

Abstract
The hepatitis C virus (HCV) is an important contributor to morbidity and mortality in patients coinfected with human immunodeficiency virus (HIV). Coinfection results in increased HCV replication and more rapid rates of liver disease progression. The effect of HIV combination antiretroviral therapy (cART) on HCV replication has not been studied in depth. To address this issue, we enrolled a small cohort of HCV/HIV coinfected patients into a cART initiation trial, and used dynamic modeling combined with evaluation of immune responses and microarray profiles to determine how effective treatment of HIV affects HCV. Treatment with cART resulted in HCV flare and alanine aminotransferase (ALT) increase (2× or more increase from baseline) in a subset of treated patients. Subjects with evidence of hepatic injury (increased ALT) were more likely to have HCV-specific immune responses directed against HCV epitopes. Over time, HCV viral loads declined. Reproducible and biologically important gene expression changes occurred in patients who underwent successful cART, particularly with respect to downregulation of genes with known antiviral roles. Our findings suggest that the effective suppression of HIV by cART initiates a cascade of early and late events in treated patients with HCV. Early events involving downregulation of interferon-stimulated genes may lead to transiently increased viral replication and hepatic injury. At later time points, HCV viral load declines to levels comparable to those seen in the setting of HCV monoinfection. These findings support early antiretroviral therapy in those with HCV/HIV coinfection.
Introduction
Hepatitis C virus (HCV) coinfection is a frequent cause of significant co-morbidities and mortality among those infected with human immunodeficiency virus (HIV). In the United States, 200,000–300,000 people have HCV/HIV coinfection, and worldwide estimates range from 4–8 million(1). HCV/HIV coinfection is not benign and is associated with higher rates of HCV replication, increased hepatocyte injury, more rapid progression of liver disease to advanced fibrosis and cirrhosis, and increased rates of development of hepatocellular carcinoma (HCC) (2). The presence of liver injury as evidenced by abnormal levels of serum transaminases (ALT, AST) can limit treatment of HIV(3).
HCV/HIV coinfection is associated with increased HCV viral load in serum/plasma(4, 5). This observation has been well-described but poorly characterized. Higher HCV viral loads are associated with a delay in HCV clearance following HCV treatment(6). Furthermore, longitudinal treatment studies demonstrate that a paradoxical increase in HCV viral load following initiation of combination antiretroviral therapy (cART) for HIV occurs in some patients. This appears to be more frequent among those with low CD4+ T cell counts at initiation of cART(7, 8). The effect of prolonged HIV suppression on HCV is also poorly characterized with clinical studies describing persistent HCV viral load increases, decreases, or return to pre-treatment set-point levels(9). In some cases, initiation of cART is accompanied by an increase in serum transaminases, often termed an ALT “flare”, which may lead clinicians to discontinue or modify cART in response to liver enzyme abnormalities that may be attributed to drug-related hepatotoxicity.
Although the phenomenon of HCV viral load increase and ALT flare following initiation of cART is well-documented in clinical trials, the significance of these flares, their relationship to specific and innate immune responses, and the outcome of effective HIV therapy in relation to HCV remain uncharacterized. To address this issue, we embarked on a study of a small prospective cohort of HCV/HIV coinfected patients treated with cART. The study was designed to characterize changes in HCV viral load and ALT following cART initiation, to determine the relationship of these HCV viral load changes to HIV decline, CD4+ T cell rebound, and HCV-specific T-cell responses, and to evaluate the broader changes in both virus-specific and cytokine-mediated immune responses that occur following cART initiation. All subjects had HIV in conjunction with HCV genotype 1 infection. They were started on efavirenz, or atazanavir/ritonavir, along with a backbone of tenofovir/emtricitabine. Viral dynamic modeling was used to characterize the relationship between HIV decline, HCV viral load change, and other immunological and virological parameters.
Results
Study Population
18 subjects gave consent for the clinical trial. One subject was classified as a screen failure and was not enrolled. 13 of the enrolled subjects were male, and 4 were female. The majority were black, non-Hispanic (83%), and the remainder were Caucasian. The mean age was 48 (24–60) years. The median CD4+ T cell count was 346 cell/mm3 (range 87–576), and the median HIV viral load was 4.47 log10 copies/mL. The HCV viral load median was 6.29 log10 IU/mL. Baseline liver biopsy revealed that 12 subjects (70.5%) had early hepatic fibrosis (Metavir F0–F2), while the remainder had bridging fibrosis (F3) disease. No patients with frank cirrhosis were enrolled. IL28B gene polymorphisms were also evaluated in patients that provided genetic testing consent (n=9); 33% had the CC genotype, which is associated with both spontaneous viral clearance and treatment-associated clearance of HCV, and the remainder had CT or TT polymorphisms.
All patients provided informed consent and the study protocols were approved by the Institutional Review Boards at all enrolling sites (University of Cincinnati, Virginia Commonwealth University, and New York University).
Increase in Serum ALT, HCV, and Relationship to Liver Histology
The doubling of serum ALT from baseline levels met the pre-study definition of a significant ALT abnormality. Overall, 7/17 subjects (41.1%) met this criterion. ALT increases ranged from 2 to 10 times baseline levels. ALT increases first appeared at a mean of 15.4 weeks after cART initiation (range 4–32 weeks). Abnormalities in this range persisted through follow-up in only 1 subject. The peak ALT levels occurred at week 16 of HIV treatment (Fig. 1). IL28B polymorphisms showed no relationship to ALT flare. A regression model was used to examine factors possibly associated with ALT increase including age, gender, race, baseline HIV and HCV viral loads, baseline and change in CD4+ T cell counts by 24 weeks and HIV clearance. Only HIV clearance was associated with HCV increase or ALT flare (p= 0.03) Seven subjects underwent repeat liver biopsy following protocol specified levels of increase in ALT or HCV RNA serum levels. Liver histology was evaluated and scored in a blinded manner. The necroinflammatory score, a histologic measure of the degree of liver necrosis and inflammation) decreased from a median of 7 to 5 (Wilcoxon exact probability = 0.06). Paired comparison of individual patients is presented in Fig. 2. 57% (4/7) demonstrated an improvement of 1 or more points in the necroinflammatory component of the histologic scoring system. Two patients were unchanged, and one worsened by 1 point. A stepwise linear regression model designed to examine factors associated with the change in necroinflammatory score was developed. Following univariate evaluation of a number of factors, the model demonstrated that pre-antiretroviral titers of HCV and HIV, CD4+ T cell count, and pre-treatment response to HCV epitopes were important factors and yielded an adjusted r2= 0.83. ALT, age and baseline fibrosis, which were important univariate predictors, fell out of the regression model. High baseline HIV viral load and low pre-treatment CD4+ T cell counts were the key predictors of improved necroinflammatory liver biopsy scores with p values of 0.018 and 0.049, respectively. Although HCV specific immune responses at baseline strengthened the model, the association failed to reach statistical significance (p=0.17).
