

Abstract
The neuronal ceroid lipofuscinoses (NCLs), also known as Batten disease, are a group of autosomal recessive neurodegenerative disorders in children characterized by the progressive onset of seizures, blindness, motor and cognitive decline and premature death. Patients with mutations in CLN1 primarily manifest with infantile NCL (INCL or Haltia-Santavuori disease), which is second only to congenital NCL for its age of onset and devastating progression. CLN1 encodes a lysosomal enzyme, palmitoyl-protein thioesterase 1 (PPT1). Nonsense mutations in CLN1 account for 52.3% of all disease causing alleles in infantile NCL, the most common of which worldwide is the p.R151X mutation. Previously, we have shown how nonsense-mediated decay is involved in the degradation of CLN1 mRNA transcripts containing the p.R151X mutation in human lymphoblast cell lines. We have also shown how the read-through drugs gentamicin and ataluren (PTC124) increase CLN1 (PPT1) enzyme activity. Here, we provide the initial characterization of the novel Cln1R151X mouse model of infantile neuronal ceroid lipofuscinosis that we have generated. This nonsense mutation model recapitulates the molecular, histological and behavioral phenotypes of the human disease. Cln1R151X mice showed a significant decrease in Cln1 mRNA level and PPT1 enzyme activity, accumulation of autofluorescent storage material, astrocytosis and microglial activation in the brain. Behavioral characterization of Cln1R151X mice at 3 and 5 months of age revealed significant motor deficits as measured by the vertical pole and rotarod tests. We also show how the read-through compound ataluren (PTC124) increases PPT1 enzyme activity and protein level in Cln1R151X mice in a proof-of-principle study.
INTRODUCTION
The neuronal ceroid lipofuscinoses (NCLs), commonly referred to as Batten disease, are a group of autosomal recessive lysosomal storage disorders that collectively comprise the most common neurodegenerative disorder in children (1,2). These disorders are characterized pathologically by the accumulation of autofluorescent storage material within the lysosomes, astrocytosis and microglial activation, and significant loss of neurons within the retina and central nervous system. NCL has an estimated worldwide incidence of ∼1 in 100 000 live births, with different incidence rates depending on the country of origin (3). Currently, there are over 400 known genetic mutations in at least 14 different genes that lead to five overlapping clinical variants classified by age of onset (www.ucl.ac.uk/ncl/mutation.shtml) (4). NCL disease typically presents with visual deficits due to retinal degeneration which gradually progresses to complete irreversible blindness. Patients also suffer from seizures, motor abnormalities, ataxia and mental retardation leading to dementia. Currently, there is no effective treatment for any of the NCLs, only palliative care to relieve symptoms (5). These disorders are progressive and always result in premature death, with infantile neuronal ceroid lipofuscinosis (INCL) second only to congenital NCL with its age of onset and devastating progression.
Infantile neuronal ceroid lipofuscinosis (INCL or Haltia-Santavuori disease; alternatively referred to as classic INCL or CLN1 disease) is most common in populations of Finnish descent, with an incidence of 1:20 000 and a carrier frequency of 1 in 70 (1,6). Infantile NCL typically presents in children at 1–2 years of age with visual deficits that gradually progresses to complete blindness. Other common symptoms in infantile NCL include microcephaly, seizures, motor abnormalities, ataxia and mental retardation leading to dementia, with death usually occurring by 8 to 13 years of age. To date, there are 64 known mutations in CLN1, consisting of 27 missense, 11 nonsense, 11 splice-site defects, 7 small deletions, 4 small insertions of duplications, 1 large deletion, 1 deletion/insertion and 2 3′-UTR changes that lead to NCL (http://www.ucl.ac.uk/ncl/mutation.shtml) (4). Mutations in CLN1 most commonly lead to infantile NCL, but can also lead to other clinical variants of NCL, such as variant forms of late-infantile NCL (7–11), juvenile NCL (12,13) and adult NCL (14,15). CLN1 encodes palmitoyl-protein thioesterase 1 (PPT1), a ubiquitous lysosomal enzyme responsible for removing palmitoyl groups from modified cysteine residues in polypeptides that have been targeted for degradation in the lysosome. A recent finding also suggests that PPT1 is involved in endocannabinoid metabolism through hydrolysis of 2-arachidonoylglycerol (2-AG), PGE2-G and PGF2α-G (16). Significantly decreased PPT1 enzyme activity is the pathological hallmark of infantile NCL (17,18).
Nonsense mutations in CLN1 account for 52.3% of all disease causing alleles in infantile NCL, the most common of which worldwide is the p.R151X mutation (4,19). We have shown previously that CLN1 alleles harboring the p.R151X mutation are targeted for degradation by the nonsense-mediated decay (NMD) pathway, leading to a significant decrease in CLN1 mRNA level and concurrent decrease in CLN1 (PPT1) enzyme activity in patient-derived lymphoblast cell lines (20). In addition, we have shown how the read-through drugs ataluren (PTC124) and gentamicin increase PPT1 enzyme activity through nonsense suppression and subsequent production of functional protein (20). These in vitro studies provided a clear stepping stone towards using the CLN1 p.R151X mutation to study the use of read-through compounds in vivo. With this in mind, we generated a Cln1R151X point mutation mouse model for this purpose. Here, we show with an initial characterization how the Cln1R151X animal model recapitulates the molecular, neuropathological and behavioral phenotypes of NCL. We also show with a proof-of-principle study how the read-through compound ataluren can be used to increase PPT1 protein level and enzyme activity.
RESULTS
Generation of the Cln1R151X nonsense mutation mouse model
To duplicate the most common CLN1 p.R151X nonsense mutation in an infantile NCL mouse model, we ‘knocked-in’ a C>T genomic point mutation in exon 5 of the murine Cln1 gene using Applied StemCell, Inc. This strategy is illustrated in Figure 1 and Supplementary Material, Figure S1. Homologous recombination in embryonic stem cells introduced the point mutation into Cln1 with a downstream neo cassette flanked by tandem Frt-LoxP recombination sites, thus generating Cln1R151Xneo mice. Matings with CMV-cre mice from Jax labs then produced progeny in which the neo cassette was excised. Additional breeding isolated the cre transgene, yielding the Cln1R151X mouse model, carrying the most common infantile NCL mutation.
Figure 1.

Generation of the Cln1R151X nonsense mutation mouse model. Schematic representation of the Cln1R151X gene-targeting strategy. The top line depicts the wild-type Cln1 allele with the locations of exons 2 through 5 (black boxes). The second line depicts the LoxP-Frt flanked neo cassette (white box) targeting vector, with the C>T point mutation in exon 5 (gray arrow), and upstream and downstream Cln1 genomic DNA. The third line depicts the configuration of the correctly targeted Cln1R151Xneo allele created by homologous recombination within ES cells. The last line depicts the completed Cln1R151X allele after excision of the LoxP-Frt flanked neo cassette with Cre recombinase.
Cln1 gene expression and PPT1 enzyme activity is decreased in Cln1R151X mice
We have shown previously that the p.R151X mutation leads to a significant decrease in CLN1 mRNA expression and PPT1 enzyme activity in human lymphoblast cell lines (20). To measure endogenous levels of Cln1 gene expression, quantitative real-time PCR (qPCR) was performed using total RNA extracted from the cerebellum, cortex, striatum/thalamus, brainstem and liver of Cln1R151X mice to confirm the role of NMD targeting the p.R151X point mutation for degradation (Fig. 2A). Cln1 mRNA level was significantly decreased in all tissues from both heterozygous (Cln1WT/R151X) and homozygous (Cln1R151X/R151X) mice compared with wild type (WT). Interestingly, this decrease occurred in a stepwise manner based on the presence of one or two p.R151X mutations, such that heterozygous tissues showed a 1.42- to 1.85-fold decrease in Cln1 expression and homozygous tissues showed a 5.32- to 12.99-fold decrease in Cln1 expression.
Figure 2.

