

Abstract
Cortical networks spontaneously fluctuate between persistently active Up states and quiescent Down states. The Up states are maintained by recurrent excitation within local circuits, and can be turned on and off by synaptic input. GABAergic inhibition is believed to be important for stabilizing such persistent activity by balancing the excitation, and could have an additional role in terminating the Up state. Here, we report that GABAA and GABAB receptor-mediated inhibition have distinct and complementary roles in balancing and terminating persistent activity. In a model of Up–Down states expressed in slices of rat entorhinal cortex, the GABAA receptor antagonist, gabazine (50–500 nm), concentration-dependently decreased Up state duration, eventually leading to epileptiform bursts. In contrast, the GABAB receptor antagonist, CGP55845 (50 nm to 1 μm), increased the duration of persistent network activity, and prevented stimulus-induced Down state transitions. These results suggest that while GABAA receptor-mediated inhibition is necessary for balancing persistent activity, activation of GABAB receptors contributes to terminating Up states.
Introduction
Persistent activity is a fundamental form of circuit dynamics, which is important for many behavioral functions, including working memory (for review, see Major and Tank, 2004). During slow-wave sleep, anesthesia, and quiet wakefulness, cortical neurons display slow synchronous fluctuations between periods of persistent activity (“Up” states) and periods of relative quiescence (“Down” states) (Steriade et al., 1993; Isomura et al., 2006; Rudolph et al., 2007). These slow oscillations are generated intrinsically in the cortex (Timofeev et al., 2000) and can be preserved in reduced cortical preparations in vitro (Sanchez-Vives and McCormick, 2000; Shu et al., 2003), including entorhinal cortex (Cunningham et al., 2006). Phase transitions between Up and Down states (UDS) can also be triggered by sensory input in vivo (Anderson et al., 2000; Petersen et al., 2003) and synaptic stimulation in vitro (Shu et al., 2003; MacLean et al., 2005; Rigas and Castro-Alamancos, 2007), and thus UDS provide a prototypical model in which to explore the mechanisms underlying the evolution and control of persistent activity in cortical circuits.
Up states are generated by recurrent excitation within local circuits and may terminate spontaneously by synaptic depression and/or build-up of activity-dependent potassium conductances (Contreras et al., 1996; Sanchez-Vives and McCormick, 2000; Timofeev et al., 2000; Compte et al., 2003; Shu et al., 2003; Compte, 2006; Steriade, 2006). During cortical Up states, neurons also receive barrages of GABAergic inputs (Sanchez-Vives and McCormick, 2000; Shu et al., 2003; Hasenstaub et al., 2005), which produce an inhibitory conductance that scales proportionally with the degree of recurrent excitation (Shu et al., 2003; Haider et al., 2006). It has been suggested that this precise balance between excitation and inhibition is critical for stabilizing persistent activity (Compte et al., 2003), and that afferent stimuli can turn off persistent activity by tipping the balance in favor of inhibition (Shu et al., 2003). GABAergic inhibition is mediated via two distinct types of receptor, GABAA and GABAB receptors, responsible for fast and slow inhibition, respectively (Nicoll et al., 1990). The importance of fast GABAergic inhibition in controlling persistent activity is supported by the fact that blocking GABAA receptors transforms Up states into epileptiform bursts (Sanchez-Vives and McCormick, 2000; Shu et al., 2003). However, the roles of distinct forms of inhibition during cortical UDS have not previously been investigated, and, because blocking inhibition leads to epileptiform activity, it has not yet been possible to test directly the role of GABAA receptor-mediated inhibition in the stimulus-induced termination of persistent activity.
Here, we examined the roles of GABAA and GABAB receptor-mediated inhibition in controlling Up states recorded in layer III principal neurons of the rat entorhinal cortex in vitro. Our data suggest that, while GABAA receptor-mediated inhibition is critical for balancing Up states, GABAergic inhibition via GABAB receptors provides a powerful mechanism for the afferent termination of persistent network activity.
Materials and Methods
Slice preparation and electrophysiology.
