

Abstract
Breast cancer is one of the most common causes of cancer-related deaths in women. The estrogen receptor (ERα) is well known for having growth promoting effects in breast cancer. Recently, we have identified DC-SCRIPT (ZNF366) as a co-suppressor of ERα and as a strong and independent prognostic marker in ESR1 (ERα gene)-positive breast cancer patients. In this study, we further investigated the molecular mechanism on how DC-SCRIPT inhibits breast cancer cell growth. DC-SCRIPT mRNA levels from 190 primary ESR1-positive breast tumors were related to global gene expression, followed by gene ontology and pathway analysis. The effect of DC-SCRIPT on breast cancer cell growth and cell cycle arrest was investigated using novel DC-SCRIPT-inducible MCF7 breast cancer cell lines. Genome-wide expression profiling of DC-SCRIPT-expressing MCF7 cells was performed to investigate the effect of DC-SCRIPT on cell cycle-related gene expression. Findings were validated by real-time PCR in a cohort of 1,132 ESR1-positive breast cancer patients. In the primary ESR1-positive breast tumors, DC-SCRIPT expression negatively correlated with several cell cycle gene ontologies and pathways. DC-SCRIPT expression strongly reduced breast cancer cell growth in vitro, breast tumor growth in vivo, and induced cell cycle arrest. In addition, in the presence of DC-SCRIPT, multiple cell cycles related genes were differentially expressed including the tumor suppressor gene CDKN2B. Moreover, in 1,132 primary ESR1-positive breast tumors, DC-SCRIPT expression also correlated with CDKN2B expression. Collectively, these data show that DC-SCRIPT acts as a novel regulator of CDKN2B and induces cell cycle arrest in ESR1-positive breast cancer cells.
Electronic supplementary material
The online version of this article (doi:10.1007/s10549-015-3281-y) contains supplementary material, which is available to authorized users.
Keywords: DC-SCRIPT, ZNF366, CDKN2B, Breast cancer, Cell cycle, G1 arrest
Introduction
Breast cancer is the second most common malignancy in women, affecting one in eight in the US [1]. Improved knowledge on the molecular basis for breast cancer development has led to the identification of prognostic markers, enabling personalized treatment regimens beneficial for the patient [2]. Recently, we found that the nuclear receptor co-regulator dendritic cell specific transcript [DC-SCRIPT, also known as zinc finger protein 366 (ZNF366)] has statistically significant prognostic value for patients with estrogen receptor alpha (ERα)-positive and/or progesterone receptor (PR)-positive tumors [3, 4]. It was shown that high DC-SCRIPT mRNA expression represents a solid prognostic marker as it predicts—independently of current clinical prognostic markers such as age, menopausal status, grade, tumor size and receptor status, increased disease-free, metastasis-free, and overall survival in patients who did not receive any adjuvant systemic treatment [3, 4]. Yet, little is known about the molecular mechanism on how DC-SCRIPT can inhibit breast cancer progression.
DC-SCRIPT was identified in 2006 and has been shown to play a versatile role in dendritic cells (DC) as well as cancer biology [3–11]. Within the immune system, DC-SCRIPT expression is limited to DCs [1] where it co-regulates Toll-like receptor-induced cytokine production [6]. In addition, we and others have shown that DC-SCRIPT acts as a co-regulator of multiple nuclear receptors (NRs) [3, 7, 8, 11]. NRs are ligand-inducible transcription factors that can directly bind to DNA and thereby mediate transcription of genes involved in diverse biological processes, such as proliferation, differentiation, and metabolism [12–15]. DC-SCRIPT was shown to interact with: the type I NRs ERα, PR, glucocorticoid receptor, and the androgen receptor; the type II NRs peroxisome proliferator-activator receptor (PPAR), retinoic acid receptor (RAR); and the vitamin D3 receptor [3, 7, 8]. While most NR co-regulators have a distinct activating or repressing function, co-repressors and co-activators can occasionally switch roles, depending on the promoter context [16–18]. Interestingly, we have shown that DC-SCRIPT also acts as a NR co-regulator having both repressing and activating capabilities [3]. Transcription mediated by the type I NRs was inhibited by DC-SCRIPT, while the activity of the type II NRs was enhanced by DC-SCRIPT. Remarkably, although several co-regulators exist that have a dual effect on NR function, to our knowledge, there are currently no co-regulators known to have such a distinct effect on type I and type II NR mediated transcription.
In line with the finding that DC-SCRIPT exhibits a repressive activity on the pro-proliferative type I NRs and enhances the mainly anti-proliferative activity of type II NRs, DC-SCRIPT expression in breast cancer cells results in the inhibition of breast cancer cell proliferation [3]. Proliferation of cells is a tightly regulated process to avoid malignant transformation. Cellular transformation has often been linked to lack of cell cycle control, and in general, three regulatory mechanisms are responsible for cell cycle progression; (1) the production and degradation of cyclins in oscillating cycles, (2) the (de)-phosphorylation of cyclin-dependent protein kinases (CDKs) and cyclins, and (3) the interaction of different CDK inhibitory proteins from the INK4 and Cip/Kip protein families with CDK/cyclin-complexes. While the Cip/Kip family members have a wide range of CDK/cyclin-complex specificities, the INK4 family members more specifically inhibit CDK/cyclin D complexes, thereby arresting cells in the G1 phase of cell cycle progression. Interestingly, many of the INK4/Cip/Kip family members have been shown to play an important role in breast cancer [19–21].
Here, we show that DC-SCRIPT expression in ESR1+ (estrogen receptor 1, gene encoding ERα) breast cancer cells induces G1 cell cycle arrest and acts as a novel regulator of the gene CDKN2B encoding the tumor suppressor protein p15ink4b. Moreover, we show that DC-SCRIPT correlates with CDKN2B expression in a cohort of 1,132 ESR1+ -positive primary tumors of breast cancer patients.
Methods
Patients
The protocol to study biological markers associated with disease outcome was approved by the medical ethics committee of the Erasmus Medical Center Rotterdam, The Netherlands (MEC 02.953). The study was performed in accordance with the Code of Conduct of the Federation of Medical Scientific Societies in the Netherlands (www.federa.org), and consent was not required. This retrospective study used 1,505 blind-coded freshly frozen primary tumor tissues of female patients with operable breast cancer from 1978 through 2000. The primary breast tumors were from patients with detailed clinical follow-up as previously described [4].
