

Abstract
Primary cilia are sensory organelles present on most mammalian cells. The assembly and maintenance of primary cilia are facilitated by intraflagellar transport (IFT), a bidirectional protein trafficking along the cilium. Mutations in genes coding for IFT components have been associated with a group of diseases called ciliopathies. These genetic disorders can affect a variety of organs including the retina. Using whole exome sequencing in three families, we identified mutations in Intraflagellar Transport 172 Homolog [IFT172 (Chlamydomonas)] that underlie an isolated retinal degeneration and Bardet–Biedl syndrome. Extensive functional analyses of the identified mutations in cell culture, rat retina and in zebrafish demonstrated their hypomorphic or null nature. It has recently been reported that mutations in IFT172 cause a severe ciliopathy syndrome involving skeletal, renal, hepatic and retinal abnormalities (Jeune and Mainzer-Saldino syndromes). Here, we report for the first time that mutations in this gene can also lead to an isolated form of retinal degeneration. The functional data for the mutations can partially explain milder phenotypes; however, the involvement of modifying alleles in the IFT172-associated phenotypes cannot be excluded. These findings expand the spectrum of disease associated with mutations in IFT172 and suggest that mutations in genes originally reported to be associated with syndromic ciliopathies should also be considered in subjects with non-syndromic retinal dystrophy.
INTRODUCTION
Most mammalian cells contain primary cilia, non-motile microtubule-based organelles, which are involved in cellular signaling. Mutations in genes coding for ciliary proteins lead to ciliopathies, rare genetic disorders that may affect one or more of the following organs: retina, central nervous system, olfactory epithelium, heart, liver, kidney, skeletal system, gonads and adipose tissue (1). Based on the presence of particular symptoms, ciliopathies are further subdivided into subtypes. Severe manifestations of ciliopathy include Mainzer-Saldino syndrome (MZSDS [MIM 266920]) and Jeune asphyxiating thoracic dystrophy (JATD [MIM 208500]). Individuals with these disorders have severe skeletal abnormalities including constricted thoracic cage in addition to renal, hepatic, brain and retinal anomalies. Respiratory insufficiency caused by the narrow thorax and/or major organ failures often lead to early-childhood death in these individuals (2). A milder form of ciliopathy, Bardet–Biedl syndrome (BBS [MIM 209900]) is characterized by a retinal degeneration, post-axial polydactyly, obesity, mental retardation, renal dysfunction and hypogonadism in males (3). Secondary features such as liver and heart dysfunction, diabetes or speech anomalies may additionally be present (1,3). Certain cases of isolated retinal degenerations may also be regarded as ciliopathies if the genes that are mutated encode proteins functioning in photoreceptor sensory cilia, such as RP1 (MIM 603937) (4,5), RPGR (MIM 312610) (6), LCA5 (MIM 611408) (7), RPGRIP1 (MIM 605446) (8), FAM161A (MIM 613596) (9,10) or KIZ (MIM 615757) (11), among others.
Most of the genes involved in ciliopathies code for proteins expressed at the basal body and transition zone of the cilia or are involved in intraflagellar transport (IFT) (1,12). IFT is a bidirectional transport process essential for the assembly and the maintenance of cilia as well as protein trafficking between the cell body and cilia. Kinesin-2 and a 14-protein IFT-complex B are thought to mediate anterograde transport (base to tip); whereas dynein-2 and a 6-protein IFT-complex A drive retrograde transport (tip to base) (13–15). Mutations in all complex A and two complex B proteins, including the recently published IFT172 (MIM 607386) (16), are associated with various severe skeleto-renal disorders with retinal involvement in some cases (17–24).
Here, we report four affected subjects from three families with mutations in IFT172 who have a non-syndromic retinitis pigmentosa (RP, MIM 268000), also known as rod-cone dystrophy, or BBS. We describe functional studies of the identified variants in cell culture and in vivo, giving evidence for the pathogenicity of these new mutations. These findings broaden the IFT172-disease spectrum, show that genes associated with syndromic ciliopathies should be considered in subjects with non-syndromic disease and also suggest that the primary IFT172 mutations alone are not sufficient to explain the wide range of phenotypes.
RESULTS
Whole exome sequencing identifies mutations in IFT172 causing Bardet–Biedl like ciliopathy and non-syndromic RP
Two affected sisters from family 1 (Fig. 1A, subjects II.1 and II.2) presented at the Ophthalmology-Genetics Clinic at The Children's Hospital of Philadelphia with retinal degeneration (Fig. 1B–E, and Supplementary Material, Fig. S1) and other systemic features suggesting a Bardet–Biedl like ciliopathy. Both sisters had elevated body-mass index (BMI) indicating obesity (BMI of 43.2 in II.1 and 36.9 in II.2), and significantly higher than their parents (BMI of 29.3 in I.1 and 29.2 in I.2). Both subjects had a history of delayed speech development and elevated liver transaminases. One of the sisters (subject II.1) in addition had bilateral post-axial cutaneous polydactyly and pancreatitis (Table 1). No skeletal malformations were observed in the two subjects. After excluding mutations in 12 BBS genes known at the time (BBS1-12), the affected sisters and the unaffected parents were investigated genetically by whole exome sequencing (WES). After applying stringent variant filtering (minor allele frequency of <0.15%), we found two likely disease-causing mutations in IFT172 (c.1525-1G>A; c.4701C>A p.H1567Q) co-segregating in the family (Fig. 1A). The p.H1567Q missense change had never been seen before in the human genetic variant databases of dbSNP and Exome Variant Server (EVS) (25) and the c.1525-1G>A splice-site change had been seen in 1 out of 13 005 alleles (Table 2). The c.1525-1G>A change affects the essential splice acceptor site of exon 16 (Fig. 2), which is predicted to lead to the abolition of the splice site and exon skipping (26).
Figure 1.
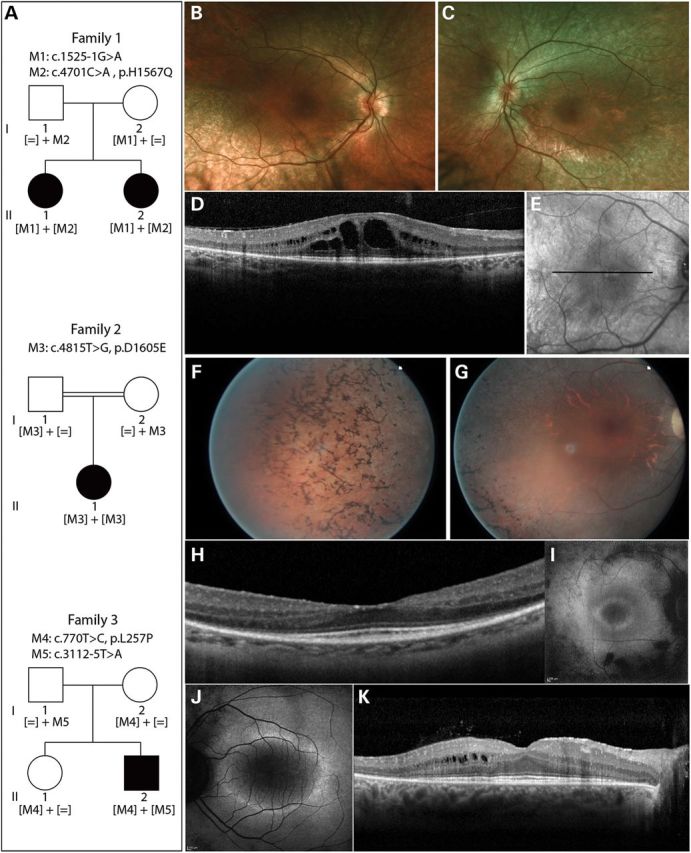
Pedigrees of subjects with IFT172 mutations and their phenotypes. (A) Pedigree information and segregation of IFT172 variants in the three families. (B and C) Fundus photographs of subject II.2 from family 1, indicating attenuation of blood vessels and RPE atrophy. (D) Horizontal optical coherence tomography (OCT) scan image through the center of the macula of subject II.2 (Family 1), showing thinning of the outer retina and macular cystic edema. (E) Infrared image of the fundus depicting the area where the OCT image was taken in E. (F and G) Fundus photographs of the right eye of subject II.1 from family 2, showing bone-spicule pigmentation in the periphery of the retina (F), attenuated blood vessels and optic disc pallor (G). (H) Horizontal OCT scan image through the center of the macula of the right eye of subject II.1 (family 2), showing thinning of the outer retina. (I and J) Fundus autofluorescence image of subject II.1 from family 2 (I) and subject II.2 from family 3 (J), depicting a perifoveal ring of increased autofluorescence and loss of autofluorescence outside the vascular arcades. (K) Horizontal OCT scan photograph of subject II.2 from family 3, showing an absent photoreceptor layer outside the macular region and cysts in the inner nuclear layer.
Table 1.
