61:4258–65. [PubMed] [Google Scholar]
- 17.Hatle KM, Neveu W, Dienz O, et al. Methylation-controlled J protein promotes c-Jun degradation to prevent ABCB1 transporter expression. Mol Cell Biol. 2007;27:2952–66. doi: 10.1128/MCB.01804-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olson C, Bates T, Izadi H, et al. Local production of IFN-gamma by invariant NKT cells modulates acute lyme carditis. J Immunol. 2009;182:3728–34. doi: 10.4049/jimmunol.0804111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dunham-Ems SM, Caimano MJ, Pal U, et al. Live imaging reveals a biphasic mode of dissemination of Borrelia burgdorferi within ticks. J Clin Invest. 2009;119:3652–65. doi: 10.1172/JCI39401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hawley KL, Olson CM, Jr., Iglesias-Pedraz JM, et al. CD14 cooperates with complement receptor 3 to mediate MyD88-independent phagocytosis of Borrelia burgdorferi. Proc Natl Acad Sci U S A. 2012;109:1228–32. doi: 10.1073/pnas.1112078109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Som A, Navasa N, Percher A, Scott RW, Tew GN, Anguita J. Identification of synthetic host defense peptide mimics that exert dual antimicrobial and anti-inflammatory activities. Clin Vaccine Immunol. 2012;19:1784–91. doi: 10.1128/CVI.00291-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moore MW, Cruz AR, Lavake CJ, et al. Phagocytosis of Borrelia burgdorferi and Treponema pallidum potentiates innate immune activation and induces IFN-{gamma} production. Infect Immun. 2007;75:2046–62. doi: 10.1128/IAI.01666-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cervantes JL, Dunham-Ems SM, La Vake CJ, et al. Phagosomal signaling by Borrelia burgdorferi in human monocytes involves Toll-like receptor (TLR) 2 and TLR8 cooperativity and TLR8-mediated induction of IFN-{beta} Proc Natl Acad Sci U S A. 2011;108:3683–8. doi: 10.1073/pnas.1013776108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petzke MM, Brooks A, Krupna MA, Mordue D, Schwartz I. Recognition of Borrelia burgdorferi, the Lyme disease spirochete, by TLR7 and TLR9 induces a type I IFN response by human immune cells. J Immunol. 2009;183:5279–92. doi: 10.4049/jimmunol.0901390. [DOI] [PubMed] [Google Scholar]
- 25.Black RA, Rauch CT, Kozlosky CJ, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–33. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 26.Moss ML, Jin SL, Milla ME, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. 1997;385:733–6. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- 27.Amour A, Slocombe PM, Webster A, et al. TNF-alpha converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett. 1998;435:39–44. doi: 10.1016/s0014-5793(98)01031-x. [DOI] [PubMed] [Google Scholar]
- 28.Guinea-Viniegra J, Zenz R, Scheuch H, et al. TNF-alpha shedding and epidermal inflammation are controlled by Jun proteins. Genes Dev. 2009;23:2663–74. doi: 10.1101/gad.543109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fuchs SY, Dolan L, Davis RJ, Ronai Z. Phosphorylation-dependent targeting of c-Jun ubiquitination by Jun N-kinase. Oncogene. 1996;13:1531–5. [PubMed] [Google Scholar]
- 30.Dong C, Davis RJ, Flavell RA. MAP kinases in the immune response. Annu Rev Immunol. 2002;20:55–72. doi: 10.1146/annurev.immunol.20.091301.131133. [DOI] [PubMed] [Google Scholar]
- 31.Tattoli I, Carneiro LA, Jehanno M, et al. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-kappaB and JNK pathways by inducing reactive oxygen species production. EMBO Rep. 2008;9:293–300. doi: 10.1038/sj.embor.7401161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mignon A, Rouquet N, Fabre M, et al. LPS challenge in D-galactosamine-sensitized mice accounts for caspase-dependent fulminant hepatitis, not for septic shock. Am J Respir Crit Care Med. 1999;159:1308–15. doi: 10.1164/ajrccm.159.4.9712012. [DOI] [PubMed] [Google Scholar]
- 33.Naik E, Dixit VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med. 2011;208:417–20. doi: 10.1084/jem.20110367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muthusamy V, Duraisamy S, Bradbury CM, et al. Epigenetic silencing of novel tumor suppressors in malignant melanoma. Cancer Res. 2006;66:11187–93. doi: 10.1158/0008-5472.CAN-06-1274. [DOI] [PubMed] [Google Scholar]