

Abstract
Renal fibrosis is the hallmark of virtually all progressive kidney diseases and strongly correlates with the deterioration of kidney function. The renin-angiotensin-aldosterone system blockade is central to the current treatment of patients with chronic kidney disease (CKD) for the renoprotective effects aimed to prevent or slow progression to end-stage renal disease (ESRD). However, the incidence of CKD is still increasing, and there is a critical need for new therapeutics. Here, we review novel strategies targeting various components implicated in the fibrogenic pathway to inhibit or retard the loss of kidney function. We focus, in particular, on anti-fibrotic approaches that target transforming growth factor (TGF)-β1, a key mediator of kidney fibrosis, and exciting new data on the role of autophagy. Bone morphogenetic protein (BMP)-7 and connective tissue growth factor (CTGF) are highlighted as modulators of pro-fibrotic TGF-β activity. BMP-7 has a protective role against TGF-β1 in kidney fibrosis, whereas CTGF enhances TGF-β-mediated fibrosis. We also discuss recent advances in the development of additional strategies for anti-fibrotic therapy. These include strategies targeting chemokine pathways via CC chemokine receptor 1 and 2 to modulate the inflammatory response, inhibition of phosphodiesterase to restore nitric oxide (NO)-cyclic 3′,5′ guanosine monophosphate (cGMP) function, inhibition of NADPH oxidase 1 (Nox1) and 4 (Nox4) to suppress reactive oxygen species production, as well as inhibition of endothelin-1 or tumor necrosis factor-α to ameliorate progressive renal fibrosis. Furthermore, a brief overview of some of the biomarkers of kidney fibrosis currently being explored that may improve the ability to monitor anti-fibrotic therapies. It is hoped that evidence based on the preclinical and clinical data discussed in this review leads to novel anti-fibrotic therapies effective in patients with CKD to prevent or delay progression to ESRD.
Keywords: Kidney fibrosis, transforming growth factor-β, autophagy, bone morphogenic protein-7, connective tissue growth factor, C-C motif chemokine, chemokine receptor, NADPH oxidase, phosphodiesterase, endothelin-1, tumor necrosis factor-α, chronic kidney disease
Introduction
Development of renal fibrosis is the hallmark of most progressive chronic kidney disease (CKD), irrespective of the cause, and is thought to represent the final common response to injury.1 Pathogenesis of renal fibrosis is characterized by the relentless accumulation of extracellular matrix (ECM) proteins such as fibronectin and collagens, accompanied by tubular atrophy and alterations in the renal vasculature.2 These pathologic changes lead to irreversible loss of tissue and impaired kidney function and, ultimately, end-stage kidney failure. In addition to the development of end stage renal disease (ESRD) requiring renal replacement therapies, important adverse outcomes of CKD include cardiovascular complications.3 Furthermore, CKD has increasingly become a major global public health concern and portends high rates of morbidity and mortality.3 Therefore, treatment strategies for CKD aimed at preventing or slowing its devastating sequelae and progression to ESRD are of utmost importance. Central to the current treatment for patients with CKD is the blockade of the renin-angiotensin-aldosterone system (RAAS) by angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) whose renoprotective effects have been impactful in retarding progression of many CKDs.4-6 However, the incidence of CKD is still increasing worldwide and the number of patients with CKD who ultimately develop ESRD remains high. Hence, there is a critical need for new therapeutics.
Studies to understand the mechanisms mediating pathogenesis of fibrosis in the kidney have been the focus of intensive research. Development of renal fibrosis underlies virtually all progressive kidney diseases and strongly correlates with deterioration of kidney function.7 Thus, targeting the various components of the fibrogenic pathways represents attractive therapeutic strategies to inhibit or retard the progressive loss of kidney function in patients with CKD, regardless of the underlying cause of kidney injury. Here, we review recent advances in anti-fibrotic therapies based on recent preclinical and clinical evidence. We focus, in particular, on potential antifibrotic approaches that target transforming growth factor (TGF)-β1, a key mediator of tissue fibrosis. A growing body of evidence demonstrates that TGF-β1 plays a pivotal role in the pathogenesis of renal fibrosis associated with progressive kidney diseases.8,9 We highlight several major pathways that either modulate or are modulated by TGF-β1, such as autophagy and bone morphogenetic protein (BMP)-7 having protective roles against TGF-β1 in kidney fibrosis, and connective tissue growth factor (CTGF) which enhances TGF-β-mediated fibrosis. Targeting CC chemokine receptors CCR1 and CCR2 to modulate the inflammatory response, inhibiting phosphodiesterase (PDE) to restores NO-cGMP function may also achieve anti-fibrotic effects. Additional strategies for inhibiting progressive renal fibrosis include inhibiting Nox1/Nox4 to suppress the production of reactive oxygen species (ROS), as well as inhibiting endothelin-1 (ET-1) or tumor necrosis factor-α (TNF-α) to ameliorate progressive renal fibrosis. This review focuses on fibrosis of the native kidney. Fibrosis in transplanted kidneys may involve other mechanisms, such as ischemic-reperfusion injury, preexisting fibrosis in the donated kidney, nephrotoxic effects of immunosuppressant medications, and allogenic immune response.10,11
TGF-β1 and signaling
Three mammalian isoforms of TGF-β exist, namely TGF-β1, -β2, and -β3. They belong to the TGF superfamily of cytokines that is composed of more than 30 structurally related polypeptide growth factors including the TGF-βs, activins, inhibins, growth differentiation factors (GDFs), and BMPs.12 TGF-β1 represents the predominant isoform that is ubiquitously expressed and is a prototypic multifunctional cytokine regulating a wide variety of cellular functions. TGF-β1 is synthesized as a 390-amino acid protein consisting of the signal peptide, the latency-associated peptide (LAP), and the mature peptide. The LAP confers latency to the mature peptide and stays noncovalently bound as small latent complex (SLC) and can become associated with a latent TGF-β binding protein (LTBP) to form the large latent complex (LLC).12 TGF-β1 is secreted as latent complexes and requires activation in the presence of low pH, ROS, thrombospodin-1, or by protease or metalloprotease cleavage to exert biological activity.9,13 TGF-β1 actions are mediated through the heteromeric interaction of its two signaling receptor serine/threonine kinases. TGF-β type II receptor (TβRII) is a constitutively active serine/threonine kinase, which binds TGF-β1 ligand and in turn induces recruitment of TGF-β type I receptor (TβRI) and heteromeric complex formation. TβRII subsequently phosphorylates TβRI in the cytoplasmic glycine serine rich domain called the GS domain, resulting in the activation of a number of intracellular signaling cascades in a cell-specific and context-specific manner to mediate the diverse biological functions of TGF-β1.9,13 The canonical TGF-β1 signaling pathway involves activation of receptor-regulated Smads (R-Smads), namely Smad2 and Smad3. The R-Smads are recruited to the receptor complex, mediated by auxiliary proteins such as Smad anchor for receptor activation (SARA), and are directly phosphorylated by activated TβRI. Phosphorylated Smad2/3 form an oligomeric complex with the common Smad, known as Smad4. The complex then translocates into the nucleus where it can interact with various transcription factors, co-activators or co-repressors to regulate the transcription of various TGF-β1 target genes.14,15 Inhibitory Smads, specifically Smad7, negatively regulate TGF-β1 signaling by competing with R-Smads for association with TβRI, thus preventing the recruitment and phosphorylation of R-Smads.15
The Smad signaling pathway is widely accepted as the canonical pathway activated by TGF-β1. However, a large body of evidence demonstrates that TGF-β1 also activates Smad-independent signaling pathways to mediate diverse actions of TGF-β1. The non-Smad signaling pathways include the mitogen-activated protein kinases (MAPKs), namely extracellular signal-regulated kinases 1/2 (ERK1/2), c-Jun N-terminal kinase (JNK), and p38 MAPK, phosphatidylinositol-3-kinase (PI3K)/AKT, Rho-like GTPases (RhoA), and protein phosphatase 2A (PP2A).9,16 TGF-β-activated kinase 1 (TAK1) is a serine/threonine kinase, originally identified as a member of the MAPK kinase kinase (MKKK) family, that is rapidly activated by TGF-β1. Phosphorylation of Thr-187 and Ser-192 in the activation loop of TAK1 results in TAK1 activation and subsequently activates several downstream signaling cascades, including MKK4/7–JNK, MKK3/6–p38 MAPK, and nuclear factor-kappa B (NF-κB)-inducing kinase-IκB kinase.9,17 An overview of the signal transduction pathways induced by TGF-β1 is shown in Fig. 1. Taken together, the complexity of TGF-β1 signaling pathways enables TGF-β1 to elicit diverse and context-dependent biological functions.
Fig. 1. TGF-β1 signaling via Smad and non-Smad pathways.
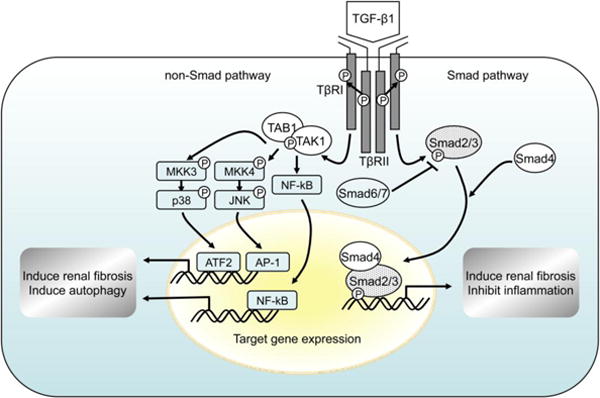
Initiation of the TGF-β signaling cascade occurs upon ligand binding to TβRII and subsequent TβRI-TβRII hetero-tetrameric complex formation. The canonical Smad pathway involves activation of Smad2/3 through recruitment and phosphorylation by activated TβRI, and requires kinase activity of TβRI. TGF-β1 also activates various non-Smad pathways, including TAK1 and subsequently activates several downstream signaling cascades, including MKK4/7–JNK, MKK3/6–p38 MAPK, and nuclear factor-kappa B (NF-κB)-inducing kinase-IκB kinase. TGF-β1, transforming growth factor-β1; TβRI, TGF-β type I receptor; TβRII, TGF-β type 2 receptor; TAK1, TGF-β-activated kinase 1; TAB1, TAK1-binding protein 1; MKK, mitogen-activated protein kinase kinase; JNK, c-Jun N-terminal kinase.
TGF-β1 and renal fibrosis
TGF-β1 is generally regarded as the most potent pro-fibrogenic cytokine and a central mediator of renal fibrosis. Dysregulated expression and activation of TGF-β1 stimulates ECM production through both Smad3-dependent and Smad-independent mechanisms that lead to the development of glomerulosclerosis and tubulointerstitial fibrosis. Importantly, TGF-β1 is one of the most important ECM regulators, both as a potent inducer of ECM synthesis and an inhibitor of the degradation of ECM components, by directly suppressing matrix metalloproteinases (MMPs) and inducing tissue inhibitors of metalloproteinases (TIMPs), resulting in a net accumulation of ECM.2 TGF-β1 can also induce tubular epithelial-to-mesenchymal transition (EMT) and directly contribute to the myofibroblast pool responsible for interstitial matrix production. However, the evidence for this was based largely on in vitro cell culture studies, and recently challenged by studies using genetic fate mapping techniques in mouse fibrosis models that argue against EMT as a direct contributor to the kidney myofibroblast population and renal fibrosis.18
Numerous studies in both human and experimental models have demonstrated that TGF-β1 is significantly up-regulated in fibrotic kidney diseases.19,20 Overexpression of mature TGF-β1 in rodent liver promotes progressive renal fibrosis.21,22 Furthermore, blockade of TGF-β1 with neutralizing TGF-β antibodies or antisense oligonucleotides significantly ameliorates renal fibrosis in vivo and in vitro.23 Others and we have shown that TAK1 functions as a major upstream signaling molecule in TGF-β1-induced type I collagen and fibronectin expression through activation of the MKK3-p38 and MKK4-JNK signaling cascades, respectively.8,24,25 Studies have also shown that TGF-β1 is a key mediator in experimental models of diabetic kidney disease as well as in patients with diabetic nephropathy.26 Recently, TGF-β1 has also been implicated in the pathogenesis of focal segmental glomerular sclerosis (FSGS) with increased TGF-β/Smad signaling activity by hyperplastic podocytes leading to mesangial cell matrix overproduction and podocyte apoptosis and development of glomerulosclerosis.27 Podocyte depletion through apoptosis is a major feature of FSGS and constitutes a central manifestation of chronic progressive glomerular diseases. Studies in TGF-β1 transgenic mice show that podocytes undergo apoptosis and progressive depletion of podocytes precedes mesangial expansion in the course of progressive glomerulosclerosis.28 In other models of glomerular diseases, including diabetic nephropathy and experimental glomerulonephritis, the activation of TGF-β1 promotes podocyte apoptosis and the development of glomerulosclerosis.29 Together, these studies indicate that TGF-β1 is a potent regulator of ECM production and that increased TGF-β signaling is a critical contributor to the progression of fibrotic kidney diseases. Hence, targeting TGF-β1 and its signaling pathway is considered a promising therapeutic strategy for the treatment of CKD.
