

Integrated flux balance analysis accurately reconstructs phototrophic and heterotrophic metabolism in Chlorella protothecoides.
Abstract
Integrated and genome-based flux balance analysis, metabolomics, and 13C-label profiling of phototrophic and heterotrophic metabolism in Chlorella protothecoides, an oleaginous green alga for biofuel. The green alga Chlorella protothecoides, capable of autotrophic and heterotrophic growth with rapid lipid synthesis, is a promising candidate for biofuel production. Based on the newly available genome knowledge of the alga, we reconstructed the compartmentalized metabolic network consisting of 272 metabolic reactions, 270 enzymes, and 461 encoding genes and simulated the growth in different cultivation conditions with flux balance analysis. Phenotype-phase plane analysis shows conditions achieving theoretical maximum of the biomass and corresponding fatty acid-producing rate for phototrophic cells (the ratio of photon uptake rate to CO2 uptake rate equals 8.4) and heterotrophic ones (the glucose uptake rate to O2 consumption rate reaches 2.4), respectively. Isotope-assisted liquid chromatography-mass spectrometry/mass spectrometry reveals higher metabolite concentrations in the glycolytic pathway and the tricarboxylic acid cycle in heterotrophic cells compared with autotrophic cells. We also observed enhanced levels of ATP, nicotinamide adenine dinucleotide (phosphate), reduced, acetyl-Coenzyme A, and malonyl-Coenzyme A in heterotrophic cells consistently, consistent with a strong activity of lipid synthesis. To profile the flux map in experimental conditions, we applied nonstationary 13C metabolic flux analysis as a complementing strategy to flux balance analysis. The result reveals negligible photorespiratory fluxes and a metabolically low active tricarboxylic acid cycle in phototrophic C. protothecoides. In comparison, high throughput of amphibolic reactions and the tricarboxylic acid cycle with no glyoxylate shunt activities were measured for heterotrophic cells. Taken together, the metabolic network modeling assisted by experimental metabolomics and 13C labeling better our understanding on global metabolism of oleaginous alga, paving the way to the systematic engineering of the microalga for biofuel production.
Green algae in the genus Chlorella spp. are a large group of eukaryotic, unicellular, and photosynthetic microorganisms that are widely distributed in freshwater environments. Chlorella spp. can grow photoautotrophically and were used as a model system in early research on photosynthetic CO2 fixation (Bassham et al., 1950; Barker et al., 1956; Calvin, 1956). They are also among the very few algal groups capable of using organic carbon for heterotrophic growth, which endows Chlorella spp. metabolic flexibility in response to environmental perturbation. Because of its robust and various metabolic capacities, Chlorella spp. has aroused widespread interest as a potential alga for industrial production of biomass (Lee, 2001), biofuel (Xu et al., 2006), and value-added chemicals (Pulz and Gross, 2004).
Chlorella spp. are among the best oil feedstock microorganisms for the production of biofuel (Gouveia and Oliveira, 2009). It was frequently reported that, under nitrogen-limited environments, carbon overflow (e.g. nitrogen depletion or organic carbon feeding) allows Chlorella spp. to accumulate a high percentage of neutral lipids that can be processed for biodiesel production (Miao and Wu, 2006). Biogenesis of neutral lipids was found in Chlorella spp. cells undergoing glucose bleaching, in which the growth is switched from photoautotrophic to heterotrophic mode and accompanied by chlorophyll degradation. This metabolic transition has been incorporated into a highly efficient biofuel production process (Xiong et al., 2010a), whereas many global changes in metabolism, such as degeneration of the chloroplast, redistribution of carbon flux, and reprogrammed nitrogen assimilation, remain poorly understood.
In our previous studies, we initiated metabolic analysis of Chlorella spp. by steady-state 13C analysis of proteinogenic amino acids and profiling of metabolic fluxes for heterotrophic Chlorella protothecoides (Xiong et al., 2010b). The quantitative metabolic information can be further expanded to the complete metabolome by means of recent development in isotope-associated mass spectroscopy (MS), which enables the accurate measurement of both isotopic labeling kinetics and intracellular metabolite concentrations (Bennett et al., 2008; Seifar et al., 2008). Intracellular fluxes can be estimated by computational modeling of dynamic isotopic labeling patterns (Young et al., 2011). This methodology allowed for mapping fluxes under autotrophic growth conditions (Young et al., 2011), because it overcame the limitation of traditional steady-state 13C metabolic flux analysis (MFA), which fails to resolve the flux from a uniformly steady 13C labeling pattern because of the assimilation of CO2 as carbon source of autotrophic organisms.
The metabolic information may also be interfaced with genomic knowledge to generate a bird’s eye view of the metabolic properties in system level. Metabolic pathways can be reassembled stoichiometrically and simulated by constraint-based flux balance analysis (FBA), which uses a linear programming approach (Lee et al., 2006) to accomplish the optimal solutions of the objective function (usually maximizing the yield of biomass or a specific metabolic product; Edwards et al., 2002; Oberhardt et al., 2009). The reconstructed metabolic models based on multidimensional genome annotation are of great value in guiding metabolic engineering and genetic improvement of photosynthetic microorganisms, such as Synechocystis sp. PCC 6803 (Shastri and Morgan, 2005; Fu, 2009; Knoop et al., 2010; Montagud et al., 2010; Nogales et al., 2012), Arthrospira platensis (Cogne et al., 2003; Klanchui et al., 2012), and Chlamydomonas reinhardtii (Boyle and Morgan, 2009; Chang et al., 2011; Dal’Molin et al., 2011).
The goal of our study is to systematically investigate C. protothecoides metabolism. Our research procedures are outlined in Figure 1, combining the computational and experimental knowledge by in silico reconstruction of the metabolic network. Specifically, based on functional annotation of the genome (Gao et al., 2014), we mapped primary metabolism of C. protothecoides and adopted FBA to simulate the optimal status for cell growth and lipid accumulation. To validate the FBA model, we quantitatively measured core metabolites involved in the metabolic model by liquid chromatography (LC) -MS. Kinetic isotopic tracing of intracellular metabolites was performed to quantify intracellular fluxes. Comparative analysis of 13C MFA measurement and FBA prediction suggests distinct properties of C. protothecoides in autotrophic and heterotrophic metabolism. Altogether, the studies presented here will detail the Chlorella spp. metabolism and pave the way to biofuel production from this microalga with the predictive ability of a constructed metabolic model.
Figure 1.

Schematic MFA workflow of C. protothecoides using computational and experimental approaches. Briefly, we reconstructed the primary metabolic network of the oleaginous microalga C. protothecoides based on the recently available gene functional annotation (Gao et al., 2014). Intracellular fluxes under different cultivation conditions were simulated, and the optimal solutions were predicted using cell growth and lipid accumulation as the objective functions. The genome-based metabolic model was further constrained by experimental metabolomics and dynamic isotope labeling for INST-MFA (Young, 2014). Results from 13C MFA measurement and FBA prediction are comparably analyzed.