Horizontal slices of entorhinal cortex (400 μm) were prepared from postnatal day 13–21 Wistar rats of both sexes after decapitation under deep isoflurane-induced anesthesia, in accordance with British Home Office regulations. Slices were maintained initially in an interface chamber, between humidified carbogen gas (95% O2, 5% CO2) and standard artificial CSF (aCSF) containing (in mm): 126 NaCl, 3–3.5 KCl, 1.25 NaH2PO4, 2 MgSO4, 2 CaCl2, 26 NaHCO3, and 10 glucose, pH 7.2–7.4. After recovering at room temperature (20−25°C) for at least 1 h, slices were mounted on coverslips (coated with 0.1% poly-l-lysine in ultrapure H2O), covered with 400 μl of aCSF, and placed in a chamber saturated with humidified carbogen gas at 32°C for at least 1 h. Coverslips were used throughout to anchor the slices, without the need for weights being applied to the upper surface of the slice to help preserve cortical connectivity and promote laminar flow across the slice during superfusion. For recordings, the coverslip with slice was transferred to a submerged style recording chamber at 32°C, and superfused with aCSF, bubbled with carbogen gas, under conditions that allow for oxygenation sufficient to maintain spontaneous network activity (Hájos et al., 2009).
Whole-cell patch-clamp recordings were performed with glass pipettes, pulled from standard borosilicate glass, containing (in mm): 70 d-gluconic acid, 10 CsCl, 5 NaCl, 10 BAPTA, 10 HEPES, 10 QX-314, 0.3 GTP, 4 Mg-ATP, adjusted to pH 7.25 with CsOH for voltage-clamp, and 110 potassium-gluconate, 40 HEPES, 2 ATP-Mg, 0.3 GTP, 4 NaCl for current-clamp experiments, respectively (pH 7.2–7.3; osmolarity 270–285 mosmol l−1). Voltage-clamp experiments were initiated more than 10 min after whole-cell access was achieved to allow diffusion of the Cs+ into the dendrites for improved space clamp. Cells were rejected if series resistance changed by >15%. Membrane potential values are given without correction for junction potential.
Data acquisition and analysis.
Data were low-pass filtered at 2 kHz and acquired at 5 kHz with a Molecular Devices Axon Multiclamp 700B amplifier. The signal was recorded on a PC using the Axon pClamp 9 acquisition software. All further analysis was performed using custom-made procedures in Igor Pro (Wavemetrics).
For Up state detection, the intracellular voltage trace was median filtered (20 ms window), and the membrane potential noise was calculated as the running SD over a 400 ms window. Up states were detected as increases in membrane noise ≥5 SD above baseline mean (20 s period during Down state excluding any after-hyperpolarization) for a duration longer than 400 ms. Up state transitions were defined as the preceding point at which the membrane noise increased to >1 SD above the baseline mean. Reductions in the membrane noise below the same threshold for >400 ms were used to detect Down states, with the Down state transition defined as the nearest point of hyperpolarization below the pre-Up state membrane potential. With gabazine concentrations >250 nm, procedures were adjusted to accommodate events shorter than 400 ms. Average spike densities were calculated by convolving the cumulative spike train from five consecutive Up states with a unity area Gaussian for each spike.
Statistical comparisons were made using repeated-measures (RM) ANOVA and one- and two-sample two-tailed Student's t test. Up and Down state delay distributions were compared using the Kolmogorov–Smirnov (K–S) test. p values <0.05 were considered significant. Data are presented as mean ± SEM.
Drugs and chemicals.
SR95531 (gabazine) and CGP55845 were purchased from Tocris Bioscience. All other drugs and chemicals were obtained from Sigma-Aldrich.