Anti-TGF-β1 therapies
Different strategies have been explored to reduce excessive TGF-β signaling activity in chronic kidney diseases (Tables 1 and 2). One such therapeutic approach is the use of neutralizing antibodies against TGF-β that has been shown to inhibit early characteristic features of diabetic nephropathy, including hypertrophy and increased matrix expression in experimental diabetes induced by streptozotocin in mice30 and in db/db diabetic mice,31 which are models of type 1 and type 2 diabetes, respectively. Moreover, administration of neutralizing anti-TGF-β antibodies partially reverses the glomerular basement membrane thickening and mesangial matrix expansion in the db/db diabetic mice.32 Anti-TGF-β antibody treatment also has been shown to be beneficial in reducing renal fibrosis in other models such as hypertension-induced renal injury in Dahl S rats fed a high-salt diet.33,34
Table 1.
Approaches to anti-fibrotic therapies in preclinical studies.
| Type | Target | References |
|---|---|---|
|
| ||
| TGF-β antagonists | Anti TGF-β antibodies | stz-induced diabetes (30), db/db mice (31, 32), salt-sensitive hypertension (33, 34) |
| Pirfenidone (inhibits TGF-β synthesis) | UUO (38) subtotal nephrectomy (39-41) db/db mice (42) |
|
| Tranilast analogs: FT011, FT061 | stz-induced diabetes (52,53) subtotal nephrectomy (53) UUO (58) |
|
| Smad7 overexpression (gene transfer) | crescentic glomerulonephritis (59) subtotal nephrectomy (60) |
|
| TAK1 inhibition | Tak 1 deficient mice (61) | |
| TGF-β receptor antagonist:GW788388 | db/db mice (63) | |
| TβR-ll siRNA | UUO (64) | |
|
| ||
| BMP-7 agonists | rhBMP-7 | nephrotoxic serum nephritis (85) UUO (86, 87) stz-induced diabetes (88, 89) MRL/MpJ/pr/lpr lupus mice (90) |
| BMPR agonists: THR-123, THR-184 | nephrotoxic nephritis, stz-induced diabetes (95, 96) | |
| Kielin/chordin-like protein | UUO, folic acid-induced ATN (97) | |
| Inhibition of USAG-1 or gremlin | UUO, model of Alport syndrome, stz-induced diabetes (98-101) | |
|
| ||
| CTGF inhibitors | Antisense oligonucleotides | UUO (110) TGF-β transgenic mice (111) |
| CTGF siRNA | stz-induced diabetes, db/db mice (113) | |
| Anti-CTGF antibody (FG-3019) | stz-induced diabetes, db/db mice (115) | |
|
| ||
| CCL2-CCR2 and CCR1 inhibitors | CCR2 antagonists: propagermanium, RS-504393 | stz-induced diabetes (124) db/db mice (125) UUO (127) |
| CCR2 antagonist: CCX140-B | high-fat fed and db/db mice (126) | |
| CCR1 antagonist: BX471 | nephrotic syndrome, FSGS (132) lupus-like nephritis (133) UUO (134) |
|
|
| ||
| PDE inhibitors | Pentoxifylline | UUO (135) 5/6 subtotal nephrectomy (136) crescentic glomerulonephritis (137) |
| Vardenafil | alloxan-induced diabetic rabbits (141) | |
| Sildenafil | stz-induced diabetes (142) | |
|
| ||
| Nox1/4 inhibitor | GKT137831 | db/db mice (144) |
|
| ||
| ET-1 antagonists | ETA receptor blocker: FR139317 | hypertensive rat (156) |
| ETA receptor blocker: atrasentan | hypertensive Ren-2 transgenic rats (157) | |
| Non selective ET blocker: bosentan | L-NAME treated mice (158) | |
|
| ||
| TNF-α antagonists | Anti-TNF-α antibody | crescentic glomerulonephritis (162) |
Abbreviations; ATN, acute tubular necrosis; BMP, bone morphogenic protein; BMPR, BMP receptor; CCL2-CCR2, CC motif chemokine ligand 2- CC receptor type 2; CCR1, CC receptor type 1 ; CTGF, connective tissue growth factor; ET, endothelin; FSGS, focal segmental glomerular sclerosis; L-NAME, NG-nitro-L-arginine methylester; Nox, NADPH oxidase ; PDE, phosphodiesterase ; stz, streptozotocin; TGF-β, transforming growth factor-β; TNF-α, tumor necrosis factor-α.
Table 2.
Anti-fibrotic therapies in clinical trials for kidney diseases.
| Therapeutic approaches | Drug | Type | Target | Disease | Stage | Clinical trial identifier www.clinicaltrials.gov |
|---|---|---|---|---|---|---|
|
| ||||||
| TGF-β antagonists | Fresolimumab (GC-1008) | Antibody | Anti-TGF-β1,2, 3 | Focal Segmental Glomerular Sclerosis | Phase 2 | NCT01665391 |
| LY2382770 | Antibody | Anti-TGF-β1 | Diabetic nephropathy | Phase 2 | NCT01113801 | |
| Pirfenidone | Small-molecule | TGF-β synthesis inhibitor | Focal Segmental Glomerular Sclerosis | Phase 2 | NCT00001959 *completed (50) | |
| Diabetic nephropathy | Phase1/2 | NCT00063583 *completed (51) | ||||
|
| ||||||
| BMP-7 agonists | THR-184 | Small peptide | BMPR agonist | Cardiac Surgery Associated-Acute Kidney Injury | Phase 2 | NCT01830920 |
|
| ||||||
| CTGF inhibitors | FG-3019 | Antibody | Anti-CTGF | Diabetic nephropathy | Phase 1 | NCT00102297 *completed |
| NCT00754143 *completed (115) | ||||||
|
| ||||||
| CCL2-CCR2 inhibitor | CCX140-B | Small-molecule | CCR2 antagonist | Diabetic nephropathy | Phase 2 | NCT01028963 *completed (131) |
| Diabetic nephropathy | Phase 2 | NCT01440257 | ||||
| Diabetic nephropathy | Phase 2 | NCT01447147 | ||||
|
| ||||||
| PDE inhibitors | Pentoxifylline | Small-molecule | Nonselective PDE inhibitor | Chronic kidney disease | Phase 3 | NCT00285298 *completed (138) |
| Diabetic nephropathy | Phase 4 | NCT01382303 | ||||
| Chronic kidney disease | Phase 4 | NCT01377285 | ||||
| PF00489791 | Small-molecule | PDE5 inhibitor | Diabetic nephropathy | Phase 2 | NCT01200394 | |
|
| ||||||
| Nox1/4 inhibitor | GKT137831 | Small-molecule | Nox1/4 inhibitor | Diabetic nephropathy | Phase 2 | NCT02010242 |
|
| ||||||
| ET-1 antagonists | Avosentan | Peptide | ETA receptor antagonist | Diabetic nephropathy | Phase 3 | NCT00120328 *terminated (160) |
|
| ||||||
| TNF-α antagonists | Adalimumab | Antibody | Anti-TNF-α | Focal Segmental Glomerular Sclerosis | Phase 2 | NCT00814255 *completed (163) |
Abbreviations; BMP, bone morphogenic protein; BMPR, BMP receptor; CCL2-CCR2, CC motif chemokine ligand 2- CC receptor type 2; CTGF, connective tissue growth factor; ET, endothelin; Nox, NADPH oxidase ; PDE, phosphodiesterase ; TGF-β, transforming growth factor-β; TNF-α, tumor necrosis factor-α.
There are several clinical trials underway using anti-TGF-β antibodies. Fresolimumab (GC-1008, Genzyme), a member of the G4 immunoglobulin subclass, is a human monoclonal antibody that neutralizes all three isoforms of TGF-β. A phase 1 multicenter, open-label study conducted in patients with treatment-resistant primary FSGS revealed that a single-dose infusion of fresolimumab up to 4mg/kg was relatively safe and well tolerated.35 A phase 2 multicenter, double-blind, parallel dosing, randomized study of fresolimumab in patients with steroid-resistant primary FSGS is currently ongoing (clinicaltrials.gov:NCT01665391). LY2382770 (Eli Lilly), an antibody that neutralizes the bioactivity of TGF-β1, is also being investigated in a phase 2 clinical trial in patients with type 1 or type 2 diabetes to evaluate whether it is more effective than placebo at slowing the progression of diabetic kidney disease in patients treated with an ACE inhibitor or an ARB (clinicaltrials.gov:NCT01113801). A recent preclinical study using an experimental model of diabetic nephropathy induced by streptozotocin injection in uninephrectomized rats has suggested that administration of anti-TGF-β antibodies in combination with either an ACE inhibitor or an ARB resulted in better outcomes than either alone.36
Pirfenidone (5-methyl-1-phenyl-2(1H)-pyridone) is a small-molecular weight synthetic molecule that has garnered much attention and excitement as an orally active, anti-fibrotic agent. The compound exerts anti-fibrotic and anti-inflammatory activities in a variety of animal and cell-based models, and its effects are, in part, mediated by inhibition of TGF-β1 synthesis.37 The effect of pirfenidone in reducing renal fibrosis has been demonstrated in several experimental models including unilateral ureteral obstruction (UUO),38 subtotal nephrectomy,39-41 and diabetic nephropathy.42 The antifibrotic effects of pirfenidone have also been demonstrated in other models of tissue fibrosis such as bleomycin-induced pulmonary fibrosis.43 Pirfenidone has been evaluated clinically for its safety and efficacy in numerous disorders, including idiopathic pulmonary fibrosis (IPF),44 multiple sclerosis,45 and liver fibrosis associated with chronic hepatitis C virus infection.46 In 2011, pirfenidone was approved for use in Europe for treatment of patients with IPF under the trade name Esbriet® (InterMune).47 Four phase 3 trials comparing pirfenidone with placebo in patients with IPF have been completed. One study was conducted in Japan and three were multinational studies conducted in Europe, Australia, and the US.48,49 In three of four studies, pirfenidone reduced disease progression, as measured by the decline in forced vital capacity or vital capacity.48,49 Moreover, mortality analyses in the three multinational trials showed significant between-group differences favoring pirfenidone treatment.49 However, pirfenidone has not yet received an approval by the Food and Drug Administration in the US.
Several clinical trials have also examined the efficacy of pirfenidone in the treatment of CKD. In an open-label, single-center pilot study of patients with FSGS, pirfenidone significantly slowed the decline in kidney function with a median improvement of 25% in the estimated glomerular filtration rate (eGFR) decline rate in patients who have moderate to severe CKD and are already being treated with angiotensin antagonists.50 Although the results were very encouraging, there were limitations of this clinical study including the small-scale, exploratory nature and the lack of a placebo control group. Therefore, it is possible that unidentified interventions or natural variability in GFR decline rate during the pirfenidone treatment period may have contributed to the improved outcome. A recently completed, randomized, placebo-controlled trial further demonstrated the beneficial effect of pirfenidone in improving kidney function in 77 patients with overt diabetic nephropathy.51 The mean creatinine-based eGFR increased in the pirfenidone (1200 mg/d) group (+3.3 ± 8.5 ml/min per 1.73 m2); whereas, the mean eGFR decreased in the placebo group (-2.2 ± 4.8 ml/min per 1.73 m2). The results from both clinical trials suggest that pirfenidone might be an effective agent to slow renal function decline associated with CKD. However, it is important to note that in the above studies, assessment of kidney function was by eGFR using the Modification of Diet in Renal Disease (MDRD) equation, and not by direct measurement of GFR, and serial kidney biopsies were not performed to assess whether there was histologic evidence of regression of renal fibrosis.
With similarity to pirfenidone as a promising anti-fibrotic agent, tranilast and its cinnamoylanthranilate derivatives are being investigated for their ability to inhibit TGF-β1-induced collagen production and the potential renoprotective effects. Tranilast was originally marketed in Japan and South Korea to treat allergic disorders since 1982 and more recently for the treatment of hypertrophic scars and scleroderma.52 For optimization of the anti-fibrotic effects of this compound, several cinnamoylanthranilate derivatives of tranilast have been generated with purported superior potency and reduced cellular toxicity relative to tranilast. FT011 and FT061 are orally-active analogs that have been demonstrated to attenuate TGF-β1-stimulated collagen production in cultured renal mesangial cells and reduce albuminuria in rodent models of progressive diabetic nephropathy.52,53 In addition to inhibiting TGF-β1-induced collagen production, FT011 was found to also inhibit the pro-fibrotic and pro-proliferative effects of platelet-derived growth factor (PDGF) and significantly reduced renal fibrosis and interstitial macrophage infiltration and attenuated the decline in renal function.53 FT011 (Fibrotech Therapeutics, Melbourne, Australia) is currently in clinical development for the treatment of diabetic nephropathy.
Peroxisome proliferator-activated receptor γ (PPARγ), a ligand-activated transcription factor of the nuclear receptor superfamily, has received particular attention in recent years. Activation of PPARγ by natural and synthetic agonists effectively inhibits TGF-β-induced pro-fibrotic effects in many organs.54 Studies in experimental models and clinical studies suggest that glitazones, which are PPARγ agonists, may inhibit the progression of renal disease.55 However, this class of drugs carries an increased risk of developing cardiovascular diseases, and thus, there is much interest in developing selective PPARγ modulators for safer therapeutic PPARγ activation.55-57
Inhibition of TGF-β signaling by Smad 7 is an alternative therapeutic approach that has also been explored. Overexpression of renal Smad7 by gene transfer therapy blocks activation of TGF-β1-induced Smad2/3 signaling and attenuates renal fibrosis and inflammation, as evidenced in studies using animal models of obstructive nephropathy induced by UUO,58 autoimmune crescentic glomerulonephritis,59 and remnant kidney.60 Targeting the TGF-β1-induced TAK1 signaling pathway is an attractive strategy that targets the major pro-inflammatory, pro-apoptotic, and pro-fibrotic pathways, such as the MMK3-p38 MAPK or JNK.24,61 A recently reported preclinical study demonstrates that the administration of LYTAK1, a selective small-molecule inhibitor of TAK1, plus gemcitabine significantly reduced tumor burden in nude mice with human pancreatic tumor xenografts and prolonged survival duration.62
Additional strategies to inhibit TGF-β signaling activity include antisense oligonucleotides to reduce TGF-β expression, soluble TGF-β receptors that act as decoys to sequester TGF-β, small-molecule TGF-β receptor antagonists (GW788388), and small interfering RNA (siRNA) for TGF-β receptor.63,64 However, these strategies remain at preclinical stages and therapeutic potential in the treatment of patients with CKD has yet to be tested in clinical trials.
