

Summary
The feasibility of performing broad and deep tumour genome sequencing has shed new light into tumour heterogeneity and provided important insights into the evolution of metastases arising from different clones1,2. To add an additional layer of complexity, tumour evolution may be influenced by selective pressure provided by therapy, in a similar fashion as it occurs in infectious diseases. Here, we have studied the tumour genomic evolution in a patient with metastatic breast cancer bearing an activating PIK3CA mutation. The patient was treated with the PI3Kα inhibitor BYL719 and achieved a lasting clinical response, although eventually progressed to treatment and died shortly thereafter. A rapid autopsy was performed and a total of 14 metastatic sites were collected and sequenced. All metastatic lesions, when compared to the pre-treatment tumour, had a copy loss of PTEN, and those lesions that became refractory to BYL719 had additional and different PTEN genetic alterations, resulting in the loss of PTEN expression. Acquired bi-allelic loss of PTEN was found in one additional patient treated with BYL719 whereas in two patients PIK3CA mutations present in the primary tumour were no longer detected at the time of progression. To functionally characterize our findings, inducible PTEN knockdown in sensitive cells resulted in resistance to BYL719, while simultaneous PI3Kp110β blockade reverted this resistance phenotype, both in cell lines and in PTEN-null xenografts derived from our patient. We conclude that parallel genetic evolution of separate sites with different PTEN genomic alterations leads to a convergent PTEN- null phenotype resistant to PI3Kα inhibition.
We are currently engaged in testing the antitumour activity of a novel PI3Kα inhibitor, BYL719, in patients with tumours harbouring activating PI3K p110α mutations3. The PI3K pathway is essential for cell growth, proliferation, survival, and metabolism4,5. The PI3K family of enzymes is divided into three main classes (I to III), with class I being the most often implicated in human cancer6. Class IA PI3K is a heterodimer composed of a catalytic subunit (p110α, β, or δ) and a regulatory subunit 7,8. PIK3CA, the gene encoding p110α, is mutated in up to 40% of oestrogen receptor (ER) and/or HER2 positive breast tumours9,10. In our ongoing phase I clinical study of BYL719 we have observed clinical responses in breast, head and neck, and other tumours3, providing proof of principle that PI3Kα targeting is active against tumours harbouring PIK3CA mutation.
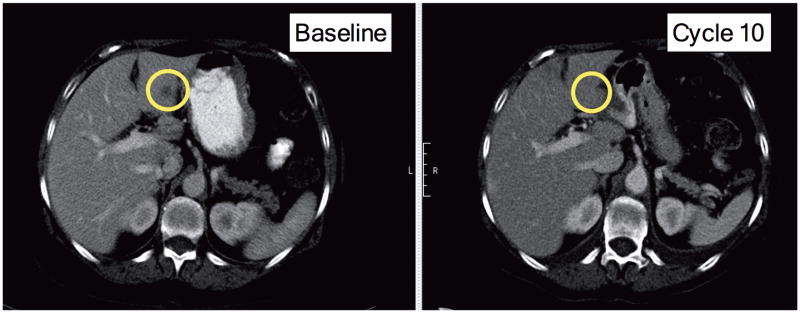
We are presenting the case of a 60-year-old breast cancer patient diagnosed with invasive ductal carcinoma that underwent surgery followed by adjuvant treatment with chemotherapy and the aromatase inhibitor examestane. Four years later, the patient developed bone metastases and started therapy with the oestrogen receptor antagonist fulvestrant, achieving stable disease. After eighteen months on therapy, her disease progressed in the liver, bone and lymph nodes. The archival tissue of the primary tumour was subjected to PCR-based genetic analysis11 and a hot spot mutation in PIK3CA (E542K) was detected. This finding led to the patient's enrolment in a phase I clinical trial of BYL719 (NCT01219699). The patient rapidly achieved a confirmed partial response according to the RECIST 1.0 criteria12 that lasted 9.5 months (Table 1 and Extended Data Fig. 1). At that point, while the tumour remained stable in multiple sites including a peri-aortic lymph node location, progression occurred in the lungs (Fig. 1) and consequently therapy with BYL719 was discontinued. The clinical status of the patient deteriorated rapidly and she died two months after termination of the BYL719 treatment.
Table 1. Patient information.
| Patient | Site |
PIK3CA baseline |
Dose (mg) |
Response (RECIST) |
DOT (days) |
PTEN Post-T |
PIK3CA Post-T |
|---|---|---|---|---|---|---|---|
| Index | Breast | E542K | 400 | PR (-52.4%) | 285 | Loss | E542 |
| 1 | Breast | H1047R | 400 | SD* | 181 | Unch | H1047R |
| 2 | Breast | H1047R | 400 | SD (-26.3%) | 424 | Loss | H1047R |
| 3 | Breast | H1047R | 400 | SD (-28.5%) | 179 | Unch | WT |
| 4 | Breast | H1047L | 400 | SD (-11.3%) | 504 | Unch | H1047L |
| 5 | Breast | H1047R | 400 | SD (-24.9%) | 110 | Unch | H1047R |
| 6 | Salivary | E545K | 400 | SD (-17%) | 112 | Unch | WT |
With interval decrease in left breast and left chest wall skin thickening). DOT= duration of treatment. Unch= unchanged. Mut= mutated. WT= wild-type. Amp=amplified. Post-T= post-treatment.