Results
Up states in the layer III principal neurons in vitro are network driven
Whole-cell current-clamp recordings from layer III principal cells of medial entorhinal cortex revealed spontaneous episodes of persistent activity (Up duration 6.4 ± 0.7 s, Down duration 45.4 ± 6.5 s, Vm Up −54.4 ± 1.2 mV, Vm Down −62.0 ± 1.2 mV, n = 24 slices) (Fig. 1A,B). Concurrent recordings from neighboring neurons (<200 μm apart) (Fig. 1B) showed highly synchronous Up and Down state transitions (cross-correlation peak 0.19 ± 0.07 s, n = 5) (Fig. 1C) that depended on synaptic excitation (blocked by CNQX; data not shown). Up state transitions showed a smaller delay between cells than Down state transitions (Up 0.29 ± 0.09 s, Down 0.48 ± 0.16 s, n = 4) (Fig. 1D,E) and were more tightly distributed than transitions into the Down state (p < 0.001, K–S test). The interval between Up state transitions was variable (coefficient of variation 0.59 ± 0.07, n = 9), and Down state duration did not seem to affect subsequent Up state duration (Fig. 1F). Similar to UDS reported in neocortical slices (Sanchez-Vives and McCormick, 2000; Shu et al., 2003; Rigas and Castro-Alamancos, 2007), Up states were followed by a transient after-hyperpolarization (−4.0 ± 0.9 mV, n = 18), occurred at a lower rate than in vivo (Steriade et al., 1993) (Up frequency 0.029 ± 0.004 Hz, n = 24 slices), and could be evoked by local synaptic stimulation in layer III (stimulation strength 50–250 μA, 50–150 μs, Up duration 2.8 ± 0.3 s, n = 14 slices). These properties suggest that the Up states recorded in layer III principal neurons are network-driven events that are supported by both synaptic and intrinsic mechanisms (Shu et al., 2003; Major and Tank, 2004) and distinct from persistent activity mediated purely by intrinsic conductances (Egorov et al., 2002).
Figure 1.

Up states are network-driven events. A, B, Dual current clamp recordings from LIII medial entorhinal cortex cells (<200 μm apart; A) show simultaneous Up and Down state transitions reflected in a bimodal distribution of the membrane potential (time shown on logarithmic scale; B). Spikes are truncated at 0 mV. C, The Vm cross-correlation shows an average lag of 0.19 ± 0.07 s for five dual recordings. Average in black, colored traces from individual recordings. D, E, Pooled data from four dual recordings show that mean Up state transition delays (0.29 ± 0.09 s, D) are not significantly different from mean Down state transition delays (0.48 ± 0.16 s, E). However, comparison of the collated delay histograms shows a significantly tighter distribution of Up delays than Down delays (K–S test, p < 0.001). Top panel shows individual delays. Vertical bars indicate mean ± SEM. F, The return plot does not show any dependency between the durations of 30 consecutive Up and Down states. “S” and “E” indicate start and end of the recordings; LI, layer I; LIII, layer III.
Reducing GABAA receptor-mediated inhibition compresses Up states
Completely blocking GABAA receptors transforms neocortical UDS into epileptiform bursts (Sanchez-Vives and McCormick, 2000; Shu et al., 2003). Because GABAA receptor-mediated inhibition has been proposed to be involved in both stabilizing and terminating Up states, one might predict that partial disinhibition could reveal the balance between these competitive processes. Accordingly, we investigated the concentration-dependent effects of GABAA receptor antagonist, gabazine (50–500 nm) (Wermuth et al., 1987), on the pattern of activity during entorhinal Up states. We measured Up state duration, and used the mean interspike interval (ISI) during Up states to quantify Up state intensity. Increasing concentrations of gabazine progressively and monotonically shortened the duration (control: 4.2 ± 1.9 s vs 500 nm gabazine: 0.4 ± 0.1 s; F(5,20) = 3.93; p < 0.05; RM ANOVA) and increased the intensity (control: 4.0 ± 0.7 s−1 vs 500 nm gabazine: 97.4 ± 30.2 s−1; F(5,20) = 8.86; p < 0.001) of both spontaneous and synaptically triggered Up states (Fig. 2A,B,D,E), and ultimately led to epileptiform activity (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). This confirms that the activation of GABAA receptors is essential for balancing Up states. In addition to balancing Up states, synaptic inhibition has been reported to determine membrane potential dynamics and control action potential generation during slow-wave sleep in vivo (Rudolph et al., 2007). Consistent with this study, we observed that inhibitory conductances contribute strongly to the membrane potential noise during Up states. The increase in membrane potential SD during Up states (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material) was significantly reduced by gabazine application (supplemental Fig. S2B, available at www.jneurosci.org as supplemental material), associated with a reduction in the inhibitory synaptic noise recorded in voltage clamp (supplemental Fig. S2C, available at www.jneurosci.org as supplemental material).
