

Abstract
We report the first two genetically confirmed cases of primary renal sclerosing epithelioid fibrosarcoma (SEF), occurring in a 17 year-old male and a 61 year-old female. In both cases, the tumors demonstrated the typical epithelioid clear cell morphology associated with extensive hyalinizing fibrosis, raising the differential diagnosis of solitary fibrous tumor, metanephric stromal tumor, and the sclerosing variant of clear cell sarcoma of the kidney. Both neoplasms demonstrated diffuse immunoreactivity for MUC4, a highly specific marker for SEF, and both demonstrated evidence of rearrangement of both the EWSR1 and CREB3L1 genes which have recently shown to be fused in this entity. Both neoplasms presented with metastatic disease. Primary renal SEF represents yet another translocation-associated sarcoma now shown to arise primarily in the kidney.
Keywords: Renal Neoplasm, Fibrosarcoma, Translocation
Introduction
Given the vast potential of Wilms tumor (nephroblastoma) to demonstrate mesenchymal differentiation, the existence of primary renal sarcomas in children was historically difficult to accept. Due in large part to the meticulous, classic clinicopathologic studies of Dr. J. Bruce Beckwith, three major primary renal sarcomas were recognized: rhabdoid tumor of the kidney (RTK)1,2, clear sarcoma of the kidney (CCSK)3,4,5,6, and congenital mesoblastic nephoma (CMN)7,8. The distinctive nature of these lesions was later corroborated by the demonstration of distinctive, characteristic genetic alterations in each of them; specifically, deletion of the SMARCB1 gene and SMARCB1 protein loss in RTK9,10, the ETV6-NTRK3 gene fusion in cellular CMN11,12,13, and the YWHAE-FAM2214 gene fusion in a subset of CCSK.
Over the past 20 years, a variety of translocation-associated soft tissue sarcomas have been documented to arise primarily within the kidney. In each case, the existence of these distinctive entities has been proven by molecular assays documenting the presence of a characteristic specific gene fusion product. Examples of these sarcomas arising in the kidney include primitive neuroectodermal tumor (PNET)15,16, synovial sarcoma,17 intra-abdominal desmoplastic small round cell tumor18, and a rare case of clear cell sarcoma of tendon sheath (melanoma of soft parts)19. Some of these entities were likely previously confused with other established lesions, contributing to misinformation regarding the behavior of the latter. For example, primary renal PNET15, 16 was historically often confused with blastemal Wilms tumor in young adults, contributing to the perception that adult Wilms tumors had a particularly poor prognosis. Primary renal synovial sarcomas were often confused with sarcomas arising in cystic nephroma, some of which are now thought to belong to the family of DICER1- related sarcomas20. Hence, delineation of novel specific morphologic and molecular entities in the kidney has also helped clarify the clinicopathologic features of existing ones.
We report in the first two genetically confirmed cases of primary renal sclerosing epithelioid fibrosarcoma. In both cases, the diagnosis was confirmed by fluorescence in situ hybridization (FISH) showing the characteristic EWSR1-CREB3L1 gene fusion, which has recently been found to be highly prevalent in this entity. Both patients presented with large renal masses and evidence of metastatic disease. Both cases raised the morphologic differential diagnosis of a variety of mesenchymal lesions including the sclerosing variant of clear cell sarcoma of the kidney.
Materials and Methods
Cases
Both cases were sent in consultation to one of the authors (PA), with the differential diagnosis of clear cell sarcoma of the kidney (CCSK). In each case, H&E stained slides and paraffin tissue blocks were available for immunohistochemistry and fluorescence in situ hybridization (FISH) as described below.
Immunohistochemistry
At Johns Hopkins University, immunohistochemical labeling was performed on the Benchmark XT autostainer (Ventana Medical Systems Inc, Tucson, AZ) using the I-View detection kit. The standard antibodies used, vendors, pretreatments, and dilutions were as follows: desmin (Dako M0760, clone D33, steam, 1:100), cytokeratin AE1/3 (Chemicon, steam, 1:4000) vimentin (Ventana, 790-2917, prediluted), EMA (Ventana, 760-4259, steam, prediluted), CD99 (Leica, Clone 12E7, steam, prediluted), Bcl2 (Leica, ORG-8714, steam, prediluted), S100 protein (Ventana, 760-2914, stream, prediluted), and CD34 (Immunotech/Coulter, catalog 0786, steam, prediluted). MUC4 immunohistochemical staining was performed at Memorial Sloan Kettering Cancer Center using anti-human mouse monoclonal antibody clone 8G7 at a dilution of 0.1 ug/ml (Santa Cruz Biotechnology, Santa Cruz, CA).