Clinical table of subjects with IFT172 mutations
| Subject | Clinical center | Genotype | Disease onset | Age at the last visit | Retinal disease | Polydactyly/skeletal anomalies | Liver disease | Obesity | Other features |
|---|---|---|---|---|---|---|---|---|---|
| Family 1, II.1 | CHOP, USA | c.1525-1G>A | Night blindness in the first decade | 15 | RP with atrophic changes in the macula | Bilateral post-axial cutaneous polydactyly | Elevated transaminases |
Yes (BMI = 43.2) | Hypercholesterolemia, pancreatitis, speech abnormalities in childhood (initial consonant omission) |
| c.4701C>A p.H1567Q | |||||||||
| Family 1, II.2 | CHOP, MEEI, USA | c.1525-1G>A | Night blindness in the first decade | 22 | RP with granularities and cysts in the macula | ND | Elevated transaminases |
Yes (BMI = 36.9) | Speech abnormalities in childhood (initial consonant omission) |
| c.4701C>A p.H1567Q | |||||||||
| Family 2, II.1 | Hôpital XV-XX, France | Homozygous c.4815T>G p.D1605E | Night blindness in the second decade | 33 | RP with lamellar macular hole on the left and optic nerve drusen visible on the right eye | ND | NT | No | CD4 lymphopenia from unknown origin with recurrent pneunompathies and ENT infections regularly treated with antibiotics |
| Family 3, II.2 | REH, Netherlands | c.770T>C; p.L257P | Night blindness in the first decade, RP diagnosis at 23 | 38 | RP with preserved foveal lamination, epiretinal membrane, cysts in the macula and optic nerve drusen | History of scoliosis at age 11–12, never treated, no back problems | NT | No | None |
| c.3112-5T>A |
Subjects were seen in four clinical centers: CHOP (Ophthalmology-Genetics Clinic at the Children's Hospital of Philadelphia); MEEI (Massachusetts Eye and Ear Infirmary); Hôpital XV-XX CIC503 (Centre d'Investigation Clinique 503); REH (Rotterdam Eye Hospital). Clinical diagnosis of subjects II.1 and II.2 from family 1 were consistent with BBS. All subjects were of Caucasian origin and none of them showed any signs of renal disease. The mutation nomenclature is based on transcript NM_015662.1 and A from the ATG initiation codon is designated as position 1. ND, not detected; NT, not tested.
Table 2.
New mutations in IFT172
| cDNA change | Effect | EVS frequency | Polyphen-2 | SIFT | Provean | Mutation Taster | PhyloP | PhastCons | Protein domain | Splicing prediction/outcome |
|---|---|---|---|---|---|---|---|---|---|---|
| c.770T>C | p.L257P | Absent | Probably damaging | Damaging | Deleterious | Disease causing | 4.903 | 0.996 | WD40 repeat | – |
| c.1525-1G>A | Essential splice site | 1/13005 | – | – | – | Disease causing | 5.789 | 1.000 | – | Exon skipping |
| c.3112-5T>A | Extended splice site | Absent | – | – | Deleterious (Q insertion) | Disease causing (Q insertion) | 1.018 | 0.097 | TPR domain | Insertion of Q in a subset of transcripts (p.K1037_E1038insQ, lymphocytes) or intron retention (mini-gene) |
| c.4701C>A | p.H1567Q | Absent | Probably damaging | Damaging | Deleterious | Disease causing | 4.94 | 1.000 | TPR domain | – |
| c.4815T>G | p.D1605E | Absent | Benign | Tolerated | Neutral | Disease causing | −0.019 | 0.99 | – | – |
The mutation nomenclature is based on transcript NM_015662.1 and A from the ATG initiation codon is designated as position 1. EVS frequency denotes the frequency of the mutant alleles in the Exome Variant Server made available by NHLBI Exome Sequencing Project (http://eversusgs.washington.edu/EVS/). SIFT and Provean mutation prediction tools were accessed at http://provean.jcvi.org/index.php, and Polyphen-2 at http://genetics.bwh.harvard.edu/pph2/index.shtml, MutationTaster at http://www.mutationtaster.org/index.html. PhyloP and phastCons scores for the nucleotide substitutions were taken from MutationTaster. All the databases were accessed in May 2014.
Figure 2.
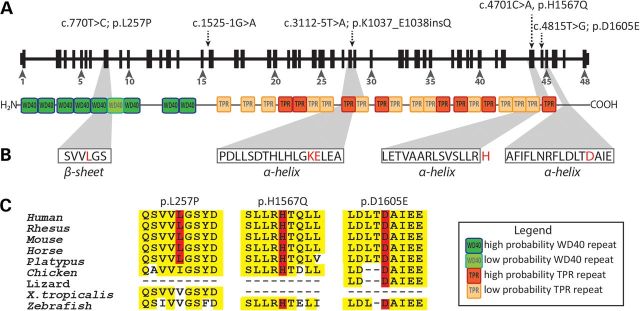
IFT172 mutations in BBS and rod-cone dystrophy subjects. (A) New mutations indicated on the gene and protein structures. Protein domains were taken from Taschner et al. (13) (WD40: WD40 repeat domain, TPR: tetratricopeptide repeats). (B) IFT172 protein secondary structures predicted by REPPER program (PSIPRED and COILS algorithms) in the Bioinformatics toolkit at the Max-Plank Institute for Developmental Biology (http://toolkit.tuebingen.mpg.de/repper). The mutated residues are indicated in red. Histidine (H) lies just outside of a predicted α-helix. (C) Conservation of missense changes found in this study, the sequences were taken from UCSC genome browser (http://genome.ucsc.edu/).
The affected subject from a consanguineous family 2 presented at the Center of Clinical Investigation at the Quinze-Vingts Hospital in Paris with RP of moderate severity and no other systemic symptoms (Table 1 and Fig. 1A, F–I). After exclusion of known mutations with arRP mutation microarray (ASPER Ophthalmics, Tartu, Estonia) (27,28) and Sanger sequencing of a major RP-associated gene, EYS (29) in the index subject, the three available family members (index and his unaffected parents) were analyzed by homozygosity mapping (30). The index subject's genome contained 23 homozygous regions ranging from 0.54 to 34.85 Mb, summing up to the total of 211 Mb. Six genes known to be implicated in RP were present in the homozygous regions [RPE65 (MIM 180069) (31,32), ABCA4 (MIM 601691) (33–35), C2orf71 (MIM 613425) (36,37), ZNF513 (MIM 613598) (38), CLRN1 (MIM 606397) (39) and PDE6A (MIM 180071) (40)], all of which were excluded for mutations in the exons and flanking intronic regions by Sanger sequencing. WES on DNA samples from all available family members and stringent filtering of the sequence data resulted in the identification of the homozygous c.4815T>G variant in exon 44 of IFT172 leading to a p.D1605E substitution. This change was absent in dbSNP and EVS databases (Table 2). The IFT172 gene was located in the second largest homozygous region of 20.5 Mb on chromosome 2.
An isolated case from family 3 (Fig. 1A, subject II.2) presented with typical signs of non-syndromic RP (Fig. 1J and K) at the Rotterdam Eye Hospital. Only after a further clinical interview, a history of scoliosis in early teenage years was revealed, though it is not clear if the two phenotypes are related (Table 1). Previously, targeted next-generation sequencing of 110 genes known to be associated with non-syndromic retinal dystrophy had not yielded any causative variants (41). WES in the affected individual, followed by stringent filtering of variants identified compound heterozygous mutations in IFT172 to be the most likely variants underlying RP in this individual (Fig. 1A). The first change is a missense variant in exon 8 (c.770T>C; p.L257P), whereas the second change is located in the 3′- splice site of exon 29 (c.3112-5T>A). Both changes were absent in genomic variant databases (Table 2). According to the online prediction program BayNAGNAG (42) and Alamut® (Interactive Biosoftware, Rouen, France), the c.3112-5T>A variant will affect splicing by creating a new splice acceptor site three nucleotides upstream of the original one (Supplementary Material, Fig. S2A). To confirm this splicing defect in the affected subject, RNA was isolated from fresh blood for reverse transcription polymerase chain reaction (RT-PCR) analysis. Following cDNA synthesis, amplification of IFT172 cDNA, cloning of the PCR products and Sanger sequence analysis of the clones, two mRNA products were identified that corresponded to the use of both the original and the new splice site generated by the c.3112-5T>A change (Supplementary Material, Fig. S2B). Of the sequenced clones, 20% indicated the usage of the alternative splice site, which was due to the presence of transcripts from the second allele and also to the fact that the alternative splice site was used only in a subset of transcripts harboring the c.3112-5T>A change. The same analysis of IFT172 transcripts from control lymphoblasts showed normal splicing in all analyzed clones. At the protein level, the use of the alternative splice site generated by the c.3112-5T>A change is predicted to lead to the insertion of one amino acid (p.K1037_E1038insQ). In parallel, to check the effect of the c.3112-5T>A variant on splicing, a mini-gene splicing assay was performed using a mini-gene construct of IFT172 exons 28–30 expressed in the human embryonic kidney (HEK) 293 cells. The results obtained from this study indicated that the majority of the mini-gene transcripts harboring the c.3112-5T>A change retained intron 28, in contrast to wild-type (WT) mini-gene transcripts which were spliced correctly in the majority of cases (Supplementary Material, Fig. S2C).
With a combination of Sanger sequencing and next-generation sequencing (NGS) approaches, 343 additional patients with syndromic and non-syndromic RP were screened for mutations in IFT172. In this screen, we have not identified additional patients with mutations in this gene.