Figure 1. Clinical response of index patient treated with BYL719.
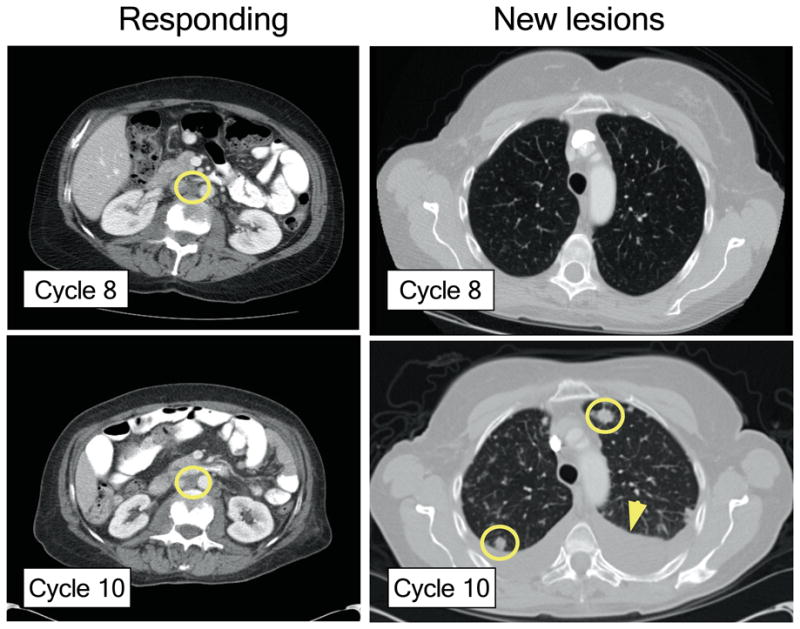
CT scans showing stable peri-aortic lymph node metastasis (yellow circle) and the appearance of new lung metastases (yellow circles) after the completion of the tenth cycle of BYL719 therapy. Arrow: pleural effusion.
A rapid autopsy was performed three hours after death and a total of 14 metastases with tumour cells present were identified and collected for sequencing (Extended Data Table 1).
In order to proceed systematically to identify possible genetic determinants of acquired resistance to PI3K p110α inhibition, we took a three-step approach. First, we examined both the primary tumour (before BYL719 treatment) and the new lung metastasis by whole genome sequencing. Although both samples shared many somatic genetic aberrations (Fig. 2a and Extended Data Fig. 2), only a PTEN copy number loss was detected in the lung metastasis (Fig 2b). Then, we analysed the primary tumour, lung metastasis, and the peri-aortic lesion that remained stable (responding) at the time of progression to BYL719 therapy by whole exome sequencing (Fig. 2c). This analysis revealed that both peri-aortic and lung lesions harboured mutations in PIK3CA, ESR1, and BRCA2, and single copy loss of PTEN. Importantly, in addition to the PTEN copy number loss, we identified a PTEN del339FS mutation only in the lung metastasis (Fig. 2c). By immunohistochemistry (IHC), we observed that PTEN protein expression was lost in the lung metastasis but was present in both the primary tumour and periaortic lesion (Fig. 2d).
Figure 2. Loss of PTEN upon BYL719 resistance.