Cln1 mRNA expression and PPT1 enzyme activity is decreased in Cln1R151X mice. (A) Endogenous expression of Cln1 in the cerebellum (Cereb), cortex (Cortex), striatum/thalamus (StrThal), brainstem (BrStem) and liver from 5-month-old homozygous (white bars) and heterozygous (hashed bars) Cln1R151X mice is decreased compared with wild type (grey bars). Quantitative real-time PCR (qPCR) was used to measure endogenous expression of Cln1. The relative expression of Cln1 was normalized with four reference genes (B2m, Gapdh, Gusb, Hgprt). (B) Endogenous PPT1 enzyme activity in the cerebellum, cortex, striatum/thalamus, brainstem and liver from 5-month-old homozygous (white bars) and heterozygous (hashed bars) Cln1R151X mice is decreased compared with wild type (grey bars). All samples were compared with age- and sex-matched wild-type controls. Four technical replicates from each of three biological replicates were used for each genotype. All values are represented as mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001.
To determine the effect of the p.R151X mutation on protein function, a fluorogenic enzyme assay was utilized to measure PPT1 activity using total protein isolated from the cerebellum, cortex, striatum/thalamus, brainstem and liver of Cln1R151X mice (Fig. 2B) (21–23). PPT1 enzyme activity was significantly decreased in all tissues from both heterozygous and homozygous Cln1R151X mice compared with wild type. PPT1 activity decreased in a stepwise manner similar to Cln1 mRNA expression in these tissues, with heterozygous tissues showing a 29.5–56.3% of wild-type activity and homozygous tissues showing a 1.7–3.1% of wild-type activity.
Autofluorescent storage material accumulates in the brain of Cln1R151X mice
The accumulation of autofluorescent storage material within the brain is one of the most classic findings of NCL and has been seen in the human disease as well as the Cln1−/− (Ppt1−/−) mouse model (1,2,24). To measure autofluorescent storage material, unstained coronal sections were imaged and analyzed from the cortex, thalamus and hippocampus of 5-month-old wild type and Cln1R151X male mice. A representative comparison of these brain regions from a Cln1R151X and wild-type animal at 5 months of age is shown in Figure 3. Storage material fluoresced over a wide range of filter combinations, including Texas Red, FITC and DAPI, and was distributed widely throughout the brain of Cln1R151X mice (data not shown).
Figure 3.
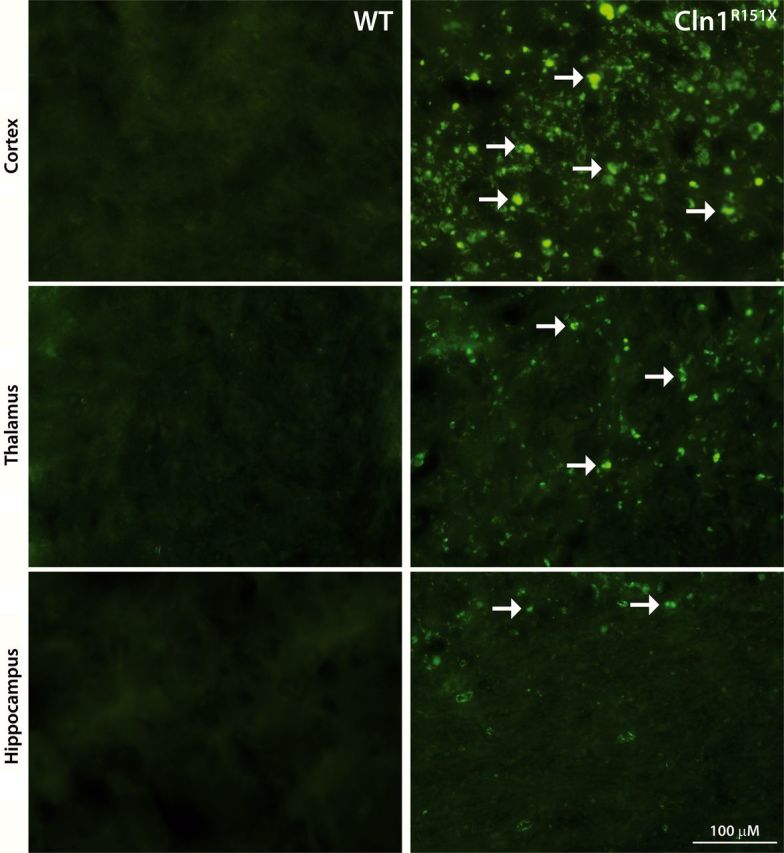
Accumulation of autofluorescent storage material in Cln1R151X mice. Representative images of unstained sections from the cortex, thalamus and hippocampus viewed by epifluorescence using the FITC filter set show the abundant intracellular accumulation of autofluorescent storage material (white arrows) in 5-month-old Cln1R151X mice (right images) compared with age- and sex-matched wild-type controls (left images).
Cln1R151X mice show widespread astrocytosis and microglial activation throughout the brain
Neuropathological changes within the brain can be tested by measuring glial fibrillary acidic protein (GFAP) and CD68 immunoreactivity. Increased GFAP immunoreactivity is an indicator of astrocytosis and increased CD68 immunoreactivity is an indicator of reactive microglia, both signs of pathology within the brain. To measure the glial cell response in Cln1R151X mice, we used thresholding image analysis to quantify the immunoreactivity of GFAP (astrocytes) and CD68 (quiescent and activated microglia) as previously described (25,26). The immunoreactivity of these markers was assessed in the cortex, thalamus and hippocampus. A significant and widespread increase of both GFAP and CD68 was found in Cln1R151X mice versus wild-type controls (Figs 4 and 5). GFAP-positive astrocytes appeared hypertrophied with enlarged cell soma and thickened processes throughout the brain (Fig. 4), and showed a significant 2.03-fold increase in GFAP immunoreactivity compared with controls in the cortex (P = 0.0017 by unpaired t-test), a 1.69-fold increase in GFAP immunoreactivity in the thalamus (P = 0.0006 by unpaired t-test), and a 1.72-fold increase in GFAP immunoreactivity in the hippocampus (P = 0.0030 by unpaired t-test) (Fig. 4). CD68-positive cells exhibited a macrophage-like appearance with rounded cell bodies and shortened processes in the cortex, thalamus and hippocampus (Fig. 5). There was a significant 4.98-fold increase in CD68 immunoreactivity compared with controls in the cortex (P = 0.0007 by unpaired t-test), a 3.09-fold increase in CD68 immunoreactivity in the thalamus (P = 0.0030 by unpaired t-test), and a 1.76-fold increase in CD68 immunoreactivity in the hippocampus (P = 0.0010 by unpaired t-test) (Fig. 5).
Figure 4.

Astrocytosis in Cln1R151X mice. Immunohistochemical staining for GFAP reveals activation of astrocytes, which are hypertrophied with numerous thickened processes, in the cortex, thalamus and hippocampus of Cln1R151X mice. Quantitative image analysis reveals the significantly increased expression level of GFAP within the cortex, thalamus and hippocampus of Cln1R151X mice compared with age- and sex-matched wild-type controls. Statistical significance was determined by unpaired t-test with Welch's correction. Data are plotted as the mean sum immunoreactivity of each genotype as a fold-change compared with wild type (30 fields per animal; 3 animals per genotype) ± SEM.
Figure 5.

Microglial activation in Cln1R151X mice. Immunohistochemical staining for CD68 reveal microglial reactivity, which exhibit macrophage-like morphology, in the cortex, thalamus and hippocampus of Cln1R151X mice. Quantitative image analysis reveals the significantly increased expression level of CD68 within the cortex, thalamus and hippocampus of Cln1R151X mice compared with age- and sex-matched wild-type controls. Statistical significance was determined by unpaired t-test with Welch's correction. Data are plotted as the mean sum immunoreactivity of each genotype as a fold-change compared with wild type (30 fields per animal; 3 animals per genotype) ± SEM.
