

Abstract
Congenital left-sided lesions (LSLs) are serious, heritable malformations of the heart. However, little is known about the genetic causes of LSLs. This study was undertaken to identify common variants acting through the genotype of the affected individual (i.e. case) or the mother (e.g. via an in utero effect) that influence the risk of LSLs. A genome-wide association study (GWAS) was performed using data from 377 LSL case-parent triads, with follow-up studies in an independent sample of 224 triads and analysis of the combined data. Associations with both the case and maternal genotypes were assessed using log-linear analyses under an additive model. An association between LSLs and the case genotype for one intergenic SNP on chromosome 16 achieved genome-wide significance in the combined data (rs8061121, combined P = 4.0 × 10−9; relative risk to heterozygote: 2.6, 95% CI: 1.9–3.7). In the combined data, there was also suggestive evidence of association between LSLs and the case genotype for a variant in the synaptoporin gene (rs1975649, combined P = 3.4 × 10−7; relative risk to heterozygote: 1.6, 95% CI: 1.4–2.0) and between LSLs and the maternal genotype for an intergenic SNP on chromosome 10 (rs11008222, combined P = 6.3 × 10−7; relative risk to heterozygote: 1.6, 95% CI: 1.4–2.0). This is the first GWAS of LSLs to evaluate associations with both the case and maternal genotypes. The results of this study identify three candidate LSL susceptibility loci, including one that appears to be associated with the risk of LSLs via the maternal genotype.
INTRODUCTION
Congenital heart defects are the most common, serious type of birth defect (1–3) and are the leading cause of birth defect-related infant mortality (4). In addition, as survival of affected infants has improved, congenital heart defects have increasingly become an adult healthcare issue (5, 6). In general, congenital heart defects are thought to be complex conditions that are influenced by the genotype of the developing embryo as well as in utero exposures, which may be determined or influenced by the maternal genotype [e.g. (7–10)]. However, since congenital heart defects include a wide phenotypic spectrum, studies of potential risk factors are generally focused on phenotypic subsets for which there is evidence of a shared etiology (11).
Congenital left-sided lesions (LSLs), which include hypoplastic left heart syndrome, aortic valve stenosis, coarctation of the aorta, mitral valve anomalies and bicuspid aortic valve are a common and often serious subgroup of congenital heart defects. Although a growing body of evidence suggests that rare de novo mutations and copy number variants contribute to the burden of congenital heart defects, including LSLs [e.g. (12–16)], family studies demonstrate that LSLs are highly heritable (17). Hence, inherited genetic variation must also contribute to the risk of LSLs, and both linkage analyses (18, 19) and candidate-gene association studies (20) have identified LSL-related genes and regions. However, little is known about the specific genetic causes of LSLs, or the extent to which the maternal genotype may influence the development of these conditions (e.g. via an effect on the in utero environment).
To identify novel susceptibility loci that are associated with LSLs, we conducted a case-parent triad genome-wide association study (GWAS) with follow-up of top associations in an independent sample of LSL case-parent triads and, analysis of the combined data. Because the risk of congenital malformations in offspring may be influenced by the maternal genotype [e.g. (10, 21–24)] as well as the genotype of the child and because failure to account for maternal genetic effects can bias estimates of association with the child's genotype (25), our study design and analytical strategy were selected to allow for the evaluation of associations between LSLs and both the genotypes of affected individuals (i.e. cases) and their mothers.
RESULTS
Stage 1 association analysis
After making exclusions based on standard quality control metrics, data were available for 486,840 genotyped SNPs with a minor allele frequency (MAF) >5% in 923 individuals, representing 377 triads (Stage 1). The demographic and clinical characteristics of the cases used in Stage 1 are summarized in Table 1. The genomic inflation factor was 1.06 for associations with the case genotype and 1.00 for associations with the maternal genotype and quartile–quartile (Q–Q) plots provided no evidence of a systematic deviation from the expected distribution for the test statistics (see Supplementary material online, Figures S1A and B, last accessed on 25 August 2014). Since tests involving the case genotype using triad data are not subject to bias due to population stratification (26), no attempt was made to reduce the genomic inflation factor (e.g. by adjusting for principal components) for the associations with the case genotype. Log-linear analyses of these data identified five case genotypes and two maternal genotypes with association P < 10−5 (see Supplementary material online, Figures S2A and B and Table S1, last accessed on 25 August 2014).
Table 1.
Characteristics of cases with left-sided lesions
| Stage 1 | Stage 2 | |
|---|---|---|
| n (%) | n (%) | |
| Race/ethnicity | ||
| Non-Hispanic Caucasian | 284 (75.3) | 216 (96.4) |
| Other | 93 (24.7) | 8 (3.6) |
| Sex | ||
| Male | 240 (63.7) | 151 (67.4) |
| Female | 137 (36.3) | 72 (32.1) |
| Lesion | ||
| Hypoplastic left heart syndrome | 198 (52.5) | 64 (28.6) |
| Coarctation of the aorta | 111 (29.4) | 93 (41.5) |
| Aortic stenosis | 68 (18.0) | 65 (29.0) |
| Mitral valve anomalies | 0 (0.0) | 2 (0.9) |
| Total | 377 (100.0) | 224 (100.0) |
Using the subset of non-Hispanic Caucasian triads, data for non-genotyped autosomal variants were imputed. After making exclusions based on standard quality control metrics, data were available for 1,712,604 imputed SNPs with a MAF >5% in 716 individuals, representing 284 non-Hispanic Caucasian triads. The genomic inflation factor for genotyped and imputed SNPs was 1.01 for associations with the case genotype and 1.00 for associations with the maternal genotype and Q–Q plots provided no evidence of a systematic deviation from the expected distribution for the test statistics (see Supplementary material online, Figures S1C and D, last accessed on 25 August 2014). Log-linear analyses of the genotyped and imputed variants in this subset of triads identified an additional 23 case and 47 maternal genotypes with association P < 10−5 (see Supplementary material online, Figures S2C and D and Table S1, last accessed on 25 August 2014). As P < 10−5 was used to identify suggestive associations in both the analyses of genotyped SNPs only, and analyses of genotyped plus imputed SNPs, it is not surprising that more suggestive associations were identified in the latter than in the former.