Fluorescence in situ hybridization (FISH)
FISH was performed in the laboratory of one of the authors (CRA) at Memorial Sloan Kettering Cancer Center as previously described21. Briefly, FISH on interphase nuclei from paraffin embedded 4-micron sections was performed applying custom probes using bacterial artificial chromosomes (BAC), covering and flanking the EWSR1 and CREB3L1 genes. BAC clones were chosen according to USCS genome browser (http://genome.uscs.edu) (see Supplemental Table 1, Supplemental Digital Content 1, http://links.lww.com/PAS/A238). The BAC clones were obtained from BACPAC sources of Children's Hospital of Oakland Research Institute (CHORI) (Oakland, CA) (http://bacpac.chori.org). DNA from individual BACs was isolated according to the manufacturer's instructions, labeled with different fluorochromes in a nick translation reaction, denatured, and hybridized to pretreated slides. Slides were then incubated, washed, and mounted with DAPI in an antifade solution. The genomic location of each BAC set was verified by hybridizing them to normal metaphase chromosomes. Two hundred successive nuclei were examined using a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany), controlled by Isis 5 software (Metasystems). A positive score was interpreted when at least 20% of the nuclei showed a break apart signal. Nuclei with incomplete set of signals were omitted from the score.
Results
Case Histories
Case 1
Patient one was a 17 year-old white male who presented with left flank, back and abdominal pain, 40 pound weight loss over six months, dysuria and decreased appetite. He was found on imaging to have a 25 × 15 × 20 cm tumor arising in the lower pole of the left kidney, along with evidence of metastases involving the right fifth rib, vertebrae, epidural spinal cord, and liver (Figure 1). Following tumor embolization, the patient underwent left radical nephrectomy, with excision of a liver lesion and epidural spinal cord tumor. Beginning one month following resection, the patient received 10 fractions of radiotherapy with a total dose of 30Gy over a period of 3 weeks. However, he then developed volvulus and closed small bowel obstruction secondary to adhesions, which were treated surgically. Soon after he developed abdominal pain and was found to have free air in the abdomen. On surgical exploration, he was found to have extensive ischemic necrosis involving his abdominal organs, not thought to be compatible with continuation of life. The patient expired without further intervention.
Figure 1.
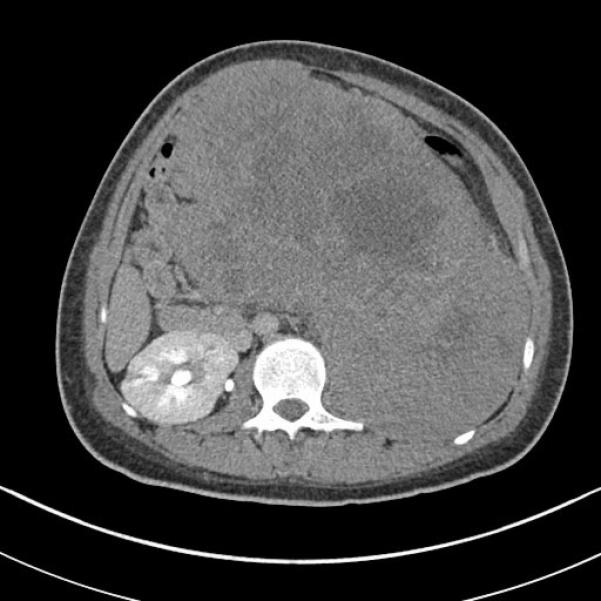
(Case 1) Computerized Tomography scan of the abdomen demonstrates a large tumor effacing the left kidney.
Case 2
The patient was a 61 year-old female who presented with rib pain. She was found to have a 5cm left renal mass with metastatic lesions involving the ribs, lung, bone, and lymph nodes. A nephrectomy was performed. The patient's disease progressed and she was placed in hospice 6 months after diagnosis.
Pathology
On gross examination, both lesions were described as firm, white tumors centered on the kidney. In case one, the tumor grossly penetrated the renal capsule to involve perirenal soft tissue (Figure 2A). In case 2, the tumor abutted the renal capsule (Figure 2B) and protruded into the renal pelvis.
Figure 2.

