

Abstract
Importance
Numerically, the most important genetic risk factor for the development of Parkinson disease (PD) is the presence of a glucocerebrosidase gene (GBA) mutation.
Objective
The purpose of this study was the longitudinal clinical evaluation of a GBA mutation positive cohort and the evolution of the prodromal features of PD.
Design
Individuals were participants in a study of the aetiology and prodrome of PD and have been re-evaluated in this 2 year follow-up report.
Setting
Clinic-based.
Participants
Type 1 GD patients and heterozygous GBA mutation positive carriers were recruited in 2010 from the Lysosomal Storage Disorder Unit at the Royal Free Hospital, London. Thirty previously diagnosed Type 1 GD patients, twenty-eight heterozygous GBA mutation carriers and twenty-six genetically unrelated controls were included. For both GD and carrier subjects, exclusion criteria included a diagnosis of PD or dementia and for controls, any existing neurological disease.
Main Outcome(s) and Measure(s)
Assessment was performed for clinical markers including hyposmia, rapid eye movement sleep behaviour disorder (RBD), depression, autonomic dysfunction, cognitive function and parkinsonian motor signs (UPDRS part III).
Results
Over 2 years, depression scores were significantly worse in heterozygotes (P = ·01), RBD scores were significantly worse in GD patients (P < ·001) and heterozygotes (P < ·001), and UPDRS III scores were significantly worse in GD patients (P < ·001) and heterozygotes (P < ·001). In controls, there was a small but significant deterioration in the UPDRS II score (P = ·006). At 2 years, olfactory and cognitive assessment scores were lower in GD patients and heterozygotes compared to controls, but did not differ significantly from baseline. When the results from GD patients and heterozygotes were combined, there was a significant deterioration from baseline in RBD, BDI, UPDRS II and III scores (in all, P < ·01), and at 2 years, significant differences in UPSIT, UMSARS, MMSE, MoCA, UPDRS II and UPDRS III scores when compared to controls (in all, P < ·05).
Conclusions and Relevance
This study indicates that as a group, GBA mutation positive individuals show deterioration in clinical markers consistent with the prodrome of PD. Within this group, 10% appear to be evolving at a more rapid rate.
Keywords: Parkinson’s disease, Gaucher disease, neurodegeneration
INTRODUCTION
Homozygous GBA mutations cause Gaucher disease (GD), a lysosomal storage disorder. It is presently estimated that homozygous or heterozygous GBA mutations confer an increased risk for Parkinson disease (PD) of 20-30 fold1,2 and at least 7% of PD patients have GBA mutations2,3, and this is higher in the Ashkenazi Jewish population4. The penetrance of GBA mutation carriers to develop PD has been estimated as 13·7% at age 60 years and 29·7% at age 80 years5, and so a method to determine individual risk for PD expression in this population would be very valuable. In addition, those with dementia with Lewy bodies (DLB) are 8 times more likely to carry a mutation in GBA than healthy controls, suggesting a role for GBA mutations in other Lewy body disorders6.
For any neuroprotective treatment or disease modifying therapy to be most effective, PD should be detected at as early a stage as possible. The deposition of α-synuclein is not restricted to the brain, with deposits found in the olfactory bulb, peripheral nervous system, enteric nervous system (ENS), cardiac, and pelvic plexuses, etc7. This pathology probably underlies the early non-motor manifestations of PD, which may precede the onset of more typical PD motor symptoms by several years8.
Candidate biomarkers have been proposed, and may be useful objective measures for the early detection of PD9,10. In this study, we have used early clinical markers to quantify non-motor symptoms such as hyposmia, rapid eye movement sleep behaviour disorder (RBD), depression, cognition, and autonomic dysfunction.
The aim of this study was to provide longitudinal data on a GBA positive cohort at high risk for the development of PD, and to identify biomarkers or symptoms indicating progression to early PD. The first clinical evaluation of this cohort has been published previously11 and the results presented here represent the two year follow up.
METHODS
Participants
Type 1 GD patients were recruited from the Lysosomal Storage Disorder Unit at the Royal Free London NHS Foundation Trust in 2010. Potential heterozygous GBA mutation positive carrier relatives (parents: 78·6%; siblings: 10·7%; children >21 years: 10·7%) and genetically unrelated controls (spouses/partners) were identified by taking a detailed family history from each GD patient, and recruited with consent. Individuals were also recruited from the UK Gaucher Disease Association. In all, this unique cohort included one hundred and thirty-five participants. Among them, ninety participants have been followed longitudinally with target follow-up assessments at two year intervals beginning in 2012. For both GD and carrier subjects, exclusion criteria included a diagnosis of PD or dementia and for controls, any existing neurological disease. The diagnosis of PD was made according to the UK Parkinson’s Disease Society Brain Bank Criteria12. Dementia was diagnosed according to DSM-IV criteria in patients with a Mini-Mental State Examination score of ≤24. The GBA mutation status in all participants was confirmed by Sanger sequencing of the GBA gene, as previously described11. The senior researcher was blinded to genotype. The study was approved by the Hampstead Research Ethics Committee (reference number 10/H0720/21). All individuals provided written informed consent.