Stage 2 association analysis
We genotyped 77 SNPs with association P-values <10−5 (for associations of LSLs with either the case or maternal genotype) in an additional 224, predominantly non-Hispanic Caucasian, LSL triads (Stage 2). The demographic and clinical characteristics of the cases used in Stage 2 are summarized in Table 1. Log-linear analyses of the variants that passed the quality control metrics identified two case and one maternal genotypes that were associated with LSLs with unadjusted P < 0.05. In the combined data, the P-values for the association between LSLs and these three genotypes were smaller than the corresponding P-values in Stage 1 and the case genotype for an intergenic variant on chromosome 16 achieved genome-wide significance (rs8061121, P = 4.0 × 10−9; relative risk to heterozygote: 2.7, 95% CI: 1.9–3.7) (Table 2). The estimated sibling relative risk (λS) attributable to this variant was 1.20. There was also suggestive evidence, in the combined data, of an association between LSLs and the case genotype for a SNP in the synaptoporin (SYNPR) gene (rs1975649, P = 3.4 × 10−7; relative risk to heterozygote: 1.7, 95% CI: 1.4–2.0, λS = 1.06) and the maternal genotype for an intergenic SNP on chromosome 10 (rs11008222, P = 6.3 × 10−7; relative risk to heterozygote: 1.6, 95% CI: 1.3–2.0) (Table 2).
Table 2.
Summary data for variants rs1975649, rs11008222 and rs8061121 in left-sided lesions in Stages 1 and 2
| SNP (genotyped/imputed in Stage 1) | Chra | MAFb | Positionc (bp) | Gened | Function |
P-values |
Genotype relative riske (95% confidence interval) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Stage 1 | Stage 2 | Stage 1 + 2 | Stage 1 | Stage 2 | Stage 1 + 2 | ||||||
| Inherited effects | |||||||||||
| rs8061121 (imputed)f | 16 | 0.11 | 85750585 | (AC136285.1) | Intergenic | 2.44 × 10−7 | 0.005 | 4.04 × 10−9 | 2.94 (1.91–4.54) | 2.16 (1.24–3.76) | 2.65 (1.89–3.71) |
| rs1975649 (genotyped) | 3 | 0.33 | 63413926 | SYNPR | Intron | 1.08 × 10−6 | 0.03 | 3.38 × 10−7 | 1.87 (1.45–2.43) | 1.38 (1.03–1.87) | 1.65 (1.36–2.01) |
| Maternal effects | |||||||||||
| rs11008222 (genotyped) | 10 | 0.49 | 31082750 | (ZNF438) | Intergenic | 9.60 × 10−6 | 0.02 | 6.27 × 10−7 | 1.68 (1.32–2.13) | 1.55 (1.06–2.26) | 1.64 (1.34–2.00) |
aChromosome.
bMAF among study participant founders (i.e. mother and father).
cHg18/NCBI build 36.
dFor SNPs mapping within genes, gene names are listed, and for intergenic SNPs, the nearest gene is listed in parentheses.
eRelative risk estimate for carrying one copy of the high-risk allele compared to no copies, and corresponding 95% confidence interval.
fP-value for this variant in Stage 1 is based on the analyses of the subgroup of non-Hispanic Caucasian triads.
In Stage 1, genotypes for two of the top SNPs (rs1975649 and rs1100822) were derived from the array data (see Supplementary material online, Figures S3A and B for cluster plots, last accessed on 25 August 2014) and genotypes for the third SNP (rs8061121) were based on imputation. In the samples from Stage 1 that were successfully genotyped in Stage 2, genotype concordance was ≥97% (concordance for rs8061121, which was imputed in Stage 1, was 100%). Further, regional association plots demonstrate that SNPs in linkage disequilibrium with these three variants (i.e. rs8061121, rs1975649 and rs11008222) were also nominally associated (P < 0.05) with LSLs in Stage 1 (Fig. 1A–C). In particular, the maternal genotype for several SNPs in linkage disequilibrium with rs11008222 had association P-values between 0.001 and 0.0001. Although no other SNPs were strongly associated with LSLs in the regions surrounding rs8061121 and rs1975649 (Figs 1A and C), there were also no genotyped or imputed SNPs in strong linkage disequilibrium with these variants. Further, as no proxies for rs8061121 and rs1975649 were identified using SNAP (http://www.broadinstitute.org/mpg/snap/ldsearch.php, default settings), imputation of a denser SNP panel would be unlikely to provide additional evidence either for or against an association in these regions.
Figure 1.
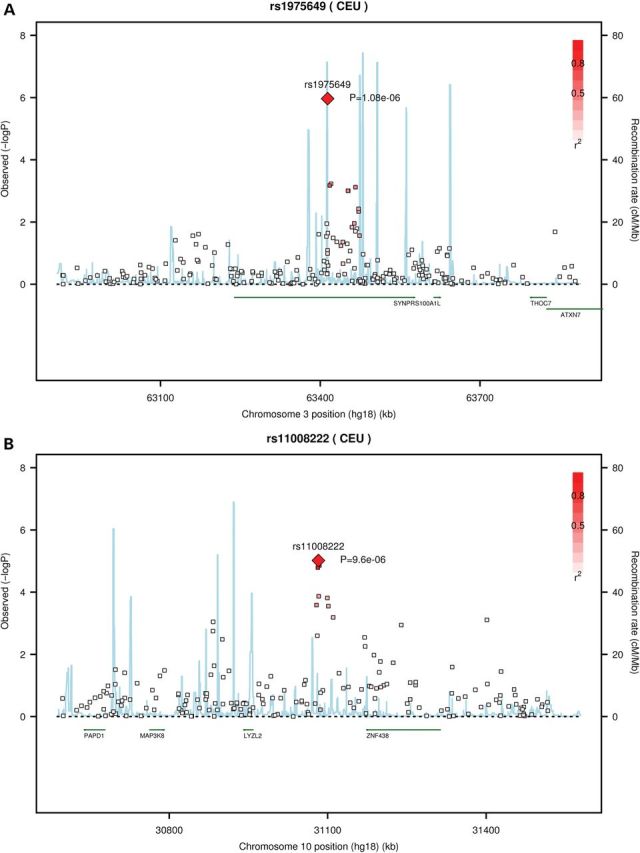

Regional association plots for three loci with genome-wide significant or suggestive associations with LSLs. Each pane: (A) rs1975649 (B) rs11008222 (C) rs8061121, shows the association statistic (−log10 P for the likelihood ratio test) from Stage 1 for the variant (diamond) and nearby markers (squares) on the left y-axis. The red shading indicates linkage disequilibrium (r2) between the variant and nearby markers. The light blue lines indicate recombination rates across each region in 1000 Genomes Pilot 1 CEU data (right y-axis). Chromosome position (hg18) and nearby genes are shown on the x-axis.