Cln1R151X mice show significant motor deficits as measured by the vertical pole test and the rotarod test
In the behavioral characterization, wild-type and Cln1R151X male mice were compared in the dish test, modified vertical pole test, tail suspension test, and rotarod test at 3 and 5 months of age. In the dish test, which assesses the exploratory behavior, Cln1R151X mice showed significantly decreased exploration; they stayed in a large Petri dish considerably longer than WT mice at both ages (Supplementary Material, Fig. S2). The depressive-like behavior of WT and Cln1R151X mice in the tail suspension test was similar at 3 and 5 months of age (Supplementary Material, Fig. S3). Besides the total time of immobility (in 6 min), the time course of immobility was also very similar (Supplementary Material, Fig. S3). The motor skill tests revealed marked motor deficits of Cln1R151X mice at both ages. In the modified vertical pole test, which measures the balance, spatial orientation and motor coordination, mutant mice climbed down on the pole drastically slower, and turned downward on the pole significantly slower than WT mice (Fig. 6A). In the rotarod test, which measures balance and motor coordination, 3-month-old Cln1R151X mice fell from the rotating rod 31 s sooner than WT mice (Fig. 6B). At the age of 5 months, however, the rotarod performance of WT and Cln1R151X mice was similar (Fig. 6B). Our findings that 5-month-old Cln1R151X mice display a motor deficit in the vertical pole test, but not in the rotarod test (Fig. 6) show, in agreement with previous studies (27,28), that the vertical pole test is more sensitive than the rotarod test to detect motor deficits.
Figure 6.
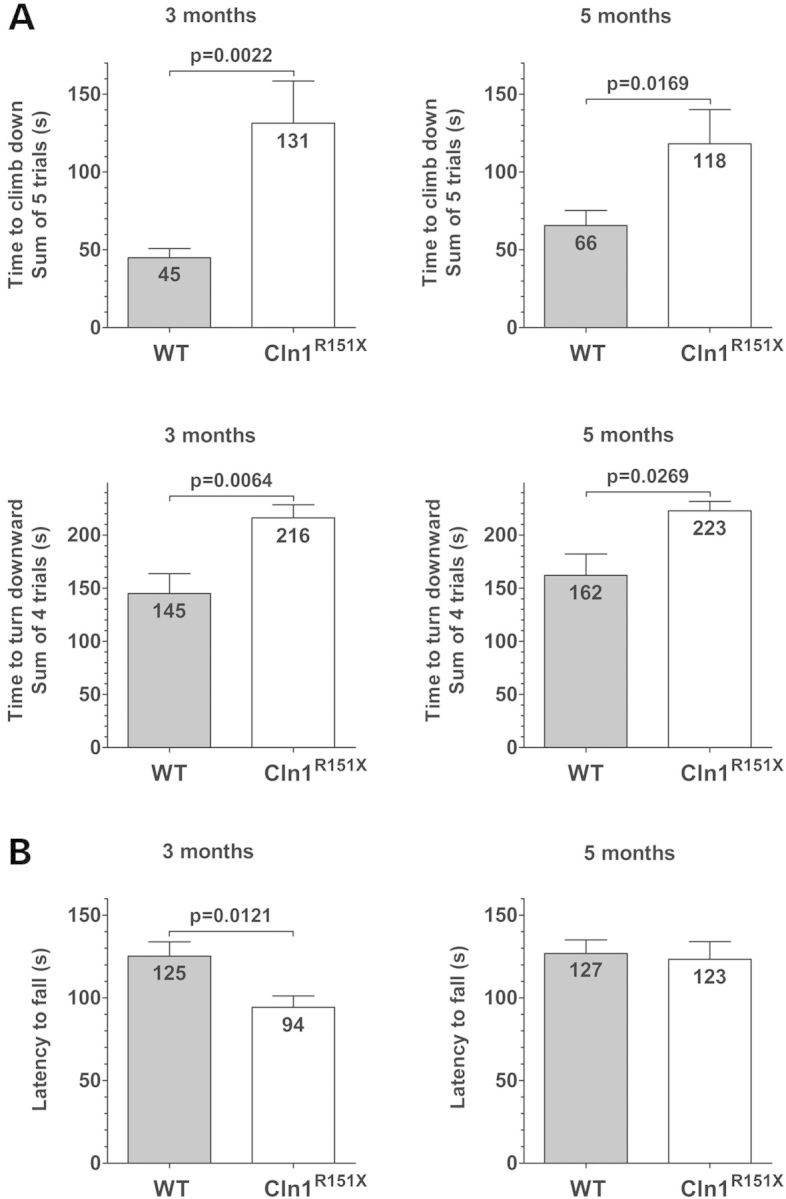
Cln1R151X mice have motor deficits as measured by the vertical pole test and rotarod test. The motor skills of WT and Cln1R151X male mice were examined at 3 and 5 months of age. (A) In the modified vertical pole test, Cln1R151X mice climbed down on the pole drastically slower (upper 2 graphs), and turned downward on the pole significantly slower (lower 2 graphs) than WT mice. Columns and bars represent mean ± SEM. The same WT and Cln1R151X mice were tested at 3 and 5 months of age (14 WT and 12 Cln1R151X mice). Statistical significance was determined by the non-parametric Mann–Whitney test. (B) In an accelerating rotarod test (0.2 rpm/s starting from 0 rpm), 3-month-old Cln1R151X mice fell from the rotating rod 31 s sooner than WT mice (left graph). At the age of 5 months, the rotarod performance of Cln1R151X and WT mice was similar (right graph). Columns and bars represent mean ± SEM. The same WT and Cln1R151X mice were tested at 3 and 5 months of age (14 WT and 12 Cln1R151X mice). Statistical significance was determined by unpaired t-test.
For weight comparison, the same WT and Cln1R151X mice were weighed at 3 and 5 months of age, 30–40 min before starting the behavioral tests with them. At the age of 3 months, Cln1R151X mice (33.0 ± 1.9 g; n = 12) were significantly heavier than WT mice (28.6 ± 3.0 g; n = 16); P = 0.0002 with unpaired t-test. By the age of 5 months, however, the weight difference disappeared: WT (n = 16) and Cln1R151X (n = 12) mice weighed 35.4 ± 5.7 g and 36.9 ± 2.1 g, respectively. Since the weight may affect the performance in behavioral tests, particularly in motor skill tests, we carried out correlation analyses between the weight and the behavioral test results. The weight of 3- and 5-month-old Cln1R151X mice did not correlate with their performance in any of the behavioral tests. In the case of WT mice, a negative correlation was found between the weight and the time to climb down on the vertical pole at the age of 3 months: heavier mice climbed down faster (P = 0.0091). At the age of 5 months, the weight of WT mice positively correlated with the time to turn downward on the vertical pole: heavier mice turned downward slower (P = 0.005). Other behavioral test results did not show a correlation with the weight of WT mice. Though Cln1R151X mice were significantly heavier than WT mice at 3 months of age, the correlation analyses between weight and behavioral test results clearly show that the weight difference did not contribute to the observed behavioral deficits of 3-month-old Cln1R151X mice.