Figure 2.

Distinct roles of GABAA and GABAB receptors in balancing and terminating spontaneous Up states. A, Whole-cell recordings were made from layer III principal cells in control (top), 500 nm gabazine (middle), and 1 μm CGP55845 (bottom). Insets show equivalent Up states triggered by layer III stimulation in the same cells. B, C, Spike density over time from the start of Up state in control condition and during different concentrations of gabazine (B) and CGP55845 (C). Corresponding spike rasters have been plotted above each graph (color coded). The average Up state duration is indicated with a vertical gray bar for each condition. Insets show representative traces. D, E, Up state duration (D) and spike density (1/ISI) (E) as a function of concentration of gabazine and CGP55845. Error bars indicate SEM. LIII, Layer III; Stim, stimulation.
Blocking GABAB receptors increases Up state duration
GABAB receptors mediate inhibitory currents with slow kinetics (Dutar and Nicoll, 1988), which could potentially act in conjunction with GABAA receptors to balance Up states or to compensate for their blockade. With GABAA receptor-mediated inhibition intact, the GABAB receptor antagonist, CGP55845 (50 nm to 1 μm) (Davies et al., 1993), was actually found to prolong the Up state (control: 2.9 ± 0.1 s vs 1 μm CGP55845: 3.8 ± 0.6 s; F(5,20) = 2.82; p < 0.05; RM ANOVA) (Fig. 2A,C,D). This increase in duration was associated with an increase in Up state intensity (control: 3.9 ± 0.4 s−1 vs 1 μm CGP55845: 5.3 ± 0.7 s−1; F(5,20) = 5.56; p < 0.01), but the effect of CGP55845 on Up state intensity was minimal compared with the effect of gabazine (Fig. 2E). Moreover, even in the presence of concentrations of gabazine that induced burst activity, application of CGP55845 prolonged the Up state-like membrane depolarizations that could still be seen (supplemental Fig. S3, available at www.jneurosci.org as supplemental material) (n = 4). Thus, these results suggest that, while GABAA receptors are essential for balancing Up states, GABAB receptors do not contribute to balancing Up states, but are rather involved in terminating persistent activity.
Synaptic stimulation triggers Down state transitions via activation of GABAB receptors
Synaptic stimulation in distal dendritic regions is known to recruit a GABAB receptor-mediated response (Dutar and Nicoll, 1988; Pérez-Garci et al., 2006), whereas the postsynaptic effect of interneurons targeting the perisomatic regions of pyramidal cells appears to be mediated exclusively by GABAA receptors (Somogyi et al., 1998). We therefore tested for layer-specific differences in the ability of synaptic stimulation to terminate Up states. Up states were triggered by synaptic stimulation in layer III (50–250 μA, 50–150 μs). Then, a second stimulus was applied at varying intervals after Up state initiation (100 ms to 1 s) via the same stimulation electrode in layer III or a second stimulation electrode placed in layer I. Although very strong stimulation in layer III (> 350 μA, > 200 μs) was able to expedite Down state transitions in 3 of 19 slices, stimulation intensity at threshold for Up state initiation did not reliably trigger Down state transitions, and there was no consistent effect of the second stimulation on Up state duration across the different ISI (Fig. 3A,B). In contrast, stimulation in layer I (50–200 μA, 50–150 μs) could readily induce Down state transitions when applied after an Up state duration of at least 250 ms (Fig. 3C,D). The ability of layer I stimulation to turn off persistent activity was caused by activation of GABAB receptors, because CGP55845 completely blocked this effect of layer I stimulation Figure 4A–D). In an attempt to rescue Up state termination, we also applied burst stimulation in layer I, which similarly failed to induce Down state transition in the presence of CGP55845 (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Thus, GABAB receptors appear essential for synaptically induced termination of Up states.
Figure 3.