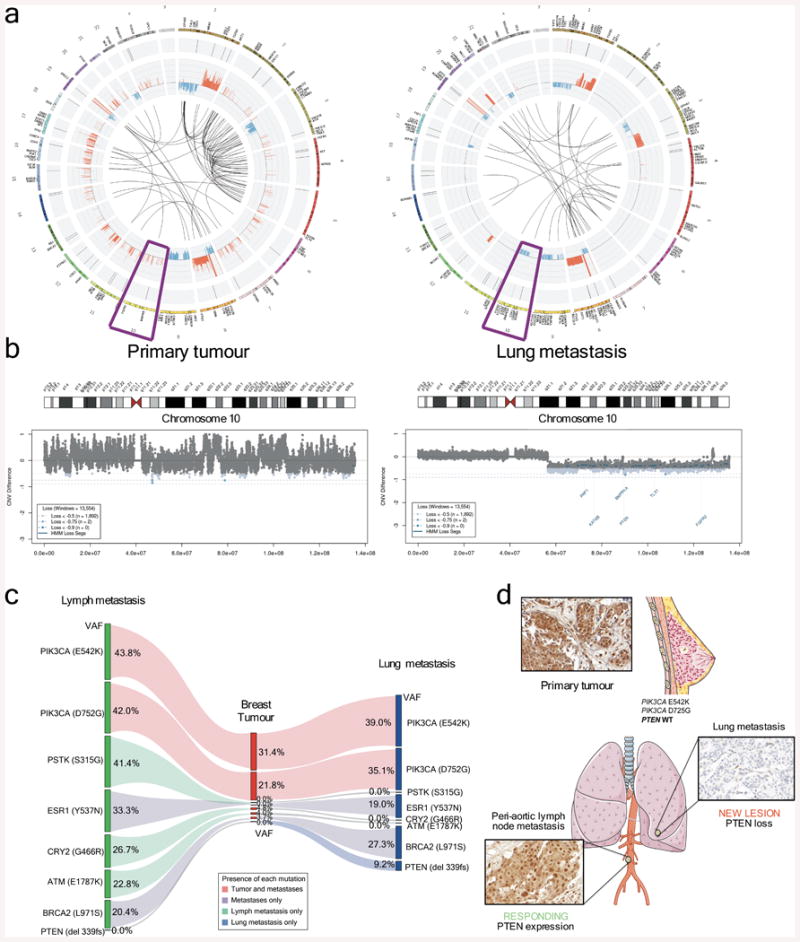
a, Circos plots from WGS analysis of primary tumour (prior to BYL719 treatment) and a lung metastasis appearing after the tenth cycle of BYL719 therapy. b, CNV of chromosome 10. c, WES of the peri-aortic lymph node showing durable stable disease during BYL719 therapy compared to both primary tumour and the progressing lung lesion. The diagram shows the variation of allele frequencies (VAF) of the listed gene mutations in the three lesions. The estimated tumour purities are 44% for the breast primary tumour, 50% for the lung metastasis, and 59% for the lymph node metastasis. d, PTEN IHC of primary tumour, peri-aortic lymph node, and lung metastasis. Images were taken from Servier Medical Art.
Finally, to confirm and expand our findings, we sequenced the primary tumour and all the metastatic lesions to >500-fold coverage using a custom targeted deep-sequencing assay, MSK-IMPACT13,14. A number of mutations were shared by the primary tumour and the metastatic sites whereas others were observed only in all or in selected metastatic lesions (Fig. 3a and Supplementary Table). We confirmed that the PIK3CA E542K mutation in the primary tumour was conserved in the metastatic samples and detected the presence of another PIK3CA mutation (D725G). Moreover, we found increased copy number of FGFR1 and EI4EBP1 in all tumour samples, consistent with the relatively frequent 8p11-12 amplification described in breast cancer15,16. ESR1 Y537N and BRCA2 L971S alterations were present in all the metastatic lesions but not in the primary tumour. We speculate that the ESR1 Y537N mutation, reported to promote ligand-independent ER activation13, was selected upon anti-oestrogen therapy received by the patient prior to BYL719 treatment. Central to our work, all metastatic lesions appeared to harbour a single copy loss of PTEN (Extended Data Fig. 3). Furthermore, we found that 10 of 14 metastatic lesions harboured additional genomic alterations within PTEN. The spectrum of PTEN alterations was heterogeneous across the 10 samples and included a splice site mutation at K342, a frameshift indel at P339 (confirming the WES result), and 4 different exon-level deletions (Fig. 3a and Extended Data Fig. 4). All 10 specimens with either secondary PTEN mutations or copy number loss were confirmed negative for PTEN staining by IHC whereas the four specimens that retained a PTEN WT allele were positive for PTEN protein expression (Extended Data Fig. 5). In addition, for those lesions that were visualized by CT scan, there was a tight correlation between progression of disease and loss of PTEN expression. The peri-aortic lesion (M02) that was responding at the time of disease progression still contained one PTEN WT allele and protein expression. On the contrary, the lung lesions (M04, M06, M09, and M11) and liver lesion (M12) with documented progression to therapy had bi-allelic PTEN alteration and lack of expression.
Figure 3. Loss of PTEN by different genetic alterations.