Figure 1.
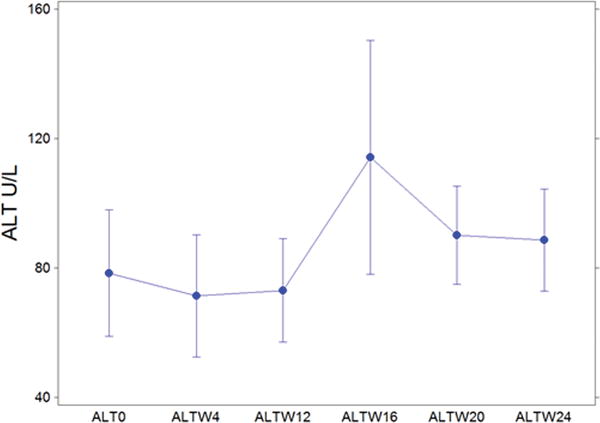
ALT levels (U/L, units per liter) for all subjects from baseline through week 24 of cART treatment. ALT is a marker of hepatocyte injury. Points show mean values for weeks 0, 4, 12, 16, 20 and 24. Bars show S.E.M.
Figure 2.
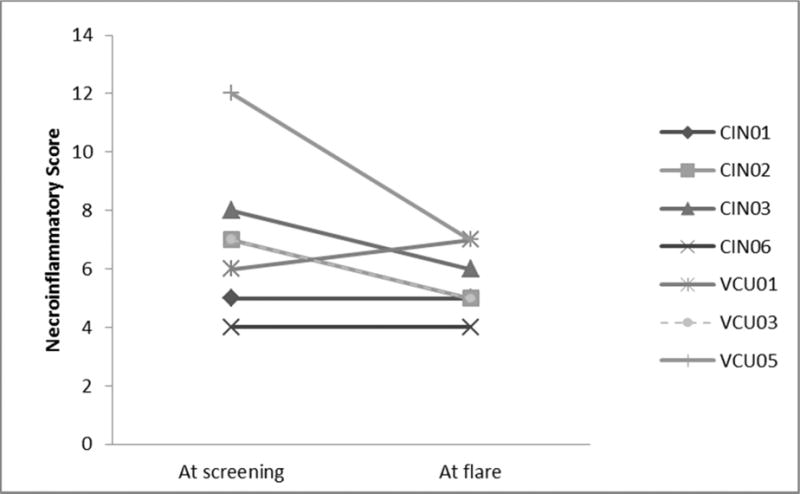
The necroinflammatory component score represents a composite measure of inflammation and injury as described by Ishak et al (J Hepatology, 1995). Paired scores show baseline activity at pretreatment screening and following biopsy after subject met protocol-specified criteria for increased HCV RNA or serum ALT.
Changes in HCV RNA
Four subjects (23.5%) met the study definition of HCV RNA flare increase (>0.5 log10 increase from baseline). The majority of these occurred at week 8 or 12, although there was one late event at week 88. The mean HCV RNA increase among those meeting the HCV flare criterion was 0.65 log10 IU/mL. One subject with an early ALT flare had a late (week 72) HCV RNA increase greater than 0.5 log10 IU/ml as well, but the protocol did not allow a second post-treatment liver biopsy. The maximal mean increase in HCV RNA was 0.33 log10 units in all subjects at any time point, generally occurring within the first 24 weeks after cART initiation. Overall, 82% (14/17) of subjects experienced an increased HCV viral load over baseline at one or more time points. This exceeded expectations of normal variation [Odds Ratio (OR)= 5.25, 95% confidence intervals (CI) 1.09–25.21]. Despite these increases, the overall trend in HCV RNA was to decline from baseline levels. Paired sample data were available for 13/17 subjects at week 72. The mean HCV RNA at HCV baseline was 6.38 log10 IU/mL (SEM 0.15). At week 72 the mean had decreased by 0.52 log10 IU/mL (Wilcoxon test p=0.025).
Kinetic Analysis of HIV Viral Decline
HIV viral load over time, denoted V(t), was fitted with the following two-phase decline model (see Materials and Methods):
| [1] |
where V0 is the baseline viral load, π0 is a constant, δ) is the death rate of short lived HIV-infected cells, δM is the death rate for long-lived HIV-infected cells, and t0 is the delay until the viral load starts to decline. We ignored an exponential term proportional to the rate of HIV clearance, c, as this only affects the viral kinetics in a minor way during the first half-day of therapy.