RESULTS
A Primary Metabolic Network of C. protothecoides
The starting point of metabolic mapping was to reconstruct the primary metabolic network based on the genome of C. protothecoides, which was sequenced recently (Gao et al., 2014). The genomic information contained in the database was reorganized into various essential pathways, such as the Calvin-Benson cycle/the pentose phosphate (PP) pathway, glycolysis/gluconeogenesis, the tricarboxylic acid cycle, biosynthetic pathways of macrobiomolecules (amino acids, nucleotides, UDP-Glc, glycerol-3-P, and fatty acids as well as chlorophyll), etc. (Fig. 2). Some catabolic pathways, such as β-oxidation of fatty acids, were also assembled. For each biochemical reaction, details are listed for gene number, gene ontology number, and enzyme name. The EC number and the Kyoto Encyclopedia of Genes and Genomes identifier for each enzyme are also provided so that the stoichiometry of the reactions can be subsequently looked up and extracted from the online database.
Figure 2.

An overview of the reconstructed primary metabolic pathways in C. protothecoides. Dashed arrows denote simplified multiple steps in a pathway. Dotted arrows denote intracellular transmembrane transport reaction. Ac, Acetate; AcCoa, acetyl CoenzymeA; AH, acetaldehyde; AKG, α-ketoglutarate; Cit, citrate; DHAP, dihydroxyacetone phosphate; E4P, erythrose-4-phosphate; EtOH, ethanol; F6P, fructose-6-phosphate; FBP, fructose-1,6-bisphosphate; Fum, fumarate; G3P, glucose-3-phosphate; G6P, glucose-6-phosphate; Glx, glyoxylate; Go3P, glycerol-3-phosphate; Gr, glycerate; Hpyr, hydroxypyruvate; Isocit, isocitrate; Lac, lactate; Mal, malate; OAA, oxaloacetate; 3PG, 3-phosphoglycerate; P5P, pentose-5-phosphate; PEP, phosphoenolpyruvate; PGI, phosphoglycolate; PGluc, 6-phosphogluconate; PI, phosphatidyl-inositol; PSP, Pyr, pyruvate; RuBP, ribulose-1,5-bisphosphate; SBP, sedoheptulose-1,7-bisphosphate; S7P, sedoheptulose-7-phosphate.
All reactions were considered to be localized into four main compartments: cytosol, chloroplast (Gao et al., 2014), mitochondria (Grant and Hommersand, 1974), and peroxisome (Codd et al., 1972; Chou et al., 2008), where most of the common metabolic reactions take place. Corresponding compounds, including gas, nutrients, and intracellular metabolites, were shuttled between compartments through transport systems or passive diffusion. The methodology for localization was described in “Materials and Methods.”
In the process of reassembling the genome-based information, in total, 270 functional enzymes coded by 461 metabolism-related genes catalyzing 272 biochemical reactions are relocated into their metabolic pathways, suggesting the completeness of primary metabolism in Chlorella spp. Absences of some reactions were identified. Gaps found in the pathways were cured using a protein query tblastn search, which confirms the possible existence of the missing enzymes in the C. protothecoides genome. These unannotated enzymes are listed in Supplemental Table S1. Of these enzymes, only homo-Ser O-acetyltransferase (EC 2.3.1.31), histidinol-phosphatase (EC 3.1.3.15), 1-deoxy-d-xylulose-5-P reductoisomerase (EC 1.1.1.267), and uroporphyrinogen-III synthase (EC 4.2.1.75) have no hits in the database. To maintain the integrity of the metabolic pathway, assumptions of their presence in the model were made.
To simulate cell growth and predict intracellular fluxes, biomass and macromolecular compositions were measured and are presented in Table I. Biomass was divided into protein, lipid, carbohydrate, DNA, and RNA, which totally account for 92% and 95% dry cell weight of autotrophic and heterotrophic cells, respectively (Fig. 3, A and B). The significant variations between the two types of cells are the predominant presence of protein in autotrophic C. protothecoides and the vast majority of lipid in heterotrophic biomass, indicating nutrition effect on cell storage. To detail lipid metabolism in our model, we also analyzed the fatty acid profile in C. protothecoides and found predominantly fatty acid chains of 18 carbons with one and two degrees of unsaturation in both autotrophic and heterotrophic cells.
Table I. Biomass components and macromolecular composition of C. protothecoides.
Biomass components of major cellular macromolecules were expressed based on mass fraction of dry cell weight. Detailed macromolecular composition was expressed on the basis of mole percentage. Data are represented as mean ± sd.
| Cellular Constituent | Autotrophic Cells | Heterotrophic Cells |
|---|---|---|
| Proportion of macromolecular components (g per 100 g dry cell wt)a | ||
| Protein | 55.89 ± 0.35 | 14.14 ± 0.20 |
| Lipid | 12.11 ± 0.12 | 53.60 ± 0.21 |
| Carbohydrate | 12.93 ± 0.63 | 15.53 ± 0.54 |
| DNA | 2.97 ± 0.23 | 3.07 ± 0.04 |
| RNA | 6.63 ± 0.32 | 7.54 ± 0.36 |
| Chlorophyll | 1.00 ± 0.10 | 0 |
| Proteinogenic amino acid composition (mol %) | ||
| Ala | 0.133 ± 0.047 | 0.131 ± 0.026 |
| Arg | 0.041 ± 0.014 | 0.050 ± 0.010 |
| Asn | 0.045 ± 0.016 | 0.044 ± 0.009 |
| Asp | 0.045 ± 0.016 | 0.044 ± 0.009 |
| Cys | 0.004 ± 0.001 | 0.005 ± 0.001 |
| Gln | 0.054 ± 0.019 | 0.050 ± 0.010 |
| Glu | 0.054 ± 0.019 | 0.050 ± 0.010 |
| Gly | 0.134 ± 0.047 | 0.099 ± 0.020 |
| His | 0.016 ± 0.006 | 0.020 ± 0.004 |
| Ile | 0.035 ± 0.012 | 0.038 ± 0.008 |
| Leu | 0.083 ± 0.029 | 0.088 ± 0.018 |
| Lys | 0.054 ± 0.019 | 0.067 ± 0.013 |
| Met | 0.007 ± 0.002 | 0.012 ± 0.002 |
| Phe | 0.036 ± 0.013 | 0.036 ± 0.007 |
| Pro | 0.059 ± 0.021 | 0.053 ± 0.011 |
| Ser | 0.069 ± 0.024 | 0.067 ± 0.013 |
| Thr | 0.056 ± 0.020 | 0.060 ± 0.012 |
| Trp | 0.000 | 0.002 ± 0.000 |
| Tyr | 0.020 ± 0.007 | 0.021 ± 0.004 |
| Val | 0.058 ± 0.020 | 0.067 ± 0.013 |
| Fatty acid composition (mol %) | ||
| C14:0 | 0.026 ± 0.003 | 0.013 ± 0.003 |
| C16:0 | 0.090 ± 0.011 | 0.129 ± 0.027 |
| C18:0 | 0.007 ± 0.001 | 0.028 ± 0.006 |
| C18:1 | 0.544 ± 0.065 | 0.608 ± 0.128 |
| C18:2 | 0.311 ± 0.037 | 0.173 ± 0.036 |
| DNA (RNA) composition (mol %)b | ||
| A | 0.186 | 0.186 |
| T(U) | 0.186 | 0.186 |
| C | 0.314 | 0.314 |
| G | 0.314 | 0.314 |
Biomass components were comparable between two types of cells and applicable for additional calculation with the unmeasured dry weights less than 10% (8% and 5% of autotrophic and heterotrophic cells, respectively).
Nucleotide composition of DNA was obtained from genome sequencing, and the same composition was assumed for RNA.
Figure 3.

