

Abstract
Interaction of the Alzheimer’s Aβ peptides with the plasma membrane of cells in culture results in chronic increases in cytosolic [Ca2+]. Such increases can cause a variety of secondary effects leading to impaired cell growth or cell degeneration. In this investigation, we made a comprehensive study of the changes in cytosolic [Ca2+] in single PC12 cells and human neurons stressed by continuous exposure to a medium containing Aβ42 for several days. The differential timing and magnitude of the Aβ42-induced increase in [Ca2+] reveal subpopulations of cells with differential sensitivity to Aβ42. These results suggest that the effect produced by Aβ on the level of cytosolic [Ca2+] depends on the type of cell being monitored. Moreover, the results obtained of using potent inhibitors of Aβ cation channels such as Zn2+ and the small peptide NA7 add further proof to the suggestion that the long-term increases in cytosolic [Ca2+] in cells stressed by continuous exposure to Aβ is the result of Aβ ion channel activity.
Keywords: Calcium, Calcium channels, Beta amyloid, Beta amyloid ion channel, Alzheimer’s disease, Alzheimer’s Aβ peptide
Introduction
Early studies reported that neuronal Ca2+ increases with aging (Khachaturian 1987; Landfield 1987), and years of research have supported the concept that disturbances of intracellular calcium homeostasis may play a pathological role in the neurodegeneration associated with Alzheimer’s disease (Khachaturian 1994; Kawahara and Kuroda 2000; LaFerla 2002; Kawahara 2004; Smith et al. 2005; Green and LaFerla 2008; Korol et al. 2008; Small et al. 2009).
A marked increase in circulating Aβ peptides is a signature feature of Alzheimer’s disease. Mattson and colleagues first reported that Aβ peptides render human cortical neurons susceptible to glutamate excitotoxicity by increasing intracellular [Ca2+] (Mattson et al. 1992). Similarly, numerous reports show that the addition of Aβ peptides to cells increases the intracellular [Ca2+] level by increasing calcium membrane permeability and consequently the Ca2+ influx (Bathia et al. 2000; Zhu et al. 2000; Arispe et al. 1994a, b; Kawahara et al. 2000, 2009; Negishi-Kato and Kawahara 2008).
The exact mechanism(s) by which Aβ increases calcium membrane permeability remains unclear (Shirwany et al. 2007; Arispe et al. 2007; Bezprozvanny 2009; Supnet and Bezprozvanny 2010). We have shown that Aβ peptides form cation channels in lipid membranes (Arispe et al. 1993). This finding led us to propose that the increase in Ca2+ permeability observed in cells exposed to Aβ results from the activity of calcium ion channels formed by Aβ in the cell surface membrane (Arispe et al. 1994a, b) (Arispe et al. 2010).
While there is a growing consensus that Aβ peptides increase membrane conductance by forming conductive pores (Aguayo et al. 2009; Parodi et al. 2010; Sepulveda et al. 2010; Schauerte et al. 2010; Johnson et al. 2011; Tofoleanu and Buchete 2012; DeMuro et al. 2011; Sciacca et al. 2012; Prangkio et al. 2012; Schauerte et al. 2010; DeMuro et al. 2011), there has not been a systematic study on how a continued exposure to Aβ affects basal cytosolic calcium over time. In the present study, we analyzed intracellular calcium concentration in single cells exposed to Aβ for long periods of time. The results show that Aβ exposure induces markedly higher intracellular calcium concentration in a short-lived subpopulation of cell, defined by the presence of plasmalemmal phosphatidyl serine (PtdSer).
The formation of this subpopulation is constrained when specific Aβ calcium channel blockers are presented in the medium simultaneously with Aβ, suggesting that any population of cells in culture do not have equal sensitivity to Aβ. There may exist identifiable cell subpopulations that display more sensitivity to Aβ and will respond to its presence with intracellular Ca2+ changes. The setting of a measurable increased basal intracellular calcium level is probably caused by the activity of new calcium channels formed after the interaction of Aβ peptides with these cells.
Methods
Cell cultures
The following cell cultures were used: immortalized cell line PC12 derived from a transplantable rat pheochromocytoma purchased from American Type Culture Collection (Manassas, VA) and isolated human neurons, as described (Ravin et al. 2012).
Intracellular free calcium measurements
Cells were plated overnight (5 K cells/well) on 96-well plates (poly-d-lysine coated) and then incubated for up to 7 days in media containing Aβ42 (Aβ42.HCl, Bachem, Bubendorf, Switzerland, and Aβ42.TFA, Peptide International Inc. Louisville, KY, USA). To measure intracellular Ca2+, cells were loaded with 2 μM FURA-2AM (Molecular Probes) calcium sensitive probe in serum-free media for 30 min. Intracellular [Ca2+] was measured under incubation buffer (in mM: 135 NaCl, 5 KCl, 2.5 CaCl2, 1.2 MgCl2, 10 glucose, 10 Hepes, pH 7.4) by recording the emission from Fura-2AM using an inverted epifluorescence/phase contrast microscope equipped with a low-light level integrating CCD camera + microphotometer assembly (InCy I/P-2 Imaging & Photometry System, Intracellular Imaging INC.). Data analysis and graphing were performed using Excel (Microsoft) and Origin 8 (Origin Lab) software.