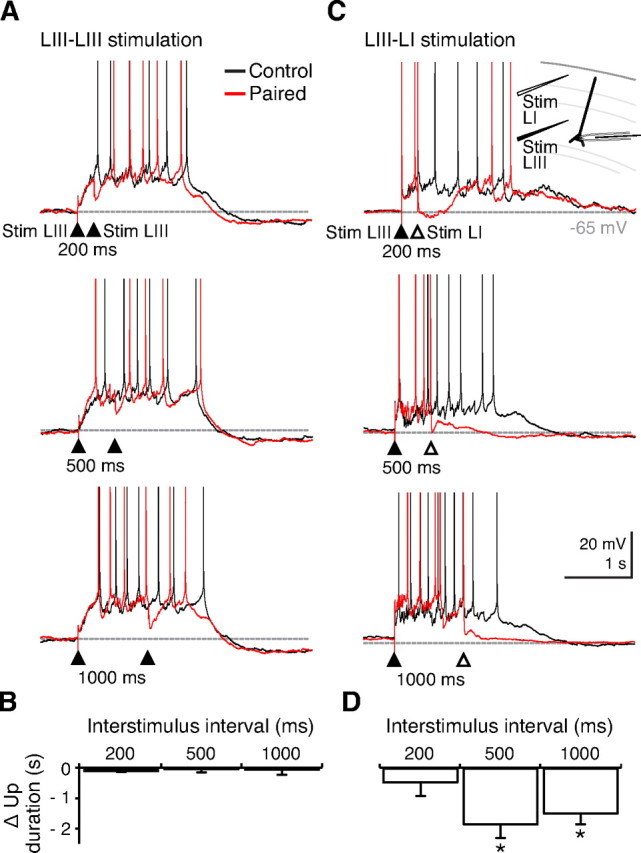
Differences in Up state shortening by layer III and layer I stimulation. A, Representative traces (black) of layer III stimulation only (filled arrowhead). Paired layer III stimulation (red) at varying interstimulus intervals (200, 500, and 1000 ms) did not significantly affect Up state duration. B, Pooled data from five slices. C, In contrast, after triggering an Up state by layer III stimulation, subsequent layer I stimulation facilitated Down state transitions. Only interstimulus intervals >250 ms triggered Down state transitions reliably. Spikes are truncated at 0 mV. Inset shows recording setup. D, Pooled data from five cells showing significant Up state shortening after layer I stimulation compared with layer III stimulation at 500 and 1000 ms after Up state initiation. Error bars indicate SEM. *p < 0.05, Student's t test. LI, Layer I; LIII, layer III; Stim, stimulation.
Figure 4.

GABAB receptor-mediated termination of Up states by layer I stimulation. A–C, Up states were initiated by stimulation in layer III (filled arrowheads; A) and terminated with subsequent stimulation in layer I (B). Termination is blocked in 1 μm CGP55845 (C). Insets show similar results on spontaneous Up states. Spikes are truncated at 0 mV. D, Summary of results, showing that Up states triggered by stimulation in layer III are significantly shortened by subsequent stimulation in layer I (interstimulus interval = 500 ms; n = 5 slices), and that this effect is blocked by CGP55845. Error bars indicate SEM. p < 0.01, Student's t test. LI, Layer I; LIII, layer III; Stim, stimulation.
Discussion
Here, we show an experimental dissociation between the roles of GABAA and GABAB receptor-mediated inhibition in controlling UDS in vitro. The activation of GABAA receptors was found to be critical for balancing Up states and preventing epileptiform activity, consistent with previous reports (Shu et al., 2003). The GABAA receptor antagonist, gabazine (50–500 nm), not only caused a concentration-dependent increase in Up state spike rate, but also a concomitant decrease in Up state duration. However, the network could still support persistent activity after partial block of GABAA receptors, because Up state duration could approach that during control conditions by addition of the GABAB receptor antagonist, CGP55845 (1 μm). CGP55845 also prolonged spontaneous and evoked Up states during control conditions, showing that activation of GABAB receptors contributes to terminating Up states. The layer and receptor specificity of these inhibitory control mechanisms suggest that different types of inhibition have distinct functions during persistently active states.
Although GABAA receptor-mediated inhibition is known to balance excitation and control spike timing during many types of physiological cortical network oscillation, defining a complementary role for GABAB receptor-mediated inhibition has proved more elusive (Scanziani, 2000). At the cellular level, dendritic GABAB receptor-mediated inhibition can have powerful effects on curbing synaptic excitation (Morrisett et al., 1991; Otmakhova and Lisman, 2004) and block regenerative calcium spikes (Pérez-Garci et al., 2006). This prompts the questions as to what functional advantage this diversity in GABA signaling pathways confers, and why GABAB receptor-mediated mechanisms do not appear to be readily engaged by local circuit activity?