Drugs that inhibit key components of the RAAS, such as ACE inhibitors and ARBs, have become a cornerstone of our current strategy to slow progression of renal disease. Evidence suggests a close link between the RAAS axis and TGF-β1 signaling pathways that further enhances renal injury. Angiotensin II (ANG II) increases the renal expression of TGF-β1 and TGF-β receptors and phosphorylation of Smad2/3, and in turn, TGF-β1 stimulates angiotensinogen expression indicating a positive feedback loop.65 Other components of the RAAS such as renin and aldosterone can also stimulate TGF-β1 signaling.65,66 Therefore, the pro-fibrotic effects from activation of the RAAS axis leading to progression of renal disease are mediated, at least in part, by induction of TGF-β1 signaling. The combination of anti-TGF-β1 therapies with RAAS inhibitors may offer a new, improved approach to halt the development of renal fibrosis and progression of renal disease.
An important caveat to consider is that although excessive TGF-β1 activity leads to fibrotic conditions, given the complexity and pleiotropic actions of TGF-β1, therapies aimed at indiscriminate blockade of TGF-β1 may not be prudent. Targeted disruption of TGF-β1 gene results in multifocal inflammatory disease in mice and growing body of evidence demonstrates cytoprotective effects of TGF-β1 to mitigate tissue injury through enhancing wound repair and tissue regeneration, as well as anti-inflammatory effects.67,68 Thus, effects of TGF-β activation in renal injury may be protective/reparative or deleterious/progressive depending on, for instance, the context and extent of injury. In this context, a more detailed understanding of the cellular and molecular mechanisms of TGF-β1 actions will not only provide a more comprehensive knowledge of the pathogenic mechanism of CKD, but may also guide in the development of therapeutic strategies that specifically target the signaling pathway responsible for the deleterious effects of TGF-β1. Thus, there is still a great need for future investigations to gain further insights into the complex TGF-β signaling mechanisms, with the ultimate goal of developing novel and more effective therapies for progressive kidney diseases.
TGF-β1 and autophagy
Macroautophagy, or commonly referred as autophagy (literally, self-eating), is a fundamental homeostatic process that cells use to degrade and recycle cellular proteins and remove damaged organelles. During the past decade, there has been a growing interest in defining the basic cellular mechanism of autophagy and its importance in health and disease.69,70 The critical role of TGF-β1 as an inducer of autophagy is just beginning to be appreciated.71 Autophagy can lead to cell death in response to stress, but it can also act as a cytoprotective mechanism for cell survival. It is plausible that the functions of TGF-β1 as both an apoptosis promoter and apoptosis suppressor may relate to its regulation of autophagy. We have reported that TGF-β1 induction of autophagy is mediated by TAK1-MKK3-p38 signaling axis, and protects renal mesangial cells from undergoing apoptosis during serum deprivation.72 Intriguingly, recent evidence has been demonstrated that two of TAK1 binding partners, TAB2 and TAB3, are endogenous inhibitors of autophagy.73,74 Under non-stimulated condition, TAB2 and TAB3 are bound with Beclin 1, an autophagy-related protein, and maintain inactive state of Beclin 1 and autophagy. In contrast, in response to pro-autophagic stimuli, TAB2 and TAB3 dissociate from Beclin 1, and subsequently bind with TAK1 to facilitate the pro-autophagic stimulation through TAK1 activation.
In recent studies, we investigated the functional role of autophagy in kidney injury and fibrosis. We uncovered a novel role of autophagy as a cytoprotective mechanism to promote intracellular degradation of collagen, and thus, negatively regulate and prevent excess collagen accumulation in the kidney.75 We also show that kidney injury following UUO potently induces autophagy and regulates TGF-β expression and that deficiency of autophagic protein LC3 (microtubule-associated protein 1 light chain 3) leads to increased collagen deposition and mature TGF-β levels in obstructed kidneys in LC3 null (LC3-/-) mice.76 These data suggest a novel intracellular mechanism by which mature TGF-β1 protein levels may be regulated through autophagic degradation and suppresses renal fibrosis. Recent studies implicate dysregulated autophagy in disorders characterized by fibrosis in various tissues, including cardiac fibrosis, liver fibrosis, and IPF.77 Our findings suggest a novel role for autophagy as a cytoprotective mechanism to negatively regulate the production of mature TGF-β proteins in renal tubular epithelial cells, and consequently limiting TGF-β secretion and suppress development of interstitial fibrosis in kidney injury. Hence, targeting autophagic pathway may be a promising therapeutic strategy.
Bone morphogenic protein (BMP)-7
BMP-7, a member of the TGF-β superfamily, is a 35-kDa homodimeric protein, also known as osteogenic protein-1 (OP-1) that was originally identified as a potent osteogenic factor purified from bone and hence the name. Among BMPs, BMP-7 is an essential signaling molecule during mouse embryogenesis and particularly important in kidney and eye development. Its genetic deletion in mice leads to impaired kidney development with a reduction in branching of the ureteric bud and loss of metanephric mesenchyme, resulting in severe renal hypoplasia, and anophthalmia.78 Similar to other members of the TGF-β superfamily, BMP-7 signals via heteromeric interactions of BMP receptors type I (BMPRI) and type II (BMPRII) to activate its R-Smads, Smad1/5/8 (Fig. 2). Phosphorylated R-Smads form complexes with common signalling component Smad4, which then translocate into the nucleus to regulate transcription of their target genes. Smad6 is an inhibitory Smad that is induced by BMPs and inhibits both TGF-β and BMP signaling. In renal mesangial cells, BMP7 reduces nuclear accumulation of Smad3 and blocks the transcriptional up-regulation of certain TGF-β/Smad3 targets such as PAI-1, which requires Smad5 and is mediated downstream, at least in part, by Smad6.79 Thus, BMP-7 opposes pro-fibrogenic effects of TGF-β1 via Smad5-dependent regulation of Smad6 expression.
Fig. 2. Opposing roles of TGF-β1 and BMP-7 signaling pathways.
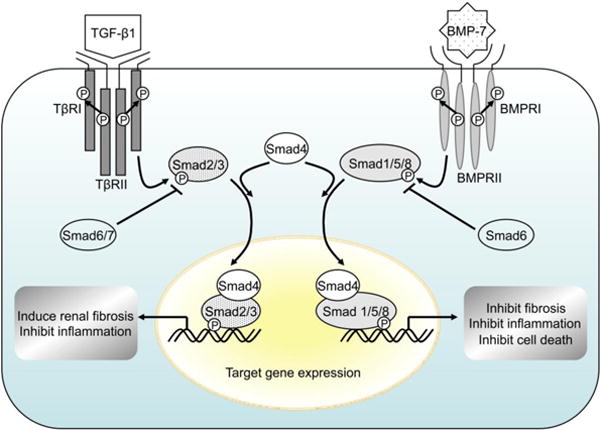
Similar to other members of the TGF-β superfamily, BMP-7 signals via heteromeric interactions of BMP receptors type I (BMPRI) and type II (BMPRII) to activate its R-Smads, Smad1/5/8. Phosphorylated R-Smads form complexes with common signalling component Smad4, which then translocate into the nucleus to regulate transcription of their target genes. Smad6 is an inhibitory Smad that is induced by BMPs and inhibits both TGF-β and BMP signaling. BMP, bone morphogenic protein; BMPR, BMP receptor; TGF-β1, transforming growth factor-β1; TβR, TGF-β receptor.
BMP-7 has a protective role against renal fibrosis in models of kidney injury. BMP-7 expression levels are reduced in the kidneys of several animal models of diabetic nephropathy and ischemia-induced acute renal failure.80-82 Significant down-regulation of renal BMP-7 expression has also been shown in human diabetic glomeruli,83 and in patients with hypertensive nephrosclerosis.84 The loss of BMP-7 activity is thought to be pro-fibrogenic and the administration of exogenous recombinant human BMP-7 (rhBMP-7) reduced renal fibrosis in rodent models of renal injury including nephrotoxic serum nephritis,85 UUO,86,87 and streptozotocin-induced diabetes.88,89 Administration of rhBMP-7 also improved kidney function, histology, and survival in mice deficient in the alpha3-chain of type IV collagen and MRL/MpJlpr/lpr lupus mice, which are two genetic models for chronic renal injury and fibrosis.90 Moreover, transgenic expression of BMP-7 in glomerular podocytes and proximal tubules prevented podocyte dropout and reductions in nephrin levels in diabetic mice, and reduced proteinuria and glomerular fibrosis and suppressed interstitial collagen accumulation as well as collagen I and fibronectin expression.91 These effects of the BMP-7 transgene were observed without changing renal TGF-β levels. It has been proposed that methylation of cytosine residues that precede a guanosine in the DNA sequence (the CpG dinucleotide) is an epigenetic event that can effectively silence transcription over multiple cell generations. Hypermethylation of the Rasal1 promoter contributes to activation of fibroblasts and progression of renal fibrosis and the anti-fibrotic effects of BMP-7 may be as a result of reversal of hypermethylation.92 Recent studies also implicate a role of histone deacetylase dependent repression of Bmp-7 transcription in the pathogenesis of renal injury due to obstructive uropathy and treatment with histone deacetylase inhibitor trichostatin A may be a potentially effective strategy that restores BMP-7 expression and its renal protective functions.93
Despite the abundant evidence in animal models of kidney disease showing that BMP-7 antagonizes TGF-β-dependent fibrosis, clinical studies in humans have been limited, in part, because of its low bioavailability in the kidney of only 0.5% of the systemically administered BMP-7.81 The need for high doses of BMP-7 increases chance of off-target effects. Thus, alternative therapeutic approaches that mimic BMP-7 activity are desirable. Renal injury is associated with increased expression of activin receptor-like kinase 3 (ALK3) which is a BMPRI that is predominantly expressed in kidney tubular epithelial cells.94 Its deletion in the tubular epithelium leads to enhanced TGF-β1-Smad3 signaling, epithelial damage and fibrosis, suggesting a protective role for ALK3-mediated signaling in the kidney.95 THR-123 (Thrasos Therapeutics, Canada) is an orally administered small-peptide agonist of BMP-7 signaling, and functions through ALK3 signaling to suppress inflammation, apoptosis, and EMT, and reverse established fibrosis in several mouse models of acute and chronic renal injury.95,96 THR-123 in combination with ACE inhibitor exerted additive therapeutic effects. Therefore, BMP-7 signaling agonists such as THR-123 represent a new line of therapeutic agents that may be an effective therapy for kidney disease. Additionally, THR-184, a structural analogue of THR-123, is currently in a phase 2 clinical trial in patients with risk for cardiac surgery-associated acute kidney injury to determine whether THR-184 when administered around the time of cardiac surgery requiring cardiopulmonary bypass can prevent or ameliorate the development of acute kidney injury (clinicaltrials.gov:NCT01830920).
Additional therapeutic approaches that are being investigated include the use of BMP agonists that enhance BMP-7 signaling activity. Kielin/chordin-like protein (KCP) is an extracellular protein with 18 cysteine-rich domains that enhances BMP-7 signaling by facilitating binding of BMP-7 to its receptors and activation of downstream Smad1-dependent transcription.97 KCP null (Kcp-/-) mice show reduced levels of phosphorylated Smad1 and are more susceptible to development of renal interstitial fibrosis following UUO injury and folic acid-induced acute tubular necrosis.97 These findings suggest that KCP through its BMP enhancing effects may be a potential therapeutic target. However, because KCP is a large, cysteine-rich protein, efforts to purify soluble, recombinant KCP have been hampered and its efficacy as an anti-fibrotic agent has not be tested directly in experimental models.
Increasing evidence indicates that reduced BMP-7 activity is pro-fibrogenic and that BMP antagonists play important roles in the pathogenesis of renal fibrotic disease. Two most extensively studied BMP-7 antagonists are the uterine sensitization-associated gene-1 (USAG-1) and gremlin. Both are secreted proteins that function through direct association with BMP, thus preventing the binding of BMP to its receptors and negatively regulate BMP signaling activity. Mice lacking USAG-1 (Usag1-/-) have been shown to be resistant to renal injury and have prolonged survival and preserved renal function in animal models of acute tubular injury from cisplatin nephrotoxicity and chronic renal injury induced by UUO.98 Renal BMP signaling was significantly enhanced in the Usag1-/- mice, and the administration of neutralizing antibody against BMP-7 abolished renoprotection. Genetic ablation of USAG-1 in a mouse model for human Alport syndrome carrying null mutation of gene encoding the α4 chain of type IV collagen (Col4α3-/-) significantly attenuated disease progression and preserved renal function.99 Gremlin plays an important role in development and mice homozygous for deletion of grem1 gene die of complete renal agenesis shortly after birth. The expression of gremlin is low in the healthy adult kidney, but is increased in kidney disease models, and allelic deletion of grem1 gene or in vivo knockdown of gremlin by siRNA attenuated or reversed diabetes-associated kidney damage in animal studies.100,101 While investigations are still limited to preclinical stage, these findings suggest that blockade of BMP antagonists such as USAG-1 and/or gremlin represents promising therapeutic approaches for renoprotective effects through maintenance of BMP-7 activity.