Cell images
PC12 cells were exposed to Aβ42 for up to 3 days. For confocal imaging, cells were loaded with the fluorescent probe Fura-2AM to test membrane integrity and treated for 10 min with Alexa Fluor 488 annexin V (Invitrogen) to detect the presence of PtdSer at the external face of the cell membrane. Two fluorofores were sequentially excited at wavelength 405 nm (fura-2AM) and 488 nm (Alexa fluor 488), respectively, and emissions were collected separately through Zeiss 710 inverted confocal laser scanning microscope. Confocal images were obtained using XYZ scan mode that a sequence of XY frames were obtained at 0.51-μm intervals in the z-(thickness) direction.
Results
Aβ42 affects only a subpopulation of PC12 cells
We have previously shown that Aβ peptides immediately interact with the cell surface resulting in measurable increases in intracellular calcium concentration followed by externalization of PtdSer (Simakova and Arispe 2006). In the present study, our objective was to assess the long-term consequences of Aβ peptide exposure on cytosolic [Ca2+]. Figure 1 shows confocal images of PC12 cells before and after incubation with 5 μM Aβ42 for 2 and 3 days. Before the images were taken, the cells were loaded with a calcium sensitive probe (Fura 2AM, red) for 30 min to detect cell integrity and exposed to a marker for exposed PtdSer (Alexa Fluor 488 annexin V, green). The images of cells that were not exposed to Aβ42 (control) show that most cells retain the Fura 2AM dye, and there is little or no annexin V labeling. After 2 days in Aβ42, most cells show noticeable increases in annexin V labeling indicating externalization of PtdSer. By day 3 in the presence of Aβ, most cells that retain Fura 2AM do not show the green fluorescence label corresponding to externally exposed PtdSer. Because cell membrane interaction with Aβ causes a loss of membrane integrity and the ability to retain fura 2AM, most of the cells remaining on day 3 posttreatment are likely to be functional Aβ-resistant.
Fig. 1.

Aβ affects only a subpopulation of PC12 cells. Confocal images of PC12 to examine the consequences of long-term exposure to Aβ42 (2 or 3 days). Control images show cells that were not exposed to Aβ. The figure shows three independent fields for each condition. Before images were taken, cells were loaded with Fura 2AM (red) for 30 min to identify viable cells and exposed to Alexa Fluor 488 annexin V (green) to label externally exposed PtdSer. Those cells that remain loaded with Fura 2 (red) preserve membrane integrity and are considered viable cells. Aβ induces externalization of PtdSer before cell degeneration. At the third day, cells that remain in the field do not show positive annexin V green fluorescence staining, preserve red fluorescence staining, and consequently are mainly functional Aβ-resistant cells
Aβ42 induces slow increases in intracellular [Ca2+] in PC12 cells
Intracellular [Ca2+] was measured in individual, viable PC12 cells (i.e., those that retain Fura 2AM) on six consecutive days to analyze the long-term effects of Aβ exposure (Fig. 2). Continuous cell division and cell degeneration by Aβ42 changed unevenly the number of viable cells available for screening. To evaluate comparatively the results for different conditions, the calcium values from each individual cell plotted in the figure were arranged from low to high corresponding to 100 representative points as analyzed by the Origin Lab software. The results show that cells normally exhibit a modest increase in intracellular [Ca2+] after several days in culture under control conditions (red lines). In contrast, cells grown in media containing Aβ42 (blue lines) exhibit a larger increase in intracellular [Ca2+]; however, the increase was not uniform for all cells. In a significant percentage of cells, the calcium levels continued to increase with time, until cell calcium homeostasis was probably overwhelmed leading to cell degeneration.
Fig. 2.
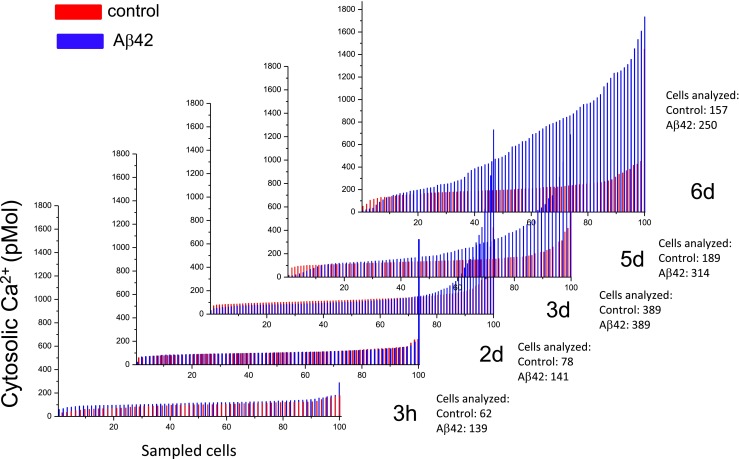
Aβ increases in intracellular calcium concentration in PC12 cells. Intracellular calcium levels from individual, viable PC12 cells measured on six consecutive days of continuous exposure to Aβ. The calcium values in individual cells were arranged and normalized from low to high and filtered to obtain 100 representative points. Cells normally exhibit a modest increase in the intracellular calcium levels after several days in culture under control conditions (red lines). In contrast, cells grown in media containing Aβ42 (blue lines) exhibit a much greater increase in intracellular [Ca2+]
Aβ42 induces slow increases in intracellular [Ca2+] in human neurons
Single-cell intracellular calcium screening was also performed in neurons isolated from human fetus (Fig. 3a). Closer examination of the results from these cells reveals that Aβ42 more rapidly induced elevation in the calcium levels and considerably reduced the density of viable neurons compared to control conditions. It is also notable that the [Ca2+] values measured at day 3 are greater than at day 7, suggesting that these cells are generally more sensitive to Aβ42 and/or less resistant to Aβ-dependent intracellular calcium increases than PC12 cells. Additionally, the frequency histogram distribution for all neurons analyzed (Fig. 3b) shows that in the presence of Aβ42, the intracellular [Ca2+] levels and the distribution of cells at day 1 and at day 7 were very similar. The two Gaussian fits to the histograms reveal peak [Ca2+] of 163 ± 1.68 and 282 ± 12.9 nM on day 1 compared to 177 ± 1.28 and 308 ± 6.5 nM on day 7. This analysis suggests that neurons remaining viable at day 7 were resistant to the deteriorating effect of Aβ42.