Data were fitted until the first data point under the limit of detection (LOD) (black dotted line in Fig. S1), and this data point was fitted in the model assuming that it was equal to half the LOD (this is why the fits are below the data point in some patients). Choosing a value at half the LOD is one recognized means of dealing with left-censored data(10). Patient CIN04 experienced HIV virologic rebound at week 4 that could not be captured by this model, and therefore data at week 4 and week 8 were not included in the fitting procedure, and the parameters δM and π0 could not be estimated for this patient. All other patients presented a biphasic viral decay that could be well fitted by our model. Viral kinetics parameters were in line with estimates from the literature for monoinfected patients (Fig. S2 and Table 1), which supports the hypothesis that infection with HCV does not significantly affect the early kinetics of HIV decline after initiation of cART(11–13). Lastly, the loss rate of short-lived HIV-infected cells, δ, was correlated with both baseline plasma HIV RNA viral levels (P=0.076) and the loss rate of long-lived HIV-infected cells, δM (P=0.08). Our model assumes that treatment is 100% effective in blocking new infections. Using this assumption, the first and the second slopes of viral decline were exactly equal to the loss rates of long- and short-lived infected cells, respectively. The correlation between the two loss rates suggested that the slopes of viral decline were also related to the effectiveness of the antiviral treatment, which may not be 100% efficient(14). More detailed analysis of HIV models showed that each slope of viral decline was roughly proportional to cART effectiveness. Interestingly, patients with a flare in ALT levels (doubling from baseline) in the first 20 weeks of treatment (N=6) tended to have lower slopes of viral decline (Table 1).
Table 1.
HIV viral kinetics estimated by fitting Eq. [1] to each patient’s viral load.
| V0 Log10(IU/ml) |
t0 days |
δ days−1 |
δM days−1 |
π0 days−1 |
ALT flare in the first 20 weeks? | |
|---|---|---|---|---|---|---|
| CIN01 | 5.05 | 0.51 | 0.56 | 0.04 | 0.98 | Yes |
| CIN02 | 4.65 | 1.10 | 1.20 | 0.03 | 0.97 | No |
| CIN03 | 5.63 | 0.75 | 0.74 | 0.02 | 0.98 | Yes |
| CIN04* | 2.14 | 0.78 | 0.22 | NA (rebound) | NA (rebound) | No |
| CIN05 | 4.28 | 0.98 | 0.73 | 0.01 | 0.97 | Yes |
| CIN06 | 5.45 | 0.99 | 0.56 | 0.03 | 0.99 | Yes |
| CIN07 | 3.77 | 0.37 | 1.00 | 0.08 | 0.96 | No |
| CIN08 | 3.98 | 0.39 | 0.99 | 0.00 | 0.98 | Yes |
| CIN09 | 4.47 | 0.18 | 0.93 | 0.07 | 0.97 | No |
| CIN10 | 4.91 | 0.89 | 1.08 | 0.10 | 0.95 | No |
| CIN11 | 3.75 | 1.00 | 1.25 | 0.10 | 0.95 | No |
| VCU01 | 3.60 | 0.68 | 0.91 | 0.09 | 0.94 | Yes |
| VCU02 | 4.30 | 1.37 | 0.88 | 0.08 | 0.94 | No |
| VCU03 | 4.74 | 0.04 | 0.82 | 0.07 | 0.98 | No |
| VCU04 | 4.71 | 0.50 | 1.24 | 0.17 | 0.95 | No |
| VCU05 | 4.70 | 0.76 | 1.31 | 0.04 | 0.97 | No |
| NYU01* | 4.91 | 0.97 | 0.68 | (last data pt at wk1) | No | |
|
| ||||||
| mean | 4.53 | 0.70 | 0.95 | 0.06 | 0.97 | |
| SD | 0.60 | 0.37 | 0.24 | 0.04 | 0.02 | |
| Mean in patients with an ALT flare | 4.67 | 0.72 | 0.75** | 0.03** | 0.97 | |
| Mean in patients without an ALT flare | 4.44 | 0.69 | 1.08 | 0.08 | 0.96 | |
CIN04 and NYU01 were considered outliers and were not included in the calculation of the means.
Both δ (P=0.026) and δM (P=0.028) were significantly different in patients with ALT flare vs no flare.
HCV and HIV Kinetics
Granger Causality: The change in HIV RNA may induce a change in HCV RNA
First, we explored a possible relationship between HIV and HCV viral kinetics. For that purpose, we introduced the notion of Granger causality originally developed in economics to quantify a causal effect from time-series observations(15). It is based on the simple idea that causes precede effects. Formally, X is said to Granger-cause Y if its past value can help to predict the future value of Y beyond what could have been done with the past value of Y only. While Granger-causality tests are now widely used in many branches of science, a positive result does not prove causality although it suggests a stronger relation between variables than simple correlation.
When restricting the analysis to data until day 7, we found that in 14 of 16 patients, HIV RNA was more likely to Granger-cause changes in HCV RNA than vice versa, and in all these patients this value was statistically significant at the level of 10% (Table S1). However, when including data until week 12, the results were not significant, possibly due to the fact that a large proportion of HIV RNA data are under the LOD and that this approach works poorly when time points are not equally spaced, as is the case in this study. Overall, this statistical approach supports the hypothesis that HIV treatment has an impact on HCV RNA. However, it does not tell us anything about the nature of this relationship. Using a modeling approach, we aimed next to characterize this correlation.
Correlation between changes in HCV viral load at different time points and HIV viral kinetics
Figure S3 displays the correlation between the parameters of HIV viral decline, namely δ and δM, and the changes in HCV RNA from baseline at different time points (weeks 1, 4, 8 for δ, weeks 4, 8, 48 for δM). These time points were chosen as they represent different periods in HCV kinetics (see changes over time in Fig. S1). A rapid initial HIV decline (i.e., high δ) was correlated with an early change in HCV RNA at week 1 and week 4 (P=0.10 and 0.048, respectively), and this change was a decline. Further, the loss rate of long-lived HIV infected cells, δM, was found to be significantly correlated with HCV RNA changes at week 4 (P=0.028). Lastly, the maximal HCV RNA decline from baseline in the first 4 weeks of treatment was significantly correlated with δ (P=0.04) but not with δM (Fig. S4). No significant correlations were found using later data points (week 12, week 40 and so on), during which changes in HCV viral load compared to baseline levels were more substantial (Fig. S3).