Cell culture
The MCF7 cell line expressing rtTA2S-M2 (ATCC, Tet-On® Advanced, Clontech) was cultured in DMEM with glutamax (Invitrogen), supplemented with 10 % heat-inactivated fetal calf serum (FCS, Greiner Bio-One), 0.5 % antibiotic–antimycotics (Invitrogen), and 100 μg/mL G418 (Gibco) to select for the rtTA2s-M2 plasmid. Cells transfected with pTRE reporter constructs (see below) were cultured with additional hygromycin B (100 μg/mL, Calbiochem).
Generation of DC-SCRIPT inducible MCF7 cells
DC-SCRIPT from pCATCH-DCSCRIPT [5] was cloned in the multiple cloning site of pTRE-tight (Clontech) vector as a BamH1/Xba1 insert. pTRE-tight was used as the control plasmid.
MCF7 cells expressing rtTA2S-M2 were transfected with pTRE-tight or pTRE-tight-DC-SCRIPT and co-transfected with a hygromycin B resistance marker using metafectene according to the manufactures protocol (Biontex). Two days after transfection the transfected cells were selected with 100 μg/mL hygromycin B and 100 μg/mL G418. After expanding the positively selected cells, single colonies were picked, expanded, and checked by PCR for complete genomic incorporation of the correct pTRE-tight vector. Clones containing the DC-SCRIPT vector were labeled MCF7SC, and control clones containing the empty vector were labeled MCF7EV. MCF7SC29, MCF7SC36, MCF7EV16, and MCF7EV37 were chosen for further studies. DC-SCRIPT protein expression in these cell lines was validated using Western blot as previously described [6].
MTT assay
MCF7SC or MCF7EV clones were plated at equal numbers in the presence of 0 (vehicle), 10, 100, or 1,000 ng/mL doxycycline at day 0, and medium was refreshed every second day with fresh doxycycline. Relative cell viability/proliferation was determined by a MTT-based colorimetric cell proliferation assay (Sigma) after 5 days of culturing.
Cell cycle distribution
MCF7 inducible cells were cultured with 0 or 100 ng/mL doxycycline and synchronized using serum starvation for 24 h. Cells were released in 10 % charcoal stripped FCS (cFCS) ± 10 nM E2 (Sigma) for 20 h, lysed in 1 g/L sodium-citrate buffer containing 0.1 mg/mL RNase A, 20 μg/mL propidium-iodide, and 0.1 % Triton, and analyzed for cell cycle distribution using bivariate flow cytometry on a FACSCalibur (BD). FlowJo software (TreeStar) was used for cell cycle position using the cell cycle algorithm.
In vivo MCF7SC/EV xenograft model
All animal experiments were approved by the Animal Experimental Committee of the Radboud UMC and were performed in accordance with institutional, national and European guidelines. 6–8-week-old female BALB/c-nude mice (Janvier Labs) were inoculated orthotopically in the mammary fat pad with 5 × 106 MCF7SC29 or MCF7EV16. On the same day, a 60-day-slow release pellet containing 17β-estradiol (0.72 mg/pellet, Innovative Research of America) was implanted s.c. using a trochar for small pellet implantation (Innovative Research of America). Upon tumor establishment, mice were treated with 2 mg/mL doxycycline in normal drinking water or vehicle control. Mice weight was monitored every second day, and no significant differences were observed between experimental groups. Tumor size was measured every second day using a caliper, and volume was calculated as described in [22]. Part of the tumor xenografts were snap-frozen and embedded in OCT embedding matrix (CellPath) and stained for DC-SCRIPT as previously described [3].
Microarray analysis
Genome-wide expression profiling of treated and untreated MCF7 cells was performed on a Illumina HumanHT-12 WG-DASL V4.0 R2 expression BeadChip according to the manufacturer’s description. For this, total RNA was isolated by lysing cells cultured in 6-well plates followed by total RNA isolation with the Quick-RNA MiniPrep kit (zymo research) and quantified using Nanodrop. Next, 500 ng total RNA was amplified in the presence of UDG and after hybridization on the BeadChip scanned on an iScan Reader using the associated BeadScan software. Scanned data were uploaded into GenomeStudio® software (v2011.1) via the gene expression module (WG-DASL Assay), after which raw bead information was further normalized and analyzed with Lumi R package [23]. All normalized and non-normalized microarray data have been submitted to the GEO database (accession number GSE59995, reviewer link: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=gxsdeysonpuhlep&acc=GSE59995 [link will be removed before publication]).
In addition, in clinical breast cancer samples, ZNF366 mRNA levels (measured as described before [4]) were compared with expression data we had available of 190 ESR1-positive LNN breast tumors that were analyzed before on the Affymetrix oligonucleotide Human U133a Gene Chips [24–26], GEO accession numbers GSE2034 and GSE5327.
Gene ontology and pathway analysis was conducted using the Functional Annotation Tool at the DAVID Bioinformatics Resources [27, 28].
RNA isolation and quantitative reverse transcriptase PCR (RT-qPCR) of MCF7
Total RNA was isolated and reverse transcribed into cDNA as described before [6]. mRNA levels for the genes of interest were determined with a CFX96 sequence detection system (Bio-Rad) with SYBR Green (Roche) as the fluorophore and gene-specific oligonucleotide primers. The primers for ZNF366 and the reference gene HMBS were described previously [3]. Other used primers are as follows: CDK6 (F-CCAGATGGCTCTAACCTCAGT, R-AACTTCCACGAAAAAGAGGCTT), and CDKN2B (F-CGAGGAGAACAAGGGCATGC, R-CTGTCGCACCTTCTCCACTAG). Reaction mixtures and program conditions were used that were recommended by the manufacturer (Bio-Rad). Quantitative PCR data were analyzed with the CFX Manager software (Bio-Rad) as described before [6], and mRNA levels were calculated according to the cycle threshold method [29].