Fig. 3.

Aβ increases in intracellular calcium concentration in primary human neurons. Intracellular calcium levels measured from human neurons, at consecutive days of continuous exposure to Aβ. a Cells normally exhibit a modest increase in the intracellular calcium levels after several days in culture under control conditions (red lines). In contrast, cells grown in media containing Aβ (blue lines) exhibit a much greater increase in intracellular [Ca2+]. The calcium values measured at day 3 are greater than at day 7. b The frequency histogram distribution for all neurons analyzed shows that the distribution of cells at day 1 and at day 7 was similar. Cells analyzed at day 7 represent the group of neurons most resistant to Aβ42
Another noteworthy observation derived from the distribution of neurons based on their intracellular calcium levels, during long exposure to Aβ42, is described in Fig. 4. On day 1, the frequency distribution of neurons was well fit by a single Gaussian around a peak intracellular calcium concentration of 176 nM. By day 3, the cell distribution is now best fit by two Gaussians. One subpopulation of cells is distributed around 206-nM calcium concentrations, while another well-distinguished large subpopulation of neurons distributed around a much higher calcium concentration of 488 nM. By day 7, this subpopulation of higher calcium concentration almost disappeared, and the remaining neurons were distributed mainly around 182 nM, which is very close to the values observed at day 1. This data suggests that the subpopulation of neurons with higher intracellular calcium concentration, clearly distinguished at day 3, represents the group of neurons most sensitive to Aβ42. By day 7, however, this subpopulation of cells is largely absent probably as a result of cell degeneration due to increase in the intracellular [Ca2+] induced by Aβ42. Because these degenerating cells would not be expected to retain the calcium fluorescent probe, they were not considered viable neurons and therefore not analyzed.
Fig. 4.

Single-cell screening of intracellular calcium levels during long exposure to Aβ42 shows a subpopulation of Aβ42-resistant cells. On day 1, cells exposed to Aβ42 were distributed mainly around a single population well fit by a single Gaussian. However, by day 2, cells begin to reveal the effect of Aβ42, and the cell distribution shows the emergence of a second subpopulation which is better defined at day 3. This subpopulation was not observed at day 7. This persistent subpopulation is presumably made of cells resistant to Aβ. The bar plot corresponds to the averaged free cytosolic calcium for days 1, 2, 3, and 7. The average free cytosolic calcium of the whole cell population does not reveal the existence of subpopulations of cells with different sensitivity to Aβ42
Because there are clearly at least two subpopulations of cells, when the effect of Aβ42 on the cytosolic calcium is expressed as the average free cytosolic calcium of the whole cell population, the numbers may not accurately reflect the increasing effect of Aβ. The bar plot in Fig. 4 illustrates this situation. At day 3, the average [Ca2+] value measured for all viable cells in the culture (308.37 ± 12.5 nM) reflects the most significant increase in cytosolic calcium induced by Aβ. However, the average cytosolic [Ca2+] value after 7 days exposed to Aβ (196.06 ± 4.2 nM) showed no significant difference (p = 0.397) compared to the value measured after day 1 (190.32 ± 5.3 nM), leaving the impression that Aβ42 had no effect on intracellular [Ca2+] after 7 days.
Aβ channel blockers prevent the slow increase in intracellular calcium concentration in neurons
We have previously shown that Zn2+ is a potent inhibitor of Aβ cation channels (Arispe et al. 1996; Kawahara et al. 1997). Small peptides that copy the sequence of the putative pore region of the Aβ channel models, such as NA7, are also efficient channel blockers (Arispe 2004; Diaz et al. 2006; Arispe et al. 2007). To determine whether the long-term increases in cytosolic calcium are the result of Aβ ion channel activity, we exposed neurons to Aβ in culture media also containing either 5 μM Zn2+ (Fig. 5) or 40 μM NA7 (Fig. 6) for 3 days. Similar results were observed with both inhibitors. Single-cell measurement of intracellular [Ca2+] levels in the cells exposed to Aβ42 alone (red lines) was notably higher compared to the levels measured in control cells (blue lines). Both channel blockers markedly blunted the increase in intracellular calcium (green lines).
Fig. 5.