IFT172 codes for the largest IFT protein, of 1749 residues with nine predicted N-terminal WD40 domains and 21 predicted C-terminal tetratricopeptide repeats (TPR) (13) (Fig. 2A). The most N-terminal mutation identified in this study (p.L257P) is predicted to affect the sixth WD40 repeat by changing the β-sheet secondary structure (PSIPRED analysis (43)) (Fig. 2B, Supplementary Material, Fig. S3), which may alter protein–protein interactions led by these domains (44–46). Leucine 257 is highly conserved in mammals but not in other species (Fig. 2C); however, this substitution was considered as damaging in pathogenicity prediction tools [Polyphen-2 (47), SIFT (48), PROVEAN (49), MutationTaster (50)] and had high PhyloP (51) and phastCons (52) conservation scores (Table 2). The insertion of glutamine from the extended splice-site mutation p.K1037_E1038insQ is predicted to affect the eighth TPR domain, altering an α-helix and is predicted to be deleterious (Table 2). The TPR domains are known to bind other proteins or small molecules (53) and structural alteration of these tandem repeats may modify the function of IFT172. The p.H1567Q change affects a residue located just outside an α-helix and in the proximity of the last TPR domain predicted by Taschner and colleagues (13); His1567 is highly conserved and the substitution to glutamine is predicted to be damaging (Table 2). The p.D1605E mutation lies in an α-helix, in the C-terminal domain of IFT172 (Fig. 2B); it is highly conserved throughout evolution (Fig. 2C), though it was predicted to be damaging only by MutationTaster prediction software (Table 2).
N-terminal p.L257P mutation leads to IFT172 mislocalization and C-terminal mutations (p.H1567Q and p.D1605E) lead to cilia shortening
For subcellular localization of the WT and mutant proteins, WT and putative mutant IFT172 cDNAs were cloned into an expression vector with an N-terminal V5 tag (54). Recombinant IFT172 proteins were then expressed in the ciliated murine inner medullary collecting duct (mIMCD3) cells stably expressing a green fluorescent protein (GFP) fused to Somatostatin Receptor 3 (SSTR3) to highlight primary cilia (54). Cells transfected with an empty vector served as a control. WT as well as p.H1567Q and p.D1605E mutant IFT172 proteins localized to primary cilia, as shown by the co-localization with the SSTR3 protein. The p.L257P mutant protein, however, failed to locate in cilia in four independent experiments (Fig. 3A). The lengths of the cilia were quantified in the mIMCD3 cells transfected with the WT, p.H1567Q and p.D1605E IFT172 constructs and the empty vector control in four independent experiments. The average lengths of the cilia in cells expressing the p.H1567Q (1.92 µm, t-test, P = 3.4 × 10−15) and p.D1605E (2.57 µm, t-test, P = 0.0019) mutant proteins were shorter than in the cells expressing the WT IFT172 construct (2.96 µm) (Fig. 3B). This effect can be explained by the high levels of expression of the transfected constructs compared with the endogenous Ift172 expression by mIMCD3 cells. Using quantitative real-time PCR with primers annealing to the mouse and human IFT172 transcripts, we established that with 20–30% of transfection efficiency, the transfected cells express over 55 times the amount of IFT172 transcript expressed by the untransfected cells or cells transfected with an empty vector (Fig. 3C). No significant difference was noted between the cilia length of the cells transfected with WT IFT172 and the empty vector control (2.69 µm, P = 0.073). Cilia lengths were not quantified for the p.L257P mutant because visibly transfected cells (cell body staining) with developed cilia were scarce, which made accurate cilia length quantifications impossible. These data, however, do suggest that the mutant p.L257P IFT172 protein affects cilia formation and/or maintenance, indicating that this mutation may disrupt protein function to a greater extent than the p.H1567Q and p.D1605E variants.
Figure 3.

Expression of WT and mutant IFT172 constructs in mIMCD3 cells. (A) Anti-V5 staining of IFT172 WT and mutant proteins (red) in mIMCD3 cells expressing somatostatin receptor 3 (SSTR3)—GFP fusion protein (the size bar represents 10 µm). Cell nuclei were visualized by Hoechst staining. (B) Quantification of cilia length in mIMCD3 cells transfected with a control pCAG-enhanced green fluorescent protein (EGFP) vector, WT or mutant constructs. The boxes represent 25–75% quintiles and the whiskers represent the 5–95% quintiles. The p.L275P mutant did not locate to the cilium and therefore this condition was excluded from the cilia length quantification (**P< 0.01, ***P < 0.0001). (C) Real-time quantitative PCR of mouse and human IFT172 transcripts in the transfected and untransfected mIMCD3 cells. The untransfected mIMCD3 cells condition was used as a calibrator sample and mIMCD3 cells transfected with the empty pCAG-EGFP served as a control.
The location of the WT and mutant IFT172 proteins was also studied in rat retinae. The WT and mutant IFT172 cDNA constructs were injected into the sub-retinal space of the newborn rat pups and the plasmids were electroporated into the photoreceptors as previously described (19,54,55). The retinae were analyzed by immunofluorescence 4 weeks after the injections. The V5 tagged WT IFT172 protein located in the photoreceptor cilia, with the characteristic punctate staining indicating both extremities of the transition zone (Fig. 4A and B). This pattern of expression was reported previously for other IFT-B complex proteins (IFT52, IFT57 and IFT88) (56). Co-staining with the anti-RP1 antibody confirmed the transition zone localization (Fig. 4C). The p.H1567Q and the p.D1605E mutant proteins showed similar localization to the WT IFT172 (Fig. 4D–G). However, the N-terminal p.L257P mutant protein showed a diffuse expression in the inner segment of the photoreceptor cells (Fig. 4H and I), consistent with the localization results from mIMCD3 cells and suggesting that the N-terminal part of the protein is involved in the localization of IFT172 to the cilia.
Figure 4.
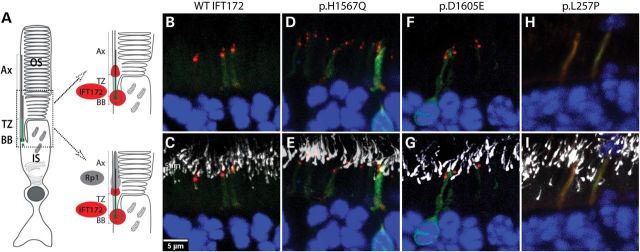
IFT172 localization in rat retinae. (A) A structure of the rod photoreceptor with cilia compartments indicated (BB: basal body, TZ: transition zone, Ax: axoneme, OS: outer segment, IS: inner segment). A magnified version of photoreceptors indicates localization of Rp1 (gray) and Ift172 (red). (B and C) Localization of WT-IFT172 without (B) and with (C) Rp1 staining. (D and E) Localization of p.H1567Q IFT172 protein without (D) and with (E) Rp1 staining. (F and G) Localization of p.D1605E IFT172 protein without (F) and with (G) Rp1 staining. (H and I) Localization of p.L257P IFT172 protein without (H) and with (I) Rp1 staining.
IFT172 mutations lead to null or hypomorphic alleles
To assess further the effects of the potential IFT172 mutations in vivo, we performed complementation studies in zebrafish embryos (57). First, we assessed the retina and body phenotypes in ift172 morphants injected with a splice blocking morpholino oligonucleotide (MO) (16) (Fig. 5A–I, Supplementary Material, Fig. S4). As previously described (16) and in accordance with the phenotype of the ift172 mutant fish (58), the ift172 morphants showed ventral curvature of the body and hydrocephaly, which were absent from the fish injected with a standard control MO (Fig. 5A and B). Semi-thin coronal sections and toluene blue staining of the zebrafish head at day post fertilization 5 (dpf5) confirmed altered cranial structure and revealed a thinner outer nuclear layer (ONL) and disorganized photoreceptor outer segments in the retina (Fig. 5C–H). Similar early eye phenotypes were also observed in the mutant ift172 zebrafish (59).
Figure 5.

Phenotype of ift172 zebrafish morphants. (A and B) Zebrafish injected with a standard control (ctrl) MO and ift172 splice block MO, showing ventral body curvature, body swelling and hydrocephaly at day post fertilization 5 (dpf5). (C and D) Coronal sections of control (C) and ift172 morphant (D) zebrafish at dpf5, depicting smaller eye size and altered cranial bone structure. (E–H) Retinae of control (E and G) and ift172 morphant (F and H) fish showing thinning of photoreceptor cell layer and shortening of the photoreceptor outer segments in the ift172 morphant fish. GCL, ganglion cell layer; INL, inner- nuclear layer; ONL; outer-nuclear layer; POS, photoreceptor outer segments. (I) RT-PCR on RNAs obtained from whole control and ift172 morphant larvae at dpf5, indicating exon 3 skipping due to the donor site blocking by the MO. The primers annealed to ift172 exon 2 and 4. RT- indicates RT-PCR negative control. (J) Zebrafish ift172 morphant phenotype rescue with total quantification of fish phenotypes induced by ift172 MO and rescue by the IFT172 WT and mutant mRNAs. Normalizing total number of morphants in the IFT172 mutant conditions by the number of WT total morphants in the same experiment shows a statistically significant difference between the complementation of the WT IFT172 and the mutants (p.H1567Q, t-test, P = 0.007; p.D1605E, P = 0.039; p.L257P, P = 0.001).
In the complementation studies, zebrafish embryos were co-injected with the ift172 MO and the IFT172 WT or mutant mRNAs. WT IFT172 mRNA was partially able to rescue the phenotype induced by the ift172 morpholino with a reduction of the total number of morphants by 17.9% across all the experiments (Fig. 5J). The mutant p.H1567Q and p.D1605E IFT172 mRNAs were not able to rescue the morphant phenotype to the same extent, obtaining a reduction of total morphant numbers of only 4.2 and 6.7% respectively, indicating the likely hypomorphic nature of each of these mutations. The p.L257P mutant failed to complement the MO-induced phenotype and therefore we consider that this mutation leads to a null allele, consistent with the results of the localization studies.