RT-qPCR of patient samples
Tissue processing, RNA isolation, cDNA synthesis, and quantitative reverse transcriptase polymerase chain reaction (RT-qPCR) were performed and normalized using the delta Cq method on the average of 3 reference genes (HMBS [F-CATGTCTGGTAACGGCAATG, R-GTACGAGGCTTTCAATGTTG], HPRT1 [F-TATTGTAAT GACCAGTCAACAG, R-GGTCCTTTTCACCAGCAAG] and TBP [F-TTCGGAGAG TTCTGGGATTG, R-ACGAAGTGCAATGGTCTTTAG) as previously described [4, 30]. Quantification of target genes was done using the following intron-spanning Taqman probe-based gene expressions assays (Applied BioSystems): CDKN2B, Hs00793225_m1; and ZNF366, Hs00403536_m1 according the manufacturer’s instructions. Other primers used to analyze mRNA expression by SYBR Green based real-time PCR in a Mx3000P Real-Time PCR System (Agilent) were as follows: CDK6: F-AGGTCTGGACTTTCTTCATTC, R-CTGGACTGGAGCAAGACTTC; ESR1: F-ATCCTACCAGACCCT TCAGTG, R-GCCAGACGAGACCAATCATC. Samples were grouped in ESR1 negative and positive according the cut off at 0.2 as described in [31].
Results
DC-SCRIPT expression in breast cancer patients negatively correlates with cell cycle genes
Previously, we reported that DC-SCRIPT is a unique NR modulator and that its mRNA expression is a strong and independent marker of favorable prognosis in ESR1-positive breast cancer [3, 4, 7, 8, 11]. To gain further insight into the functional role of DC-SCRIPT in breast cancer cells, DC-SCRIPT mRNA levels from 190 primary ESR1-positive breast tumors [4] was related with the global gene expression data that was available for these tumors. This analysis revealed that high DC-SCRIPT mRNA expression negatively correlated with several cell cycle gene ontologies and pathways (Table 1). Cell cycle related genes negatively correlating with DC-SCRIPT mRNA included CDK2, CCNB1, CCNE2, and E2F1 (Table 2). Intriguingly, a correlation with cell cycle proteins is precisely what one would expect of a protein inhibiting the activity of the pro-proliferative type I NRs ERα and PR and stimulating the activity of the mainly anti-proliferative NRs RAR and PPAR [3].
Table 1.
Gene ontology and pathways negatively correlating with DC-SCRIPT expression
| Category | GO term | # of genes | % | Benjamini p value |
|---|---|---|---|---|
| GOTERM-BP-FAT | Cell cycle | 24 | 16 | 8.87E−05 |
| GOTERM-BP-FAT | DNA metabolic process | 23 | 16 | 8.94E−07 |
| GOTERM-BP-FAT | Cell cycle process | 19 | 13 | 4.93E−04 |
| GOTERM-BP-FAT | Cell cycle phase | 18 | 12 | 5.10E−05 |
| GOTERM-BP-FAT | M phase | 17 | 11 | 1.60E−05 |
| GOTERM-BP-FAT | DNA replication | 15 | 10 | 9.34E−07 |
| GOTERM-BP-FAT | Cell division | 14 | 9 | 4.76E−04 |
| Database | Term | # of genes | % | p value |
|---|---|---|---|---|
| Reactome | Cell cycle mitotic | 18 | 12 | 3.75E−06 |
| Reactome | Cell cycle checkpoints | 10 | 7 | 6.87E−05 |
| KEGG | Cell cycle | 8 | 5 | 6.64E−04 |
| KEGG | Oxidative phosphorylation | 8 | 5 | 8.40E−04 |
Genes negatively correlating with DC-SCRIPT expression were used as input for the online Functional Annotation Tool at the DAVID Bioinformatics Resources (National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD; http://david.abcc.ncifcrf.gov). GO, BBID, Biocarta, KEGG, Panther, and Reactome databases were assayed for affected terms. From the output the top terms that had a p value < 0.001 are shown
Table 2.
Cell cycle-related genes correlating with DC-SCRIPT mRNA expression in 190 primary ESR1+ breast tumor specimens
| Gene | Spearman’s Rs | p value | Additional cell cycle-related GO annotation |
|---|---|---|---|
| ARL3 | −0.41 | ** | Cytokinesis |
| BRCA1 | −0.38 | ** | G2 DNA damage checkpoint |
| KRT18 | −0.37 | * | – |
| CDK2 | −0.35 | * | G1/S transition of mitotic cell cycle |
| MAD2L1 | −0.34 | * | Positive regulation of mitotic cell cycle spindle assembly checkpoint |
| BUB3 | −0.34 | * | Spindle assembly checkpoint |
| CCNB1 | −0.34 | * | G2/M transition of mitotic cell cycle |
| KIF11 | −0.33 | * | Spindle assembly involved in mitosis |
| ZWINT | −0.32 | * | Mitotic cell cycle checkpoint |
| E2F1 | −0.32 | * | G1/S transition of mitotic cell cycle |
| TIMELESS | −0.31 | * | Cell cycle phase transition |
| RAN | −0.31 | * | Mitotic spindle organization |
| RAD51C | −0.31 | * | Positive regulation of G2/M transition of mitotic cell cycle |
| CETN3 | −0.31 | * | Mitotic nuclear division |
| HCAP-G | −0.31 | * | Mitotic chromosome condensation |
| CHEK1 | −0.31 | * | DNA damage checkpoint |
| TUBG1 | −0.30 | * | Meiotic spindle organization |
| RAD54B | −0.30 | * | Reciprocal meiotic recombination |
| CKS2 | −0.30 | * | Cyclin-dependent protein serine/threonine kinase regulator activity |
| CKS1B | −0.30 | * | G1/S transition of mitotic cell cycle |
| CCNE2 | −0.30 | * | G1/S transition of mitotic cell cycle |
| DDA3 | −0.30 | * | Positive regulation of cyclin-dependent protein serine/threonine kinase activity |
| KIF22 | −0.29 | * | Mitotic nuclear division |
| PRC1 | −0.29 | * | Cytokinesis |
Cell cycle-related genes significantly correlating with DC-SCRIPT mRNA expression are shown
** p < 0.01, * p < 0.05
DC-SCRIPT negatively regulates cell growth in breast cancer cell lines in vitro and in vivo
Previously, we have shown that prolonged (over)expression of DC-SCRIPT in the estrogen-responsive breast cancer cell line MCF7 transiently transfected with DC-SCRIPT resulted in growth inhibition of the DC-SCRIPT expressing cells [3]. To further validate this finding, the growth inhibitory effects of DC-SCRIPT were determined in an additional estrogen-responsive cell line CAMA-1 [32]. In agreement with our previous data on MCF7 cells, also cell growth of CAMA-1 cells could be inhibited by DC-SCRIPT expression (Appendix A in supplymentary material).