Ataluren (PTC124) increases PPT1 enzyme activity and protein level in Cln1R151X mice
To induce nonsense suppression and increase PPT1 enzyme activity, the read-through drug ataluren (PTC124) was given via intraperitoneal (i.p.) injection to male Cln1R151X mice at 2 months of age. These treatments were performed four times daily for 2 consecutive days in a proof-of-principle study. Used at 10 mg/kg, ataluren increased PPT1 enzyme activity (P = 0.0001 by unpaired t-test) and protein level (P = 0.0014 by unpaired t-test) in the liver (Fig. 7A), but did not increase PPT1 enzyme activity or protein level in the cortex (Fig. 7B). This tissue-specific effect was likely due to the inability of ataluren to breach the blood brain barrier (BBB), which decreased the bioavailability of ataluren within the brain, and prevented ataluren from reaching an efficacious concentration within the therapeutic window. Therefore, we increased the dosage to 100 mg/kg, which is not within the standard range for ataluren treatment, but may improve read-through within the brain. This increased dose of ataluren caused a measureable (although biologically insignificant) increase in PPT1 enzyme activity (P = 0.0019) and protein level (P = 0.0207) in the cortex (Fig. 7B). However, this increased dosage caused a paradoxical decrease in PPT1 enzyme activity (P = 0.0012) within the liver of Cln1R151X mice (Fig. 7A). This much higher dose of ataluren may have had either toxic effects on the liver or directly inhibited activity of PPT1. Previously, it has been shown that restoring CLN1 (PPT1) enzyme activity in the brain to only 4–5% of the normal level results in a significant attenuation of neurodegeneration in the Cln1−/− mouse model (29). Although we did not achieve PPT1 activity within this range within the brain in this proof-of-principle study, we have shown the capability for nonsense suppression treatment. Longer treatments with ataluren in the Cln1R151X mouse model are needed to further investigate the effects of nonsense suppression therapy in this disease model.
Figure 7.

Nonsense suppression with Ataluren (PTC124) increases PPT1 enzyme activity and PPT1 protein level in Cln1R151X mice. Two-month-old Cln1R151X male mice were treated with ataluren (10 mg/kg or 100 mg/kg i.p.) four times a day for 2 days. Control mice were treated with the vehicle of the drug. Immediately following the last injection on the second day, tissues were collected, and PPT1 enzyme activity and protein level were measured. PPT1 activity dose- and tissue-dependently changed in the liver and cortex of ataluren-treated mice. (A) Ataluren increased PPT1 activity (left graph) and protein level (right graph) in the liver at 10 mg/kg. (B) There was no measureable increase in PPT1 activity (left graph) or protein level (right graph) in the cortex using 10 mg/kg. Due to the potential inability to breach the blood–brain barrier at the 10 mg/kg dosage, 100 mg/kg ataluren was used, which caused a measurable increase in both PPT1 activity and protein level in the cortex. However, the 100 mg/kg dosage caused a paradoxical decrease in PPT1 activity in the liver. Six to eight animals were used for each treatment group. Four technical replicates were used for each biological replicate in the PPT1 enzyme assay and three technical replicates were used for each biological replicate in the PPT1 ELISA. All data are plotted as percent of normal. Columns and bars represent mean ± SEM. Statistical significance was determined by unpaired t-test.
DISCUSSION
In this study, we have provided an initial characterization of the first Cln1R151X nonsense mutation mouse model for INCL. This mouse exhibited significant decreases in both Cln1 mRNA level and PPT1 enzyme activity within the central nervous system and liver. Immunohistochemical staining for GFAP and CD68 also revealed significant astrocytosis and microglial activation throughout the brain. There was also significant accumulation of autofluorescent storage material within the brain. Behavioral characterization of Cln1R151X mice revealed significant motor deficits as measured by the vertical pole and rotarod tests. In addition, we have shown how nonsense suppression therapy with ataluren (PTC124) increases PPT1 enzyme activity and PPT1 protein level in a tissue-specific and dose-dependent manner in the cortex and liver of Cln1R151X mice.
NMD has emerged as an important regulatory pathway that recognizes and degrades mRNA transcripts harboring nonsense mutations in genetic diseases. More importantly, several drugs can target this pathway, leading to ribosomal read-through of the premature stop codon (nonsense mutation) and subsequent production of full-length protein products. The p.R151X mutation in CLN1 accounts for the majority of nonsense mutations leading to infantile NCL. Previously, we have shown that NMD leads to a significant decrease in CLN1 mRNA level in patient-derived lymphoblast cell lines that are heterozygous and homozygous for p.R151X mutation (20). In addition, treatment of these cell lines with two read-through drugs, gentamicin and ataluren, led to significant increase in PPT1 enzyme activity (20).
Recombinant gene targeting methods were used to generate the Cln1R151X nonsense mutant mouse model for infantile NCL (Fig. 1 and Supplementary Material, Fig. S1). Quantitative real-time PCR (qPCR) showed a stepwise decrease in Cln1 mRNA level in the brain and liver of heterozygous and homozygous Cln1R151X mice compared with wild type controls (Fig. 2A). A similar trend occurred with PPT1 enzyme activity in these same tissues (Fig. 2B). This genotype-specific decrease in Cln1 expression is likely due to NMD recognizing the p.R151X nonsense mutation and targeting the mRNA transcript for degradation. The genotype-specific decrease in PPT1 enzyme activity is likely due to a combination of an overall decrease in PPT1 protein level (due to decreased mRNA level) as well as production of truncated and nonfunctional PPT1 polypeptide products from the remaining pool of Cln1 mRNA. Decreased PPT1 activity is one of the pathological hallmarks of infantile NCL, and is often used as an early diagnostic marker in children with suspected disease.
The Cln1R151X mouse model exhibits significant accumulation of autofluorescent storage material within the cortex, thalamus and hippocampus, which is one of the definitive neuropathological hallmarks of infantile NCL disease (Fig. 3) (3,30,31). In addition, immunohistochemical staining for GFAP and CD68 showed reactive changes in astrocytes and microglia cell populations, respectively (Figs 4 and 5). Astrocytosis has been reported in almost all forms of human NCL (30,32). This process mediates how neurons respond to injury, and may also influence neuronal function through direct effects upon the synaptic environment (25). There is growing evidence that the activation of astrocytes within the thalamus precedes neuronal cell loss (25). However, it is still unclear whether astrocytosis directly affects neuronal cell loss in the cortex, or if it is a downstream consequence of the disease process. Much like astrocytosis, it remains unclear whether microglial activation represents regenerative or degenerative changes in the brain of Cln1R151X mice, but these changes are considered sensitive indicators of ongoing neuronal dysfunction (26). Astrocytosis seems to be less pronounced in 5-month-old Cln1R151X mice (Fig. 3) compared with 5-month-old Cln1−/− (Ppt1−/−) mice (26), a well-characterized mouse model of infantile NCL. The levels of microglial activation in the Cln1R151X and Cln1−/− mouse models are not directly comparable since data only from 7-month-old Cln1−/− mice are available (25–26).
Behavioral characterization of the Cln1R151X mouse model revealed significantly decreased exploratory activity in the dish test (Supplementary Material, Fig. S2), and significant motor deficits in the modified vertical pole test at both 3 and 5 months of age. In the rotarod test, while the motor coordination of 3-month-old Cln1R151X mice was impaired, at 5 months of age their rotarod performance was very similar to WT mice (Fig. 6). This may be due to the fact that the same mice were tested at 3 and 5 months of age, and thus, motor learning at 3 months could improve their performance in the repeated rotarod test at 5 months. Additionally, previous studies have shown that the vertical pole test, in which Cln1R151X mice displayed motor deficits at both 3 and 5 months, is more sensitive than the rotarod test to detect motor deficits (27,28). In comparison to the Cln1−/− (Ppt1−/−) mouse model of infantile NCL, the motor dysfunction appears earlier in Cln1R151X mice. Using constant speed rotarod testing in Cln1−/− (Ppt1−/−) mice, Macauley et al. (33) did not detect motor deficits at 3 months of age. Cln1−/− mice, however, showed a progressive decline in rotarod performance starting at 5 months (33). We have not tested Cln1R151X mice older than 5 months, and the differences in motor function test results between Cln1R151X and Cln1−/− mice could be due to the different genetic backgrounds (Cln1R151X on mixed 129S6/SvEv x C57BL/6J; Cln1−/− on pure C57BL/6J) and the different testing paradigms.