Spontaneous Down state transitions have been proposed to occur because of synaptic depression (Contreras et al., 1996) or the buildup of activity-dependent potassium conductances (Sanchez-Vives and McCormick, 2000; Compte et al., 2003; Cunningham et al., 2006). We suggest that GABAB receptor-mediated conductance is a synaptically controlled potassium conductance that contributes to Down state transition. However, GABAB receptors are also found on presynaptic terminals, and could contribute to reduced synaptic drive by progressively inhibiting synaptic transmission during Up state activity. In the absence of GABAB receptor-mediated inhibition, Up states in vitro are prolonged but still finite, and thus there are clearly multiple mechanisms that can contribute to terminating persistent network activity. However, Down state transitions in vivo are remarkably synchronous between cortical regions, and may be even more precise than Up state transitions (Volgushev et al., 2006), which suggests that long-range synaptic mechanisms are also required. One suggestion has been that synchronized GABAA receptor-mediated inhibition could trigger Down states (Shu et al., 2003), but this has not been tested directly, because blocking GABAA receptors transforms persistent activity into epileptiform bursts (Sanchez-Vives and McCormick, 2000; Shu et al., 2003). One might expect Up states to be resistant to synchronized GABAA receptor-mediated inhibition, because Up states are superposed by a variety of network oscillations in vivo (Steriade, 2006), which are thought to depend on synchronized fast phasic inhibition (Hasenstaub et al., 2005). Instead, we suggest that GABAB receptor-mediated inhibition could mediate a synaptically controlled “stop” signal for persistent activity that could operate across cortical circuits, in parallel with GABAA receptor-mediated mechanisms controlling spike rate and synchrony.
The termination of persistent activity via GABAB receptor activation in the superficial cortical layers might also explain why high intensity intracortical stimulation in layer II/III in vitro (Rigas and Castro-Alamancos, 2007), and transcranial magnetic stimulation in humans subjects (Massimini et al., 2007), are effective stimuli for evoking Down states, but not Up states, in the neocortex. Nevertheless, the precise patterns of interneuronal firing that would recruit these pathways endogenously to control network activity remain unknown. Single action potentials in neurogliaform cells have been shown to be capable of evoking postsynaptic GABAB receptor-mediated inhibitory events (Tamás et al., 2003), but it is generally assumed that synaptically released GABA must be pooled or accumulated to activate GABAB receptors (Isaacson et al., 1993; Scanziani, 2000; Ulrich and Bettler, 2007). It is possible that GABAB receptors could both detect and control the patterns of activity across cortical microcircuits, and thus appear less powerful in controlling spontaneous network activity in reduced cortical preparations in vitro. Future experiments will be necessary to determine whether the mechanisms identified here for the GABAB receptor-mediated control of Down state transitions underlies their long-range synchronization in vivo.
Footnotes
This research was supported by the Wellcome Trust Ion Channels and Diseases Initiative (OXION). M.M.K. holds a Wellcome Trust Prize Studentship.