So far all cell lines analyzed were found to be essentially negative for endogenous DC-SCRIPT mRNA expression, including the above mentioned cell lines and 36 other breast carcinoma cell lines (data not shown). To circumvent the problem of the lack of DC-SCRIPT in cell lines for functional studies, DC-SCRIPT was cloned in front of the Tet-responsive promoter construct that becomes activated upon addition of doxycycline [MCF7 Tet-on advanced cell line (Clontech)]. Following transfection, multiple-independent clones expressing DC-SCRIPT (MCF7SC) upon stimulation with doxycycline or the empty control construct were isolated (MCF7EV) (data not shown). By varying the doxycycline concentration the expression levels of DC-SCRIPT can be varied and tuned toward a physiological level (Fig. 1a). Relative to its endogenous expression levels in DCs, MCF7SC29 cells treated with 100 ng/mL doxycycline show physiological DC-SCRIPT expression levels. Using 100 ng/mL doxycycline, it was determined that DC-SCRIPT has a protein half-life of 4 h following doxycycline withdrawal (Fig. 1b) and that doxycycline addition every 48 h results in the continuous expression of DC-SCRIPT in these cells (data not shown). Using an MTT assay, the effect of DC-SCRIPT expression on cell viability was assayed (Fig. 1c). Increasing DC-SCRIPT expression levels affected cell viability in two independent MCF7SC clones, whereas the viability of MCF7EV16 was not affected by increasing levels of doxycycline (Fig. 1c). Similar results were obtained from actual cell count after 6 days of DC-SCRIPT expression (Appendix B in supplymentary material).
Fig. 1.

DC-SCRIPT protein expression reduces cell viability in vitro. a MCF7SC29 was treated with increasing amounts of doxycycline. Induction of DC-SCRIPT protein expression was determined by Western blot analysis, and related to the amount of endogenously expressed DC-SCRIPT in immature DC (iDC). The negative control (MCF7EV37) treated with 500 ng/mL doxycycline is also shown. b DC-SCRIPT expression was induced with 100 ng/mL doxycycline for 16 h in MCF7SC29. Cells were washed to remove the doxycycline and cultured for the indicated time points. DC-SCRIPT protein expression was determined by Western blot analysis. The negative control MCF7EV16, is also shown. c MCF7SC clones 29 and 36, and MCF7EV16 were cultured in the presence of 0, 10, 100, and 1,000 ng/mL doxycycline. Cell viability was measured with the MTT assay at day 5 (data are expressed as the mean of three experiments ± SEM). Statistics paired two-tailed student t test)
To investigate the effect of DC-SCRIPT expression on breast tumor growth in vivo, MCF7SC29 or MCF7EV16 cells were inoculated orthotopically in the mammary fat pad of female nude mice. Simultaneously, an estradiol slow release pellet was implanted s.c. to stimulate tumor growth. 4 days after implantation when a palpable tumor was present, mice either received normal drinking water or water-containing 2 mg/mL doxycycline to induce DC-SCRIPT expression (Fig. 2). Strikingly, mice engrafted with the MCF7SC29 clone receiving doxycycline had a strongly diminished tumor growth compared to mice injected with the MCF7EV16 clone or mice receiving normal water (Fig. 2a). In line with this, DC-SCRIPT induction in mice engrafted with MCF7SC29 extended their overall survival, whereas all control mice reached their endpoint (700 mm3) before day 60, none of the mice engrafted with MCF7SC29 and receiving doxycycline reached this size before day 60 (data not shown). DC-SCRIPT expression in the tumor xenografts was confirmed by immunohistochemistry (Fig. 2b). Altogether these data demonstrate that DC-SCRIPT expression in breast cancer cells represses cell growth in vitro and inhibits breast tumor growth in vivo.
Fig. 2.
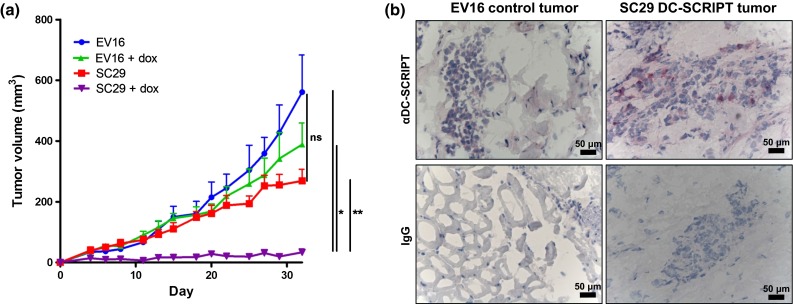
DC-SCRIPT expression diminishes tumor growth in vivo. 5 million MCF7SC29 or MCF7EV16 cells were injected into the lower mammary fat pad, and 60-day-slow release estradiol pellets (dose: 0.72 mg/pellet) were implanted subcutaneously on the back between the shoulders of nude mice. After tumor establishment, half the mice in each group were administered 2 mg/mL doxycycline in the drinking water throughout the duration of the experiment. a Tumor growth curves of the MCF7 xenografts. Data are expressed as mean ± SEM and are the representative out of three experiments (statistics unpaired two-tailed student t test assuming unequal variance, n = 4 mice/group). b Immunohistochemistry of tumor xenograft biopsies. *p < 0.05, **p < 0.01
DC-SCRIPT expression arrests breast cancer cells in G1
DC-SCRIPT mRNA expression in breast cancer patients negatively correlated with many cell cycle proteins (Table 2), suggesting that DC-SCRIPT expression inhibits cell growth by affecting cell cycle progression. To investigate this hypothesis the MCF7SC and MCF7EV cells were synchronized by serum starvation for 24 h and then released in the presence or absence of estradiol (E2) to induce cell growth, and subjected to cell cycle analysis using bivariate flow cytometry. Figure 3 shows that MCF7SC cells and the MCF7EV cells in the absence of doxycycline have a similar percentage of the cells in the G1 phase of the cell cycle. In contrast, DC-SCRIPT expression in two independent MCF7SC clones (29 and 36) results in significantly more cells in the G1 phase of the cell cycle compared to the doxycycline treated MCF7EV16. These data demonstrate that expression of DC-SCRIPT in breast carcinoma cells leads to G1 cell cycle arrest.