A. Gross photograph of the sectioned specimen from case 1 reveals a pale solid tumor emanating from the lower pole of the left kidney, and extending through the capsule into the perirenal fat. A satellite nodule of tumor deeper within the kidney is also evident. B. Gross photograph of the sectioned specimen from case 2 reveals a pale solid tumor emanating from the lower pole of the left kidney, abutting the capsule and protruding into the renal sinus
The morphologic features of the two cases were similar and therefore they are described together (Figure 3, 4). Both neoplasms were predominantly composed of cords of epithelioid cells with angulated nuclei and clear cytoplasm, in a background of hyaline sclerosis (Figure 3A-3E, 4A-4C). Cellularity was variable within the neoplasms, with frequent abrupt transitions from areas of high cellularity to highly sclerotic areas of low cellularity (Figure 3C). In areas of high cellularity, vague nodules of collagen reminiscent of those seen in hyalinizing spindle cell tumor with giant rosettes (HSCTGR) were identified (Figure 3F, 4E)22. Focally, metaplastic bone was present in each case (Figure 4D). Each neoplasm encircled native renal tubules and glomeruli (Figure 3D, 4B). In case one, a focal myxoid nodule contained bland spindle cells in a swirling pattern adjacent to a hypocellular fibrous zone, closely resembling low grade fibromyxoid sarcoma (Figure 3G). Also in case one, native renal tubules entrapped by the neoplasm demonstrate tubulopapillary hyperplasia, simulating a biphasic neoplasm (Figure 3B). The liver and epidural lesions sampled in case 1 showed metastatic disease morphologically similar to that of the primary renal tumor.
Figure 3.

(Case 1) A. Low power view shows the spindle cell neoplasm centered within the kidney and entrapping renal tubules at its periphery. B. The neoplasm consists of epithelioid cells with clear cytoplasm, associated with stromal hyalinization. The neoplasm entraps native renal tubules, which show micropapillary epithelial hyperplasia. C. In other areas the neoplasm is remarkably hypocellular and fibrous, surrounding obstructed native renal tubules. D. The neoplasm encircles a native glomerulus. E. At high power, one can appreciate the angulated nuclei, clear cytoplasm, and thin strands of collagen separating the neoplastic cells. These are typical features of SEF. F. More cellular areas of the tumor are associated with hypocellular collagenized nodules, similar to those of HSCTGR. G. Focally within the neoplasm there was a nodule of myxoid stroma with spindle cells having a whorled appearance, reminiscent of LGFMS. H. The neoplastic cells label diffusely for MUC4, while the entrapped native renal tubules are appropriately negative.
Figure 4.

(Case 2). A. This variably cellular fibroblastic neoplasm is centered in the kidney and entraps native glomeruli and renal tubules. B. The neoplasm encircles a native renal glomerulus. C. The neoplasm is composed of epithelioid clear cells in a background of dense strands of sclerotic collagen which separate individual neoplastic cells. D. Areas of metaplastic bone formation are evident. E. More cellular areas of the tumor are associated with hypocellular collagenized nodules, reminiscent of those of HSCTGR. G. Neoplastic cells are diffusely immunoreactive for MUC4.
By immunohistochemistry, both neoplasms were immunoreactive for vimentin and Bcl2, but negative for cytokeratin AE1/AE3, desmin, S100 protein, and CD34. Case one was focally positive for CD99 and negative for EMA, while case two was focally positive for EMA and negative for CD99. Both neoplasms demonstrated diffuse cytoplasmic immunoreactivity for mucin 4 (MUC4) (Figure 3H, 4F).
By fluorescence in situ hybridization, both neoplasms demonstrated evidence of rearrangement of the EWSR1 gene and the CREB3L1 gene (Figure 5). No evidence of FUS gene rearrangement was found (not shown).
Figure 5.

FISH showing rearrangement of EWSR1 and CREB3L1 genes in Cases 1 and 2. A. Case 1, CREB3L1 break apart (arrows) (red centromeric, green telomeric). B. Case 1, EWSR1 unbalanced rearrangement with loss of telomeric part (green). C. Case 2, CREB3L1 break apart (arrows)(red centromeric, green telomeric). D. Case 2, EWSR1 unbalanced rearrangement with loss of telomeric part (green).