Follow-up evaluation
Of ninety individuals who were evaluated at baseline (2010-2011)11, four participants (4·4%) were lost to follow-up because they either declined to participate (n=2) or were uncontactable (n=2). In addition, two deaths (2·2%) had occurred: one caused by pneumonia, and one by breast carcinoma. Therefore, eighty-four (93·3%) participants (thirty previously diagnosed Type 1 GD patients, twenty-eight heterozygous GBA mutation carriers, and twenty-six controls) completed the follow-up evaluation that comprised: a structured clinical work-up, a standardised clinical history, complete neurological assessment including the Unified Parkinson’s Disease Rating Scale activities of daily living and motor subscale (UPDRS parts II and III), olfactory function using the University of Pennsylvania Smell Identification Test (UPSIT), cognitive function using the Mini-Mental State Examination (MMSE) and Montreal Cognitive assessment (MoCA), RBD with the RBD Questionnaire (RBDQ), depression using the Beck’s Depression Inventory (BDI), and autonomic dysfunction using a subscale of the Unified Multiple System Atrophy Rating Scale (UMSARS). Anosmia was interpreted using age- and sex-adjusted normative scores (www.sensonics.com). All participants were examined independently by a movement disorders-trained physician (M.B.). All procedures were performed and scored identically at follow-up to those carried out at baseline. A senior neurologist expert on movement disorders (A.H.V.S.) evaluated individuals where there was a significant difference between UPDRS scores measured at follow-up and at baseline.
Statistical analysis
The data was analysed using IBM SPSS Statistics (version 21). To assess the differences between the group means across the two different time points, we performed a two-way ANCOVA with factors Group (e.g. Gaucher vs. Carrier vs. Control) and Time (Time 1 vs. Time 2). The covariates age, gender, education, and family relationship were added to the design matrix, in order to account for differences in these between the groups. Post-hoc tests were used to compare the groups at follow-up. Paired t-tests were used to compare the scores within each group before and after follow up. Differences in age, sex, and ethnicity between groups were checked using the One-way ANOVA and the Chi-squared test. We also accounted for performing multiple statistical tests across our dependent variables (UPSIT, UMSARS, RBDQ, MMSE, MoCA, UPDRS II, UPDRS III, BDI) by defining a significance threshold for statistical tests of P < ·05, and correcting this for multiple comparisons using the Benjamini-Hochberg FDR (False Discovery Rate). In brief, this procedure involves ordering all P values in ascending order and applying a sequential threshold.
RESULTS
The eighty-four participants (40 men [47·6%]) had a mean follow-up duration of 1·9±0·2 years (range, 1·5-2·3 years). The demographic, clinical and genetic characteristics, with statistical comparisons, of the cohort are shown in Table I. Participants with Type 1 GD did not differ significantly from heterozygous GBA positive carriers or controls in terms of age, sex, and ethnicity (One-way ANOVA and Chi-squared test, in all P > ·05). Both Type 1 GD patients and heterozygous GBA mutation carriers were significantly more likely to have a family history of PD than controls (P = ·03). As described previously11, the most common genotype in GD patients was N370S/L444P (11/30; 36·7%). No GD patients had features of Type III disease such as generalized seizures or progressive myoclonic epilepsy. In carriers, the most common genotype was N370S (14/28; 50%).
Table I.
Demographic, clinical and genetic characteristics of the study cohort
| Characteristic | Type 1 GD patients (n=30) | Heterozygous GBA mutation carriers (n=28) | Controls (n=26) | P value |
|---|---|---|---|---|
| Age, years | 61·0 (2·1) | 63·6 (2·0) | 61·7 (2·2) | ·19a |
| Gender (F/M) | 16/14 | 16/12 | 12/14 | ·29b |
| Ethnicity (Ashkenazi/White British) | 10/20 | 5/23 | 6/20 | ·38b |
| Family history of PD, (%) | 16·7 | 7·1 | 0·0 | ·03bc |
| Most frequent genotype | N370S/L444P | N370S | - | - |
| GD treatment (ERT/SRT/none) | 25/2/3 | - | - | - |
Results are presented as mean and SEM. Significance was taken at the 5% level.
Abbreviations: ERT Enzyme Replacement Therapy. SRT Substrate Reduction Therapy.
One-way ANOVA.
Chi-squared test.
Significant difference
GBA mutation positive individuals show significant deterioration in clinical markers
The results of prodromal clinical features of PD at baseline and follow-up are reported in Table II (see also Figure 1 and Figure 2). Please refer to Table II for the exact P values. There was a significant deterioration in RBDQ, UPDRS II and III scores for GD patients over the mean two years of follow-up. Over the same period, the GBA mutation carriers showed a significant deterioration in RBDQ, UPDRS II and III, and BDI scores. There was a marginal but significant deterioration only in the UPDRS II score in the matched controls. There was no difference between baseline and follow-up scores for all groups for assessments of olfaction, cognition and autonomic dysfunction.