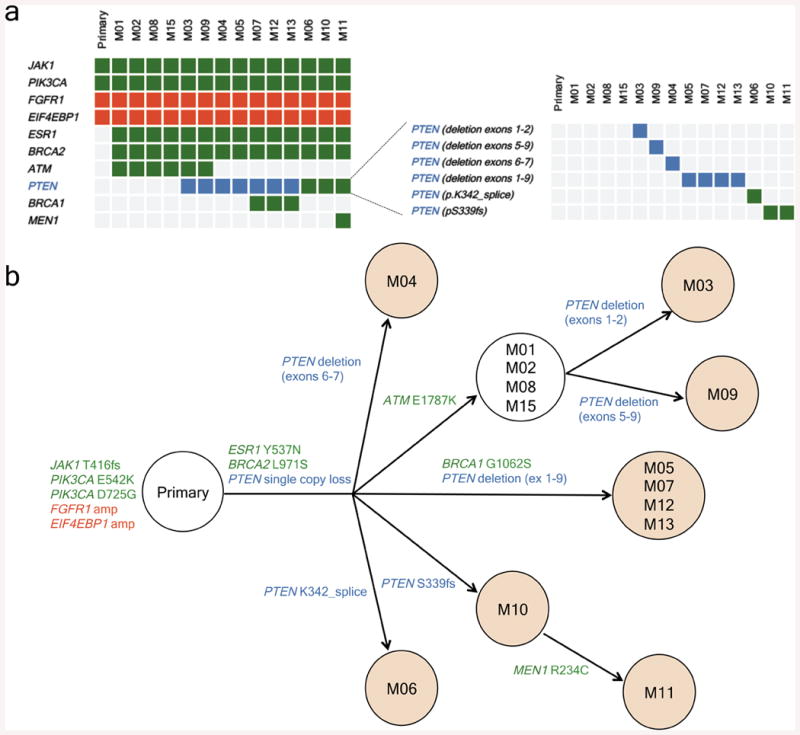
a, Heatmap of the non-silent genetic alterations across the primary tumour and the 14 metastases collected during the autopsy of the index patient. Gene mutations are depicted in green, gene amplifications in red, and gene copy number loss in blue. b, Dendrogram showing the proposed phylogenetic evolution of the metastases in the index patient. Shadowed circles represent metastatic lesions with bi-allelic loss of PTEN and lack of PTEN expression by IHC.
In an effort to integrate the genomic data from our patient, we constructed a dendrogram mapping the phylogenetic evolution of the disease. Our findings suggest that all the lesions were derived from the PTEN WT primary tumour and that there was a progressive and parallel loss of PTEN under BYL719 selective pressure (Fig. 3b). Of note, the two-month duration between progression to BYL719 and autopsy needs to be considered as well.
In order to expand our observations, we analysed paired samples (pre-treatment and at progression) from six additional patients enrolled in the same study at our institution (Table 1). Targeted exome sequencing identified homozygous loss of PTEN in a post-treatment sample of a breast cancer patient who developed resistance to BYL719 after initially experiencing a durable response to therapy (Table 1). We also confirmed lack of PTEN expression by IHC in the post-treatment sample (Extended Data Fig. 6). We found no detectable PIK3CA mutations in the post-treatment samples of two patients (Table 1). Given that the presence of PIK3CA mutations drove sensitivity to BYL719 in our cell line screens17, positive selection of clones bearing wild-type alleles of PIK3CA may explain the emergence of resistance to BYL719 in these two additional cases. These results may be an indication that in some cases PIK3CA mutations are not early founder/truncal events but branched subclonal drivers that are cleared from the tumours under the selective pressure of PI3Kα inhibition. In any case, the fact that loss of PTEN expression and emergence of PIK3CA WT clones are mutually exclusive in our patient samples indicates that both events may be important in opposing the therapeutic efficacy of BYL719.
No other alterations with an obvious connection to BYL719 resistance were found in the responding cases, with the exception of a mutation (E1490*) and an in-frame deletion in MAP3K1in one of the three patients for which neither PTEN nor PIK3CA status changed during BYL719 treatment. Further characterization is needed to determine whether these mutations lead to increased MEK and ERK signaling and limit the effects of PI3K inhibition.
PTEN encodes for a phosphatase that regulates the activity of PI3K by limiting the accumulation of phosphatidylinositol (3,4,5)-trisphosphate (PIP3), a required mediator to initiate the PI3K/AKT/mTOR signalling cascade18. In the absence of PTEN, cancer cells become dependent mostly on the activity of the p110β isoform of PI3K to propagate signalling through downstream pathway effectors19,20. In an attempt to explain this, we hypothesized that progressive decrease or loss of PTEN expression in the presence of PI3K p110α inhibition might restore PI3K/AKT signalling through PI3K p110β activity. To test our hypothesis, we established cell lines expressing either doxycycline-inducible or constitutive shRNA against PTEN mRNA in T47D and MCF7 cells, known to be intrinsically sensitive to BYL71921. As expected, induction of PTEN downregulation led to activation of AKT and the downstream effectors PRAS40, GSK3β and S6 in both T47D (Fig. 4a) and MCF7 (data not shown) cells under basal conditions. PTEN downregulation markedly limited the effects of BYL719, both at the signalling and cell viability level. On the other hand, PTEN knockdown did not result in resistance to the pan-PI3K inhibitor BKM120, which blocks all the PI3K p110 isoforms (Fig. 4b and Extended Data Fig. 7a). Similar effects were observed in another BYL719-sensitive cell line (MDA-MB-453) with constitutive PTEN knockdown (Extended Data Fig. 7b).
Figure 4. Loss of PTEN expression and sensitivity to PI3Kα and PI3Kβ blockade.