Connective tissue growth factor (CTGF)
CTGF, also known as CCN2, has emerged as an important modulator of pro-fibrotic TGF-β and anti-fibrotic BMP-7 activities. CTGF is the second member of the CCN family of matricellular proteins (cysteine-rich 61; Cyr61/CCN1, CTGF/CCN2, nephroblastoma overexpressed; Nov/CCN3).102 Like the other CCN family members, CTGF mediates ECM – cell communication by interacting with extracellular signaling molecules via its multiple distinct interaction domains instead of direct binding to unique receptors.103 CTGF contains four distinct domains: insulin-like growth factor (IGF)-binding domain, cysteine-rich (CR) domain, formerly designated as von Willebrand factor (vWF) type C repeat domain, thrombospondin type 1 (TSP-1) repeat domain, and C-terminal cystine knot (Cys-knot) domain (Fig. 3). Secretion of CTGF is controlled by the 37-amino acid signal sequence at the N-terminus, and a protease sensitive hinge region is located between the CR and TSP-1 repeat domains.
Fig. 3. A schematic overview of structure of CTGF and factors interacting with CTGF.
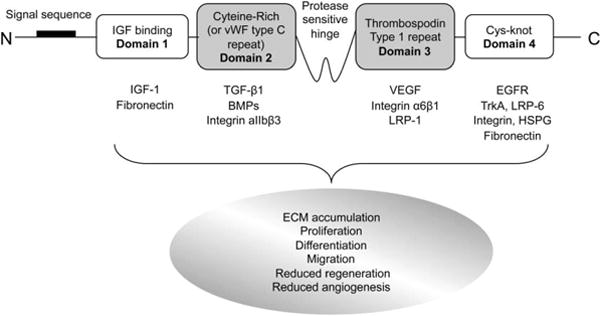
BMP, bone morphogenic protein; Cys-knot, cystine knot; ECM, extracellular matrix ; HSPG, heparan sulfate proteoglycan; IGF-1, insulin like growth factor-1; LRP, lipoprotein receptor–related protein; TGF-β1, transforming growth factor-β1; VEGF, vascular endothelial growth factor ; vWF, von Willebrand factor.
CTGF expression is increased in various human and animal models of kidney fibrosis, including diabetic nephropathy, hypertensive nephrosclerosis, and crescentic glomerulonephritis.104-106 CTGF is a direct downstream early response gene of TGF-β, but it also potentiates TGF-β signaling by directly binding TGF-β1 through its CR domain.107 These interactions lead to myofibroblast activation, de novo expression of α-smooth muscle actin, and extracellular accumulation of fibronectin to promote tissue fibrosis. CTGF also enhances TGF-β1 signaling by binding of tyrosine receptor kinase A (TrkA) through the C-terminal Cys-knot domain to induce transcription factor TGF-β-inducible early gene (TIEG)-1 and disrupt the negative feedback loop of the TGF-β signaling pathway via suppression of inhibitory Smad7.108 On the other hand, CTGF interacts with BMP-7 through its CR domain to inhibit BMP-7 signaling and repress the renoprotective effects of BMP-7.109 CTGF, by binding to BMP-7 whose own signaling pathway opposing TGF-β is inhibited, further enhances TGF-β signaling. CTGF can also bind to several other growth factors (e.g. IGF-1, epidermal growth factor (EGF), vascular endothelial growth factor (VEGF)) to modify their function, cell surface proteins (e.g. integrins, low-density lipoprotein receptor-related protein (LRP), heparan sulphate proteogycans) by which intracellular signaling may be initiated, and extracellular matrix proteins (e.g. fibronectin) that act as a sink for CCN proteins and modify their turnover (Fig. 3).
Inhibition of CTGF with antisense oligonucleotides, siRNA, or neutralizing antibodies can prevent expansion of the ECM in experimental models of kidney disease. Yokoi et al. reported that the injection of CTGF antisense nucleotides reduced expression of collagen, fibronectin, and α-smooth muscle actin, and ameliorated renal tubulointerstitial fibrosis in a 7-day UUO model.110 Another study demonstrated that kidney fibrosis was also significantly reduced by the intravenous administration of CTGF antisense oligonucleotides in transgenic mice overexpressing TGF-β.111 In a rat model of renal allograft nephropathy, in vivo delivery of CTGF siRNA prevented up-regulation of fibrotic markers.112 In addition, CTGF down-regulation attenuated progression of diabetic nephropathy in mouse models of type 1 and type 2 diabetes.113 However hemizygous deletion of CTGF in mice had limited effects on the progression of renal fibrosis in long-term streptozotocin-induced diabetic nephropathy, in a more advanced stage of obstructive nephropathy following UUO, and in high-dose aristolochic acid-induced tubulotoxic nephritis.114 These results suggest that the effectiveness of anti-CTGF therapy might be limited in severe CKD. In a small phase 1 open-label study, treatment with FG-3019 (Fibrogen), a human anti-CTGF monoclonal antibody, significantly reduced albuminuria in patients with diabetic kidney disease without significant adverse effects115 (clinicaltrials.gov:NCT00754143) These results showing changes in albuminuria are promising but will require validation in a larger prospective, randomized, blinded study. Given that CTGF holds a central position in the pathogenesis of kidney fibrosis by its interactions with factors involved in the fibrotic response, including TGF-β1 and BMP-7, reducing CTGF bioavailability by anti-CTGF therapy is an attractive anti-fibrotic drug targeting.116
CC motif chemokine receptor antagonists
Recent evidence indicates that inflammation plays a critical role in the initiation and progression of renal fibrosis. Chemokines mediate the trafficking of circulating inflammatory cells to sites of injury via activation of their seven-transmembrane G protein-coupled receptors. Monocyte chemoattractant protein-1 (MCP-1), also termed as CC motif chemokine ligand 2 (CCL2), has been shown to be a major promoter of inflammation and renal injury response that lead to renal fibrosis. Moreover, MCP-1 has been shown to induce TGF-β1 production in macrophages117 and mesangial cells,118 and in turn TGF-β1 can itself stimulate mesangial expression of MCP-1.119 Increased kidney MCP-1 production was associated with glomerular macrophage infiltration and increase in collagen IV expression in experimental models of type 1 diabetic nephropathy induced by streptozotocin120,121 and type 2 diabetic nephropathy in db/db mice.122
MCP-1 deficiency reduced glomerular deposition of fibronectin and renal expression of TGF-β1118 and was associated with marked reductions in glomerular and interstitial macrophage accumulation and renal fibrosis in streptozotocin-induced type 1 diabetic mice120 and type 2 diabetic db/db mice.123 Blockade of MCP-1 by treatment with its receptor CCR2 antagonists reduced mesangial matrix expansion, macrophage infiltration, and glomerular expression of collagen IV and TGF-β1 in streptozotocin-induced diabetic mice124 and decreased urinary albumin excretion and mesangial expansion, suppressed pro-fibrotic and pro-inflammatory cytokine synthesis, but improved insulin resistance in db/db mice.125,126 Blockade of CCR2 either by treatment with CCR2 antagonists or in CCR2 gene-targeted mice also ameliorated progressive fibrosis induced by UUO.127,128 Therefore, targeting MCP-1 by specifically disrupting CCR2 signaling pathway may be an effective anti-fibrotic strategy. In patients with diabetic nephropathy, increased urinary MCP-1 was associated with the rate of eGFR decline and progression to kidney failure, and can serve as prognostic markers for disease progression.129,130 A recently completed pilot phase 2, double-blind, randomized clinical trial evaluated CCX140-B (ChemoCentryx), an orally-administered antagonist of CCR2, in patients with type 2 diabetes with the primary objective to evaluate the safety of CCX140-B found that CCX140-B was well tolerated with no serious adverse events131 (clinicaltrials.gov:NCT01028963). Longer-term phase 2 clinical trials are currently underway to evaluate the efficacy of CCX140-B in diabetic nephropathy (clinicaltrials.gov:NCT01440257, NCT01447147). Targeting CCR1 with the small-molecule antagonist BX471 has also demonstrated that CCR1 blockade reduced interstitial inflammation and renal fibrosis in murine models of nephrotic syndrome and FSGS,132 lupus-like nephritis in MRLlpr/lpr mice,133 and after UUO in mice.134 Hence, disruption of the chemokine pathways may represent effective strategies for anti-fibrotic therapy to mitigate progressive renal fibrosis.
Phosphodiesterase (PDE) inhibitors
Cyclic nucleotide phosphodiesterases (PDEs) are promising targets for therapeutic intervention as PDE inhibitors have been shown to attenuate renal disease progression in several experimental models. Pentoxifylline is the most well documented PDE inhibitor that has been demonstrated to inhibit TGF-β1-induced CTGF expression as well as collagen type I and α-smooth muscle actin expression.135 Pentoxifylline attenuated renal fibrosis, myofibroblasts accumulation, and renal expression of TGF-β1, CTGF, and collagen in rat models of UUO,135 5/6 subtotal nephrectomy,136 and crescentic glomerulonephritis.137 In a pilot double-blind, randomized, placebo-controlled clinical trial of 40 patients with CKD, pentoxifylline slowed the eGFR decline, but had no significant effect on proteinuria after 1 year of treatment.138 However, in two separate pilot studies (one including 10 patients with idiopathic membranous nephropathy139 and the second including 40 patients with type 2 diabetes,140 pentoxifylline was shown to decrease proteinuria, and a prospective randomized placebo-controlled study is being conducted in South Korea to evaluate the effects of pentoxifylline on proteinuria in type 2 diabetic patients (clinicaltrials.gov:NCT01382303). A phase 4 multicenter, randomized, double-blind, placebo-controlled clinical trial is currently ongoing to investigate the renoprotective efficacy of combined pentoxifylline and ARB valsartan, compared with placebo and valsartan in 700 patients with CKD stages 3 and 4 (clinicaltrials.gov:NCT01377285).
Pentoxifylline is known as a nonspecific PDE inhibitor. Two selective inhibitors of PDE type 5 (PDE5) are under investigation. Vardenafil has been shown to improve kidney function and reduce proteinuria in diabetic rabbits,141 and sildenafil has anti-inflammatory activity in streptozotocin-induced diabetic rats.142 The mechanism of these actions are not well understood, however, increased activity of PDE5 in glomeruli and in cells of collecting ducts in sodium-retaining states, such as nephrotic syndrome, and diminished ability to excrete sodium can be corrected by administration of the selective PDE5 inhibitor.143 A clinical trial to evaluate the efficacy and safety of the PDE5 inhibitor, PF-00489791 (Pfizer), in a phase 2 randomized, double-blind, placebo-controlled study in patients with type 2 diabetes and overt nephropathy is in progress (clinicaltrials.gov:NCT01200394).
Nox1/Nox4 inhibitors
Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox) has normal physiological functions in the kidney, including roles in gluconeogenesis, glucose transport, tubuloglomerular feedback, hemodynamics, and electrolyte transport. On the other hand, overproduction of ROS by Nox results in oxidative stress and tissue injury such as inflammation and fibrosis. Among the five isoforms of the Nox catalytic subunit, namely Nox1, 2, 3, 4 and 5, Nox4 is highly expressed in the kidney, particularly in the proximal tubules and medullary collecting ducts, and has been implicated in renal oxidative stress and kidney injury in pathologic conditions such as diabetic nephropathy and CKD.144,145 Bondi et al. reported that Nox4 may also be a downstream activator of Smad3 in renal fibroblasts.146 Therefore, emerging evidence suggests that Nox4 is an important downstream effector of TGF-β1-mediated fibrosis, while Nox-dependent redox signaling may in turn regulate TGF-β1/Smad signaling.146,147 TGF-β1 specifically increases the expression of Nox4 and ROS production in various cell types, including smooth muscle cells, endothelial cells, hepatocytes, and fibroblasts.147 The induction of Nox4-dependent ROS by TGF-β1 is required for the conversion of cardiac fibroblasts to myofibroblasts148 and is linked to TGF-β1-induced cytoskeletal changes in human vascular endothelial cells.149 Moreover, upregulation of mitochondrial Nox4 via the TGF-β1-Smad2/3 pathway induces ROS production, mitochondrial dysfunction, and apoptosis in cultured mouse podocytes.150 Thus, pharmacological inhibition of Nox4 functions may have therapeutic implications in treatment of fibrotic disorders, particularly in the context of potentiating TGF-β1 signaling.
The pathophysiological function of Nox1 is less understood, but is thought to be involved in ANG II-mediated hypertension and Nox1-derived ROS in the kidney may participate in the regulation of the pressor response to ANG II.151 Nox1 is expressed in vascular smooth muscle cells and ANG II upregulates Nox1 expression in the kidney and induces oxidative stress, which was blocked by ANG type 1 receptor (AT1-R) antagonist candesartan.152 These studies indicate that ANG II stimulates oxidative stress and Nox1 in the kidney via AT1-R. Expression of Nox1 as well as Nox4 was increased in the diabetic milieu under high glucose and Nox1 inhibition abrogated TGF-β, and fibronectin production in mesangial cells.153 Treatment with a Nox1/4 inhibitor, GKT137831 (Genkyotex), suppressed ROS production and fibrotic gene expression and attenuated fibrosis in experimental liver fibrosis model.154 In a recent study in a mouse model of type 2 diabetes, GKT136901 treatment reduced albuminuria and renal oxidative stress, and preserved renal structure in diabetic (db/db) mice155 suggesting a renoprotective effect of the Nox1/4 inhibitor. A randomized, double-blind, placebo-controlled phase 2 clinical study is in progress to evaluate the safety and efficacy of GKT137831 in patients with type 2 diabetes and albuminuria (clinicaltrials.gov:NCT02010242).