Biomass composition of autotrophic (A) and heterotrophic (B) C. protothecoides and concentration composition of the measured intracellular metabolites of autotrophic (C) and heterotrophic (D) C. protothecoides. Biomass components of major cellular macromolecules were expressed based on mass fraction of dry cell weight. Metabolite concentrations were expressed in moles per liter, and percentages of total concentrations are in parentheses. Amino acids include Glu, Arg, Ala, Asp, Gln, Asn, Lys, Pro, Thr, Val, Gly, Ser, His, Leu, Ile, Met, Tyr, Trp, and Phe. Nucleotides include ATP, ADP, and AMP. Central carbon intermediates are γ-aminobutyric acid, (iso)citrate, malate, Glc/Fru-6-P, α-ketoglutarate, succinate, ribose-5-P, dihydroxyacetonphosphate, 6-phosphogluconate, phosphoenolpyruvate, fumarate, Hyp, and pyruvate. Redox cofactors are NAD+, NADH, NADP+, and NADPH, and coenzymes are acetyl-CoA and malonyl-CoA.
Based on the newly accessible genome knowledge as well as the biomass composition information, we reconstructed the primary metabolic network and established a fundamental model focusing on the core metabolism of C. protothecoides. This is the first genome-based metabolic network of this alga, which could serve as an in silico platform to exploit the intracellular properties of C. protothecoides metabolism. The detailed process is described in “Materials and Methods,” and the complete dataset regarding reaction network and gene-protein association of the enzymes and transporters involved in the network is presented in Supplemental Tables S2 and S3. The reconstruction data are also provided in SBML format as Supplemental Text S1 and S2.
Growth Simulation of C. protothecoides under Autotrophic and Heterotrophic Cultivation Conditions
On the basis of the reconstructed metabolic model, growth properties of C. protothecoides in different cultivation conditions were evaluated using FBA. We defined the flux of nutrients and metabolic end products in and out of cells under these circumstances as constraints to maximize biomass production as the objective function (flux balance analysis model 1 [FBA1]; described in “Materials and Methods”). The calculated specific growth rate was in good agreement with the experimentally determined one (Table II), validating the accurate determination of uptake/secretion flux in and out of cells and that no additional significant carbon assimilation reaction and secretion are missed during the cultivation. We also predicted the ideal growth rate of C. protothecoides in autotrophic and heterotrophic conditions with only the energy source specified (flux balance analysis model 2 [FBA2]; described in “Materials and Methods”). This model can explore the optimal carbon use. The result shows the potential of autotrophic and heterotrophic growth rates, which could be further improved 20% and 10%, respectively (Table II). According to the comparison of FBA1 and FBA2 (Supplemental Fig. S1), improved carbon metabolism can be realized by less carbon loss from the oxidative PP pathway and pyruvate dehydrogenase and increased carbon assimilation from phosphoenolpyruvate carboxylase as well.
Table II. Experimentally determined and predicted specific growth rate based on growth parameters of autotrophic and heterotrophic C. protothecoides.
Determined data were represented as mean ± sd. μ, Specific cell growth rate.
Phenotype-Phase Plane Analysis Predicts Optimal Growth and Fatty Acid Synthesis under Autotrophic and Heterotrophic Conditions
Based on the reconstructed network model, we predicted the optimal growth rate and corresponding productivity of targeted fuel compounds (represented by the oleic acid, which is the most abundant fatty acid in C. protothecoides) using phenotype-phase plane (PhPP) analysis (Edwards et al., 2001) with the variation of input fluxes (light absorption and CO2 uptake in autotrophic mode, and O2 consumption and Glc uptake in heterotrophic mode). For the autotrophic model, the intensity of light input was represented by absorbed photon flux. The surface of a three-dimensional PhPP corresponding to the predicted maximal growth rate and fatty acid synthesis rate was plotted as a function of the uptake photon flux (0–3 mmol g dry cell weight−1 h−1) and the CO2 uptake rate (0–0.3 mmol g dry cell weight−1 h−1; Fig. 4, A and B). It was simulated that the cells exhibited distinct phenotypes depending on the amounts of carbon fixation and light absorption. As shown, the rates for growth and fatty acid production are zero in Region I, where photon uptake is low. It is probable that light is insufficient to generate enough ATP for cell growth in this region. In Region II, the maximal cell growth is positively correlated with the light absorption; however, it is negatively correlated with the CO2 uptake. Interestingly, inhibition of photosynthesis by elevated CO2 was reported in other eukaryotic algae, such as C. reinhardtii (Yang and Gao, 2003) and Chlorococcum littorale (Satoh et al., 2001). In Region III, the growth rate is only limited by the CO2 uptake because of light saturation. In economic considerations, the optimal growth and lipid accumulation appear on the demarcation line separating Regions II and III, with the ratio of photon uptake to CO2 consumption rate around 8.4.
Figure 4.

The predicted maximal growth rate (A) and corresponding fatty acid production rate (B) as a function of light intensity and total CO2 uptake, respectively, under photoautotrophic growth. The predicted maximal growth rate (C) and fatty acid production rate (D) as a function of Glc and O2 uptake, respectively, under heterotrophic growth. The fatty acid production rate is represented by that of oleic acid, which is the most abundant fatty acid in the C. protothecoides biomass. The surface is colored relative to the value of the z axis. gCDW, Gram cell dry weight.
Similarly, prediction of the optimal growth rate and corresponding fatty acid synthesis rate in heterotrophic C. protothecoides was conducted with Glc and O2 consumption rate (ranging from 0 to 0.9 mmol g dry cell weight−1 h−1 and from 0 to 1.2 mmol g dry cell weight−1 h−1, respectively) as input variables (Fig. 4, C and D). The surface plot also shows three distinct regions. In Region I, the optimal growth rate is dependent exclusively on oxygen absorption, because oxidative phosphorylation could be repressed with low O2 availability. This is comparable with the work by Chen et al. (2011), which showed that enhanced O2 uptake could lead to higher growth rate of Escherichia coli. Cell growth and fatty acid biosynthesis are dependent on both variables in Region II at the very beginning, after which the rates of cell growth and fatty acid synthesis cease with excess O2 uptake. According to the PhPP model, one probable explanation is that excessive O2 uptake leads to the weakening of the PP pathway and greatly enhanced activity of the tricarboxylic acid cycle, which oxidizes organic carbon to CO2. The situation continues in Region III, and algal cells fail to grow with low Glc availability and high O2 absorption. Consequently, the optimal ratio of Glc uptake to O2 consumption is 2.7 for heterotrophic growth and fatty acid synthesis, which could serve as a theoretical criterion for the cultural optimization of C. protothecoides in Glc-containing media.
Metabolite Concentrations in Autotrophic and Heterotrophic Cells
To validate the genome-based metabolic network of C. protothecoides experimentally, we took advantage of the LC-MS tools to quantitatively measure the network components: intracellular metabolites throughout the central carbon metabolism. To this end, we set up an LC-MS/MS program for metabolomics that coupled hydrophilic interaction and reversed-phase HPLC by electrospray ionization to triple-quadrupole MS/MS. The optimized MS parameters for each metabolite, including the limit of quantification, coefficient of determination (R2), and relative sd, were listed in Supplemental Table S4. We also optimized the procedures for sample preparation, including cell harvesting, sample quenching, and solvent extracting. The extraction capacities of various solvent combinations were compared with a heat map analysis (Supplemental Fig. S2), and as a result, the methanol:water (50:50) mixture was shown to have major advantages for metabolite yields over other combinations.