DISCUSSION
We have identified mutations in IFT172 that lead to a recessive form of RP and BBS. This is the first report indicating that mutations in this gene can be associated with a non-syndromic or a milder systemic phenotype compared with the severe MZSDS and JATD syndromes (16). The previously reported phenotypes involved skeletal anomalies (narrowed bell-shaped thorax, short stature, brachydactyly and polydactyly), nephronophtitis leading to renal failure, liver fibrosis, obesity, retinal degeneration and in some cases cerebellar vermis hypoplasia (16). In this study, two unrelated individuals were affected with an isolated form of RP and no other systemic symptoms; and a sib-ship with BBS, where only one sister presented with cutaneous polydactyly.
The variable phenotypic severity may be due to the nature of IFT172 mutations identified in both studies or to the total mutation load in the affected subjects' genomes, as previously suggested for BBS (60–62). Through in vitro and in vivo functional analyses we showed that two missense changes identified in this study are hypomorphic in nature (p.H1567Q and p.D1605E) and one of the mutations most likely creates a null allele (p.L257P). Of the two splice-site mutations, one affected an essential site (c.1525-1G>A) and the second affected an extended site (c.3112-5T>A). Combining the in silico predictions with functional data from RT-PCR on affected subject's leukocyte mRNA and a mini-gene assay suggested that the c.3112-5T>A change may lead to a combination of events including insertion of a glutamine (p.Lys1037_Glu1038insGln), intron 28 retention and partially normal splicing. It is possible that intron 28 retention also occurs in the c.3112-5T>A harboring transcripts in lymphocytes, but is degraded by nonsense-mediated decay (63) and therefore is not detected by RT-PCR from the affected subject's RNA. In addition, we cannot be certain of the exact fate of the c.3112-5T>A carrying transcript in this person's retina; however, the above data suggest that this allele is hypomorphic or null. Because this individual is affected with an isolated form of RP, we expect that some functional protein will be produced from the c.3112-5T>A allele, especially since the p.L257P change in trans appears to be a more severe loss-of-function mutation. All of the affected subjects presented in this study carried either two hypomorphic alleles or one hypomorphic and one null allele. The 14 previously published mutations in IFT172 causing MZSDS and JATD (16) included frameshifts, stop mutations, missense and splice-site changes, and an in-frame deletion, none of which were seen in this study (Supplementary Material, Fig. S5). The more severe phenotype was not always produced by two truncating alleles, as one would expect by comparing the two studies. We therefore concluded that the published and new genotypes are not sufficient to explain the broad phenotypic spectrum of IFT172-associated disease and further functional and genetic studies will have to be undertaken. Other ciliopathy-associated gene defects also lead to pleiotropic effects. Diseases caused by mutations in OFD1 (MIM 300170) range from a severe orofaciodigital (MIM 311200) (64) and Joubert (MIM 300804) (65) syndromes to an isolated RP (66). Isolated retinal phenotypes were also seen in subjects with mutations in three other syndromic ciliopathy genes TTC8 (MIM 608132) (67,68), CEP290 (69–74) and BBS1 (75). The variable phenotype was explained by the position of the mutations within the gene (OFD1 (65)), alternative splicing in the retina (TTC8 (68)) or hypomorphic alleles due to activation of cryptic splice sites (CEP290 (69), OFD1 (66)). Epistatic effects of other alleles have also been proposed for the CEP290-associated disease (71) and other forms of ciliopathy (60,61,76). We believe that modifying alleles may also be involved in the modulation of the IFT172-related phenotypes, especially leading to more severe clinical manifestations. In the attempt to decipher possible modifying alleles in the studied patients, we performed an analysis of all the rare variants in the known IRD and ciliopathy-associated genes (Supplementary Material, Tables S1 and S2). Based on the small sample size we cannot conclude with confidence which variants may be phenotype modifiers, however, we observed that the more severely affected subject (II.1) from family 1 carries a higher mutational load than her sister (II.2). In addition, subject II.1 is compound heterozygous for mutations in WDR65 (c.3406C>T, p.Arg1136Trp; c.2678A>C, p.Lys893Thr) (MIM 614259), a gene associated with the Van der Woude syndrome (MIM 606713) (Supplementary Material, Table S1). Future studies of a larger cohort of subjects with mutations in IFT172 may shed more light on the genetic modifiers of IFT172-associated phenotypes.
IFT172 is considered as a peripheral IFT-B protein (13,77) and it plays a role in the IFT complex turnaround at the tip of the cilium, as shown by studies in Chlamydomonas IFT172 homolog fla11 temperature-sensitive mutants (78,79). The role of the IFT172 and the transition from the anterograde to the retrograde transport in the photoreceptor cilia have not been fully studied. It is known that the homozygous ift172 mutant zebrafish, apart from the severe body phenotype, also show abnormal photoreceptor development (58,59,80,81). In these fish photoreceptor cilia failed to extend, accumulating membranous structures in the inner segment (59). Mice homozygous for the Ift172 loss-of-function alleles (wim or slb) die before embryonic day 13 (82,83) and mice homozygous for a hypomorphic ift172 allele (avc1) die at birth with severe skeletal, heart and renal phenotypes (84). The study of the Ift172 mutant mice revealed the importance of IFT in Hedgehog (Hh) signaling pathway and its involvement in the specification of the ventral cell types in the neural tube during embryogenesis (82) and in the neuronal patterning of the brain (83). Given the complexity of the role of IFT172 in maintaining the primary cilia and in the embryonic development, it is possible that genes coding for other IFT components or proteins in the Hh signaling pathway could have an influence on the phenotype induced by the IFT172 mutations. Additional future studies to test this hypothesis on the genetic and functional level may help explain the broad phenotypic spectrum of IFT172-associated disease.
MATERIALS AND METHODS
Ethics statement
The human study protocols were approved by the institutional review boards in each of the investigation centers (Human Studies Committee MEEI in USA, Comité de Protection des Personnes Ile de France V in France and Wetenschaps Commissie Oogziekenhuis in the Netherlands). All the investigated affected individuals and family members provided informed written consent for the genetic study and all the protocols adhered to the Declaration of Helsinki.
The in vivo experiments performed on rats and zebrafish were performed according to protocols approved by the Animal Care Committee from the Massachusetts Eye and Ear Hospital. All procedures were performed to minimize suffering in accordance with the animal care rules in our institution in compliance with the Animal Welfare Act, the Guide for the Care and Use of Laboratory Animals and the Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Human subjects' ascertainment and clinical evaluation
The families were recruited in different clinical centers in USA (The Children's Hospital Philadelphia and Massachusetts Eye and Ear Infirmary), France (Quinze-Vingts Hospital) and the Netherlands (The Rotterdam Eye Hospital). The diagnoses of RP and BBS were based on a full ophthalmic examination and general clinical history as described previously (29,85,86).
Molecular genetics
Initial mutation screening of known genes associated with inherited retinal degenerations (IRD) was achieved through various methods involving selective exon capture, homozygosity mapping and Sanger sequencing, as described previously (29,30,41,87–89). WES was performed as before (85,90–93). Total RNA from the affected subject from family 3 was isolated from fresh blood using an RNA kit according to the manufacturer's instructions (PAXgene Blood RNA Kit; PreAnalytix, Qiagen, The Netherlands). The mutation nomenclature is based on transcript NM_015662.1 and A from the ATG initiation codon is designated as position 1. All the IFT172 variants published in this report will be deposited at the Leiden Open Variation Database (http://www.lovd.nl/3.0/home).
Gene expression reagents
Human IFT172 cDNA was amplified by RT-PCR from HEK293 cells and cloned into a pENTR/D-TOPO entry vector (Invitrogen, Life Technologies, USA). The sequence was verified by Sanger sequencing and the mutant constructs were obtained by site-directed mutagenesis (QuickChange II, Agilent Technologies, USA). The mutagenesis primers were designed using online software (http://www.genomics.agilent.com/primerDesignProgram.jsp). The WT and mutant constructs were subcloned to Gateway compatible destination vectors using LR clonase (Invitrogen). For cell expression and rat electroporations IFT172 was moved to the pCAG-V5-IRES-enhanced green fluorescent protein (EGFP) vector (55,94) adapted for Gateway cloning (54) containing an N-terminal V5 tag and EGFP separated by the internal ribosome entry site (IRES). The constructs were purified by an Endotoxin Free plasmid MaxiPrep kit (Qiagen, USA) and eluted in phosphate buffered saline (PBS) buffer (Sigma-Aldrich, USA). For zebrafish rescue experiments, the sequence of WT and mutant IFT172 cDNAs were moved to the (pCS2+GW) gateway compatible vector containing SP6 promoter (kind gift from Dr Erica Davis) and the plasmids were purified by MidiPrep kits (Qiagen).
Splicing assay
The mini-gene splicing assay was performed using a construct of IFT172 exons 28–30 obtained by gDNA PCR amplification of a healthy individual and subsequent introduction of the mutation through the site-directed mutagenesis (QuickChange II). The WT and mutant constructs were expressed in HEK293 cells for 48 h and splicing was investigated by RT-PCR amplification of RNA obtained from the transfected cells and the untransfected control. To avoid amplification of the endogenous IFT172, the primers were designed to the short adapters on exons 28 and 30.