T-cell Specific Responses and Relation to Change in HCV Viral Load
To identify the role of the antigen-specific immune responses in the induction of ALT and/or HCV flare, we evaluated HCV-specific, and non-specific immune responses in 15 of the HCV/HIV coinfected patients enrolled in the study before cART therapy, and correlated these data with the clinical and laboratory findings. We monitored HCV-specific and non-specific immune responses using IFN-γ ELISPOT assays at baseline before initiation of cART therapy. Additionally, correlations between the antigen-specific immune responses and viral loads, CD4+ T cell counts, and ALT levels were evaluated. During cART therapy, the magnitude (both mean number of antigen specific spots/106 cells and total proportion with >55 spots/106 cells) of the HCV-specific immune responses at baseline in the subjects with ALT flare were significantly higher compared to subjects who did not show ALT flare (p<0.005). (Fig S5b). A weak positive correlation was found between the HCV viral load and HCV-specific IFN-λ immune responses (r2=0.21, p= 0.0018). Additionally, no significant differences were seen in responses to positive control antigens (CEF control peptides) and non-specific T cell stimulation (anti-CD3) at baseline. (Fig S5a).
Effect of HIV Treatment on Gene Array Profiles
To evaluate patient responses to cART, we used genome-wide expression profiling of patients’ peripheral blood mononuclear cells before and after treatment initiation (baseline and week 24). RNA quality for this analysis at both timepoints was acceptable for a subset of 8 subjects. Gene expression changes were analyzed based on the change in expression of each measured transcript per patient. Genes with moderately high absolute expression (RMA>5.5, (Robust Multi-array Average)) that consistently increased or decreased in their relative expression in at least two or three patients were considered and this pool of genes was then compared statistically using both parametric and non-parametric t-tests. This approach allowed for the identification of 341 probe sets that could be mapped to 115 genes whose expression increased (“response-up”) and 157 whose expression decreased (“response-down”) irrespective of flare or HCV increase, and to the top most differentially regulated genes. (Heat map Fig. 3, Table S2). Gene set enrichment (16) and biological network-based analyses (17) (Fig. S6) revealed enrichment among downregulated genes for their functional association with interferon signaling, response, and resistance to viral infection and for genes whose promoters contained conserved motifs corresponding to interferon response factor binding elements. Genes that were upregulated following cART were enriched for gene functions associated with increased translation and normal CD4+ and CD8+ T cell responses corresponding to CD4+ T cell rebound. Analysis of serum cytokine responses revealed that cART initiation was associated with significant decreases in TNFα (p= 0.0004) and IP-10 (p< 0.0001). There was also a trend towards decreased levels of circulating interferon-α.
Figure 3.
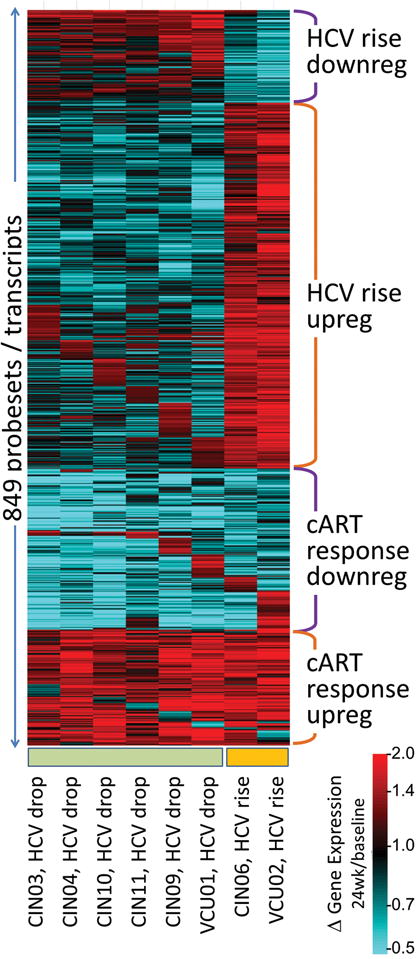
Heat map of gene expression changes that occurred following cART treatment in HIV/HCV co-infected patients. Gene expression changes are depicted as the ratio of gene expression at 24 weeks following cART compared to baseline gene expression prior to treatment. Genes that decrease from their baseline values are blue, and those that increase are red. Four clusters are indicated corresponding to genes that (i) increased or (ii) decreased following cART treatment in the patients whose HCV titers rose, (iii) that increased in response to cART for nearly all patients, and (iv) that decreased in response to cART. A change in expression pattern can be seen between the first 6 patients on the left who had a drop in HCV titer, as compared to the 2 patients on the right who had a rise in titer.
Despite the similarity of expression changes across all patients undergoing cART with respect to the above gene sets, the two individuals profiled who exhibited a rise in their HCV titers showed a remarkably different pattern of gene expression. As shown in Fig. 3, the prominent pattern was the activation of genes associated with antigen processing, vacuolar membranes, and MHC class II receptors, from screening to post therapy in the individuals whose HCV titers rose. Although there were only two individuals profiled out of 7 who exhibited this pattern, the relative connectivity of these genes was highly significant.
In an effort to link the results of the gene profiling with changes in cART associated HIV and HCV viral loads, univariate analysis followed by stepwise linear regression was performed. We focused on determining the relationship between the predominant pattern of downregulation of interferon-mediated gene expression and the observed viral load outcomes. Univariate analysis suggested correlations with ALT, age, baseline HCV and HIV titers, baseline CD4+ T cell counts, fibrosis stage at entry, interferon-related gene expression and HCV viral load over the course of the post-cART period, however, all factors except interferon-stimulated gene_ISG downregulation were not significant when linear regression analysis was employed. ISG downregulation was highly significant (p= 0.0063).