Fig. 3.
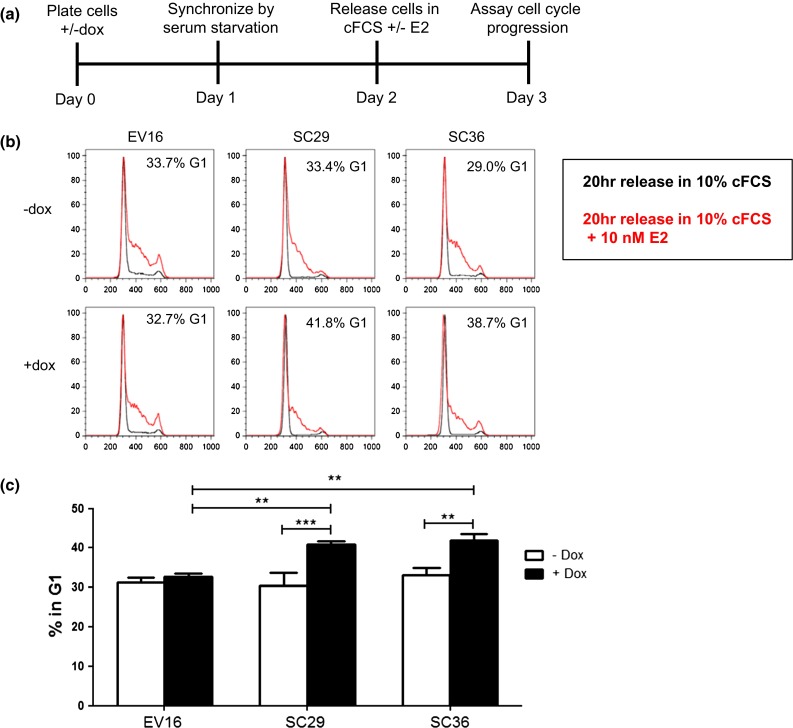
DC-SCRIPT protein expression results in G1 arrest. MCF7SC clones 29 and 36, and MCF7EV16 were cultured in the absence (−) or presence (+) of 100 ng/mL doxycycline (dox). The cells were synchronized using serum starvation for 24 h. Cells were released with 10 % charcoal-stripped cFCS or 10 % cFCS + 10 nM E2. 20 h later cells were checked for their presence in the G1 phase of the cell cycle using bivariate flow cytometry. a Schematic representation of the experiment. b Representative flow cytometry histograms. c Mean value (±SEM) of four independent experiments. Statistics repeated measures ANOVA with a Bonferroni post test
DC-SCRIPT expression induces expression of the tumor suppressor CDKN2B and its target CDK6
To obtain further insight into the G1 arrest mediated by DC-SCRIPT, a global gene profiling of the MCF7SC/EV breast cancer cell lines in the presence and absence of DC-SCRIPT was performed. Genes having a low expression level (within the 1st quartile) were filtered out, to minimize the possibility for false positive results. For the remaining 20,778 probes, MCF7SC29 and MCF7SC36 were compared to MCF7EV16 after treatment with doxycycline during 20 h release with 10 nM E2. In agreement with DC-SCRIPT expression in breast tumor specimens, also in the MCF7-DC-SCRIPT inducible model, multiple cell cycle-related genes are differentially expressed in the presence of DC-SCRIPT. In total, 22 cell cycle related genes were regulated more than 1.5 fold by DC-SCRIPT expression in MCF7 cells (Table 3 ). Interestingly, the majority of cell cycle genes were downregulated upon DC-SCRIPT expression (17 out of 22) which is in line with the negative correlation between DC-SCRIPT expression and cell cycle genes from the breast cancer specimens (Tables 1, 2). Notably, many of the 24 cell cycle genes found to be negatively correlated with DC-SCRIPT expression in the primary breast cancer specimens were also negatively regulated by DC-SCRIPT expression in the MCF7 DC-SCRIPT inducible system (Appendix C in supplymentary material). The two highest downregulated genes in the MCF7 system (MAPK13 and CYP26B1) affect cell cycle by secondary pathways via MAPK signaling [33] and retinoic acid metabolism [34, 35], respectively. Interestingly, the two highest upregulated genes, CDKN2B and CDK6, affect cell cycle directly and belong to the same pathway regulating G1 cell cycle progression [36]. Both these genes’ positive correlation with DC-SCRIPT expression was confirmed by RT-qPCR in the MCF7 system (Fig. 4), but both genes could not be reliably detected on the microarray platform used for the primary breast tumor specimens. Therefore, the CDKN2B and CDK6 mRNA expression level was assayed in the breast cancer cohort by RT-qPCR. Intriguingly, DC-SCRIPT mRNA expression levels also positively correlated with CDKN2B and CDK6 in 1,132 ESR1-positive breast cancer patients and much less in 324 ESR1-negatively breast cancer patients (Fig. 5; Table 4), which is in line with our previous data showing that DC-SCRIPT has the strongest prognostic value in the ESR1-positive tumors [3, 4]. Taken together these data indicate that DC-SCRIPT expression correlates with multiple cell cycle related genes in breast cancer tissues and is involved in the regulation of the expression of the cell cycle inhibitor CDKN2B and CDK6.
Table 3.
DC-SCRIPT mediated difference in cell cycle related genes expression in MCF7
| Upregulation | Downregulation | ||||
|---|---|---|---|---|---|
| Gene | Fold change by DC-SCRIPT-induction | Additional cell cycle-related GO annotation | Gene | Fold change by DC-SCRIPT-induction | Additional cell cycle-related GO annotation |
| CDK6 | 2.10 | G1/S transition of mitotic cell cycle | MAPK13 | −2.65 | – |
| CDKN2B | 1.77 | Negative regulation of G1/S transition of mitotic cell cycle | CYP26B1 | −2.41 | M phase |
| PCAF | 1.55 | Cell cycle arrest | GSPT1 | −1.72 | G1/S transition of mitotic cell cycle |
| LRRCC1 | 1.54 | Mitotic nuclear division | SEPT5 | −1.67 | Cytokinesis |
| CCNE1 | 1.51 | G1/S transition of mitotic cell cycle | CENPE | −1.63 | Mitotic chromosome movement toward spindle pole |
| PRR5 | −1.61 | – | |||
| NEK2 | −1.57 | Spindle assembly involved in mitosis | |||
| CCNB2 | −1.56 | G2/M transition of mitotic cell cycle | |||
| CDC14A | −1.54 | – | |||
| NEDD9 | −1.54 | Mitotic nuclear division | |||
| CENPA | −1.52 | Establishment of mitotic spindle orientation | |||
| TPD52L1 | −1.52 | G2/M transition of mitotic cell cycle | |||
| GTSE1 | −1.52 | Mitotic G2 phase | |||
| CDCA8 | −1.51 | Mitotic metaphase | |||
| DBF4B | −1.51 | Positive regulation of nuclear cell cycle DNA replication | |||
| CDC25C | −1.50 | G2/M transition of mitotic cell cycle | |||
| BUB1 | −1.50 | Mitotic spindle assembly checkpoint | |||
Genes with an altered expression level upon DC-SCRIPT-induction in MCF7 cells were assayed for gene ontology association. All genes with a cell cycle related gene ontology and a DC-SCRIPT-mediated fold change of 1.5 (up and down) are shown
Fig. 4.