Along with initial characterization of the novel Cln1R151X mouse model, using both neuropathological and behavioral testing, we show how an initial proof-of-concept treatment with the read-through drug ataluren (PTC124) increases PPT1 enzyme activity and protein level in the brain and liver (Fig. 7). These findings are consistent with previous results using nonsense suppression therapy with both gentamicin and ataluren to increase PPT1 and TPP1 enzyme activity in several infantile and late-infantile NCL lymphoblast cell lines, respectively (20,34). In addition, it has been shown in the Cln1−/− mouse model of infantile NCL that restoring CLN1 (PPT1) enzyme activity in the brain to only 4–5% of the normal level results in a significant attenuation of neurodegeneration (29). Additional experiments need to be performed to optimize ataluren dosing to reliably penetrate the blood–brain barrier and to maintain therapeutically effective drug concentrations within the brain.
This study provides the initial characterization of the novel Cln1R151X mouse model for infantile NCL using molecular, neuropathological and behavioral testing. The role that NMD plays in the disease process is highlighted by the ability to target and interrupt mRNA degradation or increase premature stop codon read-through therapeutically, thus increasing production of full-length and potentially functional protein products. Current strategies that target this pathway involve the use of read-through compounds, such as the aminoglycosides (i.e. gentamicin), ataluren (PTC124) and RTC13 (19,35–38). The Cln1R151X mouse is a disease model to test stop codon read-through drug therapy in the central nervous system, a new therapeutic approach for many neurodegenerative and neurological disorders (39). We have shown how using nonsense suppression therapy with ataluren increases PPT1 enzyme activity and protein level in vivo using Cln1R151X mice, which supports further preclinical investigation of these drugs.
MATERIALS AND METHODS
Gene targeting in embryonic stem cells and generation of Cln1R151X mice
Generation of the Cln1R151X mouse model was performed by Applied StemCell, Inc. (Menlo Park, CA, USA). To summarize, the Cln1 gene-targeting construct with the point mutation (g.12805C>T) located within exon 5 and a downstream neo cassette flanked by tandem LoxP-Frt recombination sites was created. The Cln1 gene-targeting construct was electroporated into 129S6/SvEv x C57BL/6J hybrid embryonic stem (ES) cells. Approximately 300 G418-resistant ES colonies were selected and screened by short arm PCR screening (forward primer (CLN1-N1): 5′-TGC GAG GCC AGA GGC CAC TTG TGT AG-3′; reverse primer (CLN1-A1): 5′-TGG TTG CCT TGT GGT CAT CTT CTC-3′). Several targeted ES clones were identified and insertion of the point mutation (C>T) was confirmed by long arm PCR screening and sequencing (forward primer (CLN1-SQ1): 5′-AAC AGC CAC ACG TGC TGT ATG-3′; reverse primer (CLN1-N2): 5′-TTC CTC GTG CTT TAC GGT ATC G-3′). Correctly targeted ES clones were chosen for tetraploid blastocyst microinjection to generate a line of Cln1R151Xneo mice. Generation of the Cln1R151X allele was achieved by crossing Cln1R151Xneo mice with B6.C-Tg(CMV-cre)1Cgn/J mice (The Jackson Laboratory, Bar Harbor, ME, USA; Stock# 006054). Removal of the neo cassette was confirmed by sequencing DNA extracted from tail snips. Subsequent matings of cre-negative offspring to 129S6/SvEv mice were performed and confirmed by PCR. All offspring were genotyped using a custom TaqMan SNP qPCR assay (Life Technologies) (forward primer: 5′-TCC TAC CCA GGT GTC TTT GGA-3′; reverse primer: 5′-AGT CGC AGA TGT GAG AAC TCT CT-3′; wild type reporter probe: 5′-[VIC]-TCC CCC GAT GCC C-[MGB-NFQ]-3′; mutant reporter probe: 5′-[FAM]-TCC CCT GAT GCC C-[MGB-NFQ]-3′).
Animals
Cln1R151X mice were maintained on a mixed 129S6/SvEv x C57BL/6J genetic background, and hybrid 129S6/SvEv x C57BL/6J mice served as WT controls. Mice were housed in individually vented microisolator cages (4–5 mice/cage) with ad libitum access to food and water. Mice were fed with the Teklad Global 2918 diet (Harlan Laboratories, Indianapolis, IN, USA), and their drinking water was tap water. All procedures were carried out according to the guidelines of the Animal Welfare Act and NIH policies, and were approved by the Sanford Research Animal Care and Use Committee.
Molecular testing
Sample processing and handling
For sample collection, mice were anesthetized followed by transcardial perfusion and vascular rinse using ice-cold phosphate-buffered saline (PBS). All tissue samples were collected and processed in a similar manner, and stored at −80°C no longer than 2 months before total RNA extraction or total protein isolation.
Nucleic acid extraction
Total RNA was extracted from tissue samples with a Maxwell 16 LEV simplyRNA Cells Kit (Promega, Madison, WI, USA) using a Maxwell 16 Instrument (Promega), according to the manufacturer's instructions. Sample purity and yield were determined using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). All samples had A260/A280 values between 2.051 and 2.197, and yielded between 52 and 796 ng/μl total RNA, depending on the tissue type. RNA integrity was assessed as previously described (20).
Reverse transcription
Each sample was mass-normalized using ∼1000 ng of total RNA for cDNA synthesis using a High Capacity cDNA Reverse Transcription Kit (Life Technologies, Carlsbad, CA, USA), according to the manufacturer's instructions in a 96-well plate. The reaction conditions were as follows: 25°C for 10 min, 37°C for 120 min, 85°C for 5 min. Samples were diluted with molecular grade water to 10 ng/μl following reverse transcription. Samples were assessed for DNA contamination using reactions with no reverse transcriptase added. All samples were DNA-free and stored at −20°C until use.
Quantitative real-time polymerase chain reaction
Quantitative real-time polymerase chain reaction (qPCR) was performed for the Cln1 gene using TaqMan hydrolysis assays (Life Technologies; Part.# Mm00477078_m1). qPCR was performed for reference genes using TaqMan hydrolysis assays (Life Technologies) for B2m (Part.# Mm00437762_m1), Gapdh (Part.# Mm99999915_g1), Gusb (Part.# Mm00446962_g1) and Hgprt (Part.# Mm01318741_g1). Amplification, thermal cycling and fluorescence data collection were performed as previously described (20).
qPCR data analysis
Raw fluorescence data were analyzed as previously described (20) using REST-MCS software (40,41).
Protein isolation
Approximately 25–50 mg of each tissue sample was homogenized using a Ultra-Turrax T8 homogenizer with a Dispergierstation T8.10 stand (Thermo Fisher Scientific) in 400 µl lysis buffer containing 50 mm Tris–HCl, pH 7.4, 150 mm NaCl, 0.2% Triton X-100 and 300 mm NP-40, supplemented with 1:100 protease inhibitor cocktail and 1:1 000 PMSF. This was followed by pulse sonication and 30 min incubation on ice. Following a high-speed centrifugation (12 000g) for 10 min, the supernatant was collected and the total protein concentration was measured using the Pierce 660 nm Protein Assay (Thermo Fisher Scientific).
PPT1 enzyme assay
PPT1 enzyme activity was measured as previously described (15,20–23). Total protein samples isolated from Cln1WT/WT, Cln1WT/R151X and Cln1R151X/R151X tissues were used. To summarize, total protein was extracted from the cerebellum, cortex, striatum/thalamus, brainstem and liver. 10 µg total protein per well was then incubated in a mixture containing 0.64 mm 4-methylumbelliferyl-6-thiopalmitoyl-β-glucoside (MU-6S-palm- βGlc, Moscerdam Substrates, Netherlands), 15 mm dithiothreitol (DTT) in 5.1 mg/ml bovine serum albumin and 0.02% Na-azide, 0.375% (w/v) Triton X-100 in 2/1 chloroform/methanol, 0.1 U β-glucosidase from almonds (Sigma-Aldrich, St. Louis, MO, USA) in double-concentrated McIlvains phosphate/citric-acid buffer, pH 4.0. The total protein reaction mixture was then incubated for 1 h at 37°C. The reaction was stopped with the addition of 0.5 m NaHCO3/0.5 m Na2CO3 buffer, pH 10.7 with 0.025% Triton X-100 and fluorescence was measured using a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA, USA). Relative enzyme activity was estimated using total fluorescence minus background, and analyzed with an unpaired Student's t-test or one-way ANOVA with Bonferroni's Multiple Comparison Test using GraphPad Prism software version 5.04 (GraphPad Software, Inc., La Jolla, CA, USA).