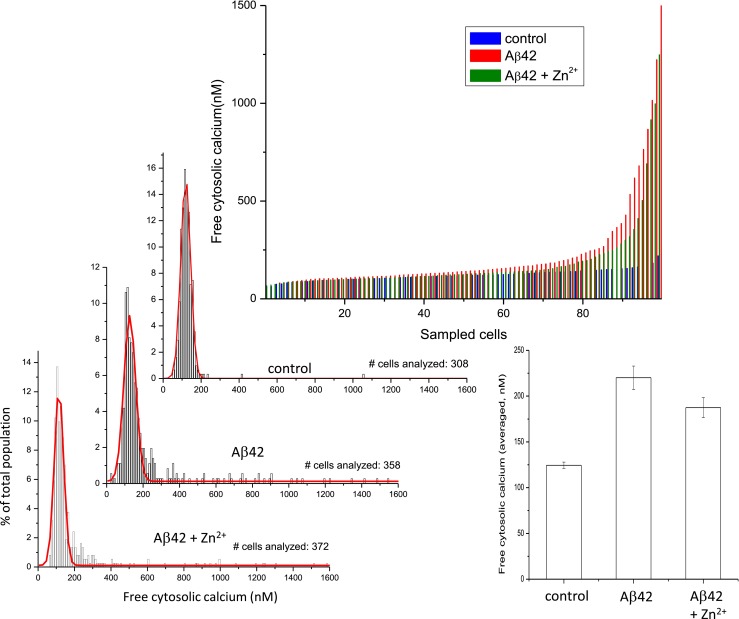
The Aβ channel blocker Zn2+ prevents the slow increase in intracellular calcium concentration in neurons. Single-cell measurement of intracellular [Ca2+] levels in the cells exposed to Aβ42 alone (red lines) and Aβ42 plus Zn2+ (blue lines) after 3 days. Continuous exposure to Aβ42 creates a subpopulation of cells with cytosolic calcium levels higher than average. The channel blocker Zn2+ markedly blunted the increase in intracellular calcium induced by Aβ in the subpopulation of Aβ42 more sensitive cells. Zn2+ also diminished the effect of calcium elevation from Aβ42. The bar plot corresponding to the averaged free cytosolic calcium of the whole cell population shows some protection by Zn2+ but does not reveal the highly effective protection by Zn2+ on the subpopulation of neurons that suffered the lowest calcium increases
Fig. 6.
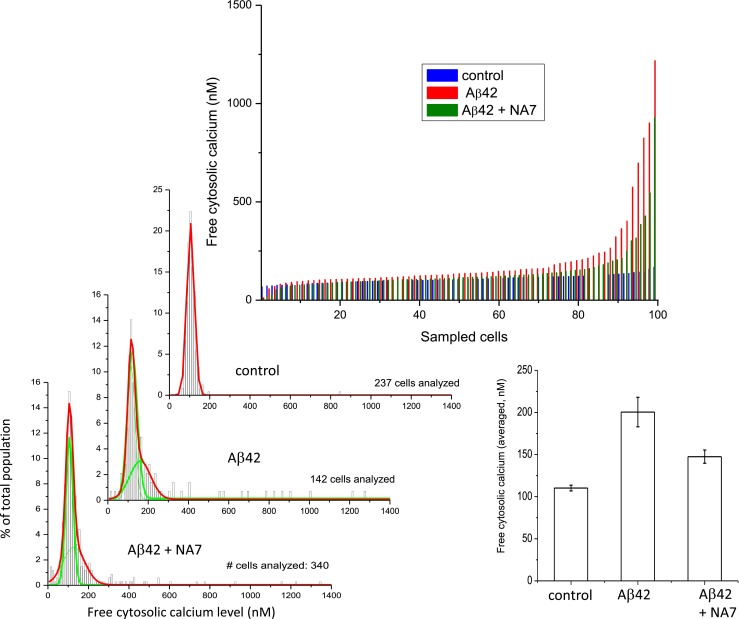
The Aβ channel blocker NA7 prevents the slow increase in intracellular calcium concentration in neurons. Single-cell measurement of intracellular [Ca2+] levels in the cells exposed to Aβ42 alone (red lines) and Aβ42 plus NA7 (blue lines) after 3 days. Continuous exposure to Aβ42 creates a subpopulation of cells with cytosolic calcium levels higher than average. NA7, a specific Aβ channel blocker small peptide, reduces the size of this subpopulation and the amplitude of calcium elevation. The bar plot of the averaged free cytosolic calcium of the whole cell population shows a significant protection by NA7 but does not reveal the absolute protection by NA7 on the subpopulation of neurons that suffered the lowest calcium increases
On closer examination, the data also shows that the protecting effect of Zn2+ was not homogeneous within the whole cell population. When zinc ions were present, approximately 70 % of total population of cells maintained the intracellular calcium at the same levels than those of control. We observed total protection by zinc ions on the subpopulation of neurons that underwent lower calcium increases and partial protection for cells that exhibited larger increases. The percentage of cells, which in the presence of Aβ42 alone underwent an intracellular calcium increase of more than 2× S.D. from the mean control of 118.5 nM, was 17.6 (63 cells of a total of 358). In the presence of zinc ions, this percentage was reduced to 12.6 meaning that in this subpopulation, zinc ions produced 28.4 % of protection. The highly effective protection by zinc ions in the total population of cells, however, was not evident when the results were considered in terms of the averaged intracellular calcium of all the cells analyzed in the culture. The bar plot at the right side of this figure shows that the treatment with Aβ42 produced an average intracellular [Ca2+] increase from 124 nM, at control condition, to 220 nM. When Aβ42 and zinc ions were simultaneously in the medium, the averaged intracellular [Ca2+] concentration was reduced to 187 nM. According to this analysis, zinc ions only produced a 34 % protection.