Table II.
Baseline and follow-up clinical markers in a group comparison between Type 1 GD patients, carriers and controls.
| Type 1 GD patients (n=30) | Heterozygous GBA mutation carriers (n=28) | Controls (n=26) |
P (between)b |
||||
|---|---|---|---|---|---|---|---|
| P1 | P2 | P3 | |||||
|
|
|||||||
| UPSIT | Baseline | 32·57 (0·96) | 31·11 (0·93) | 35·32 (0·40) | |||
| Follow-up | 31·21 (0·98) | 30·22 (1·10) | 33·95 (0·62) | ·003c | ·001c | ·52 | |
| P (within)a | ·03 | ·29 | ·13 | ||||
| UMSARS | Baseline | 0·40 (0·15) | 0·37 (0·15) | 0·08 (0·06) | |||
| Follow-up | 0·63 (0·16) | 0·53 (0·16) | 0·13 (0·07) | ·004c | ·02c | 1·00 | |
| P (within)a | ·11 | ·59 | ·32 | ||||
| RBDQ | Baseline | 0·93 (0·31) | 0·10 (0·10) | 0·25 (0·14) | |||
| Follow-up | 2·93 (0·55) | 2·30 (0·40) | 1·08 (0·30) | ·04 | 1·00 | ·23 | |
| P (within)a | <·001c | <·001c | ·07 | ||||
| MMSE | Baseline | 29·23 (0·17) | 29·23 (0·18) | 29·28 (0·16) | |||
| Follow-up | 28·40 (0·48) | 28·63 (0·32) | 29·50 (0·21) | ·01c | ·03c | 1·00 | |
| P (within)a | ·08 | ·05 | ·30 | ||||
| MoCA | Baseline | 25·93 (0·53) | 25·55 (0·58) | 27·32 (0·23) | |||
| Follow-up | 26·33 (0·75) | 26·21 (0·57) | 27·73 (0·26) | ·001c | ·001c | 1·00 | |
| P (within)a | ·07 | ·38 | ·20 | ||||
| UPDRS II | Baseline | 1·45 (0·82) | 0·33 (0·21) | 0·00 (0·00) | |||
| Follow-up | 2.72 (0·66) | 1·33 (0·30) | 0·58 (0·19) | <·003c | 1·00 | ·009c | |
| P (within)a | ·003c | <·001c | ·006c | ||||
| UPDRS III | Baseline | 4·29 (1·45) | 1·97 (0·65) | 0·21 (0·17) | |||
| Follow-up | 7·82 (1·91) | 4·50 (0·75) | 0·92 (0·37) | <·001c | ·04 | ·006c | |
| P (within)a | <·001c | <·001c | ·06 | ||||
| BDI | Baseline | 2·68 (1·78) | 0·65 (0·41) | 0·33 (0·33) | |||
| Follow-up | 5·84 (1·14) | 2·88 (0·68) | 0·58 (0·43) | ·04 | 1·00 | ·03c | |
| P (within)a | ·04 | ·01c | ·11 | ||||
Abbreviations: UPSIT Smell Identification Test, UMSARS Unified Multiple System Atrophy Rating Scale, MoCA Montreal Cognitive assessment, MMSE Mini-Mental State Examination, RBDQ Rapid Eye Movement Sleep Behaviour Disorder Questionnaire, UPDRS Unified Parkinson’s Disease Rating Scale, BDI Becks Depression Inventory.
Results are presented as mean and SEM. Significance was taken at the 5% level for all variables. Only values which survived multiple comparisons with the FDR procedure were denoted significant.
Reported P values compare the mean values for clinical markers within groups (baseline and follow-up) and between groups (Type 1 GD, carriers and controls) at follow-up.
Controls versus Type 1 GD patients.
Controls versus heterozygote GBA mutation carriers.
Type 1 GD patients versus heterozygote GBA mutation carriers.
Paired t-test.
Two-way ANCOVA with Bonferroni correction.
Statistically significant difference.
Figure 1.
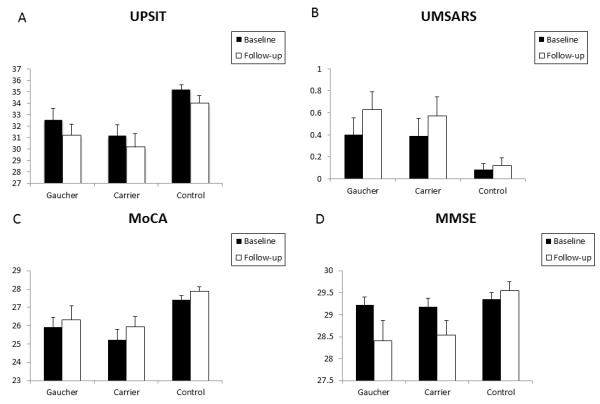
Clinical markers show progression in GBA mutation positive individuals in a two-year follow-up study.