Discussion
We report the first two genetically confirmed cases of sclerosing epithelioid fibrosarcoma of the kidney. Sclerosing epithelioid fibrosarcoma (SEF) and low grade fibromyxoid sarcoma (LGFMS) are related members of the so-called “fibrosing fibrosarcoma family”23, with SEF considered the higher grade end of this spectrum. As seen in our cases, SEF affects patients in a wide age range. SEF typically arises as a deep seated mass in the extremities, and is characterized by infiltrative, carcinoma-like cords or nests of epithelioid cells with angulated nuclei and clear cytoplasm in densely hyalinized stroma24. Many cases, including case one of this study, have small spindle cell foci which overlap with LGFMS. LGFMS typically affects young adults and similarly arises in the deep soft tissue. LGFMS typically features hypocellular fibrous zones with abrupt transitions to myxoid nodules showing increased cellularity of spindle cells with a whorling pattern, supported by curvilinear vessels25, 26. A subset of LGFMS demonstrate giant collagen rosettes, leading to the prior description of these cases as hyalinizing spindle cell tumor with giant rosettes (HSCTGR)27,28. Other cases have areas that resemble SEF. More recently, genetic studies have demonstrated the relatedness of HSCTGR, LGFMS, and SEF. First, a FUS-CREB3L2 gene fusion resulting from a t(7;16)(q33;p11) chromosome translocation was found to be present in over 90% of LGFMS, HSCTGR and hybrid forms of LGFMS/SEF29-32. An alternative FUS-CREB3L1 resulting from a t(11;16)(p11;q11) chromosome translocation was subsequently found in a minority of LGFMS33. Finally, an EWSR1-CREBL1 gene fusion resulting from a t(11;22)(p11;q12) chromosome translocation was found in the majority of SEF34, 35 and in a minority of LGFMS36. The ability of the FUS and EWSR1 genes to substitute in gene fusions is logical since they are highly related RNA binding proteins. These genes function as alternating fusion partners in three other soft tissue sarcomas: Ewing sarcoma, myxoid liposarcoma, and angiomatoid fibrous histiocytoma37. Our cases both demonstrated the EWSR1-CRB3L1 gene fusion which predominates in SEF. Further corroborating the diagnosis, both neoplasms overexpressed the mucin 4 (MUC4) protein, which has been identified as overexpressed in LGFMS by gene expression profiling, and found to be overexpressed by immunohistochemistry in LGFMS and SEF38, 39.
SEF represents the latest soft tissue sarcoma to have been documented genetically to arise primarily within the kidney, joining PNET, synovial sarcoma, intra-abdominal desmoplastic small round cell tumor, and clear cell sarcoma of the kidney. Given that the cellular variant of congenital mesoblastic nephroma shares clinical, morphologic and immunohistochemical features with infantile fibrosarcoma, and harbors the same ETV6-NTRK3 gene fusion, many would add infantile fibrosarcoma to this list.
Two other primary renal neoplasms which may be related to renal SEF have been reported. First, Rubenstein et al recently reported a case of LGFMS arising in the kidney of a 16 year old male40. This 16cm neoplasm harbored the EWSR1-CREB3L1 gene fusion that is more commonly seen in SEF (including our two cases of renal SEF) than in LGFMS of soft tissue. We note that there is a precedent for differing frequencies of usage of alternative gene fusions in a sarcoma when it arises in the kidney compared to soft tissue. In synovial sarcoma of soft tissue, the SYT-SSX1 gene fusion predominates, whereas in renal synovial sarcoma SYT-SSX2 gene fusion predominates17. Hence, it will be interesting to see if the prevalence of the EWSR1-CREB3L1 gene fusion, now identified in all three cases of fibrosing fibrosarcoma family neoplasms arising in the kidney, is greater in the kidney than it is in these same tumors in soft tissue. Second, Arbajian et al included a case of a primary renal neoplasm in a recent series of sclerosing epithelioid fibrosarcomas34. This 41 year-old female presented with a 9 cm renal mass and bone and lung metastases at diagnosis, and died of disease at 22 months. The case was not illustrated, and while FISH studies demonstrated loss of signal corresponding to the 3’ portion of the EWS gene (suggesting a rearrangement), a fusion partner was not identified.
The differential diagnosis for renal SEF is broad and includes several mesenchymal lesions previously documented to occur in the kidney; specifically, metanephric stromal tumor (MST), renal synovial sarcoma, renal solitary fibrous tumor, renal osteosarcoma, and the sclerosing variant of clear cell sarcoma of the kidney. Metanephric stromal tumor typically affects the pediatric age group, and can also be associated with extensive fibrosis/sclerosis41. Cases resembling MST with overtly malignant areas have been suggested to represent malignant counterparts to this typically benign neoplasm. However, unlike SEF, MST typically encircles entrapped native renal tubules in an onion-skinning, concentric pattern, is associated with angiodysplasia and juxtaglomerular cell hyperplasia, and often labels for CD34. Like SEF, renal synovial sarcoma may have hemangiopericytomatous vasculature and be associated with extensive collagen deposition, and typically labels for Bcl2 but not CD3417. In addition, the glandular component of synovial sarcoma can label for MUC438. However, unlike SEF, primary renal synovial sarcomas are typically monophasic spindle cell lesions lacking an epithelial component, and feature only thin, ropey, non-sclerotic strands of collagen. The neoplastic cells of monophasic synovial sarcoma are typically ovoid and and have more primitive chromatin than those of SEF. Both benign and malignant SFT have been documented to occur in the kidney42, 43, and like SEF these lesions feature extensive fibrosis/sclerosis, and hemangiopericytomatous vasculature. In contrast to SEF, SFT label for CD34 and are comprised of spindled not epithelioid cells. The presence of osteoid/bone matrix in both examples of renal SEF presented herein raises the differential diagnosis of renal osteosarcoma. Renal osteosarcoma is rare44, 45, and it is possible that some of the cases reported could represent incompletely-sampled sarcomatoid carcinomas. Of note, like the cases of renal SEF reported herein, cases of primary renal osteosarcoma have tended to present at advanced stage. Given that most cases reported as primary renal osteosarcoma were published before SEF was first described in 1995, it seems possible that some of these could in fact represent SEF. It should be noted, however, that a single case reported as a small cell osteosarcoma of bone has been found to harbor the EWSR1-CREB3L1 gene fusion of SEF, suggesting that this distinction may not always be clear-cut46.