Induction of DC-SCRIPT upregulates the tumor suppressor CDKN2B and its target CDK6 in MCF7s. Purified RNA from synchronized MCF7SC29, MCF7SC36, and MCF7EV16 cells cultured with 100 ng/mL doxycycline, 20 h after 10 nM E2 stimulation were assayed by qPCR for a CDKN2B, and b CDK6 expression. Statistics was done by Mann–Whitney test, *p < 0.05 (n = 3, error bars SEM)
Fig. 5.

Correlation between DC-SCRIPT mRNA and CDKN2B in the Rotterdam breast cancer patient cohort. Tumor specimens from the Rotterdam breast cancer patient cohort (n = 1,504 were assayed by RT-qPCR for DC-SCRIPT mRNA (ZNF366) and CDKN2B expression. Reliable expression data were correlated between DC-SCRIPT mRNA and CDKN2B using the spearman’s rank correlation coefficient (two-tailed) in a ESR1-positive (n = 1,132), and b ESR1-negative (n = 324) patients. *p < 0.05, ***p < 0.001
Table 4.
Correlation between DC-SCRIPT mRNA and CDKN2B or CDK6 in breast cancer patients assayed by qRT-PCR
| Gene | ESR1-positive | ESR1-negative | ||||
|---|---|---|---|---|---|---|
| Spearman’s Rs | p value | n | Spearman’s Rs | p value | n | |
| CDKN2B | 0.303 | *** | 1,132 | 0.047 | * | 324 |
| CDK6 | 0.381 | *** | 1,144 | 0.139 | * | 324 |
1,504 breast cancer patients were grouped according to ESR1-positivity, and the spearman correlation coefficient was calculated between DC-SCRIPT mRNA (ZNF366) and CDKN2B and CDK6
*p < 0.05, ***p < 0.001
Discussion
DC-SCRIPT expression level has been associated with a favorable prognosis in breast cancer [3, 4], but the molecular mechanism responsible for this relationship is unknown. Applying an unbiased transcriptome profiling, we now report that DC-SCRIPT expression in breast cancer patients correlates with many cell cycle related genes. Using a novel DC-SCRIPT-inducible breast cancer cell line, we further show that DC-SCRIPT expression results in growth- and cell cycle-arrest both in vitro and in vivo. Additional transcriptome profiling revealed that the presence of DC-SCRIPT induces the expression of CDKN2B, the gene encoding the tumor suppressor protein p15ink4b. Interestingly, DC-SCRIPT mRNA also positively correlates with CDKN2B expression in 1,132 ESR1-positive breast cancer patients.
The CDKN2B gene has been shown to be expressed in a range of breast cancer cell lines and normal mammary epithelial cell strains [37]. Moreover CDKN2B has previously been linked to cellular senescence and shown to act as a tumor suppressor protein [38, 39]. Our data now show that DC-SCRIPT expression in breast cancer cells induces G1 cell cycle arrest and enhances CDKN2B expression, suggesting that DC-SCRIPT inhibits breast cancer cell growth at least in part via CDKN2B up regulation.
How DC-SCRIPT regulates CDKN2B expression is currently unknown. Interestingly, another zinc-finger protein ZNF217 has also been shown to regulate CDKN2B expression. ZNF217 inhibits CDKN2B expression by binding to the CDKN2B promoter, when present in a large multi-protein complex consisting of e.g., CtBP1 and HDAC1/2 [40, 41]. DC-SCRIPT is also known to be present in large multi-protein complexes consisting of CtBP1, HDAC1/3/6 and depending on the cell context, different NRs [3, 5, 7, 11]. Another protein that has been shown to induce CDKN2B expression and cell cycle arrest in non-transformed cells is TGFβ [36, 42, 43]. Possibly via regulating TGFβ expression or activity DC-SCRIPT could indirectly regulate CDKN2B. Inhibition of the proto-oncogene and ERα target PIM-1 has also been linked to the upregulation of CDKN2B and attenuated proliferation of MCF7 cells [44]. As DC-SCRIPT is known to act as corepressor protein of ERα [3, 11], DC-SCRIPT may repress the estradiol-mediated expression of PIM-1 and thereby regulate CDKN2B expression and inhibit breast cancer cell growth. It will be extremely interesting to investigate the exact molecular mechanism, using for example, ChIP-Seq, by which DC-SCRIPT regulates directly or indirectly CDKN2B expression in breast (cancer) cells in future studies.
CDKN2B is a specific inhibitor of CDK4 and CDK6, proteins that are important in cell cycle G1 phase progression and G1/S transition. Upon binding, p15ink4b diminishes cyclin D-mediated activation of these CDKs [36]. Intriguingly, together with the DC-SCRIPT-mediated CDKN2B upregulation, we also found CDK6 upregulation. This dual upregulation of a cell-cycle inhibitor and cell cycle activator has previously been observed [45] and may represent the tightly regulated balance of cell cycle proteins present during cell proliferation. Our data have shown that DC-SCRIPT expression is associated with many cell cycle regulators in a MCF7 breast cancer cell line as well as in primary breast cancer specimens. DC-SCRIPT expression skews the balance toward G1 arrest, resulting in diminished cell growth in vitro. Moreover, turning on DC-SCRIPT expression in established MCF7 breast tumors cells in nude mice strongly inhibits the outgrowth of these tumor cells in vivo. These data emphasize the dominant role of DC-SCRIPT in breast cancer cell growth.