Pathway analysis
Using data for both genotyped and imputed SNPs in the Stage 1, Caucasian subset, we assessed enrichment of Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) terms for regions containing SNPs with association P-values <10−5 using INRICH (27). Analyses were performed separately for inherited and maternal genotypes and were repeated using regions containing SNPs with association P-values <10−4. No pathways with a corrected P-value < 0.05 were identified.
Annotation
The suggestive association with the maternal genotype involved a variant at 10p11.23 (rs11008222) that is located in the intergenic region between a predicted gene (AK302694) and coding gene ZNF438 (∼91 kb downstream), a C2H2-type zinc finger gene that is likely a transcriptional repressor (28). The predicted gene, AK302694, is highly similar to the F-actin-binding protein Supervillin and is likely to be transcribed and translated into a protein (txCdsPredict score, 1874). Upstream, the nearest known coding gene is LYZL2 (MIM 612748) (∼124 kb), which encodes lysozyme-like 2. LYZL2 is expressed in the placenta and may play a role in host defense against infection (29). There are also clusters of transcription factor-binding sites ∼600 bp upstream (binding sites for GATA-1, GATA-2, GABP, CCNT2, RNA Pol II and TAL1) and 600 bp downstream (binding sites for EBF and EBF1) of rs11008222 and areas of open chromatin at ∼11, 30 and 64 kb downstream of this variant. The area of open chromatin at 30 kb coincides with a cluster of transcription factor binding sites.
The GWAVA scores for rs11008222 range from 0.14 to 0.30, and the GWAVA-TSS score (0.30) falls at approximately the median value for GWAS SNPs that have been externally replicated. Among the SNPs in linkage disequilibrium (r2 > 0.80) with rs11008222, the highest GWAVA scores were obtained for rs10826861 (range: 0.37–0.43), which had a P-value = 0.0005 in the Stage 1 analysis in the Caucasian subset. The GWAVA-TSS score (0.43) for this variant falls within the third quartile of scores for GWAS SNPs that have been externally replicated. The relatively high scores for this variant appear to be driven by evolutionary conservation at this site (GERP score: 4.09). Expression quantitative trait analyses provided no evidence that rs11008222 is associated with gene expression.
The suggestive association with the inherited genotype involved a variant at 3p14.2 (rs1975649) that is located in intron 2 of the synaptoporin (SYNPR) gene. SYNPR is a synaptic membrane protein of synaptic vesicles and a member of the synaptophysin gene family that is involved in the regulation of neurotransmitters. In adults, SYNPR appears to be expressed in the brain (30) and the atrium (31). The GWAVA scores for this variant ranged from 0.15 to 0.18, and the GWAVA-TSS score (0.15) falls below the median value for GWAS SNPs that have been externally replicated. Expression quantitative trait analyses provided no evidence that rs1975649 is associated with gene expression.
The genome-wide significant association with the inherited genotype involved a variant at 16q24.2 (rs8061121) that is located within the predicted gene AK125749 (txCdsPredict score, 148). The nearest upstream genes (∼578 kb), FOXC2 (forkhead box protein C2, MIM:602402) and FOXL1 (forkhead box protein L1, MIM:603252), are members of the forkhead family of transcription factors that are involved in cardiac development (32–34). Deletion of these genes has been associated with multiple congenital malformations including hypoplastic left heart syndrome (35). The closest downstream gene (∼143 kb), C16orf95, has unknown function, but deletion of this gene, as part of a de novo copy number variant, has been reported in a patient with tetralogy of Fallot (12). This variant (rs8061121) is located within 50 bp of a predicted binding site for NKX2.5, a transcription factor critical to cardiac development and congenital heart disease (36, 37). Further, a cluster of transcription factor binding sites (FOS, GATA2, JunB, JunD and NR4A1) is located ∼14 kb upstream, and a FOS binding site is located ∼15 kb downstream of rs8061121. Binding sites for the transcriptional repressor, CTCF, are also located ∼14 kb upstream and 15 kb downstream of rs8061121. There are also several regions of open chromatin between rs8061121 and FOXL1 as well as downstream of this variant. Finally, a cluster of miRNA targets (miR-138/138ab, miR-455–5p, miR-338/338–3p, miR-9/9ab, miR-133abc, miR-26ab/1297/4465 and miR-143/1721/4770) is predicted to occur ∼18 kb upstream of rs8061121. The GWAVA scores for this variant ranged from 0.07 to 0.31, and the GWAVA-TSS score (0.08) falls below the median value for GWAS SNPs that have been externally replicated. Expression quantitative trait analyses were not performed for rs8061121 because this variant was not identified in the Geneva GenCord database.
DISCUSSION
Congenital heart defects occur in ∼1 in 200 live births and are associated with significant infant mortality and morbidity (1–4). LSLs are among the most common, severe congenital heart defects, accounting for ∼13% of all affected infants. Family studies, which demonstrate that the various LSL subtypes (e.g. hypoplastic left heart, coarctation of the aorta, aortic valve stenosis) co-segregate within families, indicate that the LSL subtypes are genetically related and highly heritable (17). These studies, in conjunction with emerging evidence that de novo mutations (13) and rare copy number variants (12, 14–16) are associated with LSLs, suggest that both inherited variation and de novo events contribute to the risk of LSLs. Further, epidemiological studies have identified associations between LSLs and a range of exposures [e.g.(38–41)], many of which may be determined (e.g. maternal obesity) or modulated (e.g. exposure to medications) by the genotype of the mother, suggesting that the maternal genotype may also influence the risk of LSLs in offspring.