To avoid the ion suppression caused by the matrix effect (Matuszewski et al., 1998; Annesley, 2003), which suppresses the signal of a compound in the sample relative to that of a standard at the same concentration and leads to the unreliability of determination, we used an isotope labeling approach proposed by Bennett et al. (2008) for metabolomic quantitation. Briefly, unlabeled standards in known concentrations were spiked into the extracts of cells that were completely labeled with isotopic substrate in the culture media. The amount of endogenous metabolite present in the cells was then determined by the ratio of endogenous metabolite to internal standard in the extract.
Identified metabolites with intracellular concentrations in both photoautotrophic and heterotrophic cells are summarized in Supplemental Table S5 with 95% confidence intervals. Of a total of 144 metabolites from the reconstructed draft metabolic network, 40 were identified and quantitated in the metabolome of C. protothecoides. The core metabolome, on a molar basis, consists of sugar phosphates (0.3% in autotrophic and 4.8% in heterotrophic mode), organic acids (7.7% in autotrophic and 11.3% in heterotrophic mode), amino acids (89.7% in autotrophic and 76.3% in heterotrophic mode), CoA (0.01% in autotrophic and 0.1% in heterotrophic mode), nucleotides (1.3% in autotrophic and 4.8% in heterotrophic mode), and reducing equivalents (0.9% in autotrophic and 2.4% in heterotrophic mode; Fig. 3, C and D). The most abundant metabolite in both nutritional patterns is Glu measured as 21.8 and 6.53 mm, respectively. Compared with metabolite concentrations between nutrient patterns, variations are present in the majority of metabolites (77.5% of which exhibit significant difference between autotrophic and heterotrophic cells; P < 0.05 by two-tailed Student’s t test), especially for intermediates in glycolysis and the tricarboxylic acid cycle, in accordance with the substantial impact of nutrient mode on the metabolome composition.
Metabolome of C. protothecoides mirrors its metabolic features in response to nutrient changes. For example, with respect to the metabolites of reducing equivalents and bioenergy currency, the molar ratio of NADPH to its oxidative counterpart rises from 0.66 in CO2-fed cells to 1.75 in Glc-fed cells. The NADH-NAD+ ratio also increases significantly from 0.003 in CO2-fed cells to 0.01 in cells fed with Glc. Also, intracellular concentration of ATP presents a 1.5-fold increase in heterotrophic growth, and the energy charge correspondingly increases from 0.9 to 0.92 according to ([ATP] + 0.5[ADP])/([ATP] + [ADP] + [AMP). Together, these results are in good agreement with the notion that Glc-fed condition favors energy-intensive metabolism, such as fatty acid biosynthesis (Hardie et al., 1999). This conclusion is further supported by altered levels of building blocks in the fatty acid pathway: 3-fold and as high as 97-fold increases are observed for acetyl-CoA and malonyl-CoA, respectively, in heterotrophic metabolome compared with the autotrophic one.
Flux Estimation Using Isotopically Nonstationary Metabolic Flux Analysis
To further validate the genome-based metabolic network in experimental conditions, we applied isotopically nonstationary metabolic flux analysis (INST-MFA; Young et al., 2011) to our metabolic network, tracing the kinetics of isotope labeling trajectories and quantifying the fluxes in C. protothecoides. Dynamic isotope labeling of eight metabolites in autotrophic cells and nine metabolites in heterotrophic cells is shown in Supplemental Figure S3. For autotrophic cells, 13C-bicarbonate uptake resulted in prompt labeling of metabolites involved in the Calvin-Benson cycle (i.e. 3-phosphoglycerate, erythrose-4-P, and Glc-6-P/Fru-6-P), whereas there was relatively slower turnover of the tricarboxylic acid cycle intermediates (i.e. citrate/isocitrate, succinate, fumarate, and malate), reflecting differentiated flux distribution among pathways. Note that the tricarboxylic acid cycle metabolites, such as (iso)citrate and succinate, were slowly labeled as above 50% of the unlabeled (M0) isotopomer that remained 60 min postlabeling. In contrast to this, sugar phosphates, such as 3-phosphoglycerate, have a short half-labeling time around 8 min. With respect to heterotrophic cells, feeding uniformly labeled 13C-Glc to cells led to faster labeling of sugar phosphates (i.e. 3-phosphoglycerate, erythrose-4-P, and Glc-6-P/Fru-6-P), reflecting high activity of glycolytic and the PP pathway. In terms of metabolites in the tricarboxylic acid cycle, one interesting finding is the identical labeling pattern of malate and fumarate. Prompt appearance of the three carbon-labeled (M3) isotope was observed for both of them, indicating the strong activities of malic enzyme or phosphoenolpyruvate carboxylase, which convert labeled (phosphoenol)pyruvate and unlabeled CO2 to organic acids and thus, shortcut the tricarboxylic acid cycle from the oxidative direction.
Based on least squares regression, by which the residuals between experimentally determined and simulated isotopomer distributions are minimized, we generated a quantitative flux map of C. protothecoides as displayed in Figure 5. Net fluxes are normalized to a CO2 fixation rate of 100 by Rubisco flux (0.154 mmol g dry cell weight−1 h−1) in autotrophic cells and Glc uptake rate of 100 in heterotrophic cells (Glc uptake rate: 0.303 mmol g dry cell weight−1 h−1). The metabolic activity of autotrophically growing cells is mainly derived from the chloroplast as a generator of energy and organic compounds. The energy needed for autotrophic metabolism mainly comes from the photophosphorylation in chloroplast, and glyceraldehyde-3-P, the product of photosynthesis, is transferred into the cytoplasm and serves as a precursor for the biosynthesis of biomass components (Fig. 5A). It is noticed that a much smaller flux through succinate was calculated compared with the average metabolic flow in the upstream and downstream of this node, suggesting a low respiratory activity for succinate dehydrogenase under illumination. This result is consistent with the notion that the major function of the tricarboxylic acid cycle in photosynthetic microbes is in biosynthesis instead of generating energy (Pearce et al., 1969; Steinhauser et al., 2012; Schwarz et al., 2013). According to the computation results, photorespiration, the oxygenation process of Rubisco (a bifunctional enzyme catalyzing the first step of CO2 fixation in the Calvin-Benson cycle), functions in autotrophic growth but merely accounts for 0.1% of the total Rubisco activity (Fig. 5A). Although photorespiration flux is measured low in photosynthetic microbes, such as cyanobacteria (Young et al., 2011), it is usually thought to be indispensable as glyoxylate, the intermediate in this pathway that is required for Gly and Ser biosynthesis (Knoop et al., 2010). In our reconstruction, these two amino acids can be synthesized elsewhere from 3-phosphoglycerate by phosphoglycerate dehydrogenase (EC 1.1.1.95: Cpr000053.1, Cpr000361.4, and Cpr001502.1), phospho-Ser transaminase (EC 2.6.1.52: Cpr004187.1), phospho-Ser phosphatase (EC 3.1.3.3: Cpr001503.4), and Gly hydroxymethyltransferase (EC 2.1.2.1: Cpr001327.2, Cpr003325.1, and Cpr003765.3). Meanwhile, high concentration of bicarbonate added for the 13C labeling experiment could further suppress photorespiration (Huege et al., 2011; Young et al., 2011).
Figure 5.