Figures demonstrate mean baseline and follow-up scores for olfaction (A), mean baseline and follow-up scores for autonomic dysfunction (B), mean baseline and follow-up MoCA scores (C) and mean baseline and follow-up MMSE scores (D) for Type 1 GD patients and heterozygous GBA positive carriers compared to controls. Means are plotted together with the SEM.
Figure 2.

Clinical markers show progression in GBA mutation positive individuals in a 2-year follow-up study.
Figures demonstrate a statistically significant increase in depressive symptoms for carriers at the follow-up evaluation (F), a statistically significant increase in mean follow-up RBDQ scores (E) and UPDRS III scores (H) in Type 1 GD patients and heterozygous GBA mutation positive carriers compared to controls, and a statistically significant increase in mean follow-up UPDRS II scores in Type 1 GD patients, heterozygous GBA mutation positive carriers, and controls (G). Means are plotted together with the SEM.
At 2 years follow-up, GD patients showed a significant difference in mean UPSIT, MMSE, MoCA, UPDRS II and III scores when compared to controls. Similarly, at 2 years, GBA mutation carriers showed a significant difference in mean follow-up UPSIT, MMSE, and MoCA scores when compared to controls. When the GD patients and GBA mutation carriers were compared at baseline, there was a significant difference in the mean BDI score. At 2 years follow-up, GD patients demonstrated significantly worse mean BDI, UPDRS II and III scores compared to carriers. There was no significant difference between mean UPSIT, UMSARS, MMSE, MoCA, or RBDQ scores in GD patients and carriers at follow-up.
When the results from individuals with homozygous or heterozygous mutations in GBA were combined in a secondary, pooled analysis (see Table III and eFigures 1 and 2 in the Supplemental material), there was a significant deterioration in mean RBDQ, BDI, UPDRS II and III scores in GBA mutation positive individuals over the two years of follow-up. At baseline, GBA mutation positive individuals showed significant differences in mean UPSIT and MoCA scores when compared to controls11. At 2 years follow-up, GBA mutation positive individuals showed significant differences in mean UPSIT, UMSARS, MMSE, MoCA, UPDRS II and UPDRS III scores when compared to controls.
Table III.
Baseline and follow-up clinical markers in a pooled analysis comparing all GBA mutation positive individuals versus controls.
| Type 1 GD patients and Heterozygous GBA mutation carriers combined scores (n=58) | Controls (n=26) |
P (between)b |
||
|---|---|---|---|---|
| P 1 | ||||
|
|
||||
| UPSIT | Baseline | 31·85 (0·67) | 35·32 (0·40) | |
| Follow-up | 30·71 (0·73) | 33·95 (0·62) | <·001c | |
| P (within)a | ·02 | ·13 | ||
| UMSARS | Baseline | 0·38 (0·11) | 0·08 (0·06) | |
| Follow-up | 0·58 (0·11) | 0·13 (0·07) | ·001c | |
| P (within)a | ·15 | ·32 | ||
| RBDQ | Baseline | 0·51 (0·20) | 0·25 (0·14) | |
| Follow-up | 2·63 (0·33) | 1·08 (0·30) | ·06 | |
| P (within) a | <·001c | ·07 | ||
| MMSE | Baseline | 29·23 (0·12) | 29·28 (0·16) | |
| Follow-up | 28·51 (0·27) | 29·50 (0·21) | ·002c | |
| P (within)a | ·02 | ·30 | ||
| MoCA | Baseline | 25·7 (0·38) | 27·32 (0·23) | |
| Follow-up | 26·3 (0·45) | 27·73 (0·26) | <·001c | |
| P (within)a | ·06 | ·20 | ||
| UPDRS II | Baseline | 0·88 (0·39) | 0·00 (0·00) | |
| Follow-up | 2·01 (0·36) | 0·58 (0·19) | ·02c | |
| P (within)a | <·001c | ·006c | ||
| UPDRS III | Baseline | 3·09 (0·75) | 0·21 (0·17) | |
| Follow-up | 6·10 (0·95) | 0·92 (0·37) | <·001c | |
| P (within)a | <·001c | ·06 | ||
| BDI | Baseline | 1·72 (0·94) | 0·33 (0·33) | |
| Follow-up | 4·44 (0·71) | 0·58 (0·43) | ·09 | |
| P (within)a | ·002c | ·11 | ||
Abbreviations: UPSIT Smell Identification Test, UMSARS Unified Multiple System Atrophy Rating Scale, MoCA Montreal Cognitive assessment, MMSE Mini-Mental State Examination, RBDQ Rapid Eye Movement Sleep Behaviour Disorder Questionnaire, UPDRS Unified Parkinson’s Disease Rating Scale, BDI Becks Depression Inventory.