Discussion
Several prior studies have suggested a link between initiation of HIV antiretroviral therapy and changes in HCV viral load. Most have reported transient increases in HCV RNA during the first 24 weeks of treatment(7,9). Chung et al. suggested that this paradoxical effect was most commonly observed in those with low CD4+ T cell counts prior to cART initiation. We observed transient increases in HCV RNA in a subset of treated patients. This was frequently accompanied by increases in serum ALT levels. The highest mean increase in ALT was observed at week 16. Such increases have often been classified as “drug hepatotoxicity” and may result in drug discontinuation by clinicians in clinical practice, often mirroring toxicity guidelines utilized in clinical trials of therapeutic agents(18). However, these abnormalities are rarely characterized with evaluation that includes liver histology. Historically, ALT abnormalities <5× the upper limit of normal (ULN) are ignored and those greater than 5× ULN (Grade 3 toxicity) require attribution of causality and, in some cases, drug discontinuation. Except for cases where liver biopsy was performed to evaluate putative severe drug toxicity in clinical practice, we are not aware of any trial that systematically performed liver biopsy evaluation on patients with ALT and/or HCV flare as defined in this study. The liver biopsy data was surprising and shows a strong trend towards improvement in hepatic necroinflammatory scores, supporting the concept that HIV treatment may be beneficial to the liver in the setting of HCV infection. This trend was most pronounced in those with the lowest CD4+ T cell counts and the higher HIV viral loads. This observation may appear to be counterintuitive. However, a number of studies have supported an inverse relationship between low CD4+ T cell counts and increased inflammation. Benhamou first reported that low CD4+ T cell counts were associated with greater necroinflammatory activity in HIV/HCV co-infected patients vs. those infected with HCV alone(19, 20). Pascual-Pareja et al. specifically examined the relationship of HIV suppression to necroinflammatory activity(21). They noted that the level of hepatic inflammation was strongly associated with active HIV replication in the absence of cART. While the mechanism of this is unclear, it has been suggested that gp120 may drive HCV replication, increasing hepatic injury(22). Interestingly, gp120 levels in plasma do not necessarily correlate with HIV viral load(23). An alternative hypothesis is that HIV infection directly drives production of localized proinflammatory cytokines in the liver leading to necroinflammation, and effective HIV suppression abrogates this process(24). Because liver biopsy occurred up to 2 weeks post-flare identification, it is possible that early findings of infected- hepatocyte clearance associated with HCV specific immune responses (e.g. increased apoptotic bodies) may have been missed. The overall degree of chronic inflammation that comprises the necroinflammatory score was decreased by the broad local downregulation of the proinflammatory cascade.
The observation that HIV suppression reduces inflammation and subsequent hepatic fibrosis formation is supported by large epidemiological studies showing lower liver-related morbidity and mortality among those receiving effective antiretroviral therapy(25). It is also supported by data from the HCV/HIV SLAM-C study, which failed to find fibrosis progression among a group of HIV-suppressed patients(26). These findings strongly support the early use of cART in those with HCV/HIV coinfection as a way to protect the liver from injury. Furthermore, our data suggest that ALT flare does not necessarily represent drug-related hepatotoxicity and the failure to recognize this possibility could result in cART discontinuation, which could actually be harmful to the patient in the long-term.
The weak positive but statistically significant correlation between baseline HCV-specific immune responses and the HCV viral load indicate that HCV viral load may stimulate the induction of HCV-specific immune responses. However, these immune responses are not effective in lowering HCV viral load. Kang et al. also noted that HCV-specific T-cell response recovery after cART initiation may lead to transient liver injury(27). It is possible that our observations of HCV-specific immune responses were not mirrored by intrahepatic immune responses present in the microenvironment of the liver. However, despite this caveat a clear relationship between peripheral HCV-specific responses in peripheral blood mononuclear cells PBMCs and early increases in viral load was observed. Although we studied IFN-γ ELISPOT, additional information may be gleaned by examination of other cytokine responses (e.g. IL2). Future studies will include analysis of other cytokines.
The mechanistic link between cART effectiveness and HCV RNA decline remains unclear. However, we have observed significant downregulation of ISGs following initiation of cART and postulate that this initially results in loss of interferon-mediated control of HCV replication, resulting initially in increased HCV viral loads. In the setting of a pre-existent HCV-specific cellular immune response, increased cell clearance leads to transient increases in serum ALT levels (i.e. immunologically mediated hepatic injury or immune reconstitution inflammatory syndrome- IRIS). However, global upregulation of CD4, CD8 and alpha-beta T-cell activity leads to broad suppression of HCV viral replication over time. Interestingly, the decrease in HCV RNA is roughly equivalent to the median increase in HCV titer observed in clinical cohorts with HIV infection compared to those with HCV monoinfection(4, 5, 28). While several studies have suggested that immune reconstitution following cART does not restore the immune status present prior to HIV infection, it appears that effective cART returns HCV/HIV coinfected patients to a functional HCV monoinfection state, at least in terms of HCV viral load. This has implications for HCV treatment response. We acknowledge that sample size is a limitation of this type of in-depth analysis, but believe that these data support emerging concepts that early HIV control with antiviral agents represents the best initial approach to management of HCV/HIV coinfected patients and is congruent with recent recommendations for early HIV treatment made by expert consensus groups(29).
Materials and methods
Study Design
Patients with HCV and HIV coinfection were enrolled in a clinical trial designed to intensively evaluate the effects of initiation of antiretroviral therapy on HCV replication and liver injury. Briefly, after providing informed consent, subjects greater than 18 years of age with HCV RNA Genotype 1 and HIV were enrolled. All subjects had a liver biopsy consistent with chronic hepatitis within 1 year of enrollment and were HIV antiretroviral treatment naïve or off ART for at least 3 months. They were required to be sensitive to the proposed study drugs. Key exclusion criteria included baseline hemoglobin <9 g/dL, pregnancy, concurrent infection with hepatitis B or presence of another liver disease, decompensated liver disease, active thyroid disease, creatinine clearance <50mL/min, recent use of corticosteroids or immunomodulatory drugs, current HCV therapy or any condition which might preclude study participation. Subjects were treated with efavirenz 60 mg oral once daily and tenofovir/emtricitabine once daily. Subjects unable or unwilling to take efavirenz were permitted to substitute atazanavir 300 mg/day boosted with ritonavir 100 mg/day. Study subjects were followed for 96 weeks following initiation of study medications. Early viral dynamic sampling was performed performed at hours 0 (baseline), 2, 4, 6, 9, 12, 18, 24, 48, 72, 96, and 168 (7 days) from initiation of first dose of cART Additional samples were collected upon doubling of ALT level from baseline or a viral load increase of ≥0.5 log10IU/ml of HCV RNA. Repeat liver biopsy was obtained when ALT or HCV RNA increased to specified levels.