Clear cell sarcoma of the kidney (CCSK) represents the most difficult differential diagnosis for renal SEF, specifically the sclerosing variant of CCSK6. In addition to its classic pattern featuring cords of epithelioid cells in a branching capillary vasculature, CCSK demonstrates a wide range of variant patterns including ribbons or rosettes, spindle cells, myxoid pools, sclerosis, palisading and anaplasia. The sclerosing variant of CCSK is characterized by extensive hyaline sclerosis surrounding cords and nests of epithelioid cells with clear cytoplasm, similar to the morphologic appearance of renal SEF. Like SEF, CCSK is negative for most standard immunohistochemical markers such as desmin, S100, CD34, and cytokeratin. Variable labeling for CD99 has been reported in CCSK, but most cases have proven to be negative, similar to SEF6. Like SEF, CCSK is a relatively slow growing tumor which encircles native renal tubules and characteristically incites hyperplasia of entrapped renal tubular epithelium, very similar to the hyperplasia of entrapped tubules seen in case one of this study. Finally, both CCSK and LGFMS/SEF may recur late, mandating long-term follow-up. Given this overlap, it seem possible some cases reported as sclerosing CCSK in the literature might in fact represent SEF, given that the latter was not described before the former were reported. Along these lines, most cases of CCSK present as localized disease, but a minority present with disseminated disease similar to the presentation of renal SEF seen in this study. One wonders if such “stage 4” CCKSs might in fact be renal SEF. Morphologic features that would favor CCSK over SEF include the admixture of other variant patterns with sclerosing areas, along with the characteristic fine, evenly dispersed chromatin of CCSK. MUC4 immunoreactivity could potentially help distinguish these entities, though we are unaware of MUC4 labeling being examined in CCSK. Detection of the characteristic gene fusions of SEF should distinguish this entity from CCSK, which in minority of cases harbors a distinct YWHAE-FAM22 gene fusion.
In summary, we report the first two genetically confirmed cases of renal sclerosing epithelioid fibrosarcoma. Both cases had an aggressive clinical course, presenting with advanced stage disease at diagnosis. Given the relatively recent recognition of SEF as a distinctive entity, one wonders if renal SEF in prior years were misclassified as CCSK, renal osteosarcoma, or other entities. Sclerosing epithelioid fibrosarcoma represents yet another soft tissue sarcoma documented to occur primarily within the kidney.
Supplementary Material
Acknowledgement
We thank Norman Barker MA, MS, RBP for expert photographic assistance.
Disclosures: Supported in part by: P01CA47179 (CRA), P50 CA 140146-01 (CRA), Cycle for Survival (CRA), Kristin Ann Carr Foundation (CRA).