Taken together, the data presented in this report demonstrate that DC-SCRIPT affects breast cancer cell cycle progression by displaying an effect on multiple cell cycle regulators including the upregulation of the cell cycle inhibitor CDKN2B.
Ultimately, stimulation of DC-SCRIPT expression or regulation of its downstream targets in ESR1-positive breast cancer patients may turn out to be a clinically relevant therapeutic strategy to inhibit breast cancer growth.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was financially supported by the KWF (Dutch Cancer Society) Grant KUN 2011-5229 (awarded to GJA, MA, and JWMM) from the Dutch Cancer Society. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
List of abbreviations
- CDK
Cyclin-dependent protein kinase
- cFCS
Charcoal stripped FCS
- DC
Dendritic cell
- DC-SCRIPT
Dendritic cell specific transcript
- ERα
Estrogen receptor alpha
- ESR1
Estrogen receptor 1
- FCS
Fetal calf serum
- MCF7EV
MCF7 empty vector control clone
- MCF7SC
MCF7 DC-SCRIPT-inducible clone
- NR
Nuclear receptors
- PPAR
Peroxisome proliferator-activator receptor
- PR
Progesterone receptor
- RAR
Retinoic acid receptor
- RT-qPCR
Quantitative reverse transcriptase PCR
- ZNF366
Zinc finger protein 366
Footnotes
Marleen Ansems and Jonas Nørskov Søndergaard have equally contributed to this study.
Contributor Information
Marleen Ansems, Email: Marleen.Ansems@radboudumc.nl.
Jonas Nørskov Søndergaard, Email: Jonas.Sondergaard@radboudumc.nl.
Anieta M. Sieuwerts, Email: a.sieuwerts@erasmusmc.nl
Maaike W. G. Looman, Email: Maaike.Looman@radboudumc.nl
Marcel Smid, Email: m.smid@erasmusmc.nl.
Annemarie M. A. de Graaf, Email: Annemarie.deGraaf@radboudumc.nl
Vanja de Weerd, Email: v.deweerd@erasmusmc.nl.
Malou Zuidscherwoude, Email: Malou.Zuidscherwoude@radboudumc.nl.
John A. Foekens, Email: j.foekens@erasmusmc.nl
John W. M. Martens, Email: j.martens@erasmusmc.nl
Gosse J. Adema, Phone: +31 243617600, Email: gosse.adema@radboudumc.nl
References
- 1.American Cancer Society . Cancer facts and figures 2014. Atlanda: American Cancer Society; 2014. [Google Scholar]
- 2.Mehta S, Shelling A, Muthukaruppan A, et al. Predictive and prognostic molecular markers for cancer medicine. Ther Adv Med Oncol. 2010;2:125–148. doi: 10.1177/1758834009360519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ansems M, Hontelez S, Looman MWG, et al. DC-SCRIPT: nuclear receptor modulation and prognostic significance in primary breast cancer. J Natl Cancer Inst. 2010;102:54–68. doi: 10.1093/jnci/djp441. [DOI] [PubMed] [Google Scholar]
- 4.Sieuwerts AM, Ansems M, Look MP, et al. Clinical significance of the nuclear receptor co-regulator DC-SCRIPT in breast cancer: an independent retrospective validation study. Breast Cancer Res. 2010;12:R103. doi: 10.1186/bcr2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Triantis V, Trancikova DE, Maaike WG, et al. Identification and characterization of DC-SCRIPT, a novel dendritic cell-expressed member of the zinc finger family of transcriptional regulators. J Immunol. 2006;176:1081–1089. doi: 10.4049/jimmunol.176.2.1081. [DOI] [PubMed] [Google Scholar]
- 6.Hontelez S, Ansems M, Karthaus N, et al. Dendritic cell-specific transcript: dendritic cell marker and regulator of TLR-induced cytokine production. J Immunol. 2012;189:138–145. doi: 10.4049/jimmunol.1103709. [DOI] [PubMed] [Google Scholar]
- 7.Ansems M, Karthaus N, Hontelez S, et al. DC-SCRIPT: AR and VDR regulator lost upon transformation of prostate epithelial cells. Prostate. 2012;72:1708–1717. doi: 10.1002/pros.22522. [DOI] [PubMed] [Google Scholar]
- 8.Hontelez S, Karthaus N, Looman MW, et al. DC-SCRIPT regulates glucocorticoid receptor function and expression of its target GILZ in dendritic cells. J Immunol. 2013 doi: 10.4049/jimmunol.1201776. [DOI] [PubMed] [Google Scholar]
- 9.Triantis V, Moulin V, Looman MWG, et al. Molecular characterization of the murine homologue of the DC-derived protein DC-SCRIPT shown by real-time quantitative polymerase chain. J Leukoc Biol. 2006;79:1083–1091. doi: 10.1189/jlb.1005588. [DOI] [PubMed] [Google Scholar]
- 10.Ansems M, Hontelez S, Karthaus N, et al. Crosstalk and DC-SCRIPT: expanding nuclear receptor modulation. Biochim Biophys Acta. 2010;1806:193–199. doi: 10.1016/j.bbcan.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 11.Lopez-Garcia J, Periyasamy M, Thomas RS, et al. ZNF366 is an estrogen receptor corepressor that acts through CtBP and histone deacetylases. Nucleic Acids Res. 2006;34:6126–6136. doi: 10.1093/nar/gkl875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ottow E, Weinmann H. Nuclear receptors as drug targets. Weinheim: Wiley; 2008. [Google Scholar]
- 13.Gronemeyer H, Gustafsson J-A, Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov. 2004;3:950–964. doi: 10.1038/nrd1551. [DOI] [PubMed] [Google Scholar]
- 14.Mangelsdorf DJ, Thummel C, Beato M, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ribeiro RC, Kushner PJ, Baxter JD. The nuclear hormone receptor gene superfamily. Annu Rev Med. 1995;46:443–453. doi: 10.1146/annurev.med.46.1.443. [DOI] [PubMed] [Google Scholar]