References
- Anderson J, Lampl I, Reichova I, Carandini M, Ferster D. Stimulus dependence of two-state fluctuations of membrane potential in cat visual cortex. Nat Neurosci. 2000;3:617–621. doi: 10.1038/75797. [DOI] [PubMed] [Google Scholar]
- Compte A. Computational and in vitro studies of persistent activity: edging towards cellular and synaptic mechanisms of working memory. Neuroscience. 2006;139:135–151. doi: 10.1016/j.neuroscience.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Compte A, Sanchez-Vives MV, McCormick DA, Wang XJ. Cellular and network mechanisms of slow oscillatory activity (<1 Hz) and wave propagations in a cortical network model. J Neurophysiol. 2003;89:2707–2725. doi: 10.1152/jn.00845.2002. [DOI] [PubMed] [Google Scholar]
- Contreras D, Timofeev I, Steriade M. Mechanisms of long-lasting hyperpolarizations underlying slow sleep oscillations in cat corticothalamic networks. J Physiol. 1996;494:251–264. doi: 10.1113/jphysiol.1996.sp021488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham MO, Pervouchine DD, Racca C, Kopell NJ, Davies CH, Jones RS, Traub RD, Whittington MA. Neuronal metabolism governs cortical network response state. Proc Natl Acad Sci U S A. 2006;103:5597–5601. doi: 10.1073/pnas.0600604103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies CH, Pozza MF, Collingridge GL. CGP 55845A: a potent antagonist of GABAB receptors in the CA1 region of rat hippocampus. Neuropharmacology. 1993;32:1071–1073. doi: 10.1016/0028-3908(93)90073-c. [DOI] [PubMed] [Google Scholar]
- Dutar P, Nicoll RA. A physiological role for GABAB receptors in the central nervous system. Nature. 1988;332:156–158. doi: 10.1038/332156a0. [DOI] [PubMed] [Google Scholar]
- Egorov AV, Hamam BN, Fransén E, Hasselmo ME, Alonso AA. Graded persistent activity in entorhinal cortex neurons. Nature. 2002;420:173–178. doi: 10.1038/nature01171. [DOI] [PubMed] [Google Scholar]
- Haider B, Duque A, Hasenstaub AR, McCormick DA. Neocortical network activity in vivo is generated through a dynamic balance of excitation and inhibition. J Neurosci. 2006;26:4535–4545. doi: 10.1523/JNEUROSCI.5297-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hájos N, Ellender TJ, Zemankovics R, Mann EO, Exley R, Cragg SJ, Freund TF, Paulsen O. Maintaining network activity in submerged hippocampal slices: importance of oxygen supply. Eur J Neurosci. 2009;29:319–327. doi: 10.1111/j.1460-9568.2008.06577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasenstaub A, Shu Y, Haider B, Kraushaar U, Duque A, McCormick DA. Inhibitory postsynaptic potentials carry synchronized frequency information in active cortical networks. Neuron. 2005;47:423–435. doi: 10.1016/j.neuron.2005.06.016. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Solís JM, Nicoll RA. Local and diffuse synaptic actions of GABA in the hippocampus. Neuron. 1993;10:165–175. doi: 10.1016/0896-6273(93)90308-e. [DOI] [PubMed] [Google Scholar]
- Isomura Y, Sirota A, Ozen S, Montgomery S, Mizuseki K, Henze DA, Buzsáki G. Integration and segregation of activity in entorhinal-hippocampal subregions by neocortical slow oscillations. Neuron. 2006;52:871–882. doi: 10.1016/j.neuron.2006.10.023. [DOI] [PubMed] [Google Scholar]
- MacLean JN, Watson BO, Aaron GB, Yuste R. Internal dynamics determine the cortical response to thalamic stimulation. Neuron. 2005;48:811–823. doi: 10.1016/j.neuron.2005.09.035. [DOI] [PubMed] [Google Scholar]
- Major G, Tank D. Persistent neural activity: prevalence and mechanisms. Curr Opin Neurobiol. 2004;14:675–684. doi: 10.1016/j.conb.2004.10.017. [DOI] [PubMed] [Google Scholar]
- Massimini M, Ferrarelli F, Esser SK, Riedner BA, Huber R, Murphy M, Peterson MJ, Tononi G. Triggering sleep slow waves by transcranial magnetic stimulation. Proc Natl Acad Sci U S A. 2007;104:8496–8501. doi: 10.1073/pnas.0702495104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrisett RA, Mott DD, Lewis DV, Swartzwelder HS, Wilson WA. GABAB-receptor-mediated inhibition of the N-methyl-d-aspartate component of synaptic transmission in the rat hippocampus. J Neurosci. 1991;11:203–209. doi: 10.1523/JNEUROSCI.11-01-00203.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA, Malenka RC, Kauer JA. Functional comparison of neurotransmitter receptor subtypes in mammalian central nervous system. Physiol Rev. 1990;70:513–565. doi: 10.1152/physrev.1990.70.2.513. [DOI] [PubMed] [Google Scholar]