The specific flux distribution estimated using INST-MFA for autotrophic (A) and heterotrophic (B) C. protothecoides. Net fluxes are normalized to CO2 fixation rate of 100 by Rubisco in autotrophic cells and Glc uptake rate of 100 in heterotrophic cells, respectively. (The actual reaction rates for Rubisco and Glc uptakes are 0.154 and 0.303 mmol g dry cell weight−1 h−1, respectively.) The thickness of the arrow is scaled proportionally to the flux value. Dashed arrows represent formation of biomass. Ac, Acetate; AcCoa, acetyl CoenzymeA; AKG, α-ketoglutarate; Cit, citrate; DHAP, dihydroxyacetone phosphate; E4P, erythrose-4-phosphate; F6P, fructose-6-phosphate; FBP, fructose-1,6-bisphosphate; Fum, fumarate; G3P, glucose-3-phosphate; G6P, glucose-6-phosphate; Ga, glycerate; Glx, glyoxylate; Mal, malate; OAA, oxaloacetate; 3PG, 3-phosphoglycerate; PEP, phosphoenolpyruvate; PGI, phosphoglycolate; R5P, ribose-5-phosphate; Ru5P, ribulose-5-phosphate; RuBP, ribulose-1,5-bisphosphate; SBP, sedoheptulose-1,7-bisphosphate; S7P, sedoheptulose-7-phosphate; X5P, xylulose-5-phosphate.
Flux map for heterotrophic cells was based on the Glc catabolism, because C. protothecoides grew upon U-13C Glc as the sole carbon source. Most of the carbon flux for heterotrophic growth is directed through the glycolytic pathway (Fig. 5B). High activity of the C4 pathway is also observed. Consistent with the rapid accumulation of malate M3 isotopomer, malic enzyme channels 36% of the flux through phosphoenolpyruvate. The glyoxylate shunt (isocitrate lyase encoding gene Cpr004092.1 and malate synthase encoding gene Cpr001903.2) was included in our model while measured to be inactive in heterotrophic cells, which agrees with our previous study (Xiong et al., 2010a, 2010b). Another noteworthy result is the high use rate of acetyl-CoA for fatty acids in heterotrophically grown C. protothecoides, exhibiting 33-fold higher flux than that in autotrophic growth (Fig. 5B). Interestingly, acetyl-CoA inputs into the tricarboxylic acid cycle also enhance greatly. In heterotrophic conditions, the activity of the tricarboxylic acid cycle depending on acetyl-CoA oxidation arises to supply the energy and reducing equivalents required for biomass formation, including the biosynthesis of fatty acids. Because acetyl-CoA is also presumably used as the building block for fatty acid synthesis, an appropriate split ratio of metabolic flux should be assigned into these two pathways for the carbon and cofactor balancing. According to our flux map, the ratio of pyruvate used for fatty acid synthesis to that directed through the tricarboxylic acid cycle is assigned as 6.8:1 in heterotrophic Chlorella spp. and correspondingly, 3.4:1 in autotrophic cells, suggesting a flexible and tunable flux distribution for fatty acid synthesis. Feasible strategies to optimize this ratio may include inhibition of citrate synthase, the first enzyme in the tricarboxylic acid cycle. It was reported to cause the accumulation of acetyl-CoA, which could be donated for fatty acid synthesis (Taylor, 1973; Coleman and Bhattacharjee, 1975; Underwood et al., 2002). Meanwhile, alternative pathways generating the reducing equivalents are desired for fatty acid synthesis.
Cofactor Balance Analysis
Based on the quantitative results of 13C MFA, the production and consumption of cofactors ATP and NAD(P)H can be analyzed in autotrophic and heterotrophic C. protothecoides. As shown in Table III, photosynthesis is principally responsible for the turnover of cofactors in autotrophic cells, because 89% ATP and 78.6% NADPH are directly derived from the light reactions and consumed for the generation of glyceraldehyde-3-P. The rest of cofactors are used in biomass synthesis for cell growth. For example, fatty acid synthesis consume 1.1% and 4.1% of the total ATP and NAD(P)H produced, respectively. In heterotrophic cells, glycolysis and tricarboxylic acid cycle are the main contributors for the formation of ATP. Note that maintenance accounts for a great percentage of ATP drain according to the quantitative model. Reducing equivalents are concomitantly produced during the formation of 3-phosphoglycerate and the decarboxylation catalyzed by pyruvate dehydrogenase as well as other reaction process in the tricarboxylic acid cycle. The abundantly formed NAD(P)H is mainly used for ATP generation through the respiratory electron transport chain and the formation of biomass components, especially the NADPH-intensive fatty acid synthesis [computationally 13.6% and 53.1% of the total ATP and NAD(P)H produced, respectively] in heterotrophic C. protothecoides.
Table III. Balance of ATP and NAD(P)H cofactors in autotrophic and heterotrophic C. protothecoides.
Stoichiometric coefficient was calculated according to net fluxes normalized to CO2 fixation rate of 100 by Rubisco (autotrophic cells) and Glc uptake rate of 100 (heterotrophic cells). Fractional contributions in percentages of different pathways to the cofactor balance are indicated in parentheses.
| Cofactor | Glycolysis/ Glyconeogenesis | Photosynthesis | PP Pathway | Pyruvate Metabolism | Tricarboxylic Acid Cycle | Anaplerotic Pathway | Biomass Formation | ATP Maintenance |
|---|---|---|---|---|---|---|---|---|
| Autotrophic cells | ||||||||
| ATP | ||||||||
| Formation | 41.6 (10.9) | 340.1 (89.0) | 0.5 (0.1) | |||||
| Consumption | 0.6 (0.2) | 300.1 (78.5) | 1.4 (0.4) | 66.5 (17.4) | 13.5 (3.5) | |||
| NAD(P)H | ||||||||
| Formation | 27.1 (11.8) | 180.4 (78.6) | 3.2 (1.4) | 7.8 (3.4) | 4.7 (2.0) | 3.0 (1.3) | 3.4 (1.5) | |
| Consumption | 200.1 (87.1) | 2.3 (1.0) | 27.2 (11.8) | |||||
| Heterotrophic cells | ||||||||
| ATP | ||||||||
| Formation | 278.0 (41.8) | 387.5 (58.2) | ||||||
| Consumption | 188.4 (27.7) | 2.0 (0.3) | 199.9 (29.4) | 290.7 (42.7) | ||||
| NAD(P)H | ||||||||
| Formation | 168.9 (44.1) | 6.8 (1.8) | 128.9 (33.6) | 60.4 (15.8) | 18.1 (4.7) | |||
| Consumption | 4.9 (1.3) | 155.0 (40.3) | 224.4 (58.4) |
DISCUSSION
Elucidation of metabolic properties and functions in a systemic level can be greatly facilitated by metabolic network modeling that integrates the genome-annotated enzymatic reactions and computational approaches to a functional entity. Most of the current reconstructed networks are assessed using FBA, which uses a linear programming approach and provides a metabolic flux distribution consistent with the optimal solution of the objective function. In this study, we reconstructed the primary metabolic network of C. protothecoides on the basis of genome information, focusing on central metabolism comprised by the Calvin-Benson cycle, glycolysis, the PP pathway, the tricarboxylic acid cycle, and the biosynthetic pathways of biomass building blocks. The model was operated with in silico calculations, which were in reasonable agreement with the measured growth rates. The application of FBA led to several conclusions: (1) completeness of metabolic functionality mined in the genome information (few gaps in the certain pathways were identified and filled, whereas only four putative enzymes missed), (2) achievement of suboptimal growth by autotrophic and heterotrophic cells, and (3) optimality of metabolic parameter for cell growth and fatty acid production. The objective function that we selected in the model, to maximize biomass formation and the inlet and outlet carbon flux through the autotrophic and heterotrophic C. protothecoides (e.g. photon uptake and CO2 uptake rate of autotrophic cells and the CO2 excretion and Glc uptake of heterotrophic cells), can be determined. Our modeling defined by carbon influx (FBA1) resulted in agreement of calculated cell growth rates with experimental determined ones, thus validating the accuracy of our approach. FBA was then applied multiple times by varying growth conditions, and the output of objective function formed the PhPP plot, which mirrors metabolic potential of C. protothecoides. Overall, this model represents the first metabolic network draft, to our knowledge, for oleaginous microalga C. protothecoides reassembled from the newest availability of genome information. Upon presented framework, higher-resolution metabolic mapping is expected through extensive refinery of genomic information.