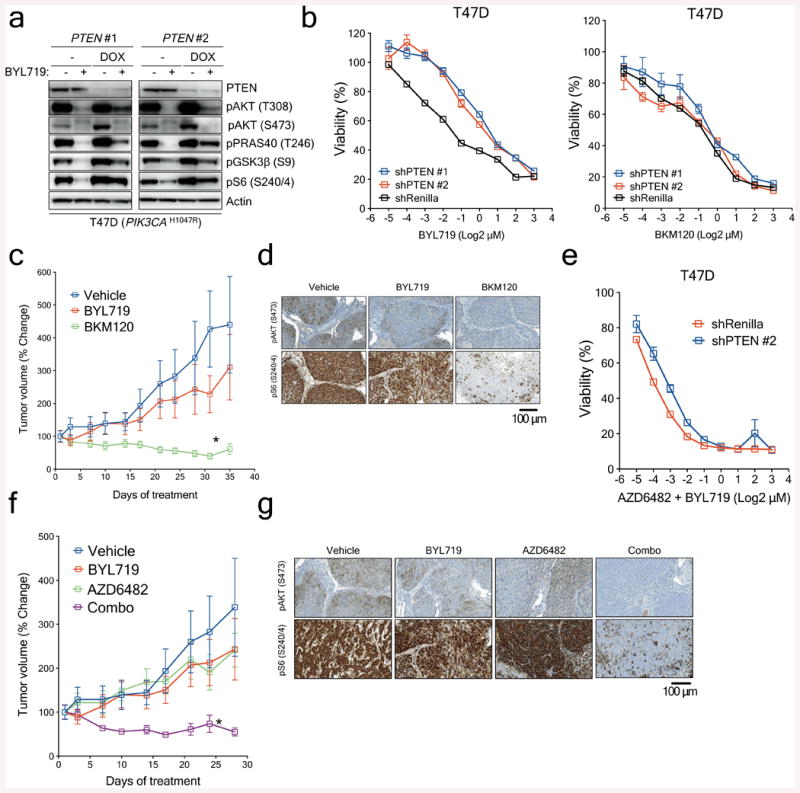
a, Western blot showing PTEN knockdown by two independent shRNAs and its effects on the PI3K/AKT/mTOR pathway. b, Cell viability assay in T47D cells with inducible PTEN knockdown treated with increasing concentrations of BYL719 or BKM120. Error bars represent standard error of the mean (SEM). c, Antitumour activity of either BYL719 (25mg/kg daily) or BKM120 (25mg/kg daily) in PDXs subcutaneously grown in nude mice (n=6 (Vehicle) and n=8 (Treatments)). Error bars represent SEM. d, Representative immunostaining for phosphorylated AKT (pAKT) and phosphorylated S6 (pS6) in PDXs treated as shown. Tumours were collected at the end of the experiment of Panel c, 2 hours after the last dosage. Scale bar represents 100μm. e, Cell viability assay in T47D cells with PTEN expression (shRenilla) or PTEN knockdown (shPTEN#2) treated with increasing concentrations of the combination of BYL719 and AZD6482. Error bars represent SEM. f, Antitumour activity of either BYL719 (25mg/kg daily) or the combination of BYL719 and AZD6482 (25mg/kg daily) in PDXs subcutaneously grown in nude mice (n=6 (Vehicle) and n=8 (Treatments)). Error bars represent SEM. g, Representative immunostaining for phosphorylated AKT (pAKT) and phosphorylated S6 (pS6) in PDXs treated as shown. Tumours were collected at the end of the experiment of panel f, 2 hours after the last dosage. Scale bar represents 100μm. * p<0.05.
From our patient's non-responding PTEN-null lung metastatic lesion, we were able to establish xenografts in nude mice. Consistent with the in vitro data, this patient-derived xenograft was resistant to BYL719 treatment but sensitive to BKM120 (Fig. 4c). The degree of inhibition of phospho-AKT and phospho-S6 was also higher with BKM120 (Fig. 4d and Extended Data Fig. 7c and d). These results were complemented by the combination of BYL719 and the PI3K p110β inhibitor AZD6482. Upon PTEN knockdown, only combined p110α and β blockade was capable of reverting the resistant phenotype (Fig. 4e and Extended Data Fig 8a). Similarly, the BYL719-resistant patient derived xenograft was insensitive to AZD6482 alone but responded to the combination of both compounds (Fig. 4f). Profound inhibition of AKT and S6 phosphorylation was achieved only upon treatment with BYL719 in combination with AZD6482 (Fig. 4g and Extended Data Fig. 8b and c). Taken together, these data indicate that inhibition of the p110β isoform is required to achieve antitumour activity in cells/tumours that lost PTEN expression and become resistant to BYL719.