Endothelin (ET)-1 antagonists
Endothelins are 21-amino acid vasoconstrictive peptides produced primarily in the endothelium and have key roles in vascular homeostasis. Endothelin (ET)-1 is the best known and predominant isoform produced in the cardiovascular system and in the kidney. ET-1 is generally not stored and released, but instead generated in response to a range of stimuli, and is a potent mitogen in vascular smooth muscle cells and mesangial cells. Gene transcription of ET-1 is enhanced during conditions of renal stress, such as hypoxia, inflammation, hyperglycemia, and acidosis, and evidence suggests the involvement of ET-1 in the pathophysiology of CKD. Endothelin antagonists have been shown to improve renal function in experimental models of kidney disease.156 Endothelin antagonism with a selective endothelin A receptor (ETA) antagonist attenuates collagen deposition and renal vascular and glomerular fibrosis in several forms of experimental hypertension.156-158 However, observations from clinical studies indicate that these antagonists also elicit volume-related side effects. This was particularly evident with the highest dose of avosentan, a predominantly ETA specific antagonist, during a large multidose phase 2a study in patients with type 2 diabetes and CKD.159 Moreover, a phase 3 randomized controlled trial involving patients with type 2 diabetes and advanced CKD, known as the ASCEND study, using high doses of avosentan, was prematurely terminated because of a three-fold increase in fluid overload and heart failure compared with that of placebo.160 The high doses of selective ETA antagonists may have induced fluid retention by blocking the endothelin B receptor, which is known to influcence sodium excretion.161 Nevertheless, avosentan significantly reduced the albumin to creatinine ratio in treatment groups compared with the control group in the ASCEND study.159 These results suggest that endothelin antagonists have a relatively narrow therapeutic window.
Anti-TNF-α therapy
Tumor necrosis factor-α (TNF-α) is a proinflammatory cytokine produced by many cells, including macrophages and intrinsic renal cells such as mesangial and renal tubular epithelial cells. Evidence suggests that TNF-α plays a role in glomerular inflammation and fibrosis. It stimulates the release of interleukin (IL)-1β, MCP-1, and TGF-β1, and blockade of TNF-α with a monoclonal antibody suppresses inflammation and renal fibrosis in experimental crescentic glomerulonephritis.162 Elevated TNF-α levels have been reported in patients with FSGS, and proteinuria is induced in animals exposed to TNF-α derived from mononuclear cells of patients with FSGS, while TNF-α antagonist reduces proteinuria.163 The potential for TNF-α antagonism as an anti-fibrotic therapy is under investigation in patients with primary FSGS and especially resistant FSGS in patients who fail to respond to conventional corticosteroid and immunosuppressive therapies. The Novel Therapies for Resistant Focal Segmental Glomerulosclerosis (FONT) project is a multicenter Phase 1/2 randomized clinical trial to investigate the efficacy of adalimumab, which is an anti-TNF-α monoclonal antibody, and galactose, which binds to FSGS permeability factor and inhibits its activity, and evaluated against conservative therapy consisting of the combination of lisinopril, losartan and atorvastatin.163 The FONT Phase 2 trial was recently completed, but the results have not yet been published (clinicaltrials.gov:NCT00814255).
Biomarkers of kidney fibrosis
The development of reliable biomarkers that can be assessed via non-invasive techniques in place of traditional kidney biopsy is becoming increasingly important for effective management of patients with CKD. Urinary biomarkers of kidney fibrosis are of particular interest given the ease of collection and the use of urine as a source serves as a direct conduit to the site of injury. Here, we highlight some of these biomarkers. Increases in urine TGF-β1 and CTGF levels have been reported in progressive renal diseases that may reflect development of renal fibrosis.164-166 TGF-β-inducible gene h3 (βig-h3) and plasminogen activator inhibitor-1 are both induced by TGF-β1 and can be detected in urine and used as markers of renal TGF-β1 activity.167 Urine excretion of type IV collagen correlates with glomerular ECM accumulation in animal studies and with declining renal function in patients with IgA nephropathy and diabetic nephropathy.168,169 Urinary mRNA levels of fibronectin, α-smooth muscle actin, and matrix metalloproteinase (MMP)-9 have been also proposed as biomarkers of diabetic kidney disease. Urine fibronectin levels are increased in patients with type 1 diabetes who have macroalbuminuria and in patients with type 2 diabetes who have microalbuminuria and macroalbuminuria.169,170 Urinary angiotensinogen can reflect intrarenal RAAS activity associated with deterioration of renal function in patients with CKD.171,172 Urinary vitamin D-binding protein or retinol-binding protein affected by tubular damage represents potential novel markers of renal interstitial fibrosis.173,174 Recent advances in urine proteomics with mass spectrometry are likely to lead to the identification of additional novel urinary biomarkers of kidney fibrosis.
Conclusions
Mechanism underlying virtually all CKD that ultimately progress to ESRD is the pathogenesis of renal fibrosis. Targeting the components of the fibrogenic pathways is under intense investigation as therapeutic approaches to inhibit or slow the progressive loss of kidney function in patients with CKD. TGF-β1 is a potent pro-fibrogenic cytokine and a central mediator of renal fibrosis. Increased TGF-β signaling is a critical contributor to the progression of fibrotic kidney diseases. Hence, targeting TGF-β1 and its signaling pathway as a therapeutic strategy for the treatment of CKD is well rationalized.
Various strategies that have been developed to reduce excessive TGF-β signaling activity in renal fibrosis are summarized in Tables 1 and 2. It should be noted that although excessive TGF-β1 activity leads to fibrotic conditions, therapies aimed at indiscriminate inhibition of TGF-β1 may not be most ideal and should be approached with caution, given that TGF-β1 also has potent anti-inflammatory and immunoregulatory effects and induces autophagy as cytoprotective mechanisms. There has been exciting progress in exploring additional anti-fibrotic therapies including targeting BMP-7, CTGF, CC motif chemokines, PDEs, Nox1/4, ET-1, and TNF-α. It is hoped that evidence based on the preclinical and clinical data discussed in this review leads to novel anti-fibrotic therapies effective in patients with CKD. Because many of the clinical trials in patients with CKD that have been completed to date have been small and/or uncontrolled, conclusive evidence regarding efficacy of many of the anti-fibrotic agents is limited. Larger studies are needed to better understand long-term efficacy and safety of these agents. Although a significant body of evidence in preclinical studies aimed at TGF-β1 inhibition showed great promise of anti-fibrotic effects in experimental models, limited advances have been made to date in translation to treatment in patients with CKD, perhaps in part due to limitations of many of the animal models of renal fibrosis that poorly mimic human diseases. There is still a great need for future investigations to gain further insights into the complex TGF-β signaling and mechanisms of renal fibrosis, with the ultimate goal of developing novel and more effective therapies for progressive kidney diseases.
Acknowledgments
Supported in part by the National Institutes of Health grants R01-DK57661, P01-HL114501, and R01 HL079904 to M.E.C.
Footnotes
All authors have read the journal's policy on disclosure of potential conflicts of interest.
Conflict of interest statement: none.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol. 2011;7:684–96. doi: 10.1038/nrneph.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest. 2007;117:524–9. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levey AS, Atkins R, Coresh J, et al. Chronic kidney disease as a global public health problem: approaches and initiatives - a position statement from Kidney Disease Improving Global Outcomes. Kidney Int. 2007;72:247–59. doi: 10.1038/sj.ki.5002343. [DOI] [PubMed] [Google Scholar]
- 4.Lewis EJ, Hunsicker LG, Clarke WR, et al. Renoprotective effect of the angiotensinreceptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001;345:851–60. doi: 10.1056/NEJMoa011303. [DOI] [PubMed] [Google Scholar]
- 5.Brenner BM, Cooper ME, de Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861–9. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 6.Ruggenenti P, Perna A, Gherardi G, et al. Renoprotective properties of ACE-inhibition in non-diabetic nephropathies with non-nephrotic proteinuria. Lancet. 1999;354:359–64. doi: 10.1016/S0140-6736(98)10363-X. [DOI] [PubMed] [Google Scholar]
- 7.Klahr S, Morrissey J. Obstructive nephropathy and renal fibrosis. Am J Physiol Renal Physiol. 2002;283:F861–75. doi: 10.1152/ajprenal.00362.2001. [DOI] [PubMed] [Google Scholar]
- 8.Border WA, Noble NA. Transforming growth factor β in tissue fibrosis. N Engl J Med. 1994;331:1286–92. doi: 10.1056/NEJM199411103311907. [DOI] [PubMed] [Google Scholar]
- 9.Choi ME, Ding Y, Kim SI. TGF-β signaling via TAK1 pathway: role in kidney fibrosis. Semin Nephrol. 2012;32:244–52. doi: 10.1016/j.semnephrol.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Torres IB, Moreso F, Sarro E, Meseguer A, Seron D. The Interplay between Inflammation and Fibrosis in Kidney Transplantation. Biomed Res Int. 2014;2014:750602. doi: 10.1155/2014/750602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grenda R. Steroid withdrawal in renal transplantation. Pediatr Nephrol. 2013;28:2107–12. doi: 10.1007/s00467-012-2391-6. [DOI] [PubMed] [Google Scholar]
- 12.Blobe GC, Schiemann WP, Lodish HF. Role of Transforming Growth Factor β in Human Disease. N Engl J Med. 2000;342:1350–8. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 13.Massagué J. How cells read TGF-β signals. Nature Rev. 2000;1:169–78. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 14.Massagué J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000;19:1745–54. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lan HY. Smad7 as a therapeutic agent for chronic kidney diseases. Front Biosci. 2008;13:4984–92. doi: 10.2741/3057. [DOI] [PubMed] [Google Scholar]
- 16.Moustakas A, Heldin CH. Non-Smad TGF-beta signals. J Cell Sci. 2005;118:3573–84. doi: 10.1242/jcs.02554. [DOI] [PubMed] [Google Scholar]
- 17.Singhirunnusorn P, Suzuki S, Kawasaki N, Saiki I, Sakurai H. Critical roles of threonine 187 phosphorylation in cellular stress-induced rapid and transient activation of transforming growth factor-beta-activated kinase 1 (TAK1) in a signaling complex containing TAK1-binding protein TAB1 and TAB2. J Biol Chem. 2005;280:7359–68. doi: 10.1074/jbc.M407537200. [DOI] [PubMed] [Google Scholar]
- 18.Kriz W, Kaissling B, Le Hir M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: fact or fantasy? J Clin Invest. 2011;121:468–74. doi: 10.1172/JCI44595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Böttinger EP, Bitzer M. TGF-beta signaling in renal disease. J Am Soc Nephrol. 2002;13:2600–10. doi: 10.1097/01.asn.0000033611.79556.ae. [DOI] [PubMed] [Google Scholar]
- 20.Wang W, Koka V, Lan HY. Transforming growth factor-beta and Smad signalling in kidney diseases. Nephrology (Carlton) 2005;10:48–56. doi: 10.1111/j.1440-1797.2005.00334.x. [DOI] [PubMed] [Google Scholar]
- 21.Sanderson N, Factor V, Nagy P, et al. Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc Natl Acad Sci U S A. 1995;92:2572–6. doi: 10.1073/pnas.92.7.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kopp JB, Factor VM, Mozes M, et al. Transgenic mice with increased plasma levels of TGF-beta 1 develop progressive renal disease. Lab Invest. 1996;74:991–1003. [PubMed] [Google Scholar]