Despite its wide application in metabolic network analysis, the limitation of FBA should be taken into account in that solution space needs to be further constrained to approach real flux values. To address this, we adopt two experimental strategies. (1) Validation of C. protothecoides metabolism by the metabolomic analysis. According to the absolutely quantitative measurement of metabolite concentrations using LC-MS, we compared the metabolite levels of C. protothecoides in different nutrients. Heterotrophic C. protothecoides exhibits high levels of ATP, NAD(P)H, acetyl-CoA, and malonyl-CoA, supporting a superior lipid synthesis over autotrophic cells and being in line with the FBA modeling results from the reconstructed network. (2) Application of FBA framework in nonstationary 13C MFA. For this purpose, INST-MFA was, for the first time to our knowledge, used for a eukaryotic alga, and the best estimates of intracellular fluxes were obtained. To investigate the difference of real fluxomics and optimal flux status, we compared the results of INST-MFA estimation with FBA predictions. Shown in Supplemental Figure S1 are the net fluxes resulted from FBA and INST-MFA. Taking autotrophic cells for example, INST-MFA led to similar flux distributions as FBA models. However, the main differences include a less active tricarboxylic acid cycle detected by INST-MFA, consistent with slower labeling of the tricarboxylic acid intermediates. Note that, generally, the FBA model performs optimization of linear systems with seldom experimental inputs, whereas nonstationary 13C MFA is further defined by ordinary differential equations aiming to fit kinetic 13C data, thus leading to a more reliable flux map closer to reality. According to the results from FBA1 and 13C MFA, differences of final flux values are in a few reactions in the PP pathway, photorespiration, glyoxylate shunt, and tricarboxylic acid cycle. Higher accuracy could be further realized if more dynamic isotope labeling trajectories of central carbon metabolites can be included in the system, and the genome-based FBA model, thus, offered an open framework for improved flux estimate. We also exploit this model by fitting it with the steady-state 13C labeling data generated in our previous work (Xiong et al., 2010a, 2010b). A noncompartmented and simplified model was used to investigate fluxes of heterotrophic cells, because no detailed genomic information was available at that time. Applying the isotopomer distribution data of proteinogenic amino acids to this model generates higher-resolution flux maps (data not shown). Compared with flux profiling defined by kinetic labeling data (Fig. 5B), flux distributions throughout primary pathways are fairly similar, indicating data consistency. Remaining minor discrepancies are probably caused by changed experimental conditions, different labeling strategies, and a refined flux-estimating algorithm. Nevertheless, it should be noted that our research provided a case study showing the feasibility of both stationary and nonstationary 13C flux analyses based on a large-scale compartmentalized metabolic network. These integrated studies of metabolic network coupled with metabolomics analysis shed light on the metabolism of oleaginous microalga C. protothecoides and may serve as a cutting-edge toolbox for the systematic engineering of microalgae for biofuel production.
MATERIALS AND METHODS
Abbreviations and Nomenclature
Abbreviations and nomenclature used in the figures and supplemental materials are listed in Supplemental Table S6.
Cultivation Conditions and Analytical Procedure
Microalga Chlorella protothecoides 0710 was provided by the Culture Collection of Alga at the University of Texas. The basic medium composition was the same as previously described (Xiong et al., 2008). Briefly, for autotrophic cultivation, 2 g L−1 NH4Cl was added to the basic medium as a nitrogen source with four 18-W cool-white fluorescent lamps (Philips) providing an average surface illumination intensity of 1,470 lux (namely, 19.9 μmol m−2 s−1 with the conversion factor of 0.0135); 10 g L−1 Glc and up to 1 g L−1 NH4Cl were used as carbon and nitrogen sources, respectively, for heterotrophic growth in a 1-L fermenter (Infors).
An Ultrospec 2000 UV/Visible Spectrophotometer (Pharmacia Biotech) was used to monitor alga growth by measurements of optical density at 540 nm (Xiong et al., 2010a, 2010b). Initial and residual Glc in the media during the cultivation process were determined by a BSA-40C enzymatic bioanalyzer (Shangdong Academy of Science). CO2 consumed or secreted by autotrophic and heterotrophic algae was calculated from the difference between inlet and outlet CO2 concentrations determined by a real-time tandem gas analyzer (Milligan Instrument).
To determine the macromolecular composition of C. protothecoides, the Lowry method (Holdsworth et al., 1988) was used to measure protein content, and amino acid composition was obtained with an L-8800 Amino Acid Analyzer (Hitachi); the content and composition of oil were measured by gas chromatography (GC)-MS as previously described (Xiong et al., 2010b). The 3,5-dinitrosalicylic acid method (Miller, 1959) was applied to determine intracellular carbohydrate and starch. The percentage of nucleic acid was determined according to the diphenylamine-colorimetric method (Gendimenico et al., 1988) and the method proposed by Benthin et al. (1991); chlorophyll in autotrophic algae was measured using the improved N,N-dimethylformamide extraction method according to Pan et al. (2001).
Primary Metabolic Network Reconstruction
The primary metabolic network is a foundational model focusing on the central metabolism of C. protothecoides. The reconstruction work began with the genome annotation of C. protothecoides, the genome that was recently sequenced by us (Gao et al., 2014). Textbook knowledge and constructed metabolic networks of other photosynthetic microbes, including Synechocystis sp. PCC 6803 and Chlamydomonas reinhardtii (Shastri and Morgan, 2005; Boyle and Morgan, 2009; Manichaikul et al., 2009; Knoop et al., 2010; Chang et al., 2011; Hädicke et al., 2011), were also referenced. An original list of reactions and corresponding enzymes was collated according to the pathway maps in the Kyoto Encyclopedia of Genes and Genomes (Kanehisa and Goto, 2000).
Each reaction in the network was manually checked, and those genes with hypothetical protein product were reannotated using the online blast program of UniProt (http://www.uniprot.org/blast/) and confirmed with conserved domain analysis (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). Reaction gaps resulting from the incompleteness of genome annotation were identified. Missing genes were searched for in other species of Chlorophyta (C. reinhardtii, Ostreococcus tauri, and Volvox carteri), Arabidopsis (Arabidopsis thaliana), or Saccharomyces cerevisiae, and the known nucleotide or amino acid sequences were blasted against the C. protothecoides genome database. Most of the missing enzymes had hits with acceptable e values, except for four essential enzymes, which were retained in the stoichiometric model for the completeness and functionality of the network. Reactions in a pathway without branch were lumped (e.g. routes in the biosynthesis of macromolecules) to simplify the model calculation, which was still sufficient to detail the network.