Viral kinetic modeling
Previous studies of HIV dynamics demonstrated that after cART is initiated there are two phases of HIV-1 RNA decay; an initial rapid, exponential decline of 1–2 logs, in which productively infected short-lived cells are lost, followed by the second phase, which is characterized by a slower decline, where long-lived infected cells are lost(11, 30, 31). HIV viral kinetics were thus fit using the previously developed long-lived infected cell model that considers short and long-lived productively infected cells using the equation:
| [2] |
where V0 is the baseline viral load, δ is the death rate of short-lived productively infected cells, δM is the death rate of long-lived productively infected cells, c is the rate of virion clearance, t0 is the time at which the viral load starts decaying, i.e., a pharmacological delay, and A and C are constants defined in Perelson et al.(11). We fixed the half-life of free virus, c, to the previously estimated value c=23 day−1(32). With this value of c the last exponential term in Eq. [1] rapidly becomes negligible and thus was ignored.
Statistical Methods
Parametric and non-parametric methodologies including Student’s t, Chi-square and Fisher exact were employed for comparisons as described in the Results section. Univariate analysis and stepwise linear regression models were utilized to explore relationships of factors associated with key outcomes. Data were analyzed using Statistix 9.0/10.0 (Analytical Software, Tallahassee, FL). Statistical significance was set at an alpha= 0.05 using a two-tailed hypothesis unless otherwise specified.
Evaluation of immune response using ELISPOT assay
Isolation of peripheral blood mononuclear cells (PBMC)
PBMCs were isolated by Ficoll-Hypaque density gradient centrifugation and were then used either fresh or cryopreserved in cell-freezing medium (90% fetal bovine serum [FBS; Biowhittaker, USA] and 10% DMSO [Sigma, USA]). They were stored in liquid N2 or at −80°C until used to test for cytokine production as described below. Before performing any assay with frozen samples, PBMCs were thawed, washed twice in complete RPMI-1640 medium supplemented with penicillin/streptomycin (10,000 units/mL potassium penicillin, 10,000 μg/mL streptomycin sulfate), 1% sodium pyruvate, 0.1% 2-mercaptoethanol, 10 % heat-inactivated FBS. Viability was determined by trypan blue exclusion. Fresh and frozen PBMC have provided comparable results using ELISPOT immunoassays in our laboratory as previously reported(33).
HLA class I typing for the patients was performed at Hoxworth Blood Center, Cincinnati, OH, using sequence-specific oligonucleotide probes (SSOP). Allele-level HLA typing was performed by sequence-based typing (SBT).
Antigens
HCV antigens: Four pooled HCV peptides were obtained from NIH AIDS Research & Reference Reagent program (HCV type 1a H77 (18-mer peptides) (Cat No 7620), as well as dominant HLA peptides according to the HLA class I of the patient. Each peptide was used at the final concentration of 1–10ug/mL. CEF control peptide pool (Cat No 9808) was obtained from NIH AIDS Research &Reference Reagent program), and contains pool of 32 CMV, EBV, and influenza virus dominant peptides which stimulate CD8+ T cells and were used at a concentration of 1–10ug/mL.
IFN-γ ELISPOT assay
The ELISPOT assay has the advantage of being able to specifically enumerate cells as well as quantify cytokine secretion patterns. The assay procedures are described elsewhere(33). Briefly, freshly prepared PBMCs (1×105 cell/well) were incubated in duplicate cultures in the ELISPOT plates coated with anti-IFN-γ antibody and incubated for 16 hours with or without antigens at the proper concentration in complete RMPI-1640 medium. Positive and negative controls were anti-CD3, CEF, and medium alone, respectively. The plate was incubated for 16–18 hours, developed until spots appeared in the wells, then rinsed with tap water. The number of spots per well was scored using an automated ELISPOT reader (Cellular Technology Ltd., Cleveland, USA). Our previous studies suggested that thawed PBMCs with an average number of >103 SFU/106 cells after anti-CD3 stimulation are functionally viable(34). Averaged numbers of spot forming units (SFU) in control wells were subtracted from antigen-stimulated wells to correct for spontaneous cytokine production and expressed per 106 cells. Positive HCV antigen-specific responses were defined by a mean result of > 55 SFU/106 PBMC. This cut off value was derived from the work by testing of multiple healthy controls in this study and coincides with results previously reported by our group and others (35–37).
RNA preparation for Gene-Array Profiling
PBMC samples were thawed slowly and washed with RPMI media and DPBS. Following washing, total RNA was extracted using Qiagen’s RNeasy Mini Kit spin-column technology, as outlined in the manufacturer’s protocol (Qiagen, Valencia, CA.) Samples were eluted with 30uL RNase-free water. RNA was treated with Ambion’s TURBO DNA-free kit (Ambion, Grand Island, NY) to remove DNA contamination.
Microarray and genomic data analysis
Gene expression from whole blood RNA was measured via Affymetrix PrimeView microarrays and the Affymetrix-specified 3′UTR-based labeling method. The microarray quality control factors for the samples were examined using the Affymetrix Expression Console Program. CEL files were analyzed using the RMA normalization and expression level quantitation algorithm. Probesets were filtered to identify those with absolute expression in at least two samples above RMA>5.5, and then for their differential expression in their treated versus baseline per patient using Benjamini Hochberg corrected p<0.05. The resulting list of probe sets were subjected to hierarchical clustering of relative change from baseline values per patient to identify patients and genes that exhibited similar relative gene expression change patterning (heat map). Geneset enrichment and network connectivity analyses were carried out using http://toppcluster.cchmc.org and features were selected that reflected statistically significant features within category and that maximally connected between categories. In this manner a network was generated using Cytoscape to illustrate the interactions and shared properties of upregulated and downregulated genes based on known pathways, biological processes, mutant effects phenotypes, conserved promoter cis-elements, protein-protein interactions, etc. The network figures were generated based on clustering the interactions and features that were shared among genes whose expression was decreased or increased in nearly all patients following their completion of the treatment regimen. Clusters were analyzed by group in regression models. For example the interferon-stimulated gene (ISG) cluster contained interferon-alfa, interferon induced proteins, 2,5 oligoadenylate synthase (2,5 OAS) and other markers of ISG group.