To explore the potential contribution of both inherited case and maternal genotypes to LSLs, a case-parent triad GWAS was undertaken. Using log-linear models, one genome-wide significant association (rs8061121) and two suggestive associations (rs1975649 and rs11008222) were identified, one of which was associated with LSLs via the maternal genotype (rs11008222). When we began this study, there were no reported GWAS of congenital heart defects. While three such studies have subsequently been published, none of them evaluated associations between the maternal genotype and LSLs. Further, two of these studies focused on heart defects other than LSLs [i.e. tetralogy of Fallot (42) and septal defects (43)]. The third study included a broad range of congenital heart defects (44), but conducted a subgroup analysis of LSLs (∼400 cases). However, no genome-wide significant (P < 5 × 10−8) associations were identified in the LSL subgroup. Our data provided no evidence of association between LSLs and any of the top hits in these three studies (P ≥ 0.29). Further, in the GWAS of tetralogy of Fallot (the only one of the three published GWAS to provide access to summary data from all SNPs), only one of our top hits was evaluated (rs1975649), and there was no evidence for an association between this variant and TOF (P = 0.38). Given the etiologic complexity and heterogeneity of heart defects, it is not entirely surprising that there is no overlap between the genes/regions identified in these distinct studies.
The results of this study should be interpreted in light of its limitations, including the relatively small sample sizes available for both Stages 1 and 2. However, as little is known about the specific genes that influence the risk of LSL, the identification of three loci with at least suggestive evidence of association with LSLs provides new insights regarding the potential genetic underpinnings of this condition. Further, our finding of a suggestive association with the maternal genotype highlights the importance of considering maternal genetic effects in studies of LSLs and other conditions that have their origin in utero. Nonetheless, given that genome-wide significance (i.e. P < 10−8) was attained for only one variant and only in the combined data, these findings require follow-up in additional cohorts.
This study also had several strengths, including well-phenotyped cases ascertained from a single centre. In addition, the family-based approach is robust to the potentially biasing influences of population stratification and provides unbiased assessment of associations with both the case and maternal genotypes.
In summary, our findings, as well as those from other recent genomic studies of LSLs, highlight the potential utility of genome-wide approaches for LSLs, despite the relatively small sample sizes that are generally available for this condition. Future studies should seek to confirm the associations reported here. Larger studies, with increased power to detect associations with LSLs, are also warranted and should include consideration of both the case and maternal genotypes.
MATERIALS AND METHODS
Human subjects
Cases with an LSL and their parents were recruited from the Cardiac Center at The Children's Hospital of Philadelphia with approval from the Institutional Review Board for the Protection of Human Subjects. Written informed consent and, when appropriate, assent was obtained for each study subject.
Cases used in Stage 1 (n = 405 LSL case-parent triads) were recruited between 1997 and 2007, and those used in Stage 2 (n = 236 triads), was recruited between 1997 and 2010. Cases in both stages included males and females with an LSL including: hypoplastic left heart syndrome, coarctation of the aorta with or without bicuspid aortic valve, aortic valve stenosis and isolated mitral valve anomalies. A detailed cardiac phenotype was ascertained from all available cardiac medical records, including echocardiography, cardiac MRI, cardiac catheterization and/or operative notes. If there was any discrepancy or ambiguity in the diagnosis, then original images were inspected by one reviewer (E.G.). Individuals with variants of hypoplastic left heart syndrome, such as mal-aligned atrioventricular canal defects and double outlet right ventricle with mitral valve atresia, were excluded from the study. Medical records were also reviewed to identify other medical conditions, and cases with a recognized or suspected genetic syndrome were excluded from the study. Cases in Stage 1 included individuals of all races and ethnicities, whereas cases in Stage 2 were selected to be predominantly non-Hispanic Caucasian.
GWAS genotyping and quality control
Pre-transfusion blood samples were collected from cases at the time of admission to the hospital and blood or saliva samples were collected from available parents. DNA was extracted from the study samples using standard methods (Puregene DNA isolation kit by Gentra Systems, Inc., Minneapolis, MN, USA). For Stage 1, genotyping was performed at the Center for Applied Genomics at The Children's Hospital of Philadelphia, using the Illumina Infinium™ II HumanHap550K (V3), as previously described (45). Standard quality control measures included exclusion of BeadChips with call rates <97.5% and individual SNPs with <95% genotyping yields. In addition, we excluded from statistical analyses data for: non-autosomal SNPs; SNPs with a MAF <5%; SNPs for which the genotype distribution in parents deviated from the Hardy–Weinberg equilibrium (P < 1 × 10−5); SNPs for which >1% of triads had a Mendelian inheritance errors; triads with Mendelian inheritance errors for >1% of the genotyped variants and individuals with a genotype call rate <95%.
Imputation
Imputation for non-genotyped autosomal SNPs was carried out using MACH version 1.0.16 (46) (http://www.sph.umich.edu/csg/abecasis/MACH/). Phased haplotypes for HapMap II CEU subjects (n = 60 founders) were used as the reference for imputing genotypes in the subset of Stage 1 triads in which all members were reported to be non-Hispanic and Caucasian (n = 284 triads). Imputation was based on genotyped SNPs with a MAF >10% and that met all other quality control criteria. Imputed SNPs with imputation ri2 (i.e. estimated squared correlation between imputed and actual genotypes) <0.3 were excluded. Data for imputed SNPs that were non-autosomal or that had a MAF <5% were also excluded from all statistical analyses.
SNP selection and genotyping
SNPs (genotyped or imputed) with association P< 1 × 10−5 in Stage 1 were genotyped in Staged 2. This genotyping was performed using the Illumina Veracode GoldenGate assay (47) in The Center for Applied Genomics at The Children's Hospital of Philadelphia. Samples from 24 Stage 1, case-parent triads were also genotyped using this platform, for comparisons with genotypes that were imputed in Stage 1. Quality control filters for SNPs genotyped in Stage 2 were identical to those used for genotyped SNPs in Stage 1.
Statistical analysis
The log-linear likelihood approach (26, 48), implemented using the MI-GWAS platform (49) was used to assess the association between LSLs and both the case and maternal genotypes for each variant. This approach has been widely applied in candidate gene and genome-wide association studies of birth defects and other pediatric conditions [e.g. (22, 50–56)]. Briefly, each triad is characterized by the number of high-risk alleles (0, 1, 2) in the genotypes of the father, mother and child. Asymmetry in the distribution of the case genotypes, conditional on parental genotypes, permits estimation of the case genotypic effects and asymmetry in the distribution of reciprocal mating types permits estimation of maternal genotypic effects. The likelihood ratio test comparing log-linear models with and without parameters characterizing the case genotypic effects is a generalization of the transmission disequilibrium test that is not subject to population stratification bias (although the power to detect an association in a stratified sample may be reduced if the association is not present in all sub-populations) and provides results that are comparable with PLINK (http://pngu.mgh.harvard.edu/~purcell/plink/) (49). However, unlike the analyses implemented in PLINK, the log-linear approach can be used to assess the association of the maternal genotype with the offpsring phenotype. Further, use of the expectation-maximization algorithm allows for the inclusion of data from incomplete triads (e.g. triads missing data for one parent) without invalidation of the results and relatively little loss of power compared to analyses based on a similar number of complete triads (57).