Figure 6 shows that when the Aβ channel blockers NA7 intracellular calcium for most cells were also maintained near the levels observed in control conditions. The two Gaussian fits to the cell distribution histograms corresponding to cells in Aβ42 alone revealed a main distribution of cells around a [Ca2+] peak of 116.5 nM (s.e. 0.7). When cells were in the presence of NA7, the number of cells with flagrant higher calcium values was reduced, and the averaged intracellular [Ca2+] concentration was reduced to 106.4 nM (s.e. 0.39), which is similar to the value observed in control conditions of 104.7 nM (s.e. 0.19). NA7 was also more effective to maintain the calcium levels similar to the control levels on about 70 % of all analyzed cells. This percentage corresponds to the subpopulation of neurons that suffered lower calcium increases. The percentage of cells, which in the presence of Aβ42 alone suffered an intracellular calcium increase of more than 2× S.D. from the mean control value, was 17.6 (25 cells of a total of 142). In the presence of NA7, this percentage was reduced to 9.4 %. Therefore, NA7 produced 46.6 % protection in this subpopulation. The bar plot at the right of the figure shows that the treatment with Aβ42 produced an averaged intracellular [Ca2+] increase from 110 nM, at the control condition, to 200 nM. When Aβ42 and NA7 were simultaneously in the medium, the averaged [Ca2+] increase produced by Aβ42 alone was reduced to 147 nM, indicating that the presence of NA7 effectively produced a 59 % protection.
Discussion
In this study, we present two novel findings. First, Aβ42 induces an increase in basal cytosolic calcium in both PC12 cells and human neurons. Interestingly, all cells were not equally affected by Aβ42 indicating that there are at least two subpopulations of cells—one that is Aβ42-sensitive and one that is Aβ42-insensitive. Second, the increase of cytosolic calcium is inhibited by Aβ42-cation channel blockers in Aβ42-sensitive cells,
Factors that influence the measurement of Aβ42-induced increases in cytosolic calcium
In this investigation, we made a comprehensive study of the changes in the cytosolic calcium levels that follows when cells were stressed by continuously exposed for several days to a medium containing micromolar concentrations of Aβ42. Chronic increases of intracellular calcium induced by Aβ have been observed under different experimental conditions and in various types of cells (see citations above), while others have reported no changes in basal calcium levels (Abramov et al. 2003; Chin et al. 2006).
We used two types of cells, PC12 and human neurons, to perform single-cell screening of the levels of free cytosolic calcium from hundreds of viable cells in culture. The differential timing and magnitude of the Aβ42-induced increase in calcium suggest that the effect produced by Aβ depends on the type of cell being monitored. The data displayed in Figs. 2 and 3 illustrate that measuring intracellular calcium either too early or too late could miss the induced Aβ intracellular calcium increases which has been reported by many, and it could lead to incorrectly report that Aβ has no effect.
This situation is more likely to happen when the calcium level is expressed as the averaged calcium level of the whole population of cells. Single-cell screening detects that not all cells within a culture respond to Aβ equally. Well-defined subpopulations of cells can be distinguished on the basis of their intracellular calcium levels. The subpopulations of cells with the highest calcium levels eventually degenerate and drop out of the analysis, leaving only the population of cells that is resistant to the continuing presence of Aβ. The cell-selective toxicity of Aβ has been noted before (Simakova and Arispe 2007a, b). In a well-defined subgroup of cells growing in culture medium containing Aβ, some intracellular processes, known to be sensitive to the presence of Aβ, such as lactate dehydrogenase release, caspase activation, DNA fragmentation, PtdSer externalization, have been found to remain unchanged even after multiple cell divisions (Simakova and Arispe 2007a, 2007b). It was suggested that particular characteristics or stages of the cells make them exceptionally sensitive, or resistant, to the effect of Aβ. We have also shown that a firm binding of Aβ to the cell membrane is first required for Aβ to initiate a toxic effect. In a previous work, we found that, within a wide range of cholesterol and PtdSer levels, the membrane content of these components shows a striking direct linear correlation with the capacity of the surface membrane to bind Aβ (Simakova and Arispe 2011). We noticed in the images in Fig. 1 that most viable cells after days in the continuous presence to Aβ are those that do not show positive annexin V green fluorescence staining. This finding supports the suggestion that membrane binding, regulated by externalized PtdSer, is one of the determining factors for the selective attack of Aβ on cells in culture (Lee et al. 2002; Simakova and Arispe 2007a, b, 2011). Other authors have also shown cell heterogeneity in the effect of Aβ to induce intracellular calcium increases in hippocampal neurons (Negishi-Kato and Kawahara 2008), and the regulation by cholesterol of the magnitude of the [Ca2+] increase induced by Aβ (Kawahara and Kuroda 2001). Here, we show that single-cell screening of cytosolic calcium also distinguishes cells that are resistant to Aβ.
Aβ42 cation channels and increases in cytosolic calcium
The mechanism by which Aβ increases cytosolic calcium remains a matter of debate. In the present study, Aβ channel blockers such as Zn2+ (Arispe 2004; Kawahara et al., 1997) and NA7 (Arispe et al. 2007) effectively reduced the increase in cytosolic calcium, strongly suggesting the participation of the Aβ ion channels in inducing intracellular calcium increases in cells exposed to Aβ. Both Zn2+ and NA7 completely protected about 70 % of the total population of cells and provided moderate protection to the cells which suffered larger calcium increases. This latter group of cells probably represents the most sensitive cells to Aβ, where conceivably a much larger number of Aβ channels were formed. The attenuation of the intracellular calcium increases by Aβ channel blockers compellingly suggests that exposure to Aβ provokes the formation of Aβ ion channels in the cell membrane and that calcium influx through Aβ channels is an initiating factor to other events responsible for the even more measurable cytosolic calcium increases observed in cells exposed to Aβ.