Cell culture and transfection
A murine kidney inner medullary collecting duct (mIMCD3) cell line stably expressing a GFP fused to SSTR3 (gift of Gregory J. Pazour, University of Massachusetts Medical School) were used for the IFT172 subcellular localization studies. The cells were maintained in Dulbecco's Modified Eagle Medium (DMEM): Nutrient Mixture F-12 (Gibco, Live Technologies), supplemented with 10% fetal bovine serum (Gibco). The cells grown in 6-well plates on glass coverslips to 50–60% confluence were transfected with 4 μg of DNA per well using Lipofectamine 2000 reagent (Invitrogen). After 48 h, the cells were incubated in a medium without serum to stimulate cilia growth. Seventy-two hours after transfection the cells were processed for immunostaining. For the splicing assay, HEK293 cells were used maintained in high glucose DMEM (Gibco).
mIMCD3 cell immunostaining
Following transfection, the mIMCD3-SSTR3 cells were processed in the following way: 4% paraformaldehyde (PFA) fixation for 5 min; permeabilization [0.5% Triton X100 (Roche, USA) in PBS]; blocking of non-specific sites [0.2% Triton X100, 1% bovine serum albumin (BSA) (Sigma-Aldrich) in PBS]; primary mouse anti-V5 antibody incubation (1 : 1000 dilution in blocking buffer, overnight at 4°C); secondary Alexa-555 goat anti-mouse antibody incubation (1:1000 dilution in PBS, 1 h at room temperature (RT), Invitrogen). In between the steps, the cells were washed with PBS. The nuclei were stained with Hoechst (Invitrogen) after the secondary antibody incubation (1:10 000 dilution in PBS for 5 min at RT). The coverslips were mounted on a slide with Fluoromount G (Electron Microscopy Sciences, USA) and the pictures were taken with a fluorescent microscope (Nikon Eclipse Ti, Nikon, USA). The length of the cilia was quantified with the freely available ImageJ software (95,96). The 150 largest cilia from each condition were used for the comparisons.
RNA extraction and quantitative RT-PCR
Total RNA from HEK293 and mIMCD3 cells was extracted (Trizol Reagent, Invitrogen) 48 h after transfection with the mini-gene or full-length IFT172 constructs or an empty vector. Following extraction, the RNA samples were treated with DNase I in solution (Qiagen) and the RNA was purified on RNA columns (RNAeasy mini kit, Qiagen). To obtain large complementary (c)DNA for cloning whole IFT172 gene, Superscript III reverse transcriptase was used (Invitrogen). Reverse transcription for quantitative PCR purposes was performed with M-MLV Reverse Transcriptase (Solis BioDyne, Estonia) on equal amounts of RNA (1 µg in 50 µl reaction) and quantitative PCR was performed with primers recognizing mouse and human IFT172 transcripts (forward: 5′-CAAAAGGATCTTCACTGACATG-3′ and reverse: 5′-GTTTGCCTCACTGGACTTCAC-3′) with Fast SYBR® Green Master Mix (Applied Biosystems, Life Technologies) on a qPCR machine (Mx3005P, Agilent Technologies).
In vivo rat retinae electroporation
To study location of the WT and mutant IFT172 proteins in the photoreceptors, the pCAG-V5-IFT172-IRES-EGFP constructs were injected into the subretinal space of neonatal rats at post-natal (PN) days 1.5–2.5. Each right eye was injected with 0.5 µl of endotoxin free constructs (3.3–3.5 µg/µl), which were electroporated into the photoreceptors using tweezer-type electrodes as previously described (55). Five to six pups were injected per each group. The pups were sacrificed after 4 weeks by transcardial perfusion (1% PFA), the eyes were enucleated and the corneas and lenses dissected. After cryopreservation (30% sucrose in PBS, overnight at 4°C) and embedding (OCT reagent, Tissue-Tek, Fisher Scientific, USA), the eyes were cut into 20 µm sections and the electroporated areas (showing EGFP expression) were analyzed by immunohistochemistry.
Immunohistochemistry on rat retinae
The retina sections were processed in the following way: blocking and permeabilization (0.05% Triton X100, 1% BSA in PBS, 1 h at RT); primary mouse anti-V5 antibody (1:1000 dilution) and chicken anti-Rp1 (97) (1:2000 dilution) incubation (0.3% Triton X100 in PBS, overnight at 4°C); secondary Alexa-555 conjugated goat anti-mouse (1:1000 dilution, Invitrogen) and Alexa-647 conjugated goat anti-chicken (1:2000 dilution, Invitrogen) antibodies (0.3% Triton X100 in PBS, 2 h at RT). In between the steps, the retina sections were washed with PBS. The nuclei were stained with Hoechst after the secondary antibody incubation (1:10 000 dilution in PBS for 5 min at RT). The images were acquired in a sequential manner, with pictures taken every 0.3 µm in the z-plane using confocal microscopy (SP5, Leica Microsystems, USA).
Morpholino knockdown in zebrafish embryo and rescue experiments
Wild-type zebrafish (Danio rerio) of the AB strain from the zebrafish facility at the Ocular Genomics Institute were used in the studies. After natural spawning, the embryos were collected and injected within 1.25 hpf window (1–8 cell stage) with ift172 (NM_001002312) specific splice blocker MO (5′-CTGTTCTCAGAGATACCTACCACTC-3′) (sequence kindly provided by Erica Davies) or standard control morpholino (MO) (5′-CCTCTTACCTCAGTTACAATTTATA-3′) (Gene Tools, USA). Embryos were injected with 2.3 nl of 0.4 mM MO and the phenotype was evaluated at post-fertilization day 3 (dpf3). The zebrafish semi-thin sections and toluene blue staining, performed on fish larvae at dpf5, were outsourced (Biomedical EM Services, Rose Valley, PA, USA).
For the rescue experiments ,the embryos were co-injected with the splice blocker MO and 0.4 mM and 0.5 µg/µl of mRNA generated from WT and mutant pCS2+GW-IFT172 constructs by SP6 driven in vitro transcription (mMessage mMachine SP6 Transcription Kit, Life Technologies, USA). Three independent experiments were performed for each mutant mRNA, where zebrafish embryos were injected with the following conditions: (1) standard control MO, (2) ift172 MO, (3) ift172 MO + WT IFT172 mRNA and (4) ift172 MO + mutant IFT172 mRNA (Fig. 5J). In these complementation assays, fish phenotypes were quantified at 3 dpf based on the overall body curvature: normal, morphant (ventral body curvature as shown in ift172 mutant larvae) and non-specific (monster-like) phenotypes, which were excluded from the complementation assay comparisons.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by grants from the National Eye Institute (EY012910, E.A.P), the Foundation Fighting Blindness (FFB) (E.A.P. and Q.L.), Research to Prevent Blindness (E.A.P.) and the Fleming Family Foundation (K.M.B); FFB grant CD-CL-0808-0466-CHNO (I.A.), FFB center grant C-CMM-0907-0428-INSERM04, Foundation Voir et Entendre (C.Z.), Fondation Dalloz prix ‘pour la recherche en ophtalmologie’ (C.Z.), Ville de Paris and Region Ile de France, and Labex LIFESENSES (ANR-10-LABX-65), supported by French state funds managed by the Agence Nationale de la Recherche within the Investissements d′Avenir program (ANR-11-IDEX- 0004-0). The research studies of A.M.S. and R.W.J.C. were supported by the Netherlands Organisation for Scientific Research (TOP-grant 91209047, to F.P.M.C. and A.I.d.H.) and the Stichting Wetenschappelijk Onderzoek Oogziekenhuis Prof. Dr H.J. Flieringa Foundation.