Analyses were performed using data from Stage 1 triads and repeated using data from the subset of Stage 1 triads in which both parents were self-reported non-Hispanic Caucasian. This subset was selected for analysis, a priori, because the inclusion of triads with mixed parental races/ethnicities (e.g. mother non-Hispanic Caucasian × father Hispanic) can bias estimates of maternal genetic effects (58). Data from this subset were also assessed for enrichment of GO and Kyoto Encyclopedia of Genes and Genomes (KEGG) terms. Enrichment analyses were performed separately for inherited and maternal genotypes using INRICH (27) for regions containing SNPs with association P-values <10−5 and were repeated for regions containing SNPs with association P-values <10−4. Pathways with P-values <0.05, after correction for the total number of pathways evaluated, were considered to be significantly enriched.
All log-linear analyses were performed using an additive model for the genotype being tested (e.g. case genotype) and an unrestricted model for the other (e.g. maternal) genotype. Statistical significance was assessed using a one-degree of freedom likelihood ratio test to compare models that included a parameter for the genotype being tested to models that excluded this parameter. Quartile–quartile (Q–Q) plots and estimates of lambda were generated using R version 2.15 (http://www.r-project.org/).
SNPs with association P-values <10−5 in Stage 1 were assessed in Stage 2 and SNPs with unadjusted P-values <0.05 in Stage 2 were assessed in the combined data. Pooled data from the two stages were analyzed, rather than conducting a meta-analysis, because the two sets of triads were ascertained using identical protocols at a single institution during overlapping time periods. SNPs for which the P-value in the combined analysis was lower than that in Stage 1 were considered to be potential LSL susceptibility loci. For these loci, data from Stage 1 were used to generate regional association plots with SNAP version 2.2 (http://www.broadinstitute.org/mpg/snap/ldplot.php). In addition, the relative increase in risk to a sibling attributable to each potential inherited LSL susceptibility locus (λS) was calculated (59), using the MAF among parents to estimate the frequency of the high-risk allele and parameter estimates from the additive log-linear models to estimate genotype relative risks.
Variant annotation
For each potential susceptibility locus, the UCSC genome browser (http://genome.ucsc.edu/) was used to identify coding and predicted genes flanking variants of interest and to determine TXCdsPredict scores. Visualization of transcription factor-binding sites, regions of open chromatin and miRNA-binding sites was performed using ENCODE transcription factor binding tracks, ENCODE Open Chrom Synth tracks and the TargetScan track on the UCSC genome browser. Transcription factor binding sites were also identified using TESS (http://www.cbil.upenn.edu/tess/). Genome-wide annotation of variants (GWAVA) scores (http://www.sanger.ac.uk/resources/software/gwava/), which range from 0 to 1, were used to assess the potential impact of non-coding variants on gene regulation (60). In addition, the GWAVA-TSS score, which is based on comparisons with a control set of SNPs matched for distance to the nearest transcription factor start site, was compared with the distribution of GWAVA-TSS scores for GWAS SNPs that have been externally replicated (60). When appropriate, GWAVA scores were also assessed for SNPs in linkage disequilibrium with our top hits. In addition, eQTL analyses were performed for each non-coding variant using Genevar (61). For variants associated with the risk of LSLs via the case genotype, Genevar was run using data from three cell types (fibroblast, lymphobastoid cell lines and T-cells) derived from umbilical cords of 75 Geneva GenCord individuals. For variants associated with the risk of LSLs via the maternal genotype, Genevar was run using data from three tissue types (adipose, lymphoblastoid cell lines and skin) from twin 1 of ∼90 MuTHER healthy female twin pairs. The significance of SNP–probe associations was assessed using a non-parametric permutation P-value, based on 10,000 permutations, and P values <0.05 were considered significant.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the National Institutes of Health (HL74094 to E.G. and L.E.M.) and the National Center for Research Resources (RR024134), which is now the National Center for Advancing Translational Sciences (UL1TR000003), both of which supported case ascertainment; and an Institutional Development Fund to The Center for Applied Genomics from The Children's Hospital of Philadelphia, which supported the genotyping of study subjects. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Supplementary Material
ACKNOWLEDGEMENTS
Summary results are available on dbGAP (http://www.ncbi.nlm.nih.gov/gap, accession number: phs000781.v1.p1). We thank the families for their participation, Jennifer Garbarini and Stacy Woyciechowski for case ascertainent, and Sharon Edman for data management.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Botto L.D., Correa A., Erickson J.D. Racial and temporal variations in the prevalence of heart defects. Pediatrics. 2001;107:E32. doi: 10.1542/peds.107.3.e32. [DOI] [PubMed] [Google Scholar]
- 2.Ferencz C., Rubin J.D., McCarter R.J., Brenner J.I., Neill C.A., Perry L.W., Hepner S.I., Downing J.W. Congenital heart disease: prevalence at livebirth. The Baltimore-Washington Infant Study. Am. J. Epidemiol. 1985;121:31–36. doi: 10.1093/oxfordjournals.aje.a113979. [DOI] [PubMed] [Google Scholar]
- 3.Moons P., Sluysmans T., De Wolf D., Massin M., Suys B., Benatar A., Gewillig M. Congenital heart disease in 111 225 births in Belgium: birth prevalence, treatment and survival in the 21st century. Acta Paediatr. 2009;98:472–477. doi: 10.1111/j.1651-2227.2008.01152.x. [DOI] [PubMed] [Google Scholar]