Supplementary Material
Acknowledgments
Funding. This study was supported by Grant Number R01AI065256 from the National Institute of Allergy and Infectious Diseases (KES), and supported in part by USPHS Grant #UL1 RR026314 (KES) from the National Center for Research Resources, NIH. The project was also supported by the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant 8 UL1 TR000077-04 (KES) as well as NIH grants OD011095 (ASP) and AI028433 (ASP). MC was partially supported by Grants No. FIS2009-12964-C05-03 BFU2009-08009 from the Ministerio de Ciencia e Innovacion (HMvS), FP7 PIRSES-GA-2008-230665 and PIRSES-GA-2012-317893. Portions of this work were performed under the auspices of the U.S. Department of Energy under contract DE-AC52-06NA25396 (ASP). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the NIH. Bristol-Myers Squibb and Gilead Sciences, Inc provided antiretroviral medications at no charge. They had no role in the design, performance, or interpretation of this study. We are indebted to the study subjects, and to the study coordinators at each of the sites for patient recruitment and for their participation in this project.
Footnotes
Author Contributions. KES-study concept and design; organization, analysis and interpretation of data; initial draft of manuscript; obtained funding; study supervision; JG, MC, ASP-data analysis, contributions to drafts; MTS, JTB-laboratory studies, data analysis, contributed to original draft, and critical review of manuscript revisions; SDR-oversight of study sample collections; data analysis, contributed to draft, critical review of manuscript revisions, manuscript submission; JF, RS-acquisition of data; analysis and interpretation of data; critical review of manuscript; ZG-pathological evaluation of study samples; analysis and interpretation of data; critical review of manuscript; BJA-laboratory analysis and interpretation of data; contributed to original draft; critical review of manuscript.
Competing Interests. JG consults for Gilead and has received speaking fees from Rottapharm-Madaus. JF is a Bristol Myers Squibb investigator and speaker. RS served on an advisory board for Gilead. ASP is a consultant for Bristol Myers Squibb, Gilead, Merck, Achillion and Santaris. KES has served as a consultant for Merck and as a member of a Data Monitoring Board for Janssen and Synteract)
References
- 1.Sherman K, Rouster S, Chung R, Rajicic N. Hepatitis C virus prevalence among patients infected with human immunodeficiency virus: a cross-sectional analysis of the US adult AIDS clinical trials groups. Clinical Infectious Diseases. 2002;34:831. doi: 10.1086/339042. [DOI] [PubMed] [Google Scholar]
- 2.Sherman KE, Thomas DL, Chung RT. Human immunodeficiency virus and liver disease forum 2010: conference proceedings. Hepatology. 2011 Dec;54:2245. doi: 10.1002/hep.24651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sulkowski MS. Drug-induced liver injury associated with antiretroviral therapy that includes HIV-1 protease inhibitors. Clin Infect Dis. 2004 Mar 1;38(Suppl 2):S90. doi: 10.1086/381444. [DOI] [PubMed] [Google Scholar]
- 4.Sherman KE, et al. Quantitative evaluation of hepatitis C virus RNA in patients with concurrent human immunodeficiency virus infections. Journal of Clinical Microbiology. 1993;31:2679. doi: 10.1128/jcm.31.10.2679-2682.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonacini M, et al. Patients co-infected with human immunodeficiency virus and hepatitis C virus demonstrate higher levels of hepatic HCV RNA. J Viral Hepat. 1999;6:203. doi: 10.1046/j.1365-2893.1999.00153.x. [DOI] [PubMed] [Google Scholar]
- 6.Sherman K, et al. Viral kinetics in hepatitis C or hepatitis C/human immunodeficiency virus-infected patients. Gastroenterology. 2005;128:313. doi: 10.1053/j.gastro.2004.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chung R, et al. Immune recovery is associated with persistent rise in hepatitis C RNA, infrequent liver test flares, and is not impaired by hepatitis C in co-infected subjects. AIDS. 2002;16:1915. doi: 10.1097/00002030-200209270-00008. [DOI] [PubMed] [Google Scholar]
- 8.Sherman KE, et al. Liver injury and changes in hepatitis C Virus (HCV) RNA load associated with protease inhibitor-based antiretroviral therapy for treatment-naive HCV-HIV-coinfected patients: lopinavir-ritonavir versus nelfinavir. Clin Infect Dis. 2005 Oct 15;41:1186. doi: 10.1086/444501. [DOI] [PubMed] [Google Scholar]
- 9.Ragni M, Bontempo F. Increase in hepatitis C viral load in hemophiliacs during treatment with highly active antiretroviral therapy. Journal of Infectious Diseases. 1999;180:2027. doi: 10.1086/315143. [DOI] [PubMed] [Google Scholar]
- 10.Lynn HS. Maximum likelihood inference for left-censored HIV RNA data. Stat Med. 2001 Jan 15;20:33. doi: 10.1002/1097-0258(20010115)20:1<33::aid-sim640>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 11.Perelson AS, et al. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997 May 8;387:188. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- 12.Plipat N, Ruan PK, Fenton T, Yogev R. Rapid human immunodeficiency virus decay in highly active antiretroviral therapy (HAART)-experienced children after starting mega-HAART. J Virol. 2004 Oct;78:11272. doi: 10.1128/JVI.78.20.11272-11275.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuritzkes DR, et al. Plasma HIV-1 RNA dynamics in antiretroviral-naive subjects receiving either triple-nucleoside or efavirenz-containing regimens: ACTG A5166s. J Infect Dis. 2007 Apr 15;195:1169. doi: 10.1086/512619. [DOI] [PubMed] [Google Scholar]