- Otmakhova NA, Lisman JE. Contribution of Ih and GABAB to synaptically induced afterhyperpolarizations in CA1: a brake on the NMDA response. J Neurophysiol. 2004;92:2027–2039. doi: 10.1152/jn.00427.2004. [DOI] [PubMed] [Google Scholar]
- Pérez-Garci E, Gassmann M, Bettler B, Larkum ME. The GABAB1b isoform mediates long-lasting inhibition of dendritic Ca2+ spikes in layer 5 somatosensory pyramidal neurons. Neuron. 2006;50:603–616. doi: 10.1016/j.neuron.2006.04.019. [DOI] [PubMed] [Google Scholar]
- Petersen CC, Hahn TT, Mehta M, Grinvald A, Sakmann B. Interaction of sensory responses with spontaneous depolarization in layer 2/3 barrel cortex. Proc Natl Acad Sci U S A. 2003;100:13638–13643. doi: 10.1073/pnas.2235811100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigas P, Castro-Alamancos MA. Thalamocortical Up states: differential effects of intrinsic and extrinsic cortical inputs on persistent activity. J Neurosci. 2007;27:4261–4272. doi: 10.1523/JNEUROSCI.0003-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph M, Pospischil M, Timofeev I, Destexhe A. Inhibition determines membrane potential dynamics and controls action potential generation in awake and sleeping cat cortex. J Neurosci. 2007;27:5280–5290. doi: 10.1523/JNEUROSCI.4652-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Vives MV, McCormick DA. Cellular and network mechanisms of rhythmic recurrent activity in neocortex. Nat Neurosci. 2000;3:1027–1034. doi: 10.1038/79848. [DOI] [PubMed] [Google Scholar]
- Scanziani M. GABA spillover activates postsynaptic GABAB receptors to control rhythmic hippocampal activity. Neuron. 2000;25:673–681. doi: 10.1016/s0896-6273(00)81069-7. [DOI] [PubMed] [Google Scholar]
- Shu Y, Hasenstaub A, McCormick DA. Turning on and off recurrent balanced cortical activity. Nature. 2003;423:288–293. doi: 10.1038/nature01616. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Tamás G, Lujan R, Buhl EH. Salient features of synaptic organisation in the cerebral cortex. Brain Res Brain Res Rev. 1998;26:113–135. doi: 10.1016/s0165-0173(97)00061-1. [DOI] [PubMed] [Google Scholar]
- Steriade M. Grouping of brain rhythms in corticothalamic systems. Neuroscience. 2006;137:1087–1106. doi: 10.1016/j.neuroscience.2005.10.029. [DOI] [PubMed] [Google Scholar]
- Steriade M, Nuñez A, Amzica F. A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci. 1993;13:3252–3265. doi: 10.1523/JNEUROSCI.13-08-03252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamás G, Lorincz A, Simon A, Szabadics J. Identified sources and targets of slow inhibition in the neocortex. Science. 2003;299:1902–1905. doi: 10.1126/science.1082053. [DOI] [PubMed] [Google Scholar]
- Timofeev I, Grenier F, Bazhenov M, Sejnowski TJ, Steriade M. Origin of slow cortical oscillations in deafferented cortical slabs. Cereb Cortex. 2000;10:1185–1199. doi: 10.1093/cercor/10.12.1185. [DOI] [PubMed] [Google Scholar]
- Ulrich D, Bettler B. GABAB receptors: synaptic functions and mechanisms of diversity. Curr Opin Neurobiol. 2007;17:298–303. doi: 10.1016/j.conb.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Volgushev M, Chauvette S, Mukovski M, Timofeev I. Precise long-range synchronization of activity and silence in neocortical neurons during slow-wave oscillations. J Neurosci. 2006;26:5665–5672. doi: 10.1523/JNEUROSCI.0279-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wermuth CG, Bourguignon JJ, Schlewer G, Gies JP, Schoenfelder A, Melikian A, Bouchet MJ, Chantreux D, Molimard JC, Heaulme M, et al. Synthesis and structure-activity relationships of a series of aminopyridazine derivatives of γ-aminobutyric acid acting as selective GABA-A antagonists. J Med Chem. 1987;30:239–249. doi: 10.1021/jm00385a003. [DOI] [PubMed] [Google Scholar]