Results are presented as mean and SEM. Significance was taken at the 5% level for all variables. Only values which survived multiple comparisons with the FDR procedure were denoted significant.
Reported P values compare the mean values for clinical markers within groups (baseline and follow-up) and between groups (Controls and GBA mutation positive individuals) at follow-up.
Controls versus GBA mutation positive individuals.
Paired t-test.
Two-way ANCOVA with Bonferroni correction.
Statistically significant difference.
Specific GD patients and GBA heterozygotes show parkinsonian motor signs and significant deterioration across more than one clinical marker
At baseline, three GD patients had parkinsonian motor signs, but insufficient for a diagnosis of PD. As described previously11, GD05 (male, 78 years old, Ashkenazi Jewish) had bilateral rigidity with activation manoeuvre, asymmetric bradykinesia of all limbs, and gait impairment. GD18 (male, 83 years old, Ashkenazi Jewish) had left arm rest tremor and bilateral arm rigidity with activation manoeuvre. GD27 (male, 69 years old, White British) had flexed posture, bilateral rigidity, and postural and kinetic tremor of the upper limbs. At follow-up, the parkinsonian signs present at baseline in these study subjects had worsened but did not meet the diagnostic criteria for PD13. GD05 had developed a tremor in both hands (intermittent, present at rest and worse on intention). GD18 now had bilateral rigidity without activation manoeuvre and gait impairment. GD27 had developed a head tremor and the postural and kinetic tremor of the upper limbs had worsened (now present at rest). In addition, one subject that did not have parkinsonian signs at baseline had developed them at follow-up. GD11 (male, 73 years old, Ashkenazi Jewish) had developed a very slight tremor of his right thumb (non pill rolling) present at rest but with no other features of parkinsonism.
Similarly, two GBA carriers had parkinsonian motor signs at baseline. As described previously11, C17 (female, 78 years old, White British) had bilateral rigidity, mask-like facies, and bradykinesia while C31 (male, 78 years old, White British) had masked facies, bilateral rigidity with activation manoeuvre, left arm kinetic tremor, and flexed posture. At follow-up, the parkinsonian signs present in these study subjects remained unchanged from baseline.
When specific GD patients and GBA heterozygotes with features of parkinsonism (6/58; 10·3%) were excluded, the follow-up data remained significant. The remaining GD patients and carriers (52/58; 89·7%) still showed a significant deterioration in RBDQ, UPDRS II and III, and BDI scores after two years (see eTable 1 in the supplemental material).
Premotor signs present at baseline that could predict parkinsonian motor signs
When clinical markers were compared between specific GD patients (GD05, GD11, GD18, and GD27) and GBA heterozygotes (C17 and C31) with parkinsonian motor signs and GBA mutation positive individuals without features of parkinsonism, there were significant differences in age (P = ·002) and cognition (P = ·009) at baseline (see eTable 2 and 3 in the Supplemental material). Baseline UPSIT scores were also noted to be lower in those individuals with features of parkinsonism but this difference did not reach statistical significance.
DISCUSSION
This study was designed to investigate the progression of clinical biomarkers in a cohort of individuals at high risk for PD. Our results demonstrate that clinical features associated with pre-motor PD, and motor features of PD have both evolved since the initial testing, and support the hypothesis that some GBA mutation positive individuals within this cohort are exhibiting clinical features of early neurodegeneration.
Olfactory abnormalities are found in those with PD with mutations in GBA14, but are not a reported feature of GD or its treatment. It has been proposed that the earliest α-synuclein changes occur in the dorsal motor nucleus of the vagus and olfactory bulb8, and evidence suggests that smell impairment is not simply a consequence of aging but rather is a prodromal phenomenon that may predict PD15. In the cohort studied, both Type 1 GD patients and heterozygous mutation carriers were, as a group, hyposmic at baseline11. At 2 years, follow-up olfactory scores in GD patients and heterozygous carriers remained significantly lower than those reported for controls, but unchanged from baseline. This could reflect the short length of the follow-up period, considering olfactory impairment may progress slowly.
An impaired sense of smell does appear to correlate with other modalities in the prodromal phase of PD e.g. RBD16. Due to its high specificity and long latency to clinical disease, RBD is one of the strongest clinical predictors of neurodegenerative disease, and a potential prodromal marker for preventative therapy17. The RBDQ carries a high sensitivity, and in those without existing neurological or sleep disorders, a high specificity, and therefore, represents a good tool to detect subjects with RBD18. We did identify a significantly increased frequency of symptoms of RBD at the follow-up assessment in GBA mutation positive individuals compared to controls. It is arguable whether a score of five or six should be the cut off point for a scale that is structured to determine if there is RBD or not. It should be noted, however, that the proportion of RBDQ scores greater than five was higher in GBA mutation positive individuals compared to controls at follow-up, albeit this difference did not reach significance (P = 0·39, Chi-squared test).