- 14.Ding AA, Wu H. Assessing antiviral potency of anti-HIV therapies in vivo by comparing viral decay rates in viral dynamic models. Biostatistics. 2001 Mar;2:13. doi: 10.1093/biostatistics/2.1.13. [DOI] [PubMed] [Google Scholar]
- 15.G CWJ. Investigating Causal Relations by Econometric Models and Cross-spectral Methods. Econometrica. 1969;37:424. [Google Scholar]
- 16.Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009 Jul;37:W305. doi: 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaimal V, Bardes EE, Tabar SC, Jegga AG, Aronow BJ. ToppCluster: a multiple gene list feature analyzer for comparative enrichment clustering and network-based dissection of biological systems. Nucleic Acids Res. 2010 Jul;38:W96. doi: 10.1093/nar/gkq418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mocroft A, et al. Reasons for stopping antiretrovirals used in an initial highly active antiretroviral regimen: increased incidence of stopping due to toxicity or patient/physician choice in patients with hepatitis C coinfection. AIDS Res Hum Retroviruses. 2005 Jun;21:527. doi: 10.1089/aid.2005.21.527. [DOI] [PubMed] [Google Scholar]
- 19.Benhamou Y, et al. Liver fibrosis progression in human immunodeficiency virus and hepatitis C virus coinfected patients. The Multivirc Group. Hepatology. 1999;30:1054. doi: 10.1002/hep.510300409. [DOI] [PubMed] [Google Scholar]
- 20.Benhamou Y, et al. Factors affecting liver fibrosis in human immunodeficiency virus- and hepatitis C virus-coinfected patients: impact of protease inhibitor therapy. Hepatology. 2001;34:283. doi: 10.1053/jhep.2001.26517. [DOI] [PubMed] [Google Scholar]
- 21.Pascual-Pareja JF, et al. HAART is associated with lower hepatic necroinflammatory activity in HIV-hepatitis C virus-coinfected patients with CD4 cell count of more than 350 cells/microl at the time of liver biopsy. AIDS. 2009 May 15;23:971. doi: 10.1097/qad.0b013e328329f994. [DOI] [PubMed] [Google Scholar]
- 22.Lin W, et al. HIV increases HCV replication in a TGF-beta1-dependent manner. Gastroenterology. 2008 Mar;134:803. doi: 10.1053/j.gastro.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 23.Rychert J, Strick D, Bazner S, Robinson J, Rosenberg E. Detection of HIV gp120 in plasma during early HIV infection is associated with increased proinflammatory and immunoregulatory cytokines. AIDS Res Hum Retroviruses. 2010 Oct;26:1139. doi: 10.1089/aid.2009.0290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuntzen T, et al. Intrahepatic mRNA expression in hepatitis C virus and HIV/hepatitis C virus co-infection: infiltrating cells, cytokines, and influence of HAART. AIDS. 2008 Jan 11;22:203. doi: 10.1097/QAD.0b013e3282f3553b. [DOI] [PubMed] [Google Scholar]
- 25.Qurishi N, et al. Effect of antiretroviral therapy on liver-related mortality in patients with HIV and hepatitis C virus coinfection. Lancet. 2003;362:1708. doi: 10.1016/S0140-6736(03)14844-1. [DOI] [PubMed] [Google Scholar]
- 26.Sherman KE, et al. Sustained Long-Term Antiviral Maintenance Therapy in HCV/HIV-Coinfected Patients (SLAM-C) J Acquir Immune Defic Syndr. 2010 Oct 1; doi: 10.1097/QAI.0b013e3181f6d916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang F, et al. Transient liver injury associated with the early recovery of HCV-specific T-cell responses and HCV rebound in HIV-1/HCV coinfected patients undergoing highly active antiretroviral therapy. J Acquir Immune Defic Syndr. 2013 Feb 1;62:135. doi: 10.1097/QAI.0b013e3182752d20. [DOI] [PubMed] [Google Scholar]
- 28.Eyster ME, Fried MW, Di Bisceglie AM, Goedert JJ. Increasing hepatitis C virus RNA levels in hemophiliacs: relationship to human immunodeficiency virus infection and liver disease. Multicenter Hemophilia Cohort Study. Blood. 1994;84:1020. [PubMed] [Google Scholar]
- 29.Thompson MA, et al. Antiretroviral treatment of adult HIV infection: 2012 recommendations of the International Antiviral Society-USA panel. JAMA. 2012 Jul 25;308:387. doi: 10.1001/jama.2012.7961. [DOI] [PubMed] [Google Scholar]
- 30.Ho DD, et al. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995 Jan 12;373:123. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 31.Wei X, et al. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 1995;373:117. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 32.Ramratnam B, et al. Rapid production and clearance of HIV-1 and hepatitis C virus assessed by large volume plasma apheresis. Lancet. 1999 Nov 20;354:1782. doi: 10.1016/S0140-6736(99)02035-8. [DOI] [PubMed] [Google Scholar]
- 33.Shata MT, et al. Characterization of the immune response against hepatitis C infection in recovered, and chronically infected chimpanzees. J Viral Hepat. 2002;9:400. doi: 10.1046/j.1365-2893.2002.00373.x. [DOI] [PubMed] [Google Scholar]
- 34.Hashem M, et al. Strong hepatitis C virus (HCV)-specific cell-mediated immune responses in the absence of viremia or antibodies among uninfected siblings of HCV chronically infected children. J Infect Dis. 2011 Mar 15;203:854. doi: 10.1093/infdis/jiq123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shata MT, et al. Characterization of hepatitis E-specific cell-mediated immune response using IFN-gamma ELISPOT assay. J Immunol Methods. 2007 Dec 1;328:152. doi: 10.1016/j.jim.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Al-Sherbiny M, et al. Exposure to hepatitis C virus induces cellular immune responses without detectable viremia or seroconversion. Am J Trop Med Hyg. 2005 Jul;73:44. [PubMed] [Google Scholar]
- 37.Dubey S, et al. Detection of HIV vaccine-induced cell-mediated immunity in HIV-seronegative clinical trial participants using an optimized and validated enzyme-linked immunospot assay. J Acquir Immune Defic Syndr. 2007 May 1;45:20. doi: 10.1097/QAI.0b013e3180377b5b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.