ONLINE RESOURCES USED IN THIS STUDY
Bioinformatics toolkit at the Max-Plank Institute for Developmental Biology: http://toolkit.tuebingen.mpg.de/repper; BayNAGNAG: http://www.tassdb.info/baynagnag/form.html; dbSNP: http://www.ncbi.nlm.nih.gov/SNP/; NHLBI Exome Sequencing Project Exome Variant Server: http://eversusgs.washington.edu/EVS/; NNSplice: http://www.fruitfly.org/seq_tools/splice.html; Online Mendelian Inheritance in Man (OMIM): http://www.omim.org/; Pfam: http://pfam.sanger.ac.uk/; Polyphen-2: http://genetics.bwh.harvard.edu/pph2/; RetNet Retinal Information Network: https://sph.uth.edu/retnet/; SIFT/PROVEAN: http://sift.jcvi.org/; UNIGENE: http://www.ncbi.nlm.nih.gov/unigene; Mutalyzer: https://mutalyzer.nl/; MutationTaster: http://www.mutationtaster.org/index.html; 1000 Genomes Project website: http://www.1000genomes.org/; UCSC Genome Browser: http://genome.ucsc.edu/.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank the patients and their family members for their participation in this study, Aliete Langsdorf for her help with rat retinae injections, Brian Perkins for providing us with ift172 mutant zebrafish, Erica Davis and Nicholas Katsanis for sharing with us their experience of ift172 MO knockdown in zebrafish and providing the MO sequence, Kornelia Neveling for assisting in exome sequencing data analysis and Jaclyn Lena and Ryan Hayes for their help in IFT172 constructs preparation. We are grateful to Daniel MacArthur and Brett Thomas for sharing with us the xBrowse tool to analyze WES data and to Daniel Navarro and Joe White for their help with bioinformatics analyses.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Mockel A., Perdomo Y., Stutzmann F., Letsch J., Marion V., Dollfus H. Retinal dystrophy in Bardet–Biedl syndrome and related syndromic ciliopathies. Prog. Retin. Eye Res. 2011;30:258–274. doi: 10.1016/j.preteyeres.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 2.Waters A.M., Beales P.L. Ciliopathies: an expanding disease spectrum. Pediatr. Nephrol. 2011;26:1039–1056. doi: 10.1007/s00467-010-1731-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beales P.L., Elcioglu N., Woolf A.S., Parker D., Flinter F.A. New criteria for improved diagnosis of Bardet–Biedl syndrome: results of a population survey. J. Med. Genet. 1999;36:437–446. [PMC free article] [PubMed] [Google Scholar]
- 4.Pierce E.A., Quinn T., Meehan T., McGee T.L., Berson E.L., Dryja T.P. Mutations in a gene encoding a new oxygen-regulated photoreceptor protein cause dominant retinitis pigmentosa. Nat. Genet. 1999;22:248–254. doi: 10.1038/10305. [DOI] [PubMed] [Google Scholar]
- 5.Guillonneau X., Piriev N.I., Danciger M., Kozak C.A., Cideciyan A.V., Jacobson S.G., Farber D.B. A nonsense mutation in a novel gene is associated with retinitis pigmentosa in a family linked to the RP1 locus. Hum. Mol. Genet. 1999;8:1541–1546. doi: 10.1093/hmg/8.8.1541. [DOI] [PubMed] [Google Scholar]
- 6.Meindl A., Dry K., Herrmann K., Manson F., Ciccodicola A., Edgar A., Carvalho M.R., Achatz H., Hellebrand H., Lennon A., et al. A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3) Nat. Genet. 1996;13:35–42. doi: 10.1038/ng0596-35. [DOI] [PubMed] [Google Scholar]
- 7.Den Hollander A.I., Koenekoop R.K., Mohamed M.D., Arts H.H., Boldt K., Towns K.V., Sedmak T., Beer M., Nagel-Wolfrum K., McKibbin M., et al. Mutations in LCA5, encoding the ciliary protein lebercilin, cause Leber congenital amaurosis. Nat. Genet. 2007;39:889–895. doi: 10.1038/ng2066. [DOI] [PubMed] [Google Scholar]
- 8.Dryja T.P., Adams S.M., Grimsby J.L., McGee T.L., Hong D.H., Li T., Andréasson S., Berson E.L. Null RPGRIP1 alleles in patients with Leber congenital amaurosis. Am. J. Hum. Genet. 2001;68:1295–1298. doi: 10.1086/320113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langmann T., Di Gioia S.A., Rau I., Stöhr H., Maksimovic N.S., Corbo J.C., Renner A.B., Zrenner E., Kumaramanickavel G., Karlstetter M., et al. Nonsense mutations in FAM161A cause RP28-associated recessive retinitis pigmentosa. Am. J. Hum. Genet. 2010;87:376–381. doi: 10.1016/j.ajhg.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bandah-Rozenfeld D., Mizrahi-Meissonnier L., Farhy C., Obolensky A., Chowers I., Pe'er J., Merin S., Ben-Yosef T., Ashery-Padan R., Banin E., et al. Homozygosity mapping reveals null mutations in FAM161A as a cause of autosomal-recessive retinitis pigmentosa. Am. J. Hum. Genet. 2010;87:382–391. doi: 10.1016/j.ajhg.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.El Shamieh S., Neuillé M., Terray A., Orhan E., Condroyer C., Démontant V., Michiels C., Antonio A., Boyard F., Lancelot M.-E., et al. Whole-exome sequencing identifies KIZ as a ciliary gene associated with autosomal-recessive rod-cone dystrophy. Am. J. Hum. Genet. 2014;94:625–633. doi: 10.1016/j.ajhg.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Czarnecki P.G., Shah J.V. The ciliary transition zone: from morphology and molecules to medicine. Trends Cell Biol. 2012;22:201–210. doi: 10.1016/j.tcb.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taschner M., Bhogaraju S., Lorentzen E. Architecture and function of IFT complex proteins in ciliogenesis. Differentiation. 2012;83:S12–S22. doi: 10.1016/j.diff.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosenbaum J.L., Witman G.B. Intraflagellar transport. Nat. Rev. Mol. Cell Biol. Biol. 2002;3:813–825. doi: 10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
- 15.Pedersen L.B., Rosenbaum J.L. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr. Top. Dev. Biol. 2008;85:23–61. doi: 10.1016/S0070-2153(08)00802-8. [DOI] [PubMed] [Google Scholar]
- 16.Halbritter J., Bizet A.A., Schmidts M., Porath J.D., Braun D.A., Gee H.Y., McInerney-Leo A.M., Krug P., Filhol E., Davis E.E., et al. Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am. J. Hum. Genet. 2013;93:1–11. doi: 10.1016/j.ajhg.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bredrup C., Saunier S., Oud M.M., Fiskerstrand T., Hoischen A., Brackman D., Leh S.M., Midtbø M., Filhol E., Bole-Feysot C., et al. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am. J. Hum. Genet. 2011;89:634–643. doi: 10.1016/j.ajhg.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilissen C., Arts H.H., Hoischen A., Spruijt L., Mans D.A., Arts P., van Lier B., Steehouwer M., van Reeuwijk J., Kant S.G., et al. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am. J. Hum. Genet. 2010;87:418–423. doi: 10.1016/j.ajhg.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis E.E., Zhang Q., Liu Q., Diplas B.H., Davey L.M., Hartley J., Stoetzel C., Szymanska K., Ramaswami G., Logan C.V., et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 2011;43:189–196. doi: 10.1038/ng.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perrault I., Saunier S., Hanein S., Filhol E., Bizet A.A., Collins F., Salih M.A.M., Gerber S., Delphin N., Bigot K., et al. Mainzer-Saldino syndrome is a ciliopathy caused by IFT140 mutations. Am. J. Hum. Genet. 2012;90:864–870. doi: 10.1016/j.ajhg.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arts H.H., Bongers E.M.H.F., Mans D.A., van Beersum S.E.C., Oud M.M., Bolat E., Spruijt L., Cornelissen E.A.M., Schuurs-Hoeijmakers J.H.M., de Leeuw N., et al. C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome. J. Med. Genet. 2011;48:390–395. doi: 10.1136/jmg.2011.088864. [DOI] [PubMed] [Google Scholar]
- 22.Beales P.L., Bland E., Tobin J.L., Bacchelli C., Tuysuz B., Hill J., Rix S., Pearson C.G., Kai M., Hartley J., et al. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat. Genet. 2007;39:727–729. doi: 10.1038/ng2038. [DOI] [PubMed] [Google Scholar]
- 23.Walczak-Sztulpa J., Eggenschwiler J., Osborn D., Brown D.A., Emma F., Klingenberg C., Hennekam R.C., Torre G., Garshasbi M., Tzschach A., et al. Cranioectodermal Dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am. J. Hum. Genet. 2010;86:949–956. doi: 10.1016/j.ajhg.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmidts M., Frank V., Eisenberger T., Al Turki S., Bizet A.A., Antony D., Rix S., Decker C., Bachmann N., Bald M., et al. Combined NGS approaches identify mutations in the intraflagellar transport gene IFT140 in skeletal ciliopathies with early progressive kidney Disease. Hum. Mutat. 2013;34:714–724. doi: 10.1002/humu.22294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Exome Variant Server. Seattle, WA: NHLBI GO Exome Sequencing Project (ESP); http://evs.gs.washington.edu/EVS/ (last accessed, May 2014) [Google Scholar]
- 26.Reese M., Eeckman F., Kulp D., Haussler D. Improved splice site detection in Genie. J. Comput. Biol. 1997;4:311–323. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- 27.Jaakson K., Zernant J., Külm M., Hutchinson A., Tonisson N., Glavac D., Ravnik-Glavac M., Hawlina M., Meltzer M.R., Caruso R.C., et al. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum. Mutat. 2003;22:395–403. doi: 10.1002/humu.10263. [DOI] [PubMed] [Google Scholar]