- 4.Yang Q., Chen H., Correa A., Devine O., Mathews T.J., Honein M.A. Racial differences in infant mortality attributable to birth defects in the United States, 1989–2002. Birth Defects Res. A Clin. Mol. Teratol. 2006;76:706–713. doi: 10.1002/bdra.20308. [DOI] [PubMed] [Google Scholar]
- 5.Opotowsky A.R., Siddiqi O.K., Webb G.D. Trends in hospitalizations for adults with congenital heart disease in the U.S. J. Am. Coll. Cardiol. 2009;54:460–467. doi: 10.1016/j.jacc.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 6.van der Bom T., Zomer A.C., Zwinderman A.H., Meijboom F.J., Bouma B.J., Mulder B.J. The changing epidemiology of congenital heart disease. Nat. Rev. Cardiol. 2011;8:50–60. doi: 10.1038/nrcardio.2010.166. [DOI] [PubMed] [Google Scholar]
- 7.van Beynum I.M., Kapusta L., den Heijer M., Vermeulen S.H., Kouwenberg M., Daniels O., Blom H.J. Maternal MTHFR 677C>T is a risk factor for congenital heart defects: effect modification by periconceptional folate supplementation. Eur. Heart J. 2006;27:981–987. doi: 10.1093/eurheartj/ehi815. [DOI] [PubMed] [Google Scholar]
- 8.Hobbs C.A., Cleves M.A., Karim M.A., Zhao W., MacLeod S.L. Maternal folate-related gene environment interactions and congenital heart defects. Obstet. Gynecol. 2010;116:316–322. doi: 10.1097/AOG.0b013e3181e80979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hobbs C.A., James S.J., Jernigan S., Melnyk S., Lu Y., Malik S., Cleves M.A. Congenital heart defects, maternal homocysteine, smoking, and the 677 C>T polymorphism in the methylenetetrahydrofolate reductase gene: evaluating gene-environment interactions. Am. J. Obstet. Gynecol. 2006;194:218–224. doi: 10.1016/j.ajog.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 10.Lupo P.J., Canfield M.A., Chapa C., Lu W., Agopian A.J., Mitchell L.E., Shaw G.M., Waller D.K., Olshan A.F., Finnell R.H., et al. Diabetes and obesity-related genes and the risk of neural tube defects in the national birth defects prevention study. Am. J. Epidemiol. 2012;176:1101–1109. doi: 10.1093/aje/kws190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Botto L.D., Lin A.E., Riehle-Colarusso T., Malik S., Correa A. National Birth Defects Prevention Study. Seeking causes: Classifying and evaluating congenital heart defects in etiologic studies. Birth Defects Res. A Clin. Mol. Teratol. 2007;79:714–727. doi: 10.1002/bdra.20403. [DOI] [PubMed] [Google Scholar]
- 12.Soemedi R., Wilson I.J., Bentham J., Darlay R., Topf A., Zelenika D., Cosgrove C., Setchfield K., Thornborough C., Granados-Riveron J., et al. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am. J. Hum. Genet. 2012;91:489–501. doi: 10.1016/j.ajhg.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zaidi S., Choi M., Wakimoto H., Ma L., Jiang J., Overton J.D., Romano-Adesman A., Bjornson R.D., Breitbart R.E., Brown K.K., et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature. 2013;498:220–223. doi: 10.1038/nature12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hitz M.P., Lemieux-Perreault L.P., Marshall C., Feroz-Zada Y., Davies R., Yang S.W., Lionel A.C., D'Amours G., Lemyre E., Cullum R., et al. Rare copy number variants contribute to congenital left-sided heart disease. PLoS Genet. 2012;8:e1002903. doi: 10.1371/journal.pgen.1002903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warburton D., Ronemus M., Kline J., Jobanputra V., Williams I., Anyane-Yeboa K., Chung W., Yu L., Wong N., Awad D., et al. The contribution of de novo and rare inherited copy number changes to congenital heart disease in an unselected sample of children with conotruncal defects or hypoplastic left heart disease. Hum. Genet. 2014;133:11–27. doi: 10.1007/s00439-013-1353-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carey A.S., Liang L., Edwards J., Brandt T., Mei H., Sharp A.J., Hsu D.T., Newburger J.W., Ohye R.G., Chung W.K., et al. Effect of copy number variants on outcomes for infants with single ventricle heart defects. Circ. Cardiovasc. Genet. 2013;6:444–451. doi: 10.1161/CIRCGENETICS.113.000189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McBride K.L., Pignatelli R., Lewin M., Ho T., Fernbach S., Menesses A., Lam W., Leal S.M., Kaplan N., Schliekelman P., et al. Inheritance analysis of congenital left ventricular outflow tract obstruction malformations: segregation, multiplex relative risk, and heritability. Am. J. Med. Genet. A. 2005;134A:180–186. doi: 10.1002/ajmg.a.30602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McBride K.L., Zender G.A., Fitzgerald-Butt S.M., Koehler D., Menesses-Diaz A., Fernbach S., Lee K., Towbin J.A., Leal S., Belmont J.W. Linkage analysis of left ventricular outflow tract malformations (aortic valve stenosis, coarctation of the aorta, and hypoplastic left heart syndrome) Eur. J. Hum. Genet. 2009;17:811–819. doi: 10.1038/ejhg.2008.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hinton R.B., Martin L.J., Rame-Gowda S., Tabangin M.E., Cripe L.H., Benson D.W. Hypoplastic left heart syndrome links to chromosomes 10q and 6q and is genetically related to bicuspid aortic valve. J. Am. Coll. Cardiol. 2009;53:1065–1071. doi: 10.1016/j.jacc.2008.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McBride K.L., Zender G.A., Fitzgerald-Butt S.M., Seagraves N.J., Fernbach S.D., Zapata G., Lewin M., Towbin J.A., Belmont J.W. Association of common variants in ERBB4 with congenital left ventricular outflow tract obstruction defects. Birth Defects Res. A Clin. Mol. Teratol. 2011;91:162–168. doi: 10.1002/bdra.20764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boyles A.L., DeRoo L.A., Lie R.T., Taylor J.A., Jugessur A., Murray J.C., Wilcox A.J. Maternal alcohol consumption, alcohol metabolism genes, and the risk of oral clefts: a population-based case-control study in Norway, 1996–2001. Am. J. Epidemiol. 2010;172:924–931. doi: 10.1093/aje/kwq226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van der Zanden L.F., Galesloot T.E., Feitz W.F., Brouwers M.M., Shi M., Knoers N.V., Franke B., Roeleveld N., van Rooij I.A. Exploration of gene-environment interactions, maternal effects and parent of origin effects in the etiology of hypospadias. J. Urol. 2012;188:2354–2360. doi: 10.1016/j.juro.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo Y.L., Cheng Y.L., Ye P., Wang W., Gao X.H., Chen Q. Association between MTHFR polymorphisms and orofacial clefts risk: a meta-analysis. Birth Defects Res. A Clin. Mol. Teratol. 2012;94:237–244. doi: 10.1002/bdra.23005. [DOI] [PubMed] [Google Scholar]