In summary, we report a case of parallel genetic evolution under selective therapeutic pressure leading to a progressive loss of PTEN expression and consequent gain of dependency on the PI3K p110β isoform. Parallel evolution under selective pressure has been described in conditions where treatments are highly efficacious such as in HIV22. Our case highlights that this tumour, despite its heterogeneity, was dependent on PI3K signalling probably as a result of the presence of the same activating PIK3CA mutation in all the tumour sites. Upon continued suppression of PI3Kα, diverse genomic alterations emerged, leading to PTEN loss as an alternate mechanism of PI3K activation. Moreover, our study emphasises the importance of tumour interrogation upon progression to therapy and the dynamic nature of tumour genomes under selective therapeutic pressure.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Methods
PIK3CA mutant cell lines MCF7 (E545K) and T47D (H1047R) (ATCC) were transduced with the retroviral TRMPV vector. Doxycycline (Sigma) was used to temporally activate the expression of a microRNA 30-embedded shRNA targeting Renilla luciferase (control) or PTEN mRNA. The hairpin sequences used were as follows:
Renilla Luciferase
CTCGAGAAGGTATATTGCTGTTGACAGTGAGCGCAGGAATTATAATGCTTATCTATAGTGAAGCCACAGATGTATAGATAAGCATTATAATTCCTATGCCTACTGCCTCGGAATTC
PTEN#1
CTCGAGAAGGTATATTGCTGTTGACAGTGAGCGACCAGCTAAAGGTGAAGATATATAGTGAAGCCACAGATGTATATATCTTCACCTTTAGCTGGCTGCCTACTGCCTCGGAATTC
PTEN#2
CTCGAGAAGGTATATTGCTGTTGACAGTGAGCGCCCAGATGTTAGTGACAATGAATAGTGAAGCCACAGATGTATTCATTGTCACTAACATCTGGTTGCCTACTGCCTCGGAATTC
Cell viability was assessed using the tetrazolium-based MTT assay after 6 days of treatment. All cell lines resulted negative for mycoplasma contamination. Western blotting was carried out using previously described methods21. All the in vitro experiments were performed in triplicate.
Patient-derived xenografts and IHC
Animals were maintained and treated in accordance with Institutional Guidelines of Memorial Sloan Kettering Cancer Center (Protocol number 12-10-019). Tumours were implanted subcutaneously in six-week-old female athymic NU/NU nude mice. Once the tumours reached a volume of ∼200 mm3, it was expanded in multiple mice which were then randomized to the following treatments: BYL719, BKM120 (a pan-class I PI3K inhibitor), or AZD6482 (a PI3Kβ inhibitor), each administered orally at 25 mg/kg once a day. After treatment, mice were euthanized and tumours were harvested and procured for IHC analysis. IHC was performed on a Ventana Discovery XT processor platform using standard protocols and the following antibodies:
pAKT (S473) (D9E): Cell Signaling Technology, #4060, dilution 1:70
pS6 (S240/4) (D68F8)XP: Cell Signaling Technology, #5364, dilution 1:500
PTEN (138G6): Cell Signaling Technology, #9559, dilution 1:30
All the in vivo experiments were run with at least n=8 for each treatment arm. Two-way t-test was performed using GraphPad Prism (GraphPad Software). Error bars represent the SEM. *p<0.05.
Whole genome and whole exome sequencing
For whole genome (WG) and whole exome (WE) sequencing, DNA was derived from the primary tumour, lung metastasis, and peri-aortic lymph node metastasis. DNA from the spleen was used as a normal control. WG libraries were produced as previously described23 and sequenced using the Illumina HiSeq 2500 platform as paired-end 100 base pair reads, producing ∼30-fold (primary tumour, spleen normal)-50-fold (lung metastasis) coverage for WG sequencing. By hybrid capture (Nimblegen version 3.0) of the lymph node and lung metastases, primary tumour and spleen normal, we generated ∼100-fold coverage for WE sequencing.
All patients provided written informed consent for the genetic research studies performed in accordance with protocols approved by Dana Farber/Harvard Cancer Center Institutional Review Board. Autopsy in the index patient was performed within the first three hours post mortem.
DNA sequences are deposited in the EGA database with accession number EGAS00001000991.
Targeted exome sequencing (MSK-IMPACT)
DNA derived from the primary tumour, 14 metastases, and matched normal spleen tissue was further subjected to deep-coverage targeted sequencing of key cancer-associated genes. Our assay, termed MSK-IMPACT (Integrated Mutation Profiling of Actionable Cancer Targets), involves hybridization of barcoded libraries to custom oligonucleotides (Nimblegen SeqCap) designed to capture all protein-coding exons and select introns of 279 commonly implicated oncogenes, tumour suppressor genes, and members of pathways deemed actionable by targeted therapies14. The captured pool was subsequently sequenced on an Illumina HiSeq 2500 as pairedend 75-base pair reads, producing 513-fold coverage per tumour. Sequence data were analysed to identify three classes of somatic alterations: single-nucleotide variants, small insertions/deletions (indels), and copy number alterations.
Barcoded sequence libraries were prepared using 250 ng genomic DNA (Kapa Biosystems) and combined in a single equimolar pool. Sequence data were demutliplexed using CASAVA, and reads were aligned to the reference human genome (hg19) using BWA and postprocessed using the Genome Analysis Toolkit (GATK) according to GATK best practices24,25.
MuTect and GATK were used to call single-nucleotide variants and small indels, respectively26. Exon-level copy number gains and losses were inferred from the ratio in Tumour:Normal sequence coverage for each target region, following a loess-normalization to adjust for the dependency of coverage on GC content27. DNA sequences are deposited in the EGA database with accession number EGAS00001000991.