PPT1 ELISA
PPT1 protein level was measured using a mouse PPT1 ELISA kit (Wuhan Huamei Biotech Co., Ltd, Wuhan, Hubei Province, China; Cat.#. CSB-EL018587MO) according to the manufacturer's instructions. Briefly, 1.0 μg of total protein from each sample was diluted in 100 μl of sample diluent and incubated in the coated assay plate for 2 h at 37°C, followed by brief washing and incubation with 100 μl of biotin-antibody for 1 h at 37°C. This was followed by washing and incubation with 100 μl of HRP-avidin for 1 h at 37°C and washing and incubation with 90 μl of TMB substrate for 15 min. Fifty microliters of Stop Solution was added and the plate was read at 450 nm with 540 nm subtraction using a SpectraMax M5 plate reader (Molecular Devices).
Behavioral testing
Male mice were used for behavioral testing. Mice were transported to the behavioral testing room where the lights had been dimmed. Mice were labeled on their tails for easy identification, weighed, and were allowed to accommodate to the room at least for 20 min before starting the behavioral tests. Mice were tested first in the dish test, then in the modified vertical pole test and finally in the tail suspension test. One day later the same mice were also tested in the rotarod test. All behavioral tests were carried out under dim light to keep the anxiety level of mice minimal. The same WT and Cln1R151X mice were tested at 3 and 5 months of age.
Dish test
The dish test measures the exploratory behavior of mice, though activity level and anxiety also affect the outcome of this test. The mouse was placed in a large plastic Petri dish (diameter: 15 cm; height: 1.5 cm), and the time until the mouse stepped out of the dish with all four legs was measured in a single trial. The time limit of the trial was 6 min.
Modified vertical pole test
With the vertical pole, the balance, spatial orientation and motor coordination of mice are tested. In the traditional vertical pole test [see, e.g., (42)], the mouse is placed on top of the pole head upward, and the time until it turns downward and climbs down to the base of the pole is measured. Since the majority of our disease model mice, including Cln1R151X mice, do not turn downward at all, we modified the vertical pole test so it starts with the climb down. The vertical pole was an all-thread metal rod (diameter: 1.27 cm; height: 66 cm), screwed to a 3.81-cm-thick plastic block (24.5 × 25.4 cm). The plastic block was covered with a 3.81-cm-thick green hunting seat cushion (nitrile rubber/PVC foam) to prevent the mice from injury when they fell from the pole. The height of the pole measured from the surface of the hunting seat cushion was 59 cm. The mouse was placed, head downward, on top of the pole, and the time until the mouse climbed down to the base of the pole was measured in five consecutive trials. After the 5th trial, the same mouse was placed, head upward, on top of the pole, and the time until the mouse turned completely downward was measured in four consecutive trials. Each climbing down and turning downward trial was terminated after 60 s to avoid exhaustion. If the mouse fell from the pole a trial result of 60 s was given. The time to climb down (sum of the five trials in seconds) and the time to turn downward (sum of the four trials in seconds) were calculated for each mouse.
Tail suspension test
This test measures the depressive-like behavior of mice (43). Rodents subjected to the short-term, inescapable stress of being suspended by their tail develop an immobile posture. Various antidepressant drugs reverse the immobility and promote the occurrence of escape-related behavior (44). Mice were suspended 45 cm above table level by taping their tails to Tygon tubing tightened to an extension support ring on an iron support stand. The duration of immobility (defined as the absence of all movement except for those required for respiration) was measured in 1 min bins for 6 min.
Rotarod test
The rotarod measures the ability of the mouse to maintain balance on a motor-driven, rotating rod. Thus, the fore- and hind limb motor coordination and balance can be analyzed. The rotarod test using two Rotamex-5 accelerating rotarod instruments (Columbus Instruments, Columbus, OH, USA) was performed as described previously (45), with some modifications. The start speed of the rotarod was 0 rpm and the acceleration was set to 0.2 rpm/s. The cut-off time was set at 240 s but all mice fell from the rotarod way before the set cut-off time. Mice were trained on the rotarod in three consecutive runs. Following training, mice rested for 1.5 h and then were tested on the rotarod in three test trials each consisting of three consecutive runs, with 15 min of rest between the trials. The average latency to fall from the rotating rod in the test trials (average of the 9 runs in the 3 trials) was calculated for each mouse.
Behavioral testing statistical analysis
Statistical analysis was performed using GraphPad Prism 5. Data sets from the dish test and vertical pole test did not pass the normality test (alpha level 0.05), and therefore, the non-parametric Mann–Whitney test was used for comparison in these behavioral tests. Since all data sets from the tail suspension test and rotarod test passed the normality test (alpha level 0.05), unpaired, two-tailed t-test was applied for comparison in these behavioral tests. To compare the time courses in the tail suspension test, 2-way ANOVA with Bonferroni's post-test was used. Alpha level was 0.05 in all statistical tests.
Histological processing
Tissue cryosectioning and slide preparation was performed by the Molecular Pathology Core at Sanford Research. Briefly, brain tissues were fixed in 4% paraformaldehyde and cryoprotected with 30% sucrose solution. The tissues were embedded in Tissue-Tek OCT freezing medium for sectioning, and then serially sectioned in a 1:10 series at 25 μm.
Quantification of autofluorescent storage material
Autofluorescence was measured from unstained coronal sections of wild-type and Cln1R151X mice as previously described (25,26). All RGB images were captured from the cortex, thalamus and hippocampus using the FITC filter set, with a Photometrics CoolSNAP MYO CCD camera (Tucson, AZ, USA), mounted on a Nikon Ni-E microscope (Melville, NY, USA) using a 20× objective and stored on a network server.
Immunohistochemical staining for glial markers
To assess the degree of astrocytosis and microglial activation, a one in ten series of sections was immunohistochemically stained for glial fibrillary acidic protein (GFAP, astrocytes) and the activated microglial marker CD68, using a similar method as previously published (25,26). Briefly, pre-mounted 25 µm sections were incubated overnight with the primary antibody (polyclonal rabbit anti-cow GFAP, DAKO, Carpinteria, CA, USA, 1:1000; and monoclonal rat anti-mouse CD68, AbD Serotec, Raleigh, NC, USA, 1:250). Sections were then rinsed with PBS and incubated with the appropriate secondary antibody (biotinylated goat anti-rabbit, Vector Laboratories, Burlingame, CA, USA, 1:400; and biotinylated goat anti-rat, Vector Laboratories, 1:400) followed by avidin-biotin-peroxidase complex (Vector Laboratories, PK-6100). Immunoreactivity was visualized by a standard DAB reaction (Vector Laboratories, SK-4100); slides were taken through a graded series of ethanol treatments (50, 75, 80, 95, 100, 100%), cleared with Histoclear (National Diagnostics, Atlanta, GA, USA) and coverslipped (Fisher Scientific, Cat.# 12–548-5P).
Quantitative analysis of glial phenotype
The expression of the glial markers GFAP and CD68 was measured and used to compare glial activation between wild type and Cln1R151X genotypes. A modified thresholding analysis was used according to Bible et al. (25). Briefly, 30 non-overlapping images from ten sections were captured from the cortex, thalamus and hippocampus for each animal. All RGB images were captured using brightfield microscopy with a Nikon Ni-E microscope using a 20× objective and stored on a network server. All parameters of image capture including lamp intensity, video camera setup and calibration were maintained constant. Images were subsequently analyzed using NIS-Elements AR Analysis software version 4.13. Foreground immunostaining was defined according to averaging of the highest and lowest immunoreactivities within a sample population for a given immunohistochemical marker and measured on a scale from 0 (100% transmitted light) to 255 (0% transmitted light). This threshold setting was then applied as a constant to all subsequent images analyzed per reagent used to obtain the sum intensity of the pixels from each image. Macros were used to obtain and transfer the data to a Microsoft Excel spreadsheet. Statistical significance was determined with an unpaired Student's t-test with Welch's correction using GraphPad Prism software version 5.04 (GraphPad Software, Inc.). Data were plotted graphically as the mean immunoreactivity of each genotype ± SEM.