Role of calcium in the pathogenesis of Alzheimer’s disease
The role of intracellular calcium in the pathogenesis of Alzheimer’s disease (AD) was postulated (Khachaturian 1994; Mattson 2004, 2007; Bojarski et al. 2008; Abramov et al. 2004), and growing evidence supports the proposed role of calcium and Aβ peptides in AD pathophysiology. Exogenous application of synthetic amyloid-beta or oligomeric aggregates have long been known to induce morphological alterations as well as immediate and sustained rise in intracellular calcium changes and in many types of cultured cells (Bathia et al. 2000; Zhu et al. 2000; Lin et al. 2001; Kawahara et al. 2000; Simakova and Arispe 2006; Demuro et al. 2005; Guo et al. 1999; Mattson et al. 1992, 1993; smith et al. 2005). In vivo Ca2+-imaging experiments performed with APP transgenic mice showed that resting Ca2+ levels were significantly elevated in neurites located in the immediate vicinity of Aβ plaques (Kuchibhotla et al. 2008) that contain high local concentration of Aβ oligomers (Bezprozvanny and Mattson 2008; Bezprozvanny 2009, 2010). We have proposed that the acute and in chronic increases in the cytosolic calcium concentrations observed in cultured cells was the result of direct interaction of Aβ with cells which causes permeability changes in the plasma membrane (Simakova and Arispe 2006). The data in the present study add further support for this conclusion, demonstrating that Aβ induces marked elevation of cytosolic [Ca2+], but only in a subpopulation of sensitive neurons.
Acknowledgments
This work was supported by The Alzheimer’s Association of America.
References
- Abramov AY, Canevari L, Duchen MR. Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J Neurosci. 2003;23:5088–5095. doi: 10.1523/JNEUROSCI.23-12-05088.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramov AY, Canevari L, Duchen MR. Calcium signals induced by amyloid β peptide and their consequences in neurons and astrocytes in culture. Biochim Biophys Acta. 2004;1742:81–87. doi: 10.1016/j.bbamcr.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Aguayo LG, Parodi J, Sepúlveda FJ, Opazo C (2009) Pore-forming neurotoxin-like mechanism for Abeta oligomer-induced synaptic failure. In: Current Hypotheses and research Milestones in Alzheimer’s Disease (Perry G and Maccioni RB, eds) pp13-21. Springer
- Arispe N. Architecture of the Alzheimer’s AβP ion channel pore. J Membr Biol. 2004;197:33–48. doi: 10.1007/s00232-003-0638-7. [DOI] [PubMed] [Google Scholar]
- Arispe N, Pollard HB, Rojas E. Giant multilevel cation channels formed by Alzheimer disease amyloid beta-protein [a beta P-(1–40)] in bilayer membranes. Proc Natl Acad Sci U S A. 1993;90:10573–10577. doi: 10.1073/pnas.90.22.10573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arispe N, Pollard HB, Rojas E. β-amyloid Ca2+-channel hypothesis for neuronal death in Alzheimer disease. Mol Cell Biochem. 1994;140:119–125. doi: 10.1007/BF00926750. [DOI] [PubMed] [Google Scholar]
- Arispe N, Pollard HB, Rojas E. The ability of amyloid β-protein to form Ca2+ channels provides a mechanism for neuronal death in Alzheimer’s disease. Ann N Y Acad Sci. 1994;747:256–266. doi: 10.1111/j.1749-6632.1994.tb44414.x. [DOI] [PubMed] [Google Scholar]
- Arispe N, Pollard HB, Rojas E. Zn2+ interaction with Alzheimer amyloid β protein calcium channels. Proc Natl Acad Sci U S A. 1996;93:1710–1715. doi: 10.1073/pnas.93.4.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arispe N, Diaz J, Simakova O. Aβ ion channels. Prospects for treating Alzheimer’s disease with Aβ channel blockers. Biochim Biophys Acta. 2007;1768:1952–1965. doi: 10.1016/j.bbamem.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Arispe N, Diaz J, Durell SR, Shafrir Y, Guy HR. Polyhistidine peptide inhibitor of the Aβ calcium channel potently blocks the Aβ-induced calcium response in cells. Theoretical modeling suggests a cooperative binding process. Biochemistry. 2010;49:7847–7853. doi: 10.1021/bi1006833. [DOI] [PubMed] [Google Scholar]
- Bathia R, Lin H, Lal R. Fresh and globular amyloid β protein induces rapid cellular degeneration. A possible implication for calcium-uptake via AβP-channel. FASEB J. 2000;14:1233–1243. doi: 10.1096/fasebj.14.9.1233. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I. Calcium signaling and neurodegenerative diseases. Trends Mol Med. 2009;15(3):89–100. doi: 10.1016/j.molmed.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny IB. Calcium signaling and neurodegeneration. Acta Nat. 2010;2(1):72–82. [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease.Trends in. Neurosciences. 2008;31(9):454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojarski L, Herms J, Kuznicki J. Calcium dysregulation in Alzheimer’s disease. Neurochem Int. 2008;52:621–633. doi: 10.1016/j.neuint.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Chin JH, Tse FW, Harris K, Jhamandas JH. Beta amyloid enhances intracellular calcium rises mediated by repeated activation of intracellular calcium stores and nicotinic receptors in acutely dissociated rat basal forebrain neurons. Brain Cell Biol. 2006;35(2–3):173–186. doi: 10.1007/s11068-007-9010-7. [DOI] [PubMed] [Google Scholar]
- Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Membr Biol. 2005;280:17294–17300. doi: 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- Demuro A, Smith M, Parker I. Single-channel Ca(2+) imaging implicates Aβ1-42 amyloid pores in Alzheimer’s disease pathology. J Cell Biol. 2011;195(3):515–24. doi: 10.1083/jcb.201104133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz JC, Linnhan J, Pollard HB, Arispe N. Histidines 13 and 14 in the Aβ sequence are targets for inhibition of Alzheimer’s disease Aβ ion channel and cytotoxicity. Biol Res. 2006;39:447–460. doi: 10.4067/S0716-97602006000300007. [DOI] [PubMed] [Google Scholar]
- Green KN, LaFerla FM. Linking calcium to Abeta and Alzheimer’s disease. Neuron. 2008;59(2):190–194. doi: 10.1016/j.neuron.2008.07.013. [DOI] [PubMed] [Google Scholar]
- Guo Q, Sebastian L, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP. Increased vulnerability of hippocampal neurons from presenilin-1 mutant knock-in mice to amyloid beta-peptide toxicity: central roles of superoxide production and caspase activation. J Neurochem. 1999;72(3):1019–1029. doi: 10.1046/j.1471-4159.1999.0721019.x. [DOI] [PubMed] [Google Scholar]
- Johnson RD, Schauerte JA, Wisser KC, Gafni A, Steel DG. Direct observation of single amyloid-β(1–40) oligomers on live cells: binding and growth at physiological concentrations. PLoS ONE. 2011;6(8):e23970. doi: 10.1371/journal.pone.0023970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara M. Disruption of calcium homeostasis in the pathogenesis of Alzheimer’s disease and other conformational diseases. Curr Alzheimer Res. 2004;1:87–95. doi: 10.2174/1567205043332234. [DOI] [PubMed] [Google Scholar]
- Kawahara M, Kuroda Y. Molecular mechanism of neurodegeneration induced by Alzheimer’s beta-amyloid protein: channel formation and disruption of calcium homeostasis. Brain Res Bull. 2000;53(4):389–397. doi: 10.1016/S0361-9230(00)00370-1. [DOI] [PubMed] [Google Scholar]
- Kawahara M, Kuroda Y. Intracellular calcium changes in neuronal cells induced by Alzheimer’s beta-amyloid protein are blocked by estradiol and cholesterol. Cell Mol Neurobiol. 2001;21(1):1–13. doi: 10.1023/A:1007168910582. [DOI] [PubMed] [Google Scholar]
- Kawahara M, Arispe N, Kuroda Y, Rojas E. Alzheimer’s disease amyloid β-protein forms Zn2+-sensitive cation-selective channels across excited membrane patches from hypothalamic neurons. Biophys J. 1997;73:67–75. doi: 10.1016/S0006-3495(97)78048-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara M, Kuroda Y, Arispe N, Rojas E. Alzheimer’s beta-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. J Biol Chem. 2000;275(19):14077–14083. doi: 10.1074/jbc.275.19.14077. [DOI] [PubMed] [Google Scholar]
- Kawahara M, Negishi-Kato M, Sadakane Y. Calcium dyshomeostasis and neurotoxicity of Alzheimer’s beta-amyloid protein. Expert Rev Neurother. 2009;9(5):681–93. doi: 10.1586/ern.09.28. [DOI] [PubMed] [Google Scholar]
- Khachaturian ZS. Hypothesis on the regulation of cytosol calcium concentration and the aging brain, Neurobiol. Aging. 1987;8(1987):345–346. doi: 10.1016/0197-4580(87)90073-x. [DOI] [PubMed] [Google Scholar]
- Khachaturian ZS. Calcium hypothesis of Alzheimer’s disease and brain aging. Ann N Y Acad Sci. 1994;747:1–11. doi: 10.1111/j.1749-6632.1994.tb44398.x. [DOI] [PubMed] [Google Scholar]
- Korol TY, Korol SV, Kostyuk EP, Kostyuk PG (2008) Disruption of calcium homeostasis in Alzheimer’s disease. Neurophysiology 40(5/6):457–464
- Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59:214–225. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM. Calcium dyshomeostasis and intracellular signaling in Alzheimer’s disease. Nat Rev Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- Landfield PW. ‘Increased calcium-current’ hypothesis of brain aging. Neurobiol Aging. 1987;8:346–347. doi: 10.1016/0197-4580(87)90074-1. [DOI] [PubMed] [Google Scholar]
- Lee G, Pollard HB, Arispe N. Annexin 5 and apolipoprotein E2 protect against Alzheimer’s amyloid-β-peptide cytotoxicity by competitive inhibition at a common phosphatidylserine interaction site. Peptides. 2002;23(7):1249–1263. doi: 10.1016/S0196-9781(02)00060-8. [DOI] [PubMed] [Google Scholar]