Statistical analysis
Two-way t-tests were performed using GraphPad Prism (GraphPad Software). Error bars represent the S.E.M., p values are indicated as *p<0.05. All cellular experiments were repeated at least three times. All the in vivo experiments were run with at least 6-8 tumours for each treatment arm. Samples size was chosen to detect a difference in means of 20% with a power of 90%. Animals were randomized in groups with similar average in tumour size. Investigators were blinded when assessing the outcome of the in vivo experiments.
For the cell viability graphs, non-linear regression was applied to the experimental data sets. Curves were compared using the extra-sum-of-squares F test using α=0.05. Hypothesis was rejected when non-linear models were not nested within each other and was considered statistically significant.
Extended Data
Fig 1. CT scan of index patient.
CT scan showing a liver lesion experiencing a partial response after 6 cycles of BYL719
Fig. 2. Gene copy number variation in both primary tumour and lung metastasis.

Fig. 3. Representative exon-level copy number profiles for genes on chromosome 10.

Exons in PTEN are shown in red.
Fig. 4. Loss-of-function mutations in PTEN detected by MSK-IMPACT.

Mutations were visualized by the Integrative Genomics Viewer (IGV).
Fig 5. PTEN immunostaining of the 14 metastases collected during the autopsy.

Hematoxylin and eosin (H&E) and PTEN expression detected by IHC in 14 metastases collected during the autopsy of the index patient. PTEN staining in PTEN negative samples is only present in stromal cells.
Fig 6. PTEN immunostaining in patients treated with BYL719.
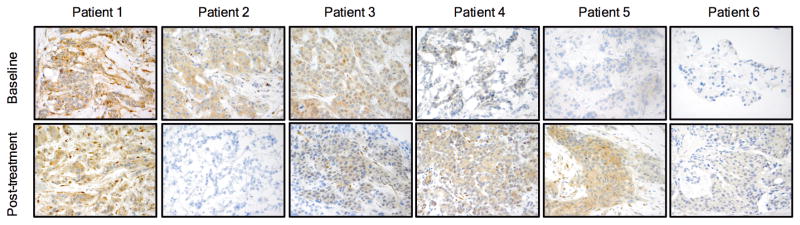
PTEN expression detected by IHC in paired samples from six additional patients treated with BYL719. Specimens before starting BYL719 therapy (baseline) and at time of disease progression (post-treatment) are compared.
Fig 7. Inducible loss of PTEN and sensitivity to BYL719 and BKM120.

a. Cell viability assay in MCF7 cells with inducible PTEN knockdown treated with increasing concentrations of either BYL719 or BKM120. Error bars indicate SEM. b. Cell viability assay in MDA-MB-453 (MDA453) cells with constitutive PTEN knockdown treated with increasing concentrations of either BYL719 or BKM120. Error bars indicate SEM. c. Quantification of pAKT (S473) and pS6 (S240/4) from Figure 4d. Student t-test was used and p values are indicated. d. Western blot from the PDXs treated as indicated.
Fig 8. Constitutive loss of PTEN and sensitivity to BYL719 and AZD6482.

a. Cell viability assay in T47D cells with inducible PTEN knockdown (#2) treated with increasing concentrations of either BYL719 or AZD6482 in the presence of doxycycline 1μg/mL. Error bars indicate SEM. b. Quantification of pAKT (S473) and pS6 (S240/4) from Figure 4g. Student t-test was used and p values are indicated. Error bars indicate SEM. c. Western blot from the PDXs treated as indicated.
Extended Data Table 1. Samples analyzed from the index patient.
Summary of the lesions collected during the autopsy of the index patient, cellularity assessment, imaging, clinical outcome, and sequencing techniques used. N=No. Y=Yes.
| Lesion | Location | Cellularity | CT Scan | Response | WGS | WES | IMPACT |
|---|---|---|---|---|---|---|---|
| Primary | Breast | Unknown | Y | Y | Y | ||
| M01 | Ovary | 75% | N | N | N | Y | |
| M02 | Periaortic lymph node | 50% | Y | Y | Y | Y | Y |
| M03 | Liver (posterior) | 65% | N | N | N | Y | |
| M04 | Left Lung (lower lobe) | 75% | Y | N | N | N | Y |
| M05 | Thoracic spine | 65% | N | N | N | Y | |
| M06 | Right Lung (upper lobe) | 55% | Y | N | N | N | Y |
| M07 | Liver (dome) | 70% | N | N | N | Y | |
| M08 | Uterus | 55% | N | N | N | Y | |
| M09 | Left Lung (upper lobe) | 75% | Y | N | N | N | Y |
| M10 | Carina (lymph node) | 70% | N | N | N | Y | |
| M11 | Right Lung (lower lobe) | 65% | Y | N | Y | Y | Y |
| M12 | Liver (left lower lobe) | 50% | Y | N | N | N | Y |
| M13 | Liver (left lobe) | 70% | N | N | N | Y | |
| M15 | Adrenal gland | 40% | N | N | N | N | Y |
Supplementary Material
Acknowledgments
We thank members of the MSKCC Diagnostic Molecular Pathology Laboratory and the MSK Genomics Core Laboratory for assistance with sequencing. We thank Marina Asher and Umeshkumar Bhanot from the MSKCC Pathology Core for assistance with tissue staining. This work was funded by a “Stand Up to Cancer” Dream Team Translational Research Grant, a Program of the Entertainment Industry Foundation (SU2C-AACR-DT0209), the Breast Cancer Research Foundation, the Geoffrey Beene Cancer Research Center, the Starr Cancer Consortium and a MMHCC grant (CA105388). DJ is funded also by a National Institute of Health Training Grant (T32 CA-71345-15) and by philanthropic support from Stephen and Kathleen Chubb.