Treatment with ataluren (PTC124)
Male mice were randomly assigned to either a treatment group or vehicle control group. Six to eight mice per group were treated with 10 or 100 mg/kg ataluren (Selleck Chemicals, Houston, TX, USA) dissolved in PBS containing DMSO (2% for 10 mg/kg and 20% for 100 mg/kg) and (2-hydroxypropyl)-β-cyclodextrin (22%) (Cat.# H5784, Sigma-Aldrich) via intraperitoneal (i.p.) injections four times daily for 2 consecutive days. Six to eight control mice were treated with the vehicle of the drug: PBS containing DMSO (2% or 20%) and (2-hydroxypropyl)-β-cyclodextrin (22%). Immediately following the last injection on the second day, tissues were collected and stored at −80°C for further use.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported in part by Hayden's Hope and Sanford Health. The Molecular Pathology Core is supported by an NIH, NIGMS, Center of Biomedical Research Excellence (COBRE) grant (1P20RR024219-01A2) awarded to the Cancer Biology Research Center at Sanford Research.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Claire Evans for preparing histological specimens for staining, and Jill Weimer and Helen Magee for advice guiding the histological processing and analysis. We also thank Sarah Radel and Sarah Brink for maintaining our mouse colony.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Haltia M. The neuronal ceroid-lipofuscinoses. J. Neuropathol. Exp. Neurol. 2003;62:1–13. doi: 10.1093/jnen/62.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Jalanko A., Braulke T. Neuronal ceroid lipofuscinoses. Biochim. Biophys. Acta. 2009;1793:697–709. doi: 10.1016/j.bbamcr.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 3.Santavuori P. Neuronal ceroid-lipofuscinoses in childhood. Brain Dev. 1988;10:80–83. doi: 10.1016/s0387-7604(88)80075-5. [DOI] [PubMed] [Google Scholar]
- 4.Kousi M., Lehesjoki A.E., Mole S.E. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum. Mutat. 2012;33:42–63. doi: 10.1002/humu.21624. [DOI] [PubMed] [Google Scholar]
- 5.Sondhi D., Rosenberg J.B., Van de Graaf B.G., Kaminsky S.M., Crystal R.G. Advances in the treatment of neuronal ceroid lipofuscinosis. Expert Opin. Orphan Drugs. 2013;1:951–975. doi: 10.1080/21678707.2019.1684258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Williams R.E., Mole S.E. New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology. 2012;79:183–191. doi: 10.1212/WNL.0b013e31825f0547. [DOI] [PubMed] [Google Scholar]
- 7.Bonsignore M., Tessa A., Di Rosa G., Piemonte F., Dionisi-Vici C., Simonati A., Calamoneri F., Tortorella G., Santorelli F.M. Novel CLN1 mutation in two Italian sibs with late infantile neuronal ceroid lipofuscinosis. Eur. J. Paediatr. Neurol. 2006;10:154–156. doi: 10.1016/j.ejpn.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 8.Das A.K., Becerra C.H., Yi W., Lu J.Y., Siakotos A.N., Wisniewski K.E., Hofmann S.L. Molecular genetics of palmitoyl-protein thioesterase deficiency in the U.S. J. Clin. Invest. 1998;102:361–370. doi: 10.1172/JCI3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simonati A., Tessa A., Bernardina B.D., Biancheri R., Veneselli E., Tozzi G., Bonsignore M., Grosso S., Piemonte F., Santorelli F.M. Variant late infantile neuronal ceroid lipofuscinosis because of CLN1 mutations. Pediatr. Neurol. 2009;40:271–276. doi: 10.1016/j.pediatrneurol.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 10.Waliany S., Das A.K., Gaben A., Wisniewski K.E., Hofmann S.L. Identification of three novel mutations of the palmitoyl-protein thioesterase-1 (PPT1) gene in children with neuronal ceroid-lipofuscinosis. Hum. Mutat. 2000;15:206–207. doi: 10.1002/(SICI)1098-1004(200002)15:2<206::AID-HUMU14>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 11.Wisniewski K.E., Connell F., Kaczmarski W., Kaczmarski A., Siakotos A., Becerra C.R., Hofmann S.L. Palmitoyl-protein thioesterase deficiency in a novel granular variant of LINCL. Pediatr. Neurol. 1998;18:119–123. doi: 10.1016/s0887-8994(97)00173-2. [DOI] [PubMed] [Google Scholar]
- 12.Mitchison H.M., Hofmann S.L., Becerra C.H., Munroe P.B., Lake B.D., Crow Y.J., Stephenson J.B., Williams R.E., Hofman I.L., Taschner P.E., et al. Mutations in the palmitoyl-protein thioesterase gene (PPT; CLN1) causing juvenile neuronal ceroid lipofuscinosis with granular osmiophilic deposits. Hum. Mol. Genet. 1998;7:291–297. doi: 10.1093/hmg/7.2.291. [DOI] [PubMed] [Google Scholar]
- 13.Perez-Poyato M.S., Mila Recansens M., Ferrer Abizanda I., Montero Sanchez R., Rodriguez-Revenga L., Cusi Sanchez V., Garcia Gonzalez M.M., Domingo Jimenez R., Camino Leon R., Velazquez Fragua R., et al. Juvenile neuronal ceroid lipofuscinosis: clinical course and genetic studies in Spanish patients. J. Inherit. Metab. Dis. 2011;34:1083–1093. doi: 10.1007/s10545-011-9323-7. [DOI] [PubMed] [Google Scholar]
- 14.Ramadan H., Al-Din A.S., Ismail A., Balen F., Varma A., Twomey A., Watts R., Jackson M., Anderson G., Green E., et al. Adult neuronal ceroid lipofuscinosis caused by deficiency in palmitoyl protein thioesterase 1. Neurology. 2007;68:387–388. doi: 10.1212/01.wnl.0000252825.85947.2f. [DOI] [PubMed] [Google Scholar]
- 15.van Diggelen O.P., Thobois S., Tilikete C., Zabot M.T., Keulemans J.L., van Bunderen P.A., Taschner P.E., Losekoot M., Voznyi Y.V. Adult neuronal ceroid lipofuscinosis with palmitoyl-protein thioesterase deficiency: first adult-onset patients of a childhood disease. Ann. Neurol. 2001;50:269–272. doi: 10.1002/ana.1103. [DOI] [PubMed] [Google Scholar]
- 16.Wang R., Borazjani A., Matthews A.T., Mangum L.C., Edelmann M.J., Ross M.K. Identification of palmitoyl protein thioesterase 1 in human THP1 monocytes and macrophages and characterization of unique biochemical activities for this enzyme. Biochemistry. 2013;52:7559–7574. doi: 10.1021/bi401138s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bellizzi J.J., III, Widom J., Kemp C., Lu J.Y., Das A.K., Hofmann S.L., Clardy J. The crystal structure of palmitoyl protein thioesterase 1 and the molecular basis of infantile neuronal ceroid lipofuscinosis. Proc. Natl Acad. Sci. USA. 2000;97:4573–4578. doi: 10.1073/pnas.080508097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ohno K., Saito S., Sugawara K., Suzuki T., Togawa T., Sakuraba H. Structural basis of neuronal ceroid lipofuscinosis 1. Brain Dev. 2010;32:524–530. doi: 10.1016/j.braindev.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 19.Brooks D.A., Muller V.J., Hopwood J.J. Stop-codon read-through for patients affected by a lysosomal storage disorder. Trends Mol. Med. 2006;12:367–373. doi: 10.1016/j.molmed.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 20.Miller J.N., Chan C.H., Pearce D.A. The role of nonsense-mediated decay in neuronal ceroid lipofuscinosis. Hum. Mol. Genet. 2013;22:2723–2734. doi: 10.1093/hmg/ddt120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Voznyi Y.V., Keulemans J.L., Mancini G.M., Catsman-Berrevoets C.E., Young E., Winchester B., Kleijer W.J., van Diggelen O.P. A new simple enzyme assay for pre- and postnatal diagnosis of infantile neuronal ceroid lipofuscinosis (INCL) and its variants. J. Med. Genet. 1999;36:471–474. [PMC free article] [PubMed] [Google Scholar]