- 16.Vadlamudi RK, Kumar R. Functional and biological properties of the nuclear receptor coregulator PELP1/MNAR. Nucl Recept Signal. 2007;5:e004. doi: 10.1621/nrs.05004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang SM, Stallcup MR. Mouse Zac1, a transcriptional coactivator and repressor for nuclear receptors. Mol Cell Biol. 2000;20:1855–1867. doi: 10.1128/MCB.20.5.1855-1867.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manavathi B, Singh K, Kumar R. MTA family of coregulators in nuclear receptor biology and pathology. Nucl Recept Signal. 2007;5:e010. doi: 10.1621/nrs.05010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abbas T, And Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400–414. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caldon CE, Daly RJ, Sutherland RL, Musgrove EA. Cell cycle control in breast cancer cells. J Cell Biochem. 2006;97:261–274. doi: 10.1002/jcb.20690. [DOI] [PubMed] [Google Scholar]
- 21.Larson PS, Schlechter BL, King C-L, et al. CDKN1C/p57kip2 is a candidate tumor suppressor gene in human breast cancer. BMC Cancer. 2008;8:68. doi: 10.1186/1471-2407-8-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Den Brok MHMGM, Sutmuller RPM, Nierkens S, et al. Efficient loading of dendritic cells following cryo and radiofrequency ablation in combination with immune modulation induces anti-tumour immunity. Br J Cancer. 2006;95:896–905. doi: 10.1038/sj.bjc.6603341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;24:1547–1548. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- 24.Smid M, Wang Y, Zhang Y, et al. Subtypes of breast cancer show preferential site of relapse. Cancer Res. 2008;68:3108–3114. doi: 10.1158/0008-5472.CAN-07-5644. [DOI] [PubMed] [Google Scholar]
- 25.Smid M, Wang Y, Klijn JGM, et al. Genes associated with breast cancer metastatic to bone. J Clin Oncol. 2006;24:2261–2267. doi: 10.1200/JCO.2005.03.8802. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Klijn JGM, Zhang Y, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- 27.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 28.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 30.Sieuwerts AM, Meijer-van Gelder ME, Timmermans M, et al. How ADAM-9 and ADAM-11 differentially from estrogen receptor predict response to tamoxifen treatment in patients with recurrent breast cancer: a retrospective study. Clin Cancer Res. 2005;11:7311–7321. doi: 10.1158/1078-0432.CCR-05-0560. [DOI] [PubMed] [Google Scholar]
- 31.Sieuwerts AM, Usher PA, Meijer-van Gelder ME, et al. Concentrations of TIMP1 mRNA splice variants and TIMP-1 protein are differentially associated with prognosis in primary breast cancer. Clin Chem. 2007;53:1280–1288. doi: 10.1373/clinchem.2006.082800. [DOI] [PubMed] [Google Scholar]
- 32.Leung BS, Qureshi S, Leung JS. Response to estrogen by the human mammary carcinoma cell line CAMA-1. Cancer Res. 1982;42:5060–5066. [PubMed] [Google Scholar]
- 33.Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005;15:11–18. doi: 10.1038/sj.cr.7290257. [DOI] [PubMed] [Google Scholar]
- 34.Ross AC, Zolfaghari R. Cytochrome P450s in the regulation of cellular retinoic acid metabolism. Annu Rev Nutr. 2011;31:65–87. doi: 10.1146/annurev-nutr-072610-145127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dimberg A, Bahram F, Karlberg I, et al. Retinoic acid-induced cell cycle arrest of human myeloid cell lines is associated with sequential down-regulation of c-Myc and cyclin E and posttranscriptional up-regulation of p27(Kip1) Blood. 2002;99:2199–2206. doi: 10.1182/blood.V99.6.2199. [DOI] [PubMed] [Google Scholar]
- 36.Hannon G, Beach D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature. 1994;371:257–261. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- 37.Musgrove E, Lilischkis R, Cornish AL, et al. Expression of the cyclin-dependent kinase inhibitors p16INK4, p15INK4B and p21WAF1/CIP1 in human breast cancer. Int J Cancer. 1995;63:584–591. doi: 10.1002/ijc.2910630420. [DOI] [PubMed] [Google Scholar]
- 38.Erickson S, Sangfelt O, Heyman M, et al. Involvement of the Ink4 proteins p16 and p15 in T-lymphocyte senescence. Oncogene. 1998;17:595–602. doi: 10.1038/sj.onc.1201965. [DOI] [PubMed] [Google Scholar]
- 39.Fuxe J, Akusjärvi G, Goike HM, et al. Adenovirus-mediated overexpression of p15INK4B inhibits human glioma cell growth, induces replicative senescence, and inhibits telomerase activity similarly to p16INK4A. Cell Growth Differ. 2000;11:373–384. [PubMed] [Google Scholar]
- 40.Thillainadesan G, Isovic M, Loney E, et al. Genome analysis identifies the p15ink4b tumor suppressor as a direct target of the ZNF217/CoREST complex. Mol Cell Biol. 2008;28:6066–6077. doi: 10.1128/MCB.00246-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thillainadesan G, Chitilian JM, Isovic M, et al. TGF-β-dependent active demethylation and expression of the p15ink4b tumor suppressor are impaired by the ZNF217/CoREST complex. Mol Cell. 2012;46:636–649. doi: 10.1016/j.molcel.2012.03.027. [DOI] [PubMed] [Google Scholar]
- 42.Iordanskaia T, Nawshad A. Mechanisms of transforming growth factor β induced cell cycle arrest in palate development. J Cell Physiol. 2011;226:1415–1424. doi: 10.1002/jcp.22477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakamura S, Kawai T, Kamakura T, Ookura T. TGF-beta3 is expressed in taste buds and inhibits proliferation of primary cultured taste epithelial cells. In Vitro Cell Dev Biol Anim. 2010;46:36–44. doi: 10.1007/s11626-009-9239-9. [DOI] [PubMed] [Google Scholar]
- 44.Malinen M, Jääskeläinen T, Pelkonen M, et al. Proto-oncogene PIM-1 is a novel estrogen receptor target associating with high grade breast tumors. Mol Cell Endocrinol. 2013;365:270–276. doi: 10.1016/j.mce.2012.10.028. [DOI] [PubMed] [Google Scholar]
- 45.Da Silva GN, de Camargo EA, Salvadori DMF. Toxicogenomic activity of gemcitabine in two TP53-mutated bladder cancer cell lines: special focus on cell cycle-related genes. Mol Biol Rep. 2012;39:10373–10382. doi: 10.1007/s11033-012-1916-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.