Depression can precede the onset of the motor symptoms of PD and is a presenting complaint in 12-22% of patients19. There was an increase in the report of depressive symptoms in GBA mutation positive individuals at follow-up. Patients with GD can exhibit moderate to severe psychological complications, similar to patients with other long-term chronic illnesses20. In addition, BDI scores of 1-10 are consistent with minimal depression and the specificity of depression alone as a clinical marker of prodromal PD is low, but may be usefully combined with other features21.
Mild cognitive impairment can occur as a prodrome to parkinsonism22 or DLB23. There are several lines of evidence now for greater cognitive impairment in those with established GBA-PD versus sporadic PD24,25,26, and this may reflect a higher burden of LB disease in GBA-related parkinsonism27,28. Interestingly, in a subgroup of six GBA mutation positive individuals with parkinsonian motor signs, mild cognitive impairment (MoCA score </=24 in 5/6, 83·3%) was the main premotor sign present at baseline that could have predicted their motor deterioration. Compared to controls, the remaining GBA mutation positive individuals demonstrated significantly lower MMSE and MoCA scores at follow-up, albeit these were unchanged from baseline and still within the normal range for cognitive function.
Control subjects showed a small but significant change in the UPDRS part II score from baseline. Particular aspects of the UPDRS II that had worsened in controls included: 2.11 (getting out of a bed, a car or a deep chair), 2.12 (walking e.g. use of a walking aid), 2.5 (dressing e.g. help with buttons). Subjective complaints of stiffness, tremors, and imbalance are associated with an increased risk for the development of PD29. However, we note that the UPDRS part II was not designed or validated as a tool for activities of daily living (ADL) in aging controls. We believe what drove the changes in the controls were a small group of individuals (n=6) who were older (mean age 70·5 years (range 62·6-77·8). When compared, a significantly higher follow-up UPDRS part II score in GD patients distinguished these individuals from age-matched controls.
There were some GBA mutation positive individuals (10%) with significant motor findings identified using the UPDRS part III, which did not overlap with normal physiology (e.g. bilateral postural tremor) or existing bone/joint abnormalities. These individuals did not meet the diagnostic criteria for PD but could represent a subgroup of GBA mutation positive individuals that are progressing towards clinical PD.
We considered the effect of concurrent medications. The majority of Type 1 GD patients (83%) were receiving enzyme replacement therapy (ERT). This does not cross the blood brain barrier and has no reported neurological side effects. Furthermore, ERT has no known impact on dysautonomia. Substrate reduction therapy (SRT) can induce memory problems30. However, only 2 GD patients were receiving SRT when evaluated at baseline and at follow-up, and neither had cognitive impairment.
Our study is the first to undertake the longitudinal follow-up of a large cohort of GBA mutation positive individuals, prior to the development of PD. Much of the work published thus far in the literature has focussed on patients with established PD. This has been essential to make important comparisons between sporadic PD and GBA-related parkinsonism and to observe subtle differences. The opportunity to follow patients prospectively within a unique at-risk cohort such as this is essential for defining the optimal time to intervene with neuroprotective therapy.
One limitation of the study was that not all investigators were blind to the genetic status of individuals. To minimise any observer bias, standardised scores were used and all follow-up data were re-examined. Other potential criticisms are the use of prodromal markers and their sensitivity, specificity, and positive and negative predictive values. The presence of clinical markers alone may be insufficient accurately to predict a neurodegenerative disorder in the majority of cases. However, clinical markers may be used in combination with other biochemical or imaging markers for prodromal PD to develop a more reliable method for PD prediction.
The data from this cohort suggest that hyposmia is the earliest and most sensitive prodromal marker. Cognitive impairment is also an early feature and this may relate to the increased cognitive impairment observed with GBA-PD. Symptoms of RBD, the most specific clinical marker, are now present in GBA mutation positive individuals. Depressive symptoms have also surfaced but must be interpreted with some caution considering their low specificity as a marker for PD. There has also and perhaps most importantly, been a significant decline on the UPDRS, which together with impaired RBD and depression, suggest that clinical markers in some individuals of this GBA mutation positive cohort have evolved, in a pattern consistent with the clinical prodrome of PD.
Supplementary Material
ACKNOWLEDGEMENTS
Study funding: The research study was funded by Wellcome Trust/MRC Joint Call in Neurodegeneration Award (WT089698) and supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre.