- 23.Border WA, Noble NA. Evidence that TGF-beta should be a therapeutic target in diabetic nephropathy. Kidney Int. 1998;54:1390–1. doi: 10.1046/j.1523-1755.1998.00127.x. [DOI] [PubMed] [Google Scholar]
- 24.Kim SI, Kwak JH, Zachariah M, He Y, Wang L, Choi ME. TGF-beta-activated kinase 1 and TAK1-binding protein 1 cooperate to mediate TGF-beta1-induced MKK3-p38 MAPK activation and stimulation of type I collagen. Am J Physiol Renal Physiol. 2007;292:F1471–8. doi: 10.1152/ajprenal.00485.2006. [DOI] [PubMed] [Google Scholar]
- 25.Hocevar BA, Prunier C, Howe PH. Disabled-2 (Dab2) mediates transforming growth factor (TGF)-stimulated fibronectin synthesis through TGF-activated kinase 1 and activation of the JNK pathway. J Biol Chem. 2005;280:25920–7. doi: 10.1074/jbc.M501150200. [DOI] [PubMed] [Google Scholar]
- 26.Sharma K, McGowan TA. TGF-beta in diabetic kidney disease: role of novel signaling pathways. Cytokine Growth Factor Rev. 2000;11:115–23. doi: 10.1016/s1359-6101(99)00035-0. [DOI] [PubMed] [Google Scholar]
- 27.Lee HS, Song CY. Effects of TGF-beta on podocyte growth and disease progression in proliferative podocytopathies. Kidney Blood Press Res. 2010;33:24–9. doi: 10.1159/000285844. [DOI] [PubMed] [Google Scholar]
- 28.Schiffer M, Bitzer M, Roberts IS, et al. Apoptosis in podocytes induced by TGF-beta and Smad7. J Clin Invest. 2001;108:807–16. doi: 10.1172/JCI12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tossidou I, Starker G, Kruger J, et al. PKC-alpha modulates TGF-beta signaling and impairs podocyte survival. Cell Physiol Biochem. 2009;24:627–34. doi: 10.1159/000257518. [DOI] [PubMed] [Google Scholar]
- 30.Sharma K, Jin Y, Guo J, Ziyadeh FN. Neutralization of TGF-beta by anti-TGF-beta antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes. 1996;45:522–30. doi: 10.2337/diab.45.4.522. [DOI] [PubMed] [Google Scholar]
- 31.Ziyadeh FN, Hoffman BB, Han DC, et al. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci U S A. 2000;97:8015–20. doi: 10.1073/pnas.120055097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen S, Iglesias-de la Cruz MC, Jim B, Hong SW, Isono M, Ziyadeh FN. Reversibility of established diabetic glomerulopathy by anti-TGF-beta antibodies in db/db mice. Biochem Biophys Res Commun. 2003;300:16–22. doi: 10.1016/s0006-291x(02)02708-0. [DOI] [PubMed] [Google Scholar]
- 33.Dahly AJ, Hoagland KM, Flasch AK, Jha S, Ledbetter SR, Roman RJ. Antihypertensive effects of chronic anti-TGF-beta antibody therapy in Dahl S rats. Am J Physiol Regul Integr Comp Physiol. 2002;283:R757–67. doi: 10.1152/ajpregu.00098.2002. [DOI] [PubMed] [Google Scholar]
- 34.Murphy SR, Dahly-Vernon AJ, Dunn KM, et al. Renoprotective effects of anti-TGF-β antibody and antihypertensive therapies in Dahl S rats. Am J Physiol Regul Integr Comp Physiol. 2012;303:R57–69. doi: 10.1152/ajpregu.00263.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trachtman H, Fervenza FC, Gipson DS, et al. A phase 1, single-dose study of fresolimumab, an anti-TGF-beta antibody, in treatment-resistant primary focal segmental glomerulosclerosis. Kidney Int. 2011;79:1236–43. doi: 10.1038/ki.2011.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benigni A, Zoja C, Corna D, et al. Add-on anti-TGF-beta antibody to ACE inhibitor arrests progressive diabetic nephropathy in the rat. J Am Soc Nephrol. 2003;14:1816–24. doi: 10.1097/01.asn.0000074238.61967.b7. [DOI] [PubMed] [Google Scholar]
- 37.Schaefer CJ, Ruhrmund DW, Pan L, Seiwert SD, Kossen K. Antifibrotic activities of pirfenidone in animal models. Eur Respir Rev. 2011;20:85–97. doi: 10.1183/09059180.00001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimizu T, Kuroda T, Hata S, Fukagawa M, Margolin SB, Kurokawa K. Pirfenidone improves renal function and fibrosis in the post-obstructed kidney. Kidney Int. 1998;54:99–109. doi: 10.1046/j.1523-1755.1998.00962.x. [DOI] [PubMed] [Google Scholar]
- 39.Chen JF, Ni HF, Pan MM, et al. Pirfenidone inhibits macrophage infiltration in 5/6 nephrectomized rats. Am J Physiol Renal Physiol. 2013;304:F676–85. doi: 10.1152/ajprenal.00507.2012. [DOI] [PubMed] [Google Scholar]
- 40.Takakura K, Fujimori A, Chikanishi T, et al. Renoprotective properties of pirfenidone in subtotally nephrectomized rats. Eur J Pharmacol. 2009;629:118–24. doi: 10.1016/j.ejphar.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 41.Shimizu T, Fukagawa M, Kuroda T, et al. Pirfenidone prevents collagen accumulation in the remnant kidney in rats with partial nephrectomy. Kidney Int Suppl. 1997;63:S239–43. [PubMed] [Google Scholar]
- 42.RamachandraRao SP, Zhu Y, Ravasi T, et al. Pirfenidone is renoprotective in diabetic kidney disease. J Am Soc Nephrol. 2009;20:1765–75. doi: 10.1681/ASN.2008090931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iyer SN, Wild JS, Schiedt MJ, Hyde DM, Margolin SB, Giri SN. Dietary intake of pirfenidone ameliorates bleomycin-induced lung fibrosis in hamsters. J Lab Clin Med. 1995;125:779–85. [PubMed] [Google Scholar]
- 44.Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377:1760–9. doi: 10.1016/S0140-6736(11)60405-4. [DOI] [PubMed] [Google Scholar]
- 45.Walker JE, Giri SN, Margolin SB. A double-blind, randomized, controlled study of oral pirfenidone for treatment of secondary progressive multiple sclerosis. Mult Scler. 2005;11:149–58. doi: 10.1191/1352458505ms1134oa. [DOI] [PubMed] [Google Scholar]
- 46.Armendáriz-Borunda J, Islas-Carbajal MC, Meza-Garcia E, et al. A pilot study in patients with established advanced liver fibrosis using pirfenidone. Gut. 2006;55:1663–5. doi: 10.1136/gut.2006.107136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kreuter M. Pirfenidone: an update on clinical trial data and insights from everyday practice. Eur Respir Rev. 2014;23:111–7. doi: 10.1183/09059180.00008513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Azuma A, Taguchi Y, Ogura T, et al. Exploratory analysis of a phase III trial of pirfenidone identifies a subpopulation of patients with idiopathic pulmonary fibrosis as benefiting from treatment. Respir Res. 2011;12:143. doi: 10.1186/1465-9921-12-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.King TE, Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–92. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 50.Cho ME, Smith DC, Branton MH, Penzak SR, Kopp JB. Pirfenidone slows renal function decline in patients with focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2007;2:906–13. doi: 10.2215/CJN.01050207. [DOI] [PubMed] [Google Scholar]
- 51.Sharma K, Ix JH, Mathew AV, et al. Pirfenidone for diabetic nephropathy. J Am Soc Nephrol. 2011;22:1144–51. doi: 10.1681/ASN.2010101049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Williams SJ, Zammit SC, Cox AJ, et al. 3′,4′-Bis-difluoromethoxycinnamoylanthranilate (FT061): an orally-active antifibrotic agent that reduces albuminuria in a rat model of progressive diabetic nephropathy. Bioorg Med Chem Lett. 2013;23:6868–73. doi: 10.1016/j.bmcl.2013.09.100. [DOI] [PubMed] [Google Scholar]
- 53.Gilbert RE, Zhang Y, Williams SJ, et al. A purpose-synthesised anti-fibrotic agent attenuates experimental kidney diseases in the rat. PLoS One. 2012;7(10):e47160. doi: 10.1371/journal.pone.0047160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wei J, Zhu H, Komura K, et al. A synthetic PPAR-gamma agonist triterpenoid ameliorates experimental fibrosis: PPAR-gamma-independent suppression of fibrotic responses. Ann Rheum Dis. 2014;73:446–54. doi: 10.1136/annrheumdis-2012-202716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bolignano D, Zoccali C. Glitazones in chronic kidney disease: potential and concerns. Nutr Metab Cardiovasc Dis. 2012;22:167–75. doi: 10.1016/j.numecd.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 56.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–71. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 57.Higgins LS, Depaoli AM. Selective peroxisome proliferator-activated receptor gamma (PPARgamma) modulation as a strategy for safer therapeutic PPARgamma activation. Am J Clin Nutr. 2010;91:267S–72S. doi: 10.3945/ajcn.2009.28449E. [DOI] [PubMed] [Google Scholar]
- 58.Lan HY, Mu W, Tomita N, et al. Inhibition of renal fibrosis by gene transfer of inducible Smad7 using ultrasound-microbubble system in rat UUO model. J Am Soc Nephrol. 2003;14:1535–48. doi: 10.1097/01.asn.0000067632.04658.b8. [DOI] [PubMed] [Google Scholar]
- 59.Ka SM, Huang XR, Lan HY, et al. Smad7 gene therapy ameliorates an autoimmune crescentic glomerulonephritis in mice. J Am Soc Nephrol. 2007;18:1777–88. doi: 10.1681/ASN.2006080901. [DOI] [PubMed] [Google Scholar]
- 60.Hou CC, Wang W, Huang XR, et al. Ultrasound-microbubble-mediated gene transfer of inducible Smad7 blocks transforming growth factor-beta signaling and fibrosis in rat remnant kidney. Am J Pathol. 2005;166:761–71. doi: 10.1016/s0002-9440(10)62297-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim SI, Lee SY, Wang Z, et al. TGF-β-Activated Kinase 1 Is Crucial in Podocyte Differentiation and Glomerular Capillary Formation. J Am Soc Nephrol. 2014 doi: 10.1681/ASN.2013030252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Melisi D, Xia Q, Paradiso G, et al. Modulation of pancreatic cancer chemoresistance by inhibition of TAK1. J Natl Cancer Inst. 2011;103:1190–204. doi: 10.1093/jnci/djr243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Petersen M, Thorikay M, Deckers M, et al. Oral administration of GW788388, an inhibitor of TGF-beta type I and II receptor kinases, decreases renal fibrosis. Kidney Int. 2008;73:705–15. doi: 10.1038/sj.ki.5002717. [DOI] [PubMed] [Google Scholar]
- 64.Kushibiki T, Nagata-Nakajima N, Sugai M, Shimizu A, Tabata Y. Delivery of plasmid DNA expressing small interference RNA for TGF-β type II receptor by cationized gelatin to prevent interstitial renal fibrosis. J Control Release. 2005;105:318–31. doi: 10.1016/j.jconrel.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 65.Wolf G. Renal injury due to renin-angiotensin-aldosterone system activation of the transforming growth factor-beta pathway. Kidney Int. 2006;70:1914–9. doi: 10.1038/sj.ki.5001846. [DOI] [PubMed] [Google Scholar]
- 66.Rüster C, Wolf G. Renin-angiotensin-aldosterone system and progression of renal disease. J Am Soc Nephrol. 2006;17:2985–91. doi: 10.1681/ASN.2006040356. [DOI] [PubMed] [Google Scholar]
- 67.Shull MM, Ormsby I, Kier AB, et al. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–39. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Majesky MW, Volkhard L, Twardzik DR, Schwartz SM, Reidy MA. Production of transforming growth factor-β1 during repair of arterial injury. J Clin Invest. 1991;88:904–10. doi: 10.1172/JCI115393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:1845–6. doi: 10.1056/NEJMc1303158. [DOI] [PubMed] [Google Scholar]
- 70.Wang Z, Choi ME. Autophagy in kidney health and disease. Antioxid Redox Signal. 2014;20:519–37. doi: 10.1089/ars.2013.5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ding Y, Choi ME. Regulation of autophagy by TGF-β: emerging role in kidney fibrosis. Semin Nephrol. 2014;34:62–71. doi: 10.1016/j.semnephrol.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ding Y, Kim JK, Kim SI, et al. TGF-beta1 protects against mesangial cell apoptosis via induction of autophagy. J Biol Chem. 2010;285:37909–19. doi: 10.1074/jbc.M109.093724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Criollo A, Niso-Santano M, Malik SA, et al. Inhibition of autophagy by TAB2 and TAB3. EMBO J. 2011;30:4908–20. doi: 10.1038/emboj.2011.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Takaesu G, Kobayashi T, Yoshimura A. TGFβ-activated kinase 1 (TAK1)-binding proteins (TAB) 2 and 3 negatively regulate autophagy. J Biochem. 2012;151:157–66. doi: 10.1093/jb/mvr123. [DOI] [PubMed] [Google Scholar]