References
- 1.Beckwith JB, Palmer NF. Histopathology and prognosis of Wilms tumors: results from the First National Wilms Tumor Study. Cancer. 1978;41:1937–48. doi: 10.1002/1097-0142(197805)41:5<1937::aid-cncr2820410538>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 2.Weeks DA, Beckwith JB, Mierau GW, et al. Rhabdoid tumor of kidney: a report of 111 cases from the National Wilms Tumor Study Pathology Center. Am J Surg Pathol. 1989;13:439–458. [PubMed] [Google Scholar]
- 3.Sandstedt BE, Delemarre JFM, Harms D, et al. Sarcomatous Wilms’ tumour with clear cells and hyalinization: a study of 38 tumours from the SIOP nephroblastoma file. Histopathology. 1987;11:273–285. doi: 10.1111/j.1365-2559.1987.tb02632.x. [DOI] [PubMed] [Google Scholar]
- 4.Marsden HB, Lawler W. Bone metastasizing renal tumor of childhood: histopathological and clinical review of 38 cases. Virchows Arch A Pathol Anat Histopathol. 1980;387:341–351. doi: 10.1007/BF00454837. [DOI] [PubMed] [Google Scholar]
- 5.Haas JE, Bonadio JF, Beckwith JB. Clear cell sarcoma of the kidney with emphasis on ultrastructural studies. Cancer. 1984;54:2978–2987. doi: 10.1002/1097-0142(19841215)54:12<2978::aid-cncr2820541228>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 6.Argani P, Perlman EJ, Breslow NE, et al. Clear cell sarcoma of the kidney: a review of 351 cases from the National Wilms Tumor Study Group Pathology Center. Am J Surg Pathol. 2000;24:4–18. doi: 10.1097/00000478-200001000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Bolande RP. Congenital mesoblastic nephroma of infancy. Perspect Pediatr Pathol. 1973;1:227–250. [PubMed] [Google Scholar]
- 8.Pettinato G, Manivel JC, Wick MR, et al. Classical and cellular (atypical) congenital mesoblastic nephroma: a clinicopathologic, ultrastructural, immunohistochemical, and flow cytometric study. Hum Pathol. 1989;20:682–690. doi: 10.1016/0046-8177(89)90156-1. [DOI] [PubMed] [Google Scholar]
- 9.Versteege I, Sevenet N, Lange J, et al. Truncating mutations of hSNF5/INI1 in aggressive pediatric cancer. Nature. 1998;394:203–206. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- 10.Judkins AR. Immunohistochemistry of INI1 expression: a new tool for old challenges in CNS and soft tissue pathology. Adv Anat Pathol. 2007;14:335–9. doi: 10.1097/PAP.0b013e3180ca8b08. [DOI] [PubMed] [Google Scholar]
- 11.Knezevich SR, Garnett MJ, Pysher TJ, et al. ETV6-NTRK3 gene fusions and trisomy 11 establish a histogenetic link between mesoblastic nephroma and congenital fibrosarcoma. Cancer Res. 1998;58:5046–5048. [PubMed] [Google Scholar]
- 12.Rubin BP, Chen CJ, Morgan TW, et al. Congenital mesoblastic nephroma t(12;15) is associated with ETV6-NTRK3 gene fusion: cytogenetic and molecular relationship to congenital (infantile) fibrosarcoma. Am J Pathol. 1998;153:1451–1458. doi: 10.1016/S0002-9440(10)65732-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Argani P, Fritsch M, Kadkol SS, et al. Detection of the ETV6-NTRK3 chimeric RNA of infantile fibrosarcoma/cellular congenital mesoblastic nephroma in paraffin-embedded tissue: application to challenging pediatric renal stromal tumors. Mod Pathol. 2000;13:29–36. doi: 10.1038/modpathol.3880006. [DOI] [PubMed] [Google Scholar]
- 14.O'Meara E, Stack D, Lee CH, Garvin J, Morris T, Argani P, Han J, Gisselson D, Leuschner I, Gessler M, Graf N, Fletcher JA, O'Sullivan MJ. Characterisation of the chromosome translocation t(10;17)(q22;p13) in clear cell sarcoma of kidney. J Pathol. 2012;227:72–80. doi: 10.1002/path.3985. [DOI] [PubMed] [Google Scholar]
- 15.Quezado M, Benjamin DR, Tsokos M. EWS/FLI-1 fusion transcripts in three peripheral primitive neuroectodermal tumors of the kidney. Hum Pathol. 1997;28:767–771. doi: 10.1016/s0046-8177(97)90147-7. [DOI] [PubMed] [Google Scholar]