- Lin H, Bhatia R, Lal R. Amyloid protein forms ion channels: implications for Alzheimer’s disease pathophysiology. FASEB J. 2001;15:2433–2444. doi: 10.1096/fj.01-0377com. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Calcium and neurodegeneration. Aging Cell. 2007;6:337–350. doi: 10.1111/j.1474-9726.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Tomaselli KJ, Rydel RE. Calcium-destabilizing and neurodegenerative effects of aggregated beta-amyloid peptide are attenuated by basic FGF. Brain Res. 1993;621(1):35–49. doi: 10.1016/0006-8993(93)90295-X. [DOI] [PubMed] [Google Scholar]
- Negishi-Kato M, Kawahara M. Neurosteroids block the increase in intracellular calcium level induced by Alzheimer’s β-amyloid protein in long-term cultured rat hippocampal neurons. Neuropsychiatr Dis Treat. 2008;4(1):209–18. doi: 10.2147/ndt.s2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parodi J, Sepúlveda FJ, Roa J, Opazo C, Inestrosa NC, Aguayo LG. Beta-amyloid causes depletion of synaptic vesicles leading to neurotransmission failure. J Biol Chem. 2010;285(4):2506–2514. doi: 10.1074/jbc.M109.030023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prangkio P, Yusko EC, Sept D, Yang J, Mayer M. Multivariate analyses of amyloid-beta oligomer populations indicate a connection between pore formation and cytotoxicity. PLoS ONE. 2012;7(10):e47261. doi: 10.1371/journal.pone.0047261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravin R, Blank PS, Steinkamp A, Rappaport SM, Ravin N, et al. Shear forces during blast, not abrupt changes in pressure alone, generate calcium activity in human brain cells. PLoS ONE. 2012;7(6):e39421. doi: 10.1371/journal.pone.0039421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauerte JA, Wong PT, Wisser KC, Ding H, Steel DG, Gafni A. Simultaneous single molecule fluorescence and conductivity studies reveal distinct classes of Aβ species on lipid bilayers. Biochemistry. 2010;49(14):3031–3039. doi: 10.1021/bi901444w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciacca MF, Kotler SA, Brender JR, Chen J, Lee DK, Ramamoorthy A. Two-step mechanism of membrane disruption by Aβ through membrane fragmentation and pore formation. Biophys J. 2012;103:702–710. doi: 10.1016/j.bpj.2012.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepulveda FJ, Parodi J, Peoples RW, Opazo C, Aguayo LG. Synaptotoxicity of Alzheimer beta amyloid can be explained by its membrane perforating property. PLoS ONE. 2010;5(7):e11820. doi: 10.1371/journal.pone.0011820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirwany NA, Payette D, Xie J, Guo Q (2007) The amyloid beta ion channel hypothesis of Alzheimer’s disease. Neuropsychiatr Dis Treat 3(5):597–612 [PMC free article] [PubMed]
- Simakova O, Arispe NJ. Early and late cytotoxic effects of external application of the Alzheimer’s Abeta result from the initial formation and function of Abeta ion channels. Biochemistry. 2006;45:5907–5915. doi: 10.1021/bi060148g. [DOI] [PubMed] [Google Scholar]
- Simakova O, Arispe NJ. Alzheimer’s disease Aβ peptide binding to cell membranes is correlated to membrane surface phosphatidylserine levels. Biophys J. 2007;92(3):612a. [Google Scholar]
- Simakova O, Arispe NJ. The cell-selective neurotoxicity of the Alzheimer’s Aβ peptide is determined by surface phosphatidylserine and cytosolic ATP levels. Membrane binding is required for Aβ toxicity,”. J Neurosci. 2007;27(50):13719–13729. doi: 10.1523/JNEUROSCI.3006-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simakova O, Arispe NJ. (2011) Fluorescent analysis of the cell-selective Alzheimer’s disease Aβ peptide surface membrane binding: influence of membrane components. Int J Alzheimers Dis. 2011;2011:917629. doi: 10.4061/2011/917629. Epub 2011 Jun 8 [DOI] [PMC free article] [PubMed]
- Small DH, Gasperini R, Vincent AJ, Hung AC, Foa L. The role of Abeta-induced calcium dysregulation in the pathogenesis of Alzheimer’s disease. J Alzheimers Dis. 2009;16(2):225–33. doi: 10.3233/JAD-2009-0951. [DOI] [PubMed] [Google Scholar]
- Smith IF, Green KN, LaFerla FM. Calcium dysregulation in Alzheimer’s disease: recent advances gained from genetically modified animals. Cell Calcium. 2005;38:427–437. doi: 10.1016/j.ceca.2005.06.021. [DOI] [PubMed] [Google Scholar]
- Supnet C, Bezprozvanny I. The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium. 2010;47:183–189. doi: 10.1016/j.ceca.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tofoleanu F, Buchete N-V. Alzheimer Aβ peptide interactions with lipid membranes: fibrils, oligomers and polymorphic amyloid channels. Prion. 2012;6(4):339–45. doi: 10.4161/pri.21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YJ, Lin H, Lal R. Fresh and nonfibrillar amyloid β protein(I-40) induces rapid cellular degeneration in aged human fibroblasts: evidence for AβP-channel-mediated cellular toxicity. FASEB J. 2000;14:1244–1254. doi: 10.1096/fasebj.14.9.1244. [DOI] [PubMed] [Google Scholar]