- 75.Kim SI, Na HJ, Ding Y, Wang Z, Lee SJ, Choi ME. Autophagy promotes intracellular degradation of type I collagen induced by Transforming Growth Factor (TGF)-β1. J Biol Chem. 2012;287:11677–88. doi: 10.1074/jbc.M111.308460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ding Y, Kim SI, Lee SY, Koo JK, Wang Z, Choi ME. Autophagy regulates TGF-β expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J Am Soc Nephrol. 2014 doi: 10.1681/ASN.2013101068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Del Principe D, Lista P, Malorni W, Giammarioli AM. Fibroblast autophagy in fibrotic disorders. J Pathol. 2013;229:208–20. doi: 10.1002/path.4115. [DOI] [PubMed] [Google Scholar]
- 78.Dudley AT, Lyons KM, Robertson EJ. A requirement for bone morphogenetic protein-7 during development of the mammalian kidney and eye. Genes Dev. 1995;9:2795–807. doi: 10.1101/gad.9.22.2795. [DOI] [PubMed] [Google Scholar]
- 79.Wang S, Hirschberg R. Bone morphogenetic protein-7 signals opposing transforming growth factor beta in mesangial cells. J Biol Chem. 2004;279:23200–6. doi: 10.1074/jbc.M311998200. [DOI] [PubMed] [Google Scholar]
- 80.Wang SN, Lapage J, Hirschberg R. Loss of tubular bone morphogenetic protein-7 in diabetic nephropathy. J Am Soc Nephrol. 2001;12:2392–9. doi: 10.1681/ASN.V12112392. [DOI] [PubMed] [Google Scholar]
- 81.Vukicevic S, Basic V, Rogic D, et al. Osteogenic protein-1 (bone morphogenetic protein-7) reduces severity of injury after ischemic acute renal failure in rat. J Clin Invest. 1998;102:202–14. doi: 10.1172/JCI2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Simon M, Maresh JG, Harris SE, et al. Expression of bone morphogenetic protein-7 mRNA in normal and ischemic adult rat kidney. Am J Physiol. 1999;276:F382–9. doi: 10.1152/ajprenal.1999.276.3.F382. [DOI] [PubMed] [Google Scholar]
- 83.De Petris L, Hruska KA, Chiechio S, Liapis H. Bone morphogenetic protein-7 delays podocyte injury due to high glucose. Nephrol Dial Transplant. 2007;22:3442–50. doi: 10.1093/ndt/gfm503. [DOI] [PubMed] [Google Scholar]
- 84.Bramlage CP, Tampe B, Koziolek M, et al. Bone morphogenetic protein (BMP)-7 expression is decreased in human hypertensive nephrosclerosis. BMC Nephrol. 2010;11:31. doi: 10.1186/1471-2369-11-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zeisberg M, Hanai J, Sugimoto H, et al. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9:964–8. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- 86.Hruska KA, Guo G, Wozniak M, et al. Osteogenic protein-1 prevents renal fibrogenesis associated with ureteral obstruction. Am J Physiol Renal Physiol. 2000;279:F130–43. doi: 10.1152/ajprenal.2000.279.1.F130. [DOI] [PubMed] [Google Scholar]
- 87.Morrissey J, Hruska K, Guo G, Wang S, Chen Q, Klahr S. Bone morphogenetic protein-7 improves renal fibrosis and accelerates the return of renal function. J Am Soc Nephrol. 2002;13(Suppl 1):S14–S21. [PubMed] [Google Scholar]
- 88.Wang S, Chen Q, Simon TC, et al. Bone morphogenic protein-7 (BMP-7), a novel therapy for diabetic nephropathy. Kidney Int. 2003;63:2037–49. doi: 10.1046/j.1523-1755.2003.00035.x. [DOI] [PubMed] [Google Scholar]
- 89.Sugimoto H, Grahovac G, Zeisberg M, Kalluri R. Renal fibrosis and glomerulosclerosis in a new mouse model of diabetic nephropathy and its regression by bone morphogenic protein-7 and advanced glycation end product inhibitors. Diabetes. 2007;56:1825–33. doi: 10.2337/db06-1226. [DOI] [PubMed] [Google Scholar]
- 90.Zeisberg M, Bottiglio C, Kumar N, et al. Bone morphogenic protein-7 inhibits progression of chronic renal fibrosis associated with two genetic mouse models. Am J Physiol Renal Physiol. 2003;285:F1060–7. doi: 10.1152/ajprenal.00191.2002. [DOI] [PubMed] [Google Scholar]
- 91.Wang S, de Caestecker M, Kopp J, Mitu G, Lapage J, Hirschberg R. Renal bone morphogenetic protein-7 protects against diabetic nephropathy. J Am Soc Nephrol. 2006;17:2504–12. doi: 10.1681/ASN.2006030278. [DOI] [PubMed] [Google Scholar]
- 92.Tampe B, Tampe D, Müller CA, et al. Tet3-mediated hydroxymethylation of epigenetically silenced genes contributes to bone morphogenic protein 7-induced reversal of kidney fibrosis. J Am Soc Nephrol. 2014;25:905–12. doi: 10.1681/ASN.2013070723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Manson SR, Song JB, Hruska KA, Austin PF. HDAC dependent transcriptional repression of Bmp-7 potentiates TGF-β mediated renal fibrosis in obstructive uropathy. J Urol. 2013;191:242–52. doi: 10.1016/j.juro.2013.06.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bosukonda D, Shih MS, Sampath KT, Vukicevic S. Characterization of receptors for osteogenic protein-1/bone morphogenetic protein-7 (OP-1/BMP-7) in rat kidneys. Kidney Int. 2000;58:1902–11. doi: 10.1111/j.1523-1755.2000.00362.x. [DOI] [PubMed] [Google Scholar]
- 95.Sugimoto H, LeBleu VS, Bosukonda D, et al. Activin-like kinase 3 is important for kidney regeneration and reversal of fibrosis. Nat Med. 2012;18:396–404. doi: 10.1038/nm.2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Motazed R, Colville-Nash P, Kwan JT, Dockrell ME. BMP-7 and proximal tubule epithelial cells: activation of multiple signaling pathways reveals a novel anti-fibrotic mechanism. Pharm Res. 2008;25:2440–6. doi: 10.1007/s11095-008-9551-1. [DOI] [PubMed] [Google Scholar]
- 97.Lin J, Patel SR, Cheng X, et al. Kielin/chordin-like protein, a novel enhancer of BMP signaling, attenuates renal fibrotic disease. Nat Med. 2005;11:387–93. doi: 10.1038/nm1217. [DOI] [PubMed] [Google Scholar]
- 98.Yanagita M, Okuda T, Endo S, et al. Uterine sensitization-associated gene-1 (USAG-1), a novel BMP antagonist expressed in the kidney, accelerates tubular injury. J Clin Invest. 2006;116:70–9. doi: 10.1172/JCI25445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tanaka M, Asada M, Higashi AY, et al. Loss of the BMP antagonist USAG-1 ameliorates disease in a mouse model of the progressive hereditary kidney disease Alport syndrome. J Clin Invest. 2010;120:768–77. doi: 10.1172/JCI39569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Roxburgh SA, Kattla JJ, Curran SP, et al. Allelic depletion of grem1 attenuates diabetic kidney disease. Diabetes. 2009;58:1641–50. doi: 10.2337/db08-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang Q, Shi Y, Wada J, et al. In vivo delivery of Gremlin siRNA plasmid reveals therapeutic potential against diabetic nephropathy by recovering bone morphogenetic protein-7. PLoS One. 2010;5(7):e11709. doi: 10.1371/journal.pone.0011709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Phanish MK, Winn SK, Dockrell ME. Connective tissue growth factor-(CTGF, CCN2) - a marker, mediator and therapeutic target for renal fibrosis. Nephron Exp Nephrol. 2010;114:e83–92. doi: 10.1159/000262316. [DOI] [PubMed] [Google Scholar]
- 103.Perbal B. CCN proteins: multifunctional signalling regulators. Lancet. 2004;363:62–4. doi: 10.1016/S0140-6736(03)15172-0. [DOI] [PubMed] [Google Scholar]
- 104.Wang S, Denichilo M, Brubaker C, Hirschberg R. Connective tissue growth factor in tubulointerstitial injury of diabetic nephropathy. Kidney Int. 2001;60:96–105. doi: 10.1046/j.1523-1755.2001.00776.x. [DOI] [PubMed] [Google Scholar]
- 105.Ito Y, Aten J, Bende RJ, et al. Expression of connective tissue growth factor in human renal fibrosis. Kidney Int. 1998;53:853–61. doi: 10.1111/j.1523-1755.1998.00820.x. [DOI] [PubMed] [Google Scholar]
- 106.Kanemoto K, Usui J, Nitta K, et al. In situ expression of connective tissue growth factor in human crescentic glomerulonephritis. Virchows Arch. 2004;444:257–63. doi: 10.1007/s00428-003-0959-z. [DOI] [PubMed] [Google Scholar]
- 107.Abreu JG, Ketpura NI, Reversade B, De Robertis EM. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta. Nat Cell Biol. 2002;4:599–604. doi: 10.1038/ncb826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wahab NA, Weston BS, Mason RM. Connective Tissue Growth Factor CCN2 Interacts with and Activates the Tyrosine Kinase Receptor TrkA. J Am Soc Nephrol. 2005;16:340–51. doi: 10.1681/ASN.2003100905. [DOI] [PubMed] [Google Scholar]
- 109.Nguyen TQ, Roestenberg P, van Nieuwenhoven FA, et al. CTGF inhibits BMP-7 signaling in diabetic nephropathy. J Am Soc Nephrol. 2008;19:2098–107. doi: 10.1681/ASN.2007111261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yokoi H, Mukoyama M, Nagae T, et al. Reduction in connective tissue growth factor by antisense treatment ameliorates renal tubulointerstitial fibrosis. J Am Soc Nephrol. 2004;15:1430–40. doi: 10.1097/01.asn.0000130565.69170.85. [DOI] [PubMed] [Google Scholar]
- 111.Okada H, Kikuta T, Kobayashi T, et al. Connective tissue growth factor expressed in tubular epithelium plays a pivotal role in renal fibrogenesis. J Am Soc Nephrol. 2005;16:133–43. doi: 10.1681/ASN.2004040339. [DOI] [PubMed] [Google Scholar]
- 112.Luo GH, Lu YP, Song J, Yang L, Shi YJ, Li YP. Inhibition of connective tissue growth factor by small interfering RNA prevents renal fibrosis in rats undergoing chronic allograft nephropathy. Transplant Proc. 2008;40:2365–9. doi: 10.1016/j.transproceed.2008.07.100. [DOI] [PubMed] [Google Scholar]
- 113.Guha M, Xu ZG, Tung D, Lanting L, Natarajan R. Specific down-regulation of connective tissue growth factor attenuates progression of nephropathy in mouse models of type 1 and type 2 diabetes. FASEB J. 2007;21:3355–68. doi: 10.1096/fj.06-6713com. [DOI] [PubMed] [Google Scholar]
- 114.Falke LL, Dendooven A, Leeuwis JW, et al. Hemizygous deletion of CTGF/CCN2 does not suffice to prevent fibrosis of the severely injured kidney. Matrix Biol. 2012;31:421–31. doi: 10.1016/j.matbio.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 115.Adler SG, Schwartz S, Williams ME, et al. Phase 1 study of anti-CTGF monoclonal antibody in patients with diabetes and microalbuminuria. Clin J Am Soc Nephrol. 2010;5:1420–8. doi: 10.2215/CJN.09321209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Falke LL, Goldschmeding R, Nguyen TQ. A perspective on anti-CCN2 therapy for chronic kidney disease. Nephrol Dial Transplant. 2014;29(Suppl 1):i30–7. doi: 10.1093/ndt/gft430. [DOI] [PubMed] [Google Scholar]
- 117.Wang SN, LaPage J, Hirschberg R. Role of glomerular ultrafiltration of growth factors in progressive interstitial fibrosis in diabetic nephropathy. Kidney Int. 2000;57:1002–14. doi: 10.1046/j.1523-1755.2000.00928.x. [DOI] [PubMed] [Google Scholar]
- 118.Giunti S, Tesch GH, Pinach S, et al. Monocyte chemoattractant protein-1 has prosclerotic effects both in vivo in experimental diabetes and in vitro in human mesangial cells. Diabetologia. 2008;51:198–207. doi: 10.1007/s00125-007-0837-3. [DOI] [PubMed] [Google Scholar]
- 119.Cheng J, Diaz Encarnacion MM, Warner GM, Gray CE, Nath KA, Grande JP. TGF-beta1 stimulates monocyte chemoattractant protein-1 expression in mesangial cells through a phosphodiesterase isoenzyme 4-dependent process. Am J Physiol Cell Physiol. 2005;289:C959–70. doi: 10.1152/ajpcell.00153.2005. [DOI] [PubMed] [Google Scholar]
- 120.Chow FY, Nikolic-Paterson DJ, Ozols E, Atkins RC, Rollin BJ, Tesch GH. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. 2006;69:73–80. doi: 10.1038/sj.ki.5000014. [DOI] [PubMed] [Google Scholar]
- 121.Sassy-Prigent C, Heudes D, Mandet C, et al. Early glomerular macrophage recruitment in streptozotocin-induced diabetic rats. Diabetes. 2000;49:466–75. doi: 10.2337/diabetes.49.3.466. [DOI] [PubMed] [Google Scholar]
- 122.Chow F, Ozols E, Nikolic-Paterson DJ, Atkins RC, Tesch GH. Macrophages in mouse type 2 diabetic nephropathy: correlation with diabetic state and progressive renal injury. Kidney Int. 2004;65:116–28. doi: 10.1111/j.1523-1755.2004.00367.x. [DOI] [PubMed] [Google Scholar]
- 123.Chow FY, Nikolic-Paterson DJ, Ma FY, Ozols E, Rollins BJ, Tesch GH. Monocyte chemoattractant protein-1-induced tissue inflammation is critical for the development of renal injury but not type 2 diabetes in obese db/db mice. Diabetologia. 2007;50:471–80. doi: 10.1007/s00125-006-0497-8. [DOI] [PubMed] [Google Scholar]
- 124.Kanamori H, Matsubara T, Mima A, et al. Inhibition of MCP-1/CCR2 pathway ameliorates the development of diabetic nephropathy. Biochem Biophys Res Commun. 2007;360:772–7. doi: 10.1016/j.bbrc.2007.06.148. [DOI] [PubMed] [Google Scholar]
- 125.Kang YS, Lee MH, Song HK, et al. CCR2 antagonism improves insulin resistance, lipid metabolism, and diabetic nephropathy in type 2 diabetic mice. Kidney Int. 2010;78:883–94. doi: 10.1038/ki.2010.263. [DOI] [PubMed] [Google Scholar]