- 24.Ouyang S., Li Y., Liu Z., Chang H., Wu J. Association between MTR A2756G and MTRR A66G polymorphisms and maternal risk for neural tube defects: a meta-analysis. Gene. 2013;515:308–312. doi: 10.1016/j.gene.2012.11.070. [DOI] [PubMed] [Google Scholar]
- 25.Buyske S. Maternal genotype effects can alias case genotype effects in case-control studies. Eur. J. Hum. Genet. 2008;16:783–785. doi: 10.1038/ejhg.2008.74. [DOI] [PubMed] [Google Scholar]
- 26.Wilcox A.J., Weinberg C.R., Lie R.T. Distinguishing the effects of maternal and offspring genes through studies of “case-parent triads”. Am. J. Epidemiol. 1998;148:893–901. doi: 10.1093/oxfordjournals.aje.a009715. [DOI] [PubMed] [Google Scholar]
- 27.Lee P.H., O'Dushlaine C., Thomas B., Purcell S.M. INRICH: interval-based enrichment analysis for genome-wide association studies. Bioinformatics. 2012;28:1797–1799. doi: 10.1093/bioinformatics/bts191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhong Z., Wan B., Qiu Y., Ni J., Tang W., Chen X., Yang Y., Shen S., Wang Y., Bai M., et al. Identification of a novel human zinc finger gene, ZNF438, with transcription inhibition activity. J. Biochem. Mol. Biol. 2007;40:517–524. doi: 10.5483/bmbrep.2007.40.4.517. [DOI] [PubMed] [Google Scholar]
- 29.Zhang K., Gao R., Zhang H., Cai X., Shen C., Wu C., Zhao S., Yu L. Molecular cloning and characterization of three novel lysozyme-like genes, predominantly expressed in the male reproductive system of humans, belonging to the c-type lysozyme/alpha-lactalbumin family. Biol. Reprod. 2005;73:1064–1071. doi: 10.1095/biolreprod.105.041889. [DOI] [PubMed] [Google Scholar]
- 30.Dai J., Ji C., Gu S., Wu Q., Wang L., Xu J., Zeng L., Ye X., Yin G., Xie Y., et al. Cloning and sequence analysis of the human cDNA encoding the synaptoporin (delta), a highly conservative synaptic vesicle protein. Mol. Biol. Rep. 2003;30:185–191. doi: 10.1023/a:1024907723000. [DOI] [PubMed] [Google Scholar]
- 31.Barth A.S., Merk S., Arnoldi E., Zwermann L., Kloos P., Gebauer M., Steinmeyer K., Bleich M., Kaab S., Pfeufer A., et al. Functional profiling of human atrial and ventricular gene expression. Pflugers Arch. 2005;450:201–208. doi: 10.1007/s00424-005-1404-8. [DOI] [PubMed] [Google Scholar]
- 32.Seo S., Kume T. Forkhead transcription factors, Foxc1 and Foxc2, are required for the morphogenesis of the cardiac outflow tract. Dev. Biol. 2006;296:421–436. doi: 10.1016/j.ydbio.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 33.Shaw-Smith C. Genetic factors in esophageal atresia, tracheo-esophageal fistula and the VACTERL association: roles for FOXF1 and the 16q24.1 FOX transcription factor gene cluster, and review of the literature. Eur. J. Med. Genet. 2010;53:6–13. doi: 10.1016/j.ejmg.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stankiewicz P., Sen P., Bhatt S.S., Storer M., Xia Z., Bejjani B.A., Ou Z., Wiszniewska J., Driscoll D.J., Maisenbacher M.K., et al. Genomic and genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am. J. Hum. Genet. 2009;84:780–791. doi: 10.1016/j.ajhg.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iascone M., Ciccone R., Galletti L., Marchetti D., Seddio F., Lincesso A.R., Pezzoli L., Vetro A., Barachetti D., Boni L., et al. Identification of de novo mutations and rare variants in hypoplastic left heart syndrome. Clin. Genet. 2012;81:542–554. doi: 10.1111/j.1399-0004.2011.01674.x. [DOI] [PubMed] [Google Scholar]
- 36.McCulley D.J., Black B.L. Transcription factor pathways and congenital heart disease. Curr. Top. Dev. Biol. 2012;100:253–277. doi: 10.1016/B978-0-12-387786-4.00008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schott J.J., Benson D.W., Basson C.T., Pease W., Silberbach G.M., Moak J.P., Maron B.J., Seidman C.E., Seidman J.G. Congenital heart disease caused by mutations in the transcription factor NKX2–5. Science. 1998;281:108–111. doi: 10.1126/science.281.5373.108. [DOI] [PubMed] [Google Scholar]
- 38.Polen K.N., Rasmussen S.A., Riehle-Colarusso T., Reefhuis J. National Birth Defects Prevention Study. Association between reported venlafaxine use in early pregnancy and birth defects, national birth defects prevention study, 1997–2007. Birth Defects Res. A Clin. Mol. Teratol. 2013;97:28–35. doi: 10.1002/bdra.23096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Broussard C.S., Rasmussen S.A., Reefhuis J., Friedman J.M., Jann M.W., Riehle-Colarusso T., Honein M.A. National Birth Defects Prevention Study. Maternal treatment with opioid analgesics and risk for birth defects. Am. J. Obstet. Gynecol. 2011;204:314. doi: 10.1016/j.ajog.2010.12.039. e311–311. [DOI] [PubMed] [Google Scholar]
- 40.Alwan S., Reefhuis J., Botto L.D., Rasmussen S.A., Correa A., Friedman J.M. National Birth Defects Prevention Study. Maternal use of bupropion and risk for congenital heart defects. Am. J. Obstet. Gynecol. 2010;203:52. doi: 10.1016/j.ajog.2010.02.015. e51–56. [DOI] [PubMed] [Google Scholar]
- 41.Gilboa S.M., Correa A., Botto L.D., Rasmussen S.A., Waller D.K., Hobbs C.A., Cleves M.A., Riehle-Colarusso T.J. National Birth Defects Prevention Study. Association between prepregnancy body mass index and congenital heart defects. Am. J. Obstet. Gynecol. 2010;202:51. doi: 10.1016/j.ajog.2009.08.005. e51–51 e10. [DOI] [PubMed] [Google Scholar]