- 16.Jimenez RE, Folpe AL, Lapham RL, et al. Primary Ewing's sarcoma/primitive neuroectodermal tumor of the kidney. A clinicopathologic and immunohistochemical analysis of 11 cases. Am J Surg Pathol. 2002;26:320–327. doi: 10.1097/00000478-200203000-00005. [DOI] [PubMed] [Google Scholar]
- 17.Argani P, Faria PA, Epstein JI, et al. Primary renal synovial sarcoma. Morphologic and molecular delineation of an entity previously included among embryonal sarcomas of the kidney. Am J Surg Pathol. 2000;24:1087–1096. doi: 10.1097/00000478-200008000-00006. [DOI] [PubMed] [Google Scholar]
- 18.Wang LL, Perlman EJ, Vujanic GM, et al. Desmoplastic small round cell tumor of the kidney in childhood. Am J Surg Pathol. 2007;31:576–584. doi: 10.1097/01.pas.0000213432.14740.14. [DOI] [PubMed] [Google Scholar]
- 19.Rubin BP, Fletcher JA, Renshaw AA. Clear cell sarcoma of soft parts: report of a case primary in the kidney with cytogenetic confirmation. Am J Surg Pathol. 1999;23:589–94. doi: 10.1097/00000478-199905000-00014. [DOI] [PubMed] [Google Scholar]
- 20.Doros L, Rossi CT, Yang J, et al. DICER1 mutations in childhood cystic nephroma and its relationship to DICER1-renal sarcoma. Mod Pathol. 2014 Jan 31; doi: 10.1038/modpathol.2013.242. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Antonescu CR, Dal Cin P, Nafa K, et al. EWSR1-CREB1 is the predominant gene fusion in angiomatoid fibrous histiocytoma. Genes Chromosomes Cancer. 2007;46:1051–60. doi: 10.1002/gcc.20491. [DOI] [PubMed] [Google Scholar]
- 22.Lane KL, Shannon RJ, Weiss SW. Hyalinizing spindle cell tumor with giant rosettes: a distinctive tumor closely resembling low-grade fibromyxoid sarcoma. Am J Surg Pathol. 1997;21:1481–8. doi: 10.1097/00000478-199712000-00011. [DOI] [PubMed] [Google Scholar]
- 23.Antonescu CR, Rosenblum MK, Pereira P, et al. Sclerosing epithelioid fibrosarcoma: a study of 16 cases and confirmation of a clinicopathologically distinct tumor. Am J Surg Pathol. 2001;25:699–709. doi: 10.1097/00000478-200106000-00001. [DOI] [PubMed] [Google Scholar]
- 24.Meis-Kindblom JM, Kindblom LG, Enzinger FM. Sclerosing epithelioid fibrosarcoma. A variant of fibrosarcoma simulating carcinoma. Am J Surg Pathol. 1995;19:979–993. doi: 10.1097/00000478-199509000-00001. [DOI] [PubMed] [Google Scholar]
- 25.Evans HL. Low-grade fibromyxoid sarcoma. A report of two metastasizing neoplasms having a deceptively benign appearance. Am J Clin Pathol. 1987;88:615–9. doi: 10.1093/ajcp/88.5.615. [DOI] [PubMed] [Google Scholar]
- 26.Evans HL. Low-grade fibromyxoid sarcoma. A report of 12 cases. Am J Surg Pathol. 1993;17:595–600. doi: 10.1097/00000478-199306000-00007. [DOI] [PubMed] [Google Scholar]
- 27.Woodruff JM, Antonescu CR, Erlandson RA, Boland PJ. Low-grade fibrosarcoma with palisaded granulomalike bodies (giant rosettes): report of a case that metastasized. Am J Surg Pathol. 1999;23:1423–8. doi: 10.1097/00000478-199911000-00015. [DOI] [PubMed] [Google Scholar]
- 28.Folpe AL, Lane KL, Paull G, Weiss SW. Low-grade fibromyxoid sarcoma and hyalinizing spindle cell tumor with giant rosettes: a clinicopathologic study of 73 cases supporting their identity and assessing the impact of high-grade areas. Am J Surg Pathol. 2000;24:1353–60. doi: 10.1097/00000478-200010000-00004. [DOI] [PubMed] [Google Scholar]
- 29.Reid R, de Silva MV, Paterson L, Ryan E, et al. Low-grade fibromyxoid sarcoma and hyalinizing spindle cell tumor with giant rosettes share a common t(7;16)(q34;p11) translocation. Am J Surg Pathol. 2003;27:1229–36. doi: 10.1097/00000478-200309000-00006. [DOI] [PubMed] [Google Scholar]
- 30.Panagopoulos I, Storlazzi CT, Fletcher CD, et al. The chimeric FUS/CREB3l2 gene is specific for low-grade fibromyxoid sarcoma. Genes Chromosomes Cancer. 2004;40:218–28. doi: 10.1002/gcc.20037. [DOI] [PubMed] [Google Scholar]