- 126.Sullivan T, Miao Z, Dairaghi DJ, et al. CCR2 antagonist CCX140-B provides renal and glycemic benefits in diabetic transgenic human CCR2 knockin mice. Am J Physiol Renal Physiol. 2013;305:F1288–97. doi: 10.1152/ajprenal.00316.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kitagawa K, Wada T, Furuichi K, et al. Blockade of CCR2 ameliorates progressive fibrosis in kidney. Am J Pathol. 2004;165:237–46. doi: 10.1016/S0002-9440(10)63292-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Xia Y, Entman ML, Wang Y. CCR2 regulates the uptake of bone marrow-derived fibroblasts in renal fibrosis. PLoS One. 2013;8(10):e77493. doi: 10.1371/journal.pone.0077493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tam FW, Riser BL, Meeran K, Rambow J, Pusey CD, Frankel AH. Urinary monocyte chemoattractant protein-1 (MCP-1) and connective tissue growth factor (CCN2) as prognostic markers for progression of diabetic nephropathy. Cytokine. 2009;47:37–42. doi: 10.1016/j.cyto.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 130.Titan SM, Vieira JM, Jr, Dominguez WV, et al. Urinary MCP-1 and RBP: independent predictors of renal outcome in macroalbuminuric diabetic nephropathy. J Diabetes Complications. 2012;26:546–53. doi: 10.1016/j.jdiacomp.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 131.Hanefeld M, Schell E, Gouni-Berthold I, et al. Orally-administered chemokine receptor CCR2 antagonist CCX140-B in type 2 diabetes: a pilot double-blind, randomized clinical trial. J Diabetes Metab. 2012;3:225. [Google Scholar]
- 132.Vielhauer V, Berning E, Eis V, et al. CCR1 blockade reduces interstitial inflammation and fibrosis in mice with glomerulosclerosis and nephrotic syndrome. Kidney Int. 2004;66:2264–78. doi: 10.1111/j.1523-1755.2004.66038.x. [DOI] [PubMed] [Google Scholar]
- 133.Anders HJ, Belemezova E, Eis V, et al. Late onset of treatment with a chemokine receptor CCR1 antagonist prevents progression of lupus nephritis in MRL-Fas(lpr) mice. J Am Soc Nephrol. 2004;15:1504–13. doi: 10.1097/01.asn.0000130082.67775.60. [DOI] [PubMed] [Google Scholar]
- 134.Anders HJ, Vielhauer V, Frink M, et al. A chemokine receptor CCR-1 antagonist reduces renal fibrosis after unilateral ureter ligation. J Clin Invest. 2002;109:251–9. doi: 10.1172/JCI14040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lin SL, Chen RH, Chen YM, et al. Pentoxifylline Attenuates Tubulointerstitial Fibrosis by Blocking Smad3/4-Activated Transcription and Profibrogenic Effects of Connective Tissue Growth Factor. J Am Soc Nephrol. 2005;16:2702–13. doi: 10.1681/ASN.2005040435. [DOI] [PubMed] [Google Scholar]
- 136.Lin SL, Chen YM, Chien CT, Chiang WC, Tsai CC, Tsai TJ. Pentoxifylline attenuated the renal disease progression in rats with remnant kidney. J Am Soc Nephrol. 2002;13:2916–29. doi: 10.1097/01.asn.0000034909.10994.8a. [DOI] [PubMed] [Google Scholar]
- 137.Ng YY, Chen YM, Tsai TJ, Lan XR, Yang WC, Lan HY. Pentoxifylline inhibits transforming growth factor-beta signaling and renal fibrosis in experimental crescentic glomerulonephritis in rats. Am J Nephrol. 2009;29:43–53. doi: 10.1159/000150600. [DOI] [PubMed] [Google Scholar]
- 138.Perkins RM, Aboudara MC, Uy AL, Olson SW, Cushner HM, Yuan CM. Effect of pentoxifylline on GFR decline in CKD: a pilot, double-blind, randomized, placebo-controlled trial. Am J Kidney Dis. 2009;53:606–16. doi: 10.1053/j.ajkd.2008.11.026. [DOI] [PubMed] [Google Scholar]
- 139.Ducloux D, Bresson-Vautrin C, Chalopin J. Use of pentoxifylline in membranous nephropathy. Lancet. 2001;357:1672–3. doi: 10.1016/s0140-6736(00)04830-3. [DOI] [PubMed] [Google Scholar]
- 140.Rodriguez-Morán M, González-González G, Bermúdez-Barba MV, et al. Effects of pentoxifylline on the urinary protein excretion profile of type 2 diabetic patients with microproteinuria: a double-blind, placebo-controlled randomized trial. Clin Nephrol. 2006;66:3–10. doi: 10.5414/cnp66003. [DOI] [PubMed] [Google Scholar]
- 141.Lau DH, Mikhailidis DP, Thompson CS. The effect of vardenafil (a PDE type 5 inhibitor) on renal function in the diabetic rabbit: a pilot study. In Vivo. 2007;21:851–4. [PubMed] [Google Scholar]
- 142.Jeong KH, Lee TW, Ihm CG, Lee SH, Moon JY, Lim SJ. Effects of sildenafil on oxidative and inflammatory injuries of the kidney in streptozotocin-induced diabetic rats. Am J Nephrol. 2009;29:274–82. doi: 10.1159/000158635. [DOI] [PubMed] [Google Scholar]
- 143.Dousa TP. Cyclic-3′,5′-nucleotide phosphodiesterase isozymes in cell biology and pathophysiology of the kidney. Kidney Int. 1999;55:29–62. doi: 10.1046/j.1523-1755.1999.00233.x. [DOI] [PubMed] [Google Scholar]
- 144.Sedeek M, Callera G, Montezano A, et al. Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: implications in type 2 diabetic nephropathy. Am J Physiol Renal Physiol. 2010;299:F1348–58. doi: 10.1152/ajprenal.00028.2010. [DOI] [PubMed] [Google Scholar]
- 145.Sedeek M, Nasrallah R, Touyz RM, Hébert RL. NADPH oxidases, reactive oxygen species, and the kidney: friend and foe. J Am Soc Nephrol. 2013;24:1512–8. doi: 10.1681/ASN.2012111112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bondi CD, Manickam N, Lee DY, et al. NAD(P)H oxidase mediates TGF-beta1-induced activation of kidney myofibroblasts. J Am Soc Nephrol. 2010;21:93–102. doi: 10.1681/ASN.2009020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Barnes JL, Gorin Y. Myofibroblast differentiation during fibrosis: role of NAD(P)H oxidases. Kidney Int. 2011;79:944–56. doi: 10.1038/ki.2010.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cucoranu I, Clempus R, Dikalova A, et al. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97:900–7. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 149.Hu T, Ramachandrarao SP, Siva S, et al. Reactive oxygen species production via NADPH oxidase mediates TGF-beta-induced cytoskeletal alterations in endothelial cells. Am J Physiol Renal Physiol. 2005;289:F816–25. doi: 10.1152/ajprenal.00024.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Das R, Xu S, Quan X, et al. Upregulation of mitochondrial Nox4 mediates TGF-β-induced apoptosis in cultured mouse podocytes. Am J Physiol Renal Physiol. 2014;306:F155–67. doi: 10.1152/ajprenal.00438.2013. [DOI] [PubMed] [Google Scholar]
- 151.Gooch JL, Gorin Y, Zhang BX, Abboud HE. Involvement of calcineurin in transforming growth factor-beta-mediated regulation of extracellular matrix accumulation. J Biol Chem. 2004;279:15561–70. doi: 10.1074/jbc.M308759200. [DOI] [PubMed] [Google Scholar]
- 152.Chabrashvili T, Kitiyakara C, Blau J, et al. Effects of ANG II type 1 and 2 receptors on oxidative stress, renal NADPH oxidase, and SOD expression. Am J Physiol Regul Integr Comp Physiol. 2003;285:R117–24. doi: 10.1152/ajpregu.00476.2002. [DOI] [PubMed] [Google Scholar]
- 153.Gao L, Huang W, Li J. NOX1 abet mesangial fibrogenesis via iNOS induction in diabetes. Mol Cell Biochem. 2013;382:185–91. doi: 10.1007/s11010-013-1733-4. [DOI] [PubMed] [Google Scholar]
- 154.Aoyama T, Paik YH, Watanabe S, et al. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology. 2012;56:2316–27. doi: 10.1002/hep.25938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sedeek M, Gutsol A, Montezano AC, et al. Renoprotective effects of a novel Nox1/4 inhibitor in a mouse model of Type 2 diabetes. Clin Sci (Lond) 2013;124:191–202. doi: 10.1042/CS20120330. [DOI] [PubMed] [Google Scholar]
- 156.Benigni A, Zoja C, Corna D, et al. A specific endothelin subtype A receptor antagonist protects against injury in renal disease progression. Kidney Int. 1993 Aug;44(2):440–4. doi: 10.1038/ki.1993.263. [DOI] [PubMed] [Google Scholar]
- 157.Opočenský M, Kramer HJ, Backer A, et al. Late-onset endothelin-A receptor blockade reduces podocyte injury in homozygous Ren-2 rats despite severe hypertension. Hypertension. 2006;48:965–71. doi: 10.1161/01.HYP.0000245117.57524.d6. [DOI] [PubMed] [Google Scholar]
- 158.Boffa JJ, Tharaux PL, Dussaule JC, Chatziantoniou C. Regression of renal vascular fibrosis by endothelin receptor antagonism. Hypertension. 2001;37(2 Pt 2):490–6. doi: 10.1161/01.hyp.37.2.490. [DOI] [PubMed] [Google Scholar]
- 159.Wenzel RR, Littke T, Kuranoff S, et al. Avosentan reduces albumin excretion in diabetics with macroalbuminuria. J Am Soc Nephrol. 2009;20:655–64. doi: 10.1681/ASN.2008050482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mann JF, Green D, Jamerson K, et al. Avosentan for overt diabetic nephropathy. J Am Soc Nephrol. 2010;21:527–35. doi: 10.1681/ASN.2009060593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.de Zeeuw D, Coll B, Andress D, et al. The endothelin antagonist atrasentan lowers residual albuminuria in patients with type 2 diabetic nephropathy. J Am Soc Nephrol. 2014;25:1083–93. doi: 10.1681/ASN.2013080830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Khan SB, Cook HT, Bhangal G, Smith J, Tam FW, Pusey CD. Antibody blockade of TNF-alpha reduces inflammation and scarring in experimental crescentic glomerulonephritis. Kidney Int. 2005;67:1812–20. doi: 10.1111/j.1523-1755.2005.00279.x. [DOI] [PubMed] [Google Scholar]
- 163.Trachtman H, Vento S, Gipson D, et al. Novel therapies for resistant focal segmental glomerulosclerosis (FONT) phase II clinical trial: study design. BMC Nephrol. 2011;12:8. doi: 10.1186/1471-2369-12-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tsakas S, Goumenos DS. Accurate measurement and clinical significance of urinary transforming growth factor-beta1. Am J Nephrol. 2006;26:186–93. doi: 10.1159/000093178. [DOI] [PubMed] [Google Scholar]
- 165.Cheng O, Thuillier R, Sampson E, et al. Connective tissue growth factor is a biomarker and mediator of kidney allograft fibrosis. Am J Transplant. 2006;6:2292–306. doi: 10.1111/j.1600-6143.2006.01493.x. [DOI] [PubMed] [Google Scholar]
- 166.Gilbert RE, Akdeniz A, Weitz S, et al. Urinary connective tissue growth factor excretion in patients with type 1 diabetes and nephropathy. Diabetes Care. 2003;26:2632–6. doi: 10.2337/diacare.26.9.2632. [DOI] [PubMed] [Google Scholar]
- 167.Tesch GH. Review: Serum and urine biomarkers of kidney disease: A pathophysiological perspective. Nephrology (Carlton) 2010;15:609–16. doi: 10.1111/j.1440-1797.2010.01361.x. [DOI] [PubMed] [Google Scholar]
- 168.Io H, Hamada C, Fukui M, Horikoshi S, Tomino Y. Relationship between levels of urinary type IV collagen and renal injuries in patients with IgA nephropathy. J Clin Lab Anal. 2004;18:14–8. doi: 10.1002/jcla.10099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Cohen MP, Lautenslager GT, Shearman CW. Increased collagen IV excretion in diabetes. A marker of compromised filtration function. Diabetes Care. 2001;24:914–8. doi: 10.2337/diacare.24.5.914. [DOI] [PubMed] [Google Scholar]
- 170.Takahashi M. Increased urinary fibronectin excretion in type II diabetic patients with microalbuminuria. Nihon Jinzo Gakkai Shi. 1995;37:336–42. [PubMed] [Google Scholar]
- 171.Nishiyama A, Konishi Y, Ohashi N, et al. Urinary angiotensinogen reflects the activity of intrarenal renin-angiotensin system in patients with IgA nephropathy. Nephrol Dial Transplant. 2011;26:170–7. doi: 10.1093/ndt/gfq371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Yamamoto T, Nakagawa T, Suzuki H, et al. Urinary angiotensinogen as a marker of intrarenal angiotensin II activity associated with deterioration of renal function in patients with chronic kidney disease. J Am Soc Nephrol. 2007;18:1558–65. doi: 10.1681/ASN.2006060554. [DOI] [PubMed] [Google Scholar]
- 173.Mirkovic K, Doorenbos CR, Dam WA, et al. Urinary vitamin D binding protein: a potential novel marker of renal interstitial inflammation and fibrosis. PLoS One. 2013;8(2):e55887. doi: 10.1371/journal.pone.0055887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Pallet N, Chauvet S, Chasse JF, et al. Urinary retinol binding protein is a marker of the extent of interstitial kidney fibrosis. PLoS One. 2014;9(1):e84708. doi: 10.1371/journal.pone.0084708. [DOI] [PMC free article] [PubMed] [Google Scholar]