- 28.Ávila-Fernández A., Cantalapiedra D., Aller E., Vallespín E., Aguirre-Lambán J., Blanco-Kelly F., Corton M., Riveiro-Álvarez R., Allikmets R., Trujillo-Tiebas M.J., et al. Mutation analysis of 272 Spanish families affected by autosomal recessive retinitis pigmentosa using a genotyping microarray. Mol. Vis. 2010;16:2550–2558. [PMC free article] [PubMed] [Google Scholar]
- 29.Audo I., Sahel J.-A., Mohand-Saïd S., Lancelot M.-E., Antonio A., Moskova-Doumanova V., Nandrot E.F., Doumanov J., Barragan I., Antinolo G., et al. EYS is a major gene for rod-cone dystrophies in France. Hum. Mutat. 2010;31:E1406–E1435. doi: 10.1002/humu.21249. [DOI] [PubMed] [Google Scholar]
- 30.Audo I., Bujakowska K., Mohand-Said S., Tronche S., Lancelot M.E., Antonio A., Germain A., Lonjou C., Carpentier W., Sahel J.A., et al. A novel DFNB31 mutation associated with Usher type 2 syndrome showing variable degrees of auditory loss in a consanguineous Portuguese family. Mol. Vis. 2011;17:1598–1606. [PMC free article] [PubMed] [Google Scholar]
- 31.Marlhens F., Bareil C., Griffoin J., Zrenner E., Amalric P., Eliaou C., Liu S.-Y., Harris E., Redmond T.M., Arnaud B., et al. Mutations in RPE65 cause Leber's congenital amaurosis. Nat. Genet. 1997;17:140–141. doi: 10.1038/ng1097-139. [DOI] [PubMed] [Google Scholar]
- 32.Gu S.M., Thompson D.A., Srikumari C.R., Lorenz B., Finckh U., Nicoletti A., Murthy K.R., Rathmann M., Kumaramanickavel G., Denton M.J., et al. Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat. Genet. 1997;17:194–197. doi: 10.1038/ng1097-194. [DOI] [PubMed] [Google Scholar]
- 33.Allikmets R., Singh N., Sun H., Shroyer N.F., Hutchinson A., Chidambaram A., Gerrard B., Baird L., Stauffer D., Peiffer A., et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet. 1997;15:236–246. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- 34.Allikmets R., Shroyer N.F., Singh N., Seddon J.M., Lewis R.A., Bernstein P.S., Peiffer A., Zabriskie N.A., Li Y., Hutchinson A., et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science. 1997;277:1805–1807. doi: 10.1126/science.277.5333.1805. [DOI] [PubMed] [Google Scholar]
- 35.Mir A.M., Paloma E., Allikmets R., Ayuso C., del Rio T., Dean M., Vilageliu L., Gonzalez-Duarte R., Balcells S. Retinitis pigmentosa caused by a homozygous inactivating mutation in the Stargardt disease gene ABCR. Nat. Genet. 1998;18:11–12. doi: 10.1038/ng0198-11. [DOI] [PubMed] [Google Scholar]
- 36.Collin R.W.J., Safieh C., Littink K.W., Shalev S.A., Garzozi H.J., Rizel L., Abbasi A.H., Cremers F.P.M., den Hollander A.I., Klevering B.J., et al. Mutations in C2ORF71 cause autosomal-recessive retinitis pigmentosa. Am. J. Hum. Genet. 2010;86:783–788. doi: 10.1016/j.ajhg.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishimura D.Y., Baye L.M., Perveen R., Searby C.C., Avila-Fernandez A., Pereiro I., Ayuso C., Valverde D., Bishop P.N., Manson F.D.C., et al. Discovery and functional analysis of a Retinitis Pigmentosa gene, C2ORF71. Am. J. Hum. Genet. 2010;86:686–695. doi: 10.1016/j.ajhg.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li L., Nakaya N., Chavali V.R.M., Ma Z., Jiao X., Sieving P.A., Riazuddin S., Tomarev S.I., Ayyagari R., Riazuddin S.A., et al. A mutation in ZNF513, a putative regulator of photoreceptor development, causes autosomal-recessive retinitis pigmentosa. Am. J. Hum. Genet. 2010;87:400–409. doi: 10.1016/j.ajhg.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joensuu T., Hämäläinen R., Yuan B., Johnson C., Tegelberg S., Gasparini P., Zelante L., Pirvola U., Pakarinen L., Lehesjoki A.E., et al. Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3. Am. J. Hum. Genet. 2001;69:673–684. doi: 10.1086/323610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang S.H., Pittler S.J., Huang X., Oliveira L., Berson E.L., Dryja T.P. Autosomal recessive retinitis pigmentosa caused by mutations in the alpha subunit of rod cGMP phosphodiesterase. Nat. Genet. 1995;11:468–471. doi: 10.1038/ng1295-468. [DOI] [PubMed] [Google Scholar]
- 41.Neveling K., Collin R.W.J., Gilissen C., van Huet R.A.C., Visser L., Kwint M.P., Gijsen S.J., Zonneveld M.N., Wieskamp N., de Ligt J., et al. Next-generation genetic testing for retinitis pigmentosa. Hum. Mutat. 2012;33:963–972. doi: 10.1002/humu.22045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sinha R., Nikolajewa S., Szafranski K., Hiller M., Jahn N., Huse K., Platzer M., Backofen R. Accurate prediction of NAGNAG alternative splicing. Nucleic Acids Res. 2009;37:3569–3579. doi: 10.1093/nar/gkp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McGuffin L.J., Bryson K., Jones D.T. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 44.Migliori V., Mapelli M., Guccione E. On WD40 proteins. Propelling our Knowledge of Transcriptional Control? Epigenetics. 2012;7:815–822. doi: 10.4161/epi.21140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stirnimann C.U., Petsalaki E., Russell R.B., Müller C.W. WD40 proteins propel cellular networks. Trends Biochem. Sci. 2010;35:565–574. doi: 10.1016/j.tibs.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 46.Xu C., Min J. Structure and function of WD40 domain proteins. Protein Cell. 2011;2:202–214. doi: 10.1007/s13238-011-1018-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ng P.C., Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choi Y., Sims G.E., Murphy S., Miller J.R., Chan A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 51.Pollard K.S., Hubisz M.J., Rosenbloom K.R., Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Javadi Y., Itzhaki L.S. Tandem-repeat proteins: regularity plus modularity equals design-ability. Curr. Opin. Struct. Biol. 2013;23:622–631. doi: 10.1016/j.sbi.2013.06.011. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Q., Liu Q., Austin C., Drummond I., Pierce E.A. Knockdown of ttc26 disrupts ciliogenesis of the photoreceptor cells and the pronephros in zebrafish. Mol. Biol. Cell. 2012;23:3069–3078. doi: 10.1091/mbc.E12-01-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matsuda T., Cepko C.L. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc. Natl. Acad. Sci. USA. 2004;101:16–22. doi: 10.1073/pnas.2235688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sedmak T., Wolfrum U. Intraflagellar transport molecules in ciliary and nonciliary cells of the retina. J. Cell Biol. 2010;189:171–186. doi: 10.1083/jcb.200911095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Niederriter A.R., Davis E.E., Golzio C., Oh E.C., Tsai I.-C., Katsanis N. In vivo modeling of the morbid human genome using Danio rerio. J. Vis. Exp. 2013;78:e50338. doi: 10.3791/50338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lunt S.C., Haynes T., Perkins B.D. Zebrafish ift57, ift88, and ift172 intraflagellar transport mutants disrupt cilia but do not affect Hedgehog signaling. Dev. Dyn. 2009;238:1744–1759. doi: 10.1002/dvdy.21999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sukumaran S., Perkins B.D. Early defects in photoreceptor outer segment morphogenesis in zebrafish ift57, ift88 and ift172 Intraflagellar Transport mutants. Vision Res. 2009;49:479–489. doi: 10.1016/j.visres.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Katsanis N. The oligogenic properties of Bardet–Biedl syndrome. Hum. Mol. Genet., 2004;13(Spec No. 1):R65–R71. doi: 10.1093/hmg/ddh092. [DOI] [PubMed] [Google Scholar]
- 61.Badano J.L., Leitch C.C., Ansley S.J., May-Simera H., Lawson S., Lewis R.A., Beales P.L., Dietz H.C., Fisher S., Katsanis N. Dissection of epistasis in oligogenic Bardet–Biedl syndrome. Nature. 2006;439:326–330. doi: 10.1038/nature04370. [DOI] [PubMed] [Google Scholar]
- 62.Badano J.L., Kim J.C., Hoskins B.E., Lewis R.A., Ansley S.J., Cutler D.J., Castellan C., Beales P.L., Leroux M.R., Katsanis N. Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet–Biedl patients with two mutations at a second BBS locus. Hum. Mol. Genet. 2003;12:1651–1659. doi: 10.1093/hmg/ddg188. [DOI] [PubMed] [Google Scholar]
- 63.Chang Y.-F.F., Imam J.S., Wilkinson M.F. The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 64.Ferrante M.I., Giorgio G., Feather S.A., Bulfone A., Wright V., Ghiani M., Selicorni A., Gammaro L., Scolari F., Woolf A.S., et al. Identification of the gene for oral-facial-digital type I syndrome. Am. J. Hum. Genet. 2001;68:569–576. doi: 10.1086/318802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Coene K.L.M., Roepman R., Doherty D., Afroze B., Kroes H.Y., Letteboer S.J.F., Ngu L.H., Budny B., van Wijk E., Gorden N.T., et al. OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-encoded lebercilin. Am. J. Hum. Genet. 2009;85:465–481. doi: 10.1016/j.ajhg.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Webb T.R., Parfitt D.A., Gardner J.C., Martinez A., Bevilacqua D., Davidson A.E., Zito I., Thiselton D.L., Ressa J.H.C., Apergi M., et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23) Hum. Mol. Genet. 2012;21:3647–3654. doi: 10.1093/hmg/dds194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ansley S.J., Badano J.L., Blacque O.E., Hill J., Hoskins B.E., Leitch C.C., Kim J.C., Ross A.J., Eichers E.R., Teslovich T.M., et al. Basal body dysfunction is a likely cause of pleiotropic Bardet–Biedl syndrome. Nature. 2003;425:628–633. doi: 10.1038/nature02030. [DOI] [PubMed] [Google Scholar]