To model the biomass formation reactions, we determined the biomass components and macromolecular composition of autotrophic and heterotrophic C. protothecoides (Table I) consisting of proteins, lipids, carbohydrates, DNA and RNA, and chlorophyll in autotrophic algae. GC content of DNA was obtained from genome sequencing, and the same GC content was assumed for RNA. Lipids were represented by diacylglycerol, and fatty acids were subdivided to tetradecanoic, hexadecanoic, and octadecanoic acids as well as unsaturated fatty acids octadecenoic (C18:1) and octadecadienoic acid (C18:2). Genes related to the nonpolar lipid synthesis of phosphatidylethanolamine, phosphatidyl-Ser, phosphatidylcholine, phosphatidic acid, phosphatidylinositol, phosphatidylglycerol, sulfoquinovosyl diacylglycerol, monogalatosyl diacylglycerol, digalactosyl diacylglycerol, and ceramide were also identified, whereas the reactions were simplified in the flux computation. Because no reliable experimental data were available, ATP demand for maintenance (EC 2.7.4.1: Cpr003988.1) and transhydrogenase (EC 1.6.1.2: Cpr004180.1) was not specified to balance the energy generation and consumption of C. protothecoides in each scenario.
Enzymes present in the network were localized into four cellular compartments (cytoplasma, mitochondria with matrix and intermembrane space [Grant and Hommersand, 1974], chloroplast with stroma and thylakoid lumen [Gao et al., 2014], and peroxisome [Codd et al., 1972; Chou et al., 2008]) primarily based on literature evidence of C. protothecoides and enzyme homologs in other species of Chlorella spp. (Chlorella pyrenoidosa, Chlorella fusca, and Chlorella sorokiniana), C. reinhardti, and Arabidopsis supplemented by PredAlgo, a multisubcellular localization predictor for algae (Tardif et al., 2012). In the absence of any evidence for localization, enzymes were assigned according to neighboring reactions for the functional feasibility of the network.
The actual photon absorption flux of autotrophic C. protothecoides by light-harvesting complexes for metabolic use is difficult because much of the incident light was reflected or scattered before it could generate the light reaction. Here, we applied a simplified approach proposed by Manichaikul et al. (2009) to estimate the photon uptake flux. The conversion rate of incident light to photon absorption flux is assumed to be constant, which is also practicable for the photon saturation point. The minimal light uptake that is sufficient for photosynthetic saturation measured as O2 evolution was calculated to be 37.72 mmol g dry cell weight−1 h−1 when autotrophic C. protothecoides grows at its maximal specific growth rate that has ever been reported (Sorokin and Krauss, 1959). The experimental photon flux saturation point was 500 μmol m−2 s−1 (Ouyang et al., 2010). Therefore, the actual flux of photon uptake can be estimated with experimentally measured light radiation.
The cyclic and noncyclic electron transport chains in photosynthesis and the ATP synthesis reaction were adopted according to Shastri and Morgan (2005). As for enzymes for which the electron donors and acceptors are unknown, NADH was assigned as the final donor, and NAD was assigned as the final acceptor (Knoop et al., 2010). NAD(H) and NADP(H) were integrated in one reaction for which both were available cofactors, and the stoichiometric numbers were set equal to minimize the consequence on FBA results.
Growth Simulation
FBA was applied in the quantification of the stoichiometric model of metabolic network using INCA 1.1 (Young, 2014) and CellNetAnalyzer 9.9 (Klamt et al., 2007) on MATLAB 7.6 (Mathworks). To simulate the growth state of C. protothecoides in autotrophic and heterotrophic conditions, we fixed photon uptake (set to zero for heterotrophic metabolism), Glc uptake (set to zero for autotrophic metabolism), and CO2 uptake/excretion rates as constraints and maximized the growth rate as the objective function under the assumption that algal cells were striving to achieve a maximal biomass production, an assumption that is reasonable for cells in the exponential growth phase (FBA1). Other constraints used for simulations are reported in Supplemental Table S7. We also made the prediction with only energy source uptake flux (photons of the autotrophic model and Glc of the heterotrophic model) constrained to predict the ideal growth of C. protothecoides (FBA2), which allowed for the optimized carbon use and distribution within cells. Other constraints were the same as those of FBA1.
Metabolite Extraction
Extraction of metabolites in C. protothecoides was according to Bennett et al. (2008) with slight modification. A 2-mL cell culture was rapidly harvested by filtering with a 47-mm-diameter round hydrophilic nylon filter (Sartorius Nylon Membrane, 0.45 μm) and quickly transferred into a prechilled dish containing 5 mL of −70°C methanol for metabolism quenching. The dishes were stored in a −70°C freezer for 2 h; then, the filters were rinsed, and the cell suspension was centrifuged. The pellets were extracted with 500 μL of methanol:water (50:50) mixture three times (various extract solvent combinations were tested, and finally, a methanol:water [50:50] mixture was selected). The supernatant was collected and combined with the remaining methanol, and it was evaporated by centrifugation under a vacuum. The resulting pellets were redissolved in 600 μL of LC mobile phase (see below) and stored at −80°C until LC-MS analysis.
LC-MS Analysis
All cell extracts were analyzed using an Agilent 1290 Analytical HPLC System coupled with an Agilent 6460 Triple-Quadrupole Mass Spectrometer (Agilent Technologies). For LC separation, we adopted a modified method by Munger et al. (2008). A Luna NH2 Column (Phenomenex) was used in positive mode. Mobile phases are identical to those by Munger et al. (2008), but the gradient was modified as follows: t = 0 min, 82% (v/v) B; t = 4 min, 80% (v/v) B; t = 11 min, 80% (v/v) B; t = 13 min, 62% (v/v) B; t = 16 min, 62% (v/v) B; t = 22 min, 0% (v/v) B; t = 30 min, 0% (v/v) B; t = 31 min, 82% (v/v) B; and t = 45 min, 82% (v/v) B. The flow rate was kept at 0.15 mL min−1. In negative mode, both the NH2 column and a Synergi Hydro-RP (C18) Column (Phenomenex) were used. The mobile phases are solvent A (5 mm ammonium acetate with 10 mm ammonium hydroxide in water), solvent B (acetonitrile for the NH2 column with identical flow rate), and solvent A (10 mm ammonium acetate with 20 mm acetic acid in water) and solvent B (methanol for C18 column with a flow rate of 0.1 mL min−1). The gradient was as follows: t = 0 min, 3% (v/v) B; t = 5 min, 3% (v/v) B; t = 8 min, 90% (v/v) B; t = 10 min, 90% (v/v) B; t = 11 min, 3% (v/v) B; and t = 20 min, 3% (v/v) B for NH2 column and t = 0 min, 0% (v/v) B; t = 2 min, 0% (v/v) B; t = 9 min, 95% (v/v) B; t = 11 min, 95% (v/v) B; t = 12 min, 0% (v/v) B; and t = 20 min, 0% (v/v) B. Chromatography runs were divided into several segments to increase the scan time. The mass spectrometer was operated in multiple reaction monitoring; metabolite information and optimized MS parameters are described in Supplemental Table S4.