Role of the funding source
The funder had no role in determining the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Full financial disclosures of all authors for the past year
Anthony Schapira.
| Employment | None | Stock Ownership | None |
| Affiliations | None | Expert Testimony | None |
| Grants/funding | Medical Research Council (UK) Wellcome Trust Parkinson UK | Royalties | Elsevier, OUP |
| Consultancies | Zambon, Lundbeck | Contracts | None |
| Honoraria/payment | Zambon, Lundbeck | Donation of medical equipment | None |
| Advisory Boards | Zambon, Lundbeck | Patents planned, pending, or issued | None |
| Speakers’ bureaus | None | Other | None |
Michelle Beavan, Alisdair McNeill, Christos Proukakis, Derralynn Hughes, and Atul Mehta:
| Employment | None | Stock Ownership | None |
| Affiliations | None | Expert Testimony | None |
| Grants/funding | None | Royalties | None |
| Consultancies | None | Contracts | None |
| Honoraria/payment | None | Donation of medical equipment | None |
| Advisory Boards | None | Patents planned, pending, or issued | None |
| Speakers’ bureaus | None | Other | None |
Statements
Michelle Beavan had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Written permission has been obtained from all persons named in the Acknowledgment section.
Footnotes
Relevant conflicts of interest/financial disclosures
All financial disclosure information for Michelle Beavan and all co-authors is accurate, complete, and up-to-date. Michelle Beavan is funded by an MRC-Wellcome Trust Strategic award for Parkinson’s disease and reports no conflicts. Alisdair McNeill, Christos Proukakis, Derralynn Hughes and Atul Mehta report no conflicts. A.H.V.S. receives research support from the UK Medical Research Council, Wellcome Trust and Parkinson’s UK, and is a NIHR senior investigator. He reports no conflicts.
REFERENCES
- 1.Bultron G, Kacena K, Pearson D, et al. The risk of Parkinson’s disease in type 1 Gaucher disease. J Inherit Metab Dis. 2010;33(2):167–73. doi: 10.1007/s10545-010-9055-0. doi:10.1007/s10545-010-9055-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McNeill A, Duran R, Hughes DA, Mehta A, Schapira AHV. A clinical and family history study of Parkinson’s disease in heterozygous glucocerebrosidase mutation carriers. J Neurol Neurosurg Psychiatry. 2012;83(8):853–4. doi: 10.1136/jnnp-2012-302402. doi:10.1136/jnnp-2012-302402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sidransky E, Nalls M, Aasly J, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361:1651–1661. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zimran A, Gelbart T, Westwood B, Grabowski GA, Beutler E. High frequency of the Gaucher disease mutation at nucleotide 1226 among Ashkenazi Jews. Am J Hum Genet. 1991;49(4):855–9. [PMC free article] [PubMed] [Google Scholar]
- 5.Anheim M, Elbaz A, Lesage S, et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology. 2012;78(6):417–20. doi: 10.1212/WNL.0b013e318245f476. doi:10.1212/WNL.0b013e318245f476. [DOI] [PubMed] [Google Scholar]
- 6.Nalls MA, Duran R, Lopez G, et al. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol. 2013;70(6):727–35. doi: 10.1001/jamaneurol.2013.1925. doi:10.1001/jamaneurol.2013.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wakabayashi K, Mori F, Tanji K, Orimo S, Takahashi H. Involvement of the peripheral nervous system in synucleinopathies, tauopathies and other neurodegenerative proteinopathies of the brain. Acta Neuropathol. 2010;120(1):1–12. doi: 10.1007/s00401-010-0706-x. doi:10.1007/s00401-010-0706-x. [DOI] [PubMed] [Google Scholar]
- 8.Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 24(2):197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 9.Postuma RB, Aarsland D, Barone P, et al. Identifying prodromal Parkinson’s disease: pre-motor disorders in Parkinson’s disease. Mov Disord. 2012;27(5):617–26. doi: 10.1002/mds.24996. doi:10.1002/mds.24996. [DOI] [PubMed] [Google Scholar]
- 10.Schapira AHV. Recent developments in biomarkers in Parkinson disease. Curr Opin Neurol. 2013;26(4):395–400. doi: 10.1097/WCO.0b013e3283633741. doi:10.1097/WCO.0b013e3283633741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McNeill A, Duran R, Proukakis C, et al. Hyposmia and cognitive impairment in Gaucher disease patients and carriers. Mov Disord. 2012;27(4):526–32. doi: 10.1002/mds.24945. doi:10.1002/mds.24945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55(3):181–184. doi: 10.1136/jnnp.55.3.181. doi:10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lees AJ, Hardy J, Revesz T. Parkinson’s disease. Lancet. 