Footnotes
Author Contributions: DJ, PC, MFB, JB and MS conceived the project, designed and analysed the experiments, and wrote the manuscript. MG, OLG, BJA, AR and ERM performed and analysed the WGS and WES data. TH, MM-K, DS, SI, AT, LE, CQ, MP, AD, RB and AH collected and analysed patients' samples. PC, HE, SE and SL performed and supervised the laboratory experiments. HHW, GI, RHS, DBS and MFB performed and supervised the MSK-IMPACT sequencing and analysis.
Author Information: Reprints and permissions information is available at www.nature.com/reprints. CQ, MP, AD, RB and AH are Novartis employers. DJ, DBS and JB consult for Novartis.
References
- 1.Gerlinger M, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res. 2012;72:4875–4882. doi: 10.1158/0008-5472.CAN-12-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Juric D. BYL719, a next generation PI3K alpha specific inhibitor: preliminary safety, PK, and efficacy results from the first-in-human study [abstract] Cancer Res. 2012;72 CT-01. [Google Scholar]
- 4.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7 doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 5.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 6.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 7.Brachmann SM, Ueki K, Engelman JA, Kahn RC, Cantley LC. Phosphoinositide 3-kinase catalytic subunit deletion and regulatory subunit deletion have opposite effects on insulin sensitivity in mice. Mol Cell Biol. 2005;25:1596–1607. doi: 10.1128/MCB.25.5.1596-1607.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao L, Vogt PK. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci USA. 2008;105:2652–2657. doi: 10.1073/pnas.0712169105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011;13:224. doi: 10.1186/bcr3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Samuels Y, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 11.Sequist LV, et al. Implementing multiplexed genotyping of non-small-cell lung cancers into routine clinical practice. Ann Oncol. 2011;22:2616–2624. doi: 10.1093/annonc/mdr489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Therasse P, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 13.Toy W, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Gen. 2013;45:1439–1445. doi: 10.1038/ng.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Won HH, Scott SN, Brannon AR, Shah RH, Berger MF. Detecting somatic genetic alterations in tumor specimens by exon capture and massively parallel sequencing. JoVE. 2013:e50710. doi: 10.3791/50710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang ZQ, Liu G, Bollig-Fischer A, Giroux CN, Ethier SP. Transforming properties of 8p11-12 amplified genes in human breast cancer. Cancer Res. 2010;70:8487–8497. doi: 10.1158/0008-5472.CAN-10-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karlsson E, et al. High-resolution genomic analysis of the 11q13 amplicon in breast cancers identifies synergy with 8p12 amplification, involving the mTOR targets S6K2 and 4EBP1. Genes Chromosomes Cancer. 2011;50:775–787. doi: 10.1002/gcc.20900. [DOI] [PubMed] [Google Scholar]
- 17.Fritsch C et al. Characterization of the novel and specific PI3Kalpha inhibitor NVPBYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther. 2014 doi: 10.1158/1535-7163.MCT-13-0865. [DOI] [PubMed] [Google Scholar]
- 18.Stambolic V, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 19.Jia S, et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008;454:776–779. doi: 10.1038/nature07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edgar KA, et al. Isoform-specific phosphoinositide 3-kinase inhibitors exert distinct effects in solid tumors. Cancer Res. 2010;70:1164–1172. doi: 10.1158/0008-5472.CAN-09-2525. [DOI] [PubMed] [Google Scholar]
- 21.Elkabets M et al. mTORC1 Inhibition Is Required for Sensitivity to PI3K p110alpha Inhibitors in PIK3CA-Mutant Breast Cancer. Sci Transl Med. 2013;5:196ra199. doi: 10.1126/scitranslmed.3005747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lemey P, et al. Molecular footprint of drug-selective pressure in a human immunodeficiency virus transmission chain. J Virol. 2005;79:11981–11989. doi: 10.1128/JVI.79.18.11981-11989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li S, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013;4:1116–1130. doi: 10.1016/j.celrep.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25 doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DePristo MA et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Gen. 2011;43 doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cibulskis K, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wagle N, et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2012;2:82–93. doi: 10.1158/2159-8290.CD-11-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.