All standards and reagents for LC-MS analysis were purchased from Sigma-Aldrich and Acros Organics with purity ≥ 95%. Chromatographic peaks from samples were identified through comparison of retention times and mass spectra with those of standards. The lowest concentration allowed for quantification has a signal to noise ratio of 10. For comparing the extract capacity with different solvent combinations, clustering was carried out with freely available software Cluster 3.0 after normalization of the means of peak areas of the replicates against data of the 6-h sample and visualized with a heat map using TreeView 1.60 (Eisen et al., 1998).
Absolute Quantitation of Intracellular Metabolite Concentrations
Heterotrophic C. protothecoides was inoculated into basic medium with [U-13C6]Glc (99% isotopic purity; Cambridge Isotope Laboratories), and the handling was repeated two times before the 13C-labeled cells were transferred into medium containing 1 g L−1 [U-13C6]Glc and 0.1 g L−1 NH4Cl. The cell cultures in early log phase were harvested as previously described with and without unlabeled standards spiked, and the quantitation was performed using the ratio of the 13C peak height to the 12C peak height obtained from MS data (Bennett et al., 2008). Because of the triple-quadrupole MS scan time, monitoring every isotopic form in 13C-fed cultures was not feasible, and therefore, equivalent unlabeled cultivations were analyzed to evaluate the completeness of labeling. The absolute intracellular metabolite concentrations were calculated according to the following formula:
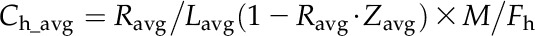 |
where 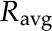 is the geometric mean of the ratio of the fully labeled peak intensity to that of the fully unlabeled peak (including spiked unlabeled standards) from three replicates,
is the geometric mean of the ratio of the fully labeled peak intensity to that of the fully unlabeled peak (including spiked unlabeled standards) from three replicates, 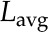 is the geometric mean of the ratio of the fully labeled peak in cells from labeled media to the fully unlabeled peak in cells from unlabeled media,
is the geometric mean of the ratio of the fully labeled peak in cells from labeled media to the fully unlabeled peak in cells from unlabeled media, 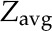 is the geometric mean of the ratio of fully unlabeled to fully labeled from cells fed with 13C Glc and extracted without spiking any standard, M is the amount of standards added to cell extract, and Fh is the total cell volume. For determination of the absolute concentrations in autotrophic algae, an equivalent of metabolite extract from green cells was mixed with that from the 13C-labeled heterotrophic culture, and the concentrations were calculated by:
is the geometric mean of the ratio of fully unlabeled to fully labeled from cells fed with 13C Glc and extracted without spiking any standard, M is the amount of standards added to cell extract, and Fh is the total cell volume. For determination of the absolute concentrations in autotrophic algae, an equivalent of metabolite extract from green cells was mixed with that from the 13C-labeled heterotrophic culture, and the concentrations were calculated by:
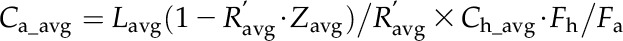 |
where 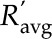 is the geometric mean of the ratio of the fully labeled peak intensity to that of the fully unlabeled peak (including spiked unlabeled compounds from autotrophic algae), and Fa is the corresponding autotrophic cell volume. se was considered by error propagation, and the final intracellular metabolite concentrations with 95% confidence intervals were calculated and reported.
is the geometric mean of the ratio of the fully labeled peak intensity to that of the fully unlabeled peak (including spiked unlabeled compounds from autotrophic algae), and Fa is the corresponding autotrophic cell volume. se was considered by error propagation, and the final intracellular metabolite concentrations with 95% confidence intervals were calculated and reported.
13C Labeling Experiment
An elementary metabolite unit-based (Antoniewicz et al., 2007) INST-MFA was used to estimate intracellular metabolic fluxes. It uses the isotope labeling kinetics of intracellular metabolites (Young et al., 2008). Exponentially growing cells were then collected by centrifugation and resuspended in basic media with no carbon or nitrogen source for 5 min. Time 0 sampling was conducted using the harvesting and extracting method previously described. The broth was transferred into a 500-mL SEBC bottle (Fisher Scientific) with BOLA Multiple Distributor in a GL 45 cap (BOLA). The bottle was sterilized with a one-way valve fitted to the cap to exhaust residual gas. The valve was then sealed to prevent unlabeled CO2 from entering the bottle. An 8-mL aliquot of 0.5 m NaH13CO3 (99% isotopic purity; Cambridge Isotope Laboratories) was injected into the bottle through a rubber septum in the cap. pH was maintain at around 7.5 by the addition of 1 m H2SO4. The broth was sampled at time points of 30 s, 90 s, 5 min, 10 min, 30 min, 60 min, and 120 min using a syringe. C-switching of the heterotrophic cells was achieved similarly by substituting uniformly 13C-labeled Glc with unlabeled carbon source, and the broth was sampled at the same time points.
MFA Based on Dynamic Isotopomer Distributions
After samples were analyzed by LC-MS, the isotopomers patterns of metabolites were monitored simultaneously (Supplemental Fig. S3). The isotopomer distributions, however, reflected mixed extracting pools of the same metabolites located in different organelles because of the compartmentalization of Chlorella spp. cells. To address this, pseudo-fluxes were introduced to simulate the pool mixing, which participates in the isotopomer balance but has no effect on the mass balance. The pseudo-fluxes were also used to account for the pool mixing of Glc-6-P and Fru-6-P, dihydroxyacetonphosphate and glyceraldehyde-3-P, and citrate and isocitrate, which were coquantitated. Dilution effect caused by the mixing of slowly labeled or metabolically inactive pools and the turnover of macromolecules was also considered with pseudo-fluxes on the principle of Isotopomer Spectral Analysis (Kelleher and Masterson, 1992). To reduce computation load and speed up the computation time, a condensed metabolic network of C. protothecoides was acquired by lumping the formation reactions of macromolecular building blocks with biomass produced directly from intermediates of primary metabolism (Supplemental Table S2). Then, the INST-MFA was applied, and the sum of squared residuals between the kinetics of isotope labeling of measured metabolites and the computationally simulated isotopomer distributions was minimized to obtain the best estimates of metabolic fluxes (other constraints were the same with FBA models) followed by calculation of ses for all estimated fluxes (Young et al., 2011). Calculation was repeated 10 times using INCA 1.1 starting from random initial guess in each scenario to obtain the global optimum. All flux estimates and ses are presented in Supplemental Table S8.
To do the cofactor balance analysis, we summarized all of the concomitant formation and consumption of ATP and NAD(P)H (NADH and NADPH were combined because of the uncertainty as cofactors of dehydrogenases and reductases) according to the INST-MFA results and categorized them into relevant pathways as shown in Table III.
Sequence data from this article can be found in the GenBank/EMBL data libraries under accession number APJO00000000.
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. Heat map of the calculated net fluxes in comparison of 13C MFA, FBA1, and FBA2.
Supplemental Figure S2. Heat map of metabolite quantification reflecting the extract capacity with different solvent combinations.
Supplemental Figure S3. Kinetics of isotope labeling of measured metabolites.
Supplemental Table S1. Unannotated enzymes confirmed using a protein query tblastn search.
Supplemental Table S2. List of reactions, catalyzing enzymes, and localization in the primary metabolic network of C. protothecoides.
Supplemental Table S3. Gene-protein association of the enzymes and transporters involved in the network.
Supplemental Table S4. MS parameters and method performance for metabolites.
Supplemental Table S5. Absolute intracellular metabolite concentrations of C. protothecoides in a 95% confidence interval