- 68.Riazuddin S.A., Iqbal M., Wang Y., Masuda T., Chen Y., Bowne S., Sullivan L.S., Waseem N.H., Bhattacharya S., Daiger S.P., et al. A splice-site mutation in a retina-specific exon of BBS8 causes nonsyndromic retinitis pigmentosa. Am. J. Hum. Genet. 2010;86:805–812. doi: 10.1016/j.ajhg.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Den Hollander A.I., Koenekoop R.K., Yzer S., Lopez I., Arends M.L., Voesenek K.E.J., Zonneveld M.N., Strom T.M., Meitinger T., Brunner H.G., et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 2006;79:556–561. doi: 10.1086/507318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Frank V., den Hollander A.I., Brüchle N.O., Zonneveld M.N., Nürnberg G., Becker C., Du Bois G., Kendziorra H., Roosing S., Senderek J., et al. Mutations of the CEP290 gene encoding a centrosomal protein cause Meckel-Gruber syndrome. Hum. Mutat. 2008;29:45–52. doi: 10.1002/humu.20614. [DOI] [PubMed] [Google Scholar]
- 71.Baala L., Audollent S., Martinovic J., Ozilou C., Babron M.-C., Sivanandamoorthy S., Saunier S., Salomon R., Gonzales M., Rattenberry E., et al. Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am. J. Hum. Genet. 2007;81:170–179. doi: 10.1086/519494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Valente E.M., Silhavy J.L., Brancati F., Barrano G., Krishnaswami S.R., Castori M., Lancaster M.A., Boltshauser E., Boccone L., Al-Gazali L., et al. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat. Genet. 2006;38:623–625. doi: 10.1038/ng1805. [DOI] [PubMed] [Google Scholar]
- 73.Sayer J.A., Otto E.A., O'Toole J.F., Nurnberg G., Kennedy M.A., Becker C., Hennies H.C., Helou J., Attanasio M., Fausett B.V., et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat. Genet. 2006;38:674–681. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- 74.Leitch C.C., Zaghloul N.A., Davis E.E., Stoetzel C., Diaz-Font A., Rix S., Alfadhel M., Al-Fadhel M., Lewis R.A., Eyaid W., et al. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet–Biedl syndrome. Nat. Genet. 2008;40:443–448. doi: 10.1038/ng.97. [DOI] [PubMed] [Google Scholar]
- 75.Estrada-Cuzcano A., Koenekoop R.K., Senechal A., De Baere E.B.W., de Ravel T., Banfi S., Kohl S., Ayuso C., Sharon D., Hoyng C.B., et al. BBS1 mutations in a wide spectrum of phenotypes ranging from nonsyndromic retinitis pigmentosa to Bardet–Biedl syndrome. Arch. Ophthalmol. 2012;130:1425–1432. doi: 10.1001/archophthalmol.2012.2434. [DOI] [PubMed] [Google Scholar]
- 76.Tory K., Lacoste T., Burglen L., Morinière V., Boddaert N., Macher M.-A., Llanas B., Nivet H., Bensman A., Niaudet P., et al. High NPHP1 and NPHP6 mutation rate in patients with Joubert syndrome and nephronophthisis: potential epistatic effect of NPHP6 and AHI1 mutations in patients with NPHP1 mutations. J. Am. Soc. Nephrol. 2007;18:1566–1575. doi: 10.1681/ASN.2006101164. [DOI] [PubMed] [Google Scholar]
- 77.Lucker B.F., Behal R.H., Qin H., Siron L.C., Taggart W.D., Rosenbaum J.L., Cole D.G. Characterization of the intraflagellar transport complex B core: direct interaction of the IFT81 and IFT74/72 subunits. J. Biol. Chem. 2005;280:27688–27696. doi: 10.1074/jbc.M505062200. [DOI] [PubMed] [Google Scholar]
- 78.Iomini C., Babaev-Khaimov V., Sassaroli M., Piperno G. Protein particles in Chlamydomonas flagella undergo a transport cycle consisting of four phases. J. Cell Biol. 2001;153:13–24. doi: 10.1083/jcb.153.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pedersen L.B., Miller M.S., Geimer S., Leitch J.M., Rosenbaum J.L., Cole D.G. Chlamydomonas IFT172 is encoded by FLA11, interacts with CrEB1, and regulates IFT at the flagellar tip. Curr. Biol. 2005;15:262–266. doi: 10.1016/j.cub.2005.01.037. [DOI] [PubMed] [Google Scholar]
- 80.Gross J.M., Perkins B.D., Amsterdam A., Egaña A., Darland T., Matsui J.I., Sciascia S., Hopkins N., Dowling J.E. Identification of zebrafish insertional mutants with defects in visual system development and function. Genetics. 2005;170:245–261. doi: 10.1534/genetics.104.039727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gross J., Perkins B. Zebrafish mutants as models for congenital ocular disorders in humans. Mol. Reprod. Dev. 2008;555:547–555. doi: 10.1002/mrd.20831. [DOI] [PubMed] [Google Scholar]
- 82.Huangfu D., Liu A., Rakeman A.S., Murcia N.S., Niswander L., Anderson K.V. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- 83.Gorivodsky M., Mukhopadhyay M., Wilsch-Braeuninger M., Phillips M., Teufel A., Kim C., Malik N., Huttner W., Westphal H. Intraflagellar transport protein 172 is essential for primary cilia formation and plays a vital role in patterning the mammalian brain. Dev. Biol. 2009;325:24–32. doi: 10.1016/j.ydbio.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Friedland-Little J.M., Hoffmann A.D., Ocbina P.J.R., Peterson M.A., Bosman J.D., Chen Y., Cheng S.Y., Anderson K.V., Moskowitz I.P. A novel murine allele of Intraflagellar Transport Protein 172 causes a syndrome including VACTERL-like features with hydrocephalus. Hum. Mol. Genet. 2011;20:3725–3737. doi: 10.1093/hmg/ddr241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Falk M.J., Zhang Q., Nakamaru-Ogiso E., Kannabiran C., Fonseca-Kelly Z., Chakarova C., Audo I., Mackay D.S., Zeitz C., Borman A.D., et al. NMNAT1 mutations cause Leber congenital amaurosis. Nat. Genet. 2012;44:1040–1045. doi: 10.1038/ng.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Siemiatkowska A.M., van den Born L.I., van Hagen P.M., Stoffels M., Neveling K., Henkes A., Kipping-Geertsema M., Hoefsloot L.H., Hoyng C.B., Simon A., et al. Mutations in the mevalonate kinase (MVK) gene cause nonsyndromic retinitis pigmentosa. Ophthalmology. 2013;120:2697–2705. doi: 10.1016/j.ophtha.2013.07.052. [DOI] [PubMed] [Google Scholar]
- 87.Audo I., Bujakowska K.M., Léveillard T., Mohand-Saïd S., Lancelot M.-E., Germain A., Antonio A., Michiels C., Saraiva J.-P., Letexier M., et al. Development and application of a next-generation-sequencing (NGS) approach to detect known and novel gene defects underlying retinal diseases. Orphanet J. Rare Dis. 2012;7:8. doi: 10.1186/1750-1172-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bujakowska K., Audo I., Mohand-Saïd S., Lancelot M.-E., Antonio A., Germain A., Leveillard T., Letexier M., Saraiva J.-P., Lonjou C., et al. CRB1 mutations in inherited retinal dystrophies. Hum. Mutat. 2012;33:306–315. doi: 10.1002/humu.21653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Audo I., Lancelot M.-E., Mohand-Saïd S., Antonio A., Germain A., Sahel J.-A., Bhattacharya S.S., Zeitz C. Novel C2orf71 mutations account for ∼1% of cases in a large French arRP cohort. Hum. Mutat. 2011;32:E2091–E2103. doi: 10.1002/humu.21460. [DOI] [PubMed] [Google Scholar]
- 90.Zeitz C., Jacobson S.G., Hamel C.P., Bujakowska K., Neuillé M., Orhan E., Zanlonghi X., Lancelot M.E., Michiels C., Schwartz S.B., et al. Whole-exome sequencing identifies LRIT3 mutations as a cause of autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet. 2013;92:67–75. doi: 10.1016/j.ajhg.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Roosing S., Rohrschneider K., Beryozkin A., Sharon D., Weisschuh N., Staller J., Kohl S., Zelinger L., Peters T.A., Neveling K., et al. Mutations in RAB28, encoding a farnesylated small GTPase, are associated with autosomal-recessive cone-rod dystrophy. Am. J. Hum. Genet. 2013;93:110–117. doi: 10.1016/j.ajhg.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Audo I., Bujakowska K., Orhan E., Poloschek C.M., Defoort-Dhellemmes S., Drumare I., Kohl S., Luu T.D., Lecompte O., Zrenner E., et al. Whole-exome sequencing identifies mutations in GPR179 leading to autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet. 2012;90:321–330. doi: 10.1016/j.ajhg.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Audo I., Bujakowska K., Orhan E., El Shamieh S., Sennlaub F., Guillonneau X., Antonio A., Michiels C., Lancelot M.-E., Letexier M., et al. The familial dementia gene revisited: a missense mutation revealed by whole-exome sequencing identifies ITM2B as a candidate gene underlying a novel autosomal dominant retinal dystrophy in a large family. Hum. Mol. Genet. 2014;23:491–501. doi: 10.1093/hmg/ddt439. [DOI] [PubMed] [Google Scholar]
- 94.Matsuda T., Cepko C.L. Controlled expression of transgenes introduced by in vivo electroporation. Proc. Natl. Acad. Sci. USA. 2007;104:1027–1032. doi: 10.1073/pnas.0610155104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Girish V., Vijayalakshmi A. Affordable image analysis using NIH Image/ImageJ. Indian J. Cancer. 2004;41:47. [PubMed] [Google Scholar]
- 97.Liu Q., Zhou J., Daiger S.P., Farber D.B., Heckenlively J.R., Smith J.E., Sullivan L.S., Zuo J., Milam A.H., Pierce E.A. Identification and subcellular localization of the RP1 protein in human and mouse photoreceptors. Invest. Ophthalmol. Vis. Sci. 2002;43:22–32. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.