- 22.van Diggelen O.P., Keulemans J.L., Winchester B., Hofman I.L., Vanhanen S.L., Santavuori P., Voznyi Y.V. A rapid fluorogenic palmitoyl-protein thioesterase assay: pre- and postnatal diagnosis of INCL. Mol. Genet. Metab. 1999;66:240–244. doi: 10.1006/mgme.1999.2809. [DOI] [PubMed] [Google Scholar]
- 23.Miller J.N., Pearce D.A. A novel c.776_777insA mutation in CLN1 leads to infantile neuronal ceroid lipofuscinosis. J. Child Neurol. 2013;28:1106–1111. doi: 10.1177/0883073813494267. [DOI] [PubMed] [Google Scholar]
- 24.Gupta P., Soyombo A.A., Atashband A., Wisniewski K.E., Shelton J.M., Richardson J.A., Hammer R.E., Hofmann S.L. Disruption of PPT1 or PPT2 causes neuronal ceroid lipofuscinosis in knockout mice. Proc. Natl Acad. Sci. USA. 2001;98:13566–13571. doi: 10.1073/pnas.251485198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bible E., Gupta P., Hofmann S.L., Cooper J.D. Regional and cellular neuropathology in the palmitoyl protein thioesterase-1 null mutant mouse model of infantile neuronal ceroid lipofuscinosis. Neurobiol. Dis. 2004;16:346–359. doi: 10.1016/j.nbd.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 26.Kielar C., Maddox L., Bible E., Pontikis C.C., Macauley S.L., Griffey M.A., Wong M., Sands M.S., Cooper J.D. Successive neuron loss in the thalamus and cortex in a mouse model of infantile neuronal ceroid lipofuscinosis. Neurobiol. Dis. 2007;25:150–162. doi: 10.1016/j.nbd.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abramow-Newerly W., Lipina T., Abramow-Newerly M., Kim D., Bechard A.R., Xie G., Clapcote S.J., Roder J.C. Methods to rapidly and accurately screen a large number of ENU mutagenized mice for abnormal motor phenotypes. Amyotroph. Lateral Scler. 2006;7:112–118. doi: 10.1080/14660820500443000. [DOI] [PubMed] [Google Scholar]
- 28.Berman A.E., Chan W.Y., Brennan A.M., Reyes R.C., Adler B.L., Suh S.W., Kauppinen T.M., Edling Y., Swanson R.A. N-acetylcysteine prevents loss of dopaminergic neurons in the EAAC1−/− mouse. Ann. Neurol. 2011;69:509–520. doi: 10.1002/ana.22162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Griffey M.A., Wozniak D., Wong M., Bible E., Johnson K., Rothman S.M., Wentz A.E., Cooper J.D., Sands M.S. CNS-directed AAV2-mediated gene therapy ameliorates functional deficits in a murine model of infantile neuronal ceroid lipofuscinosis. Mol. Ther. 2006;13:538–547. doi: 10.1016/j.ymthe.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 30.Haltia M., Rapola J., Santavuori P. Infantile type of so-called neuronal ceroid-lipofuscinosis. Histological and electron microscopic studies. Acta Neuropathol. 1973;26:157–170. doi: 10.1007/BF00697751. [DOI] [PubMed] [Google Scholar]
- 31.Santavuori P., Haltia M., Rapola J., Raitta C. Infantile type of so-called neuronal ceroid-lipofuscinosis. 1. A clinical study of 15 patients. J. Neurol. Sci. 1973;18:257–267. doi: 10.1016/0022-510x(73)90075-0. [DOI] [PubMed] [Google Scholar]
- 32.Haltia M., Rapola J., Santavuori P., Keranen A. Infantile type of so-called neuronal ceroid-lipofuscinosis. 2. Morphological and biochemical studies. J. Neurol. Sci. 1973;18:269–285. doi: 10.1016/0022-510x(73)90076-2. [DOI] [PubMed] [Google Scholar]
- 33.Macauley S.L., Wozniak D.F., Kielar C., Tan Y., Cooper J.D., Sands M.S. Cerebellar pathology and motor deficits in the palmitoyl protein thioesterase 1-deficient mouse. Exp. Neurol. 2009;217:124–135. doi: 10.1016/j.expneurol.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sarkar C., Zhang Z., Mukherjee A.B. Stop codon read-through with PTC124 induces palmitoyl-protein thioesterase-1 activity, reduces thioester load and suppresses apoptosis in cultured cells from INCL patients. Mol. Genet. Metab. 2011;104:338–345. doi: 10.1016/j.ymgme.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Finkel R.S. Read-through strategies for suppression of nonsense mutations in Duchenne/Becker muscular dystrophy: aminoglycosides and ataluren (PTC124) J. Child Neurol. 2010;25:1158–1164. doi: 10.1177/0883073810371129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee H.L., Dougherty J.P. Pharmaceutical therapies to recode nonsense mutations in inherited diseases. Pharmacol. Ther. 2012;136:227–266. doi: 10.1016/j.pharmthera.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 37.Welch E.M., Barton E.R., Zhuo J., Tomizawa Y., Friesen W.J., Trifillis P., Paushkin S., Patel M., Trotta C.R., Hwang S., et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 38.Kayali R., Ku J.M., Khitrov G., Jung M.E., Prikhodko O., Bertoni C. Read-through compound 13 restores dystrophin expression and improves muscle function in the mdx mouse model for Duchenne muscular dystrophy. Hum. Mol. Genet. 2012;21:4007–4020. doi: 10.1093/hmg/dds223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gunn G., Dai Y., Du M., Belakhov V., Kandasamy J., Schoeb T.R., Baasov T., Bedwell D.M., Keeling K.M. Long-term nonsense suppression therapy moderates MPS I-H disease progression. Mol. Genet. Metab. 2014;111:374–381. doi: 10.1016/j.ymgme.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pfaffl M.W., Horgan G.W., Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iwamoto T., Okumura S., Iwatsubo K., Kawabe J., Ohtsu K., Sakai I., Hashimoto Y., Izumitani A., Sango K., Ajiki K., et al. Motor dysfunction in type 5 adenylyl cyclase-null mice. J. Biol. Chem. 2003;278:16936–16940. doi: 10.1074/jbc.C300075200. [DOI] [PubMed] [Google Scholar]
- 43.Taniguchi S., Nakazawa T., Tanimura A., Kiyama Y., Tezuka T., Watabe A.M., Katayama N., Yokoyama K., Inoue T., Izumi-Nakaseko H., et al. Involvement of NMDAR2A tyrosine phosphorylation in depression-related behaviour. EMBO J. 2009;28:3717–3729. doi: 10.1038/emboj.2009.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cryan J.F., Mombereau C., Vassout A. The tail suspension test as a model for assessing antidepressant activity: review of pharmacological and genetic studies in mice. Neurosci. Biobehav. Rev. 2005;29:571–625. doi: 10.1016/j.neubiorev.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 45.Kovács A.D., Pearce D.A. Location- and sex-specific differences in weight and motor coordination in two commonly used mouse strains. Sci. Rep. 2013;3:2116. doi: 10.1038/srep02116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.