- 31.Guillou L, Benhattar J, Gengler C, Gallagher G, et al. Translocation-positive low-grade fibromyxoid sarcoma: clinicopathologic and molecular analysis of a series expanding the morphologic spectrum and suggesting potential relationship to sclerosing epithelioid fibrosarcoma: a study from the French Sarcoma Group. Am J Surg Pathol. 2007;31:1387–402. doi: 10.1097/PAS.0b013e3180321959. [DOI] [PubMed] [Google Scholar]
- 32.Storlazzi CT, Mertens F, Nascimento A, et al. Fusion of the FUS and BBF2H7 genes in low grade fibromyxoid sarcoma. Hum Mol Genet. 2003;12:2349–58. doi: 10.1093/hmg/ddg237. [DOI] [PubMed] [Google Scholar]
- 33.Mertens F, Fletcher CD, Antonescu CR, et al. Clinicopathologic and molecular genetic characterization of low-grade fibromyxoid sarcoma, and cloning of a novel FUS/CREB3L1 fusion gene. Lab Invest. 2005;85:408–15. doi: 10.1038/labinvest.3700230. [DOI] [PubMed] [Google Scholar]
- 34.Arbajian E, Puls F, Magnusson L, et al. Recurrent EWSR1-CREB3L1 gene fusions in sclerosing epithelioid fibrosarcoma. Am J Surg Pathol. 2014;38:801–8. doi: 10.1097/PAS.0000000000000158. [DOI] [PubMed] [Google Scholar]
- 35.Doyle LA, Hornick JL. EWSR1 rearrangement in sclerosing epithelioid fibrosarcoma. Am J Surg Pathol. 2013;37:1630–1631. doi: 10.1097/PAS.0b013e3182a05a6b. [DOI] [PubMed] [Google Scholar]
- 36.Lau PP, Lui PC, Lau GT, et al. EWSR1-CREB3L1 gene fusion: a novel alternative molecular aberration of low-grade fibromyxoid sarcoma. Am J Surg Pathol. 2013;37:734–8. doi: 10.1097/PAS.0b013e31827560f8. [DOI] [PubMed] [Google Scholar]
- 37.Antonescu CR, Dal Cin P. Promiscuous genes involved in recurrent chromosomal translocation in soft tissue tumours. Pathology. 2014;46:105–12. doi: 10.1097/PAT.0000000000000049. [DOI] [PubMed] [Google Scholar]
- 38.Doyle LA, Möller E, Dal Cin P, et al. MUC4 is a highly sensitive and specific marker for low-grade fibromyxoid sarcoma. Am J Surg Pathol. 2011;35:733–41. doi: 10.1097/PAS.0b013e318210c268. [DOI] [PubMed] [Google Scholar]
- 39.Doyle LA, Wang WL, Dal Cin P, et al. MUC4 is a sensitive and extremely useful marker for sclerosing epithelioid fibrosarcoma: association with FUS gene rearrangement. Am J Surg Pathol. 2012;36:1444–51. doi: 10.1097/PAS.0b013e3182562bf8. [DOI] [PubMed] [Google Scholar]
- 40.Rubinstein J, Visa A, Zhang L, Antonescu CR, et al. Primary low grade fibromyxoid sarcoma of the kidney in a child, with the alternative EWSR1-CREB3L1 gene fusion. Pediatr Dev Pathol. 2014 Jun 4; doi: 10.2350/14-05-1487-CR.1. [DOI] [PubMed] [Google Scholar]
- 41.Argani P, Beckwith JB. Metanephric Stromal Tumor: report of 31 cases of a distinctive pediatric renal neoplasm. Am J Surg Pathol. 2000;24:917–926. doi: 10.1097/00000478-200007000-00003. [DOI] [PubMed] [Google Scholar]
- 42.Kuroda N, Ohe C, Sakaida N, et al. Solitary fibrous tumor of the kidney with focus on clinical and pathological aspects. Int J Clin Exp Pathol. 2014;15:2737–2742. [PMC free article] [PubMed] [Google Scholar]
- 43.Fine SW, McCarthy DM, Chan TY, et al. Malignant solitary fibrous tumor of the kidney: report of a case and comprehensive review of the literature. Am J Surg Pathol. 2006;130:857–861. doi: 10.5858/2006-130-857-MSFTOT. [DOI] [PubMed] [Google Scholar]
- 44.Lopez-Beltran A, Montironi R, Carazo JL, et al. Primary renal osteosarcoma. Am J Clin Pathol. 2014;141:747–52. doi: 10.1309/AJCPM86FVHAMWJSR. [DOI] [PubMed] [Google Scholar]
- 45.Axelrod R. Primary osteosarcoma of the kidney. Cancer. 1978;41:724–727. doi: 10.1002/1097-0142(197802)41:2<724::aid-cncr2820410244>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 46.Debelenko LV, McGregory LM, Shivakumar BR, et al. A Novel EWSR1-CREB3L1 fusion transcript in a case of small cell osteosarcoma. Genes, Chromosomes & Cancer. 2011;50:1054–1062. doi: 10.1002/gcc.20923. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.