- 42.Cordell H.J., Topf A., Mamasoula C., Postma A.V., Bentham J., Zelenika D., Heath S., Blue G., Cosgrove C., Granados Riveron J., et al. Genome-wide association study identifies loci on 12q24 and 13q32 associated with tetralogy of Fallot. Hum. Mol. Genet. 2013;22:1473–1481. doi: 10.1093/hmg/dds552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu Z., Shi Y., Mo X., Xu J., Zhao B., Lin Y., Yang S., Xu Z., Dai J., Pan S., et al. A genome-wide association study identifies two risk loci for congenital heart malformations in Han Chinese populations. Nat. Genet. 2013;45:818–821. doi: 10.1038/ng.2636. [DOI] [PubMed] [Google Scholar]
- 44.Cordell H.J., Bentham J., Topf A., Zelenika D., Heath S., Mamasoula C., Cosgrove C., Blue G., Granados-Riveron J., Setchfield K., et al. Genome-wide association study of multiple congenital heart disease phenotypes identifies a susceptibility locus for atrial septal defect at chromosome 4p16. Nat. Genet. 2013;45:822–824. doi: 10.1038/ng.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hakonarson H., Grant S.F., Bradfield J.P., Marchand L., Kim C.E., Glessner J.T., Grabs R., Casalunovo T., Taback S.P., Frackelton E.C., et al. A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature. 2007;448:591–594. doi: 10.1038/nature06010. [DOI] [PubMed] [Google Scholar]
- 46.Li Y., Abecasis G. MACH 1.0: rapid haplotype reconstruction and missing genotype inference. Am. J. Hum. Genet. 2006;S79:2290. [Google Scholar]
- 47.Zhu H., Kartiko S., Finnell R.H. Importance of gene-environment interactions in the etiology of selected birth defects. Clin. Genet. 2009;75:409–423. doi: 10.1111/j.1399-0004.2009.01174.x. [DOI] [PubMed] [Google Scholar]
- 48.Weinberg C.R., Wilcox A.J., Lie R.T. A log-linear approach to case-parent-triad data: assessing effects of disease genes that act either directly or through maternal effects and that may be subject to parental imprinting. Am. J. Hum. Genet. 1998;62:969–978. doi: 10.1086/301802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agopian A.J., Mitchell L.E. MI-GWAS: a SAS platform for the analysis of inherited and maternal genetic effects in genome-wide association studies using log-linear models. BMC Bioinformatics. 2011;12:117. doi: 10.1186/1471-2105-12-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Doolin M.T., Barbaux S., McDonnell M., Hoess K., Whitehead A.S., Mitchell L.E. Maternal genetic effects, exerted by genes involved in homocysteine remethylation, influence the risk of spina bifida. Am. J. Hum. Genet. 2002;71:1222–1226. doi: 10.1086/344209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jensen L.E., Wall A.M., Cook M., Hoess K., Thorn C.F., Whitehead A.S., Mitchell L.E. A common ABCC2 promoter polymorphism is not a determinant of the risk of spina bifida. Birth Defects Res. A Clin. Mol. Teratol. 2004;70:396–399. doi: 10.1002/bdra.20023. [DOI] [PubMed] [Google Scholar]
- 52.Hancock D.B., Romieu I., Shi M., Sienra-Monge J.J., Wu H., Chiu G.Y., Li H., del Rio-Navarro B.E., Willis-Owen S.A., Weiss S.T., et al. Genome-wide association study implicates chromosome 9q21.31 as a susceptibility locus for asthma in Mexican children. PLoS Genet. 2009;5:e1000623. doi: 10.1371/journal.pgen.1000623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.James S.J., Melnyk S., Jernigan S., Pavliv O., Trusty T., Lehman S., Seidel L., Gaylor D.W., Cleves M.A. A functional polymorphism in the reduced folate carrier gene and DNA hypomethylation in mothers of children with autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010;153B:1209–1220. doi: 10.1002/ajmg.b.31094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martinelli M., Masiero E., Carinci F., Morselli P.G., Pezzetti F., Scapoli L. New evidence for the role of cystathionine beta-synthase in non-syndromic cleft lip with or without cleft palate. Eur. J. Oral Sci. 2011;119:193–197. doi: 10.1111/j.1600-0722.2011.00824.x. [DOI] [PubMed] [Google Scholar]
- 55.Lupo P.J., Nousome D., Okcu M.F., Chintagumpala M., Scheurer M.E. Maternal variation in EPHX1, a xenobiotic metabolism gene, is associated with childhood medulloblastoma: an exploratory case-parent triad study. Pediatr. Hematol. Oncol. 2012;29:679–685. doi: 10.3109/08880018.2012.722747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shi M., Murray J.C., Marazita M.L., Munger R.G., Ruczinski I., Hetmanski J.B., Wu T., Murray T., Redett R.J., Wilcox A.J., et al. Genome wide study of maternal and parent-of-origin effects on the etiology of orofacial clefts. Am. J. Med. Genet. A. 2012;158A:784–794. doi: 10.1002/ajmg.a.35257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weinberg C.R. Allowing for missing parents in genetic studies of case-parent triads. Am. J. Hum. Genet. 1999;64:1186–1193. doi: 10.1086/302337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mitchell L.E., Weinberg C.R. Evaluation of offspring and maternal genetic effects on disease risk using a family-based approach: the “pent” design. Am. J. Epidemiol. 2005;162:676–685. doi: 10.1093/aje/kwi249. [DOI] [PubMed] [Google Scholar]
- 59.James J.W. Frequency in relatives for an all-or-none trait. Ann. Hum. Genet. 1971;35:47–49. doi: 10.1111/j.1469-1809.1956.tb01377.x. [DOI] [PubMed] [Google Scholar]
- 60.Ritchie G.R., Dunham I., Zeggini E., Flicek P. Functional annotation of noncoding sequence variants. Nat. Methods. 2014;11:294–296. doi: 10.1038/nmeth.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang T.P., Beazley C., Montgomery S.B., Dimas A.S., Gutierrez-Arcelus M., Stranger B.E., Deloukas P., Dermitzakis E.T. Genevar: a database and Java application for the analysis and visualization of SNP-gene associations in eQTL studies. Bioinformatics. 2010;26:2474–2476. doi: 10.1093/bioinformatics/btq452. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.