2009;373(9680):2055–66. doi: 10.1016/S0140-6736(09)60492-X. doi:10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- 14.Saunders-Pullman R, Hagenah J, Dhawan V, et al. Gaucher disease ascertained through a Parkinson’s center: imaging and clinical characterization. Mov Disord. 2010;25(10):1364–72. doi: 10.1002/mds.23046. doi:10.1002/mds.23046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hawkes CH, Del Tredici K, Braak H. A timeline for Parkinson’s disease. Parkinsonism Relat Disord. 2010;16(2):79–84. doi: 10.1016/j.parkreldis.2009.08.007. doi:10.1016/j.parkreldis.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 16.Berg D, Lang AE, Postuma RB, et al. Changing the research criteria for the diagnosis of Parkinson’s disease: obstacles and opportunities. Lancet Neurol. 2013;12(5):514–24. doi: 10.1016/S1474-4422(13)70047-4. doi:10.1016/S1474-4422(13)70047-4. [DOI] [PubMed] [Google Scholar]
- 17.Postuma RB, Aarsland D, Barone P, et al. Identifying prodromal Parkinson’s disease: pre-motor disorders in Parkinson’s disease. Mov Disord. 2012;27(5):617–26. doi: 10.1002/mds.24996. doi:10.1002/mds.24996. [DOI] [PubMed] [Google Scholar]
- 18.Stiasny-Kolster K, Mayer G, Schäfer S, Möller JC, Heinzel-Gutenbrunner M, Oertel WH. The REM sleep behavior disorder screening questionnaire--a new diagnostic instrument. Mov Disord. 2007;22(16):2386–93. doi: 10.1002/mds.21740. doi:10.1002/mds.21740. [DOI] [PubMed] [Google Scholar]
- 19.O’Sullivan SS, Williams DR, Gallagher DA, Massey LA, Silveira-Moriyama L, Lees AJ. Nonmotor symptoms as presenting complaints in Parkinson’s disease: a clinicopathological study. Mov Disord. 2008;23(1):101–6. doi: 10.1002/mds.21813. doi:10.1002/mds.21813. [DOI] [PubMed] [Google Scholar]
- 20.Packman W, Wilson Crosbie T, Riesner A, Fairley C, Packman S. Psychological complications of patients with Gaucher disease. J Inherit Metab Dis. 2006;29(1):99–105. doi: 10.1007/s10545-006-0154-x. doi:10.1007/s10545-006-0154-x. [DOI] [PubMed] [Google Scholar]
- 21.Liepelt-Scarfone I, Behnke S, Godau J, et al. Relation of risk factors and putative premotor markers for Parkinson’s disease. J Neural Transm. 2011;118(4):579–85. doi: 10.1007/s00702-010-0553-x. doi:10.1007/s00702-010-0553-x. [DOI] [PubMed] [Google Scholar]
- 22.Dalrymple-Alford JC, MacAskill MR, Nakas CT, et al. The MoCA: well-suited screen for cognitive impairment in Parkinson disease. Neurology. 2010;75(19):1717–25. doi: 10.1212/WNL.0b013e3181fc29c9. doi:10.1212/WNL.0b013e3181fc29c9. [DOI] [PubMed] [Google Scholar]
- 23.Williams SS, Williams J, Combrinck M, Christie S, Smith AD, McShane R. Olfactory impairment is more marked in patients with mild dementia with Lewy bodies than those with mild Alzheimer disease. J Neurol Neurosurg Psychiatry. 2009;80(6):667–70. doi: 10.1136/jnnp.2008.155895. doi:10.1136/jnnp.2008.155895. [DOI] [PubMed] [Google Scholar]
- 24.Alcalay RN, Caccappolo E, Mejia-Santana H, et al. Cognitive performance of GBA mutation carriers with early-onset PD: the CORE-PD study. Neurology. 2012;78(18):1434–40. doi: 10.1212/WNL.0b013e318253d54b. doi:10.1212/WNL.0b013e318253d54b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brockmann K, Srulijes K, Hauser AK, et al. GBA-associated PD presents with nonmotor characteristics. Neurology. 2011;77(3):276–80. doi: 10.1212/WNL.0b013e318225ab77. doi:10.1212/WNL.0b013e318225ab77. [DOI] [PubMed] [Google Scholar]
- 26.Zokaei N, McNeill A, Proukakis C, et al. Visual short-term memory deficits associated with GBA mutation and Parkinson’s disease. Brain. 2014 doi: 10.1093/brain/awu143. doi:10.1093/brain/awu143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clark LN, Kartsaklis LA, Wolf Gilbert R, et al. Association of glucocerebrosidase mutations with dementia with lewy bodies. Arch Neurol. 2009;66(5):578–83. doi: 10.1001/archneurol.2009.54. doi:10.1001/archneurol.2009.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neumann J, Bras J, Deas E, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain. 2009;132(Pt 7):1783–94. doi: 10.1093/brain/awp044. doi:10.1093/brain/awp044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Lau LML, Koudstaal PJ, Hofman A, Breteler MMB. Subjective complaints precede Parkinson disease: the rotterdam study. Arch Neurol. 2006;63(3):362–5. doi: 10.1001/archneur.63.3.noc50312. doi:10.1001/archneur.63.3.noc50312. [DOI] [PubMed] [Google Scholar]
- 30.Elstein D, Guedalia J, Doniger GM, et al. Computerized cognitive testing in patients with type I Gaucher disease: effects of enzyme replacement and substrate reduction. Genet Med. 2005;7(2):124–30. doi: 10.1097/01.gim.0000153666.23707.ba. doi:10.109701.GIM.0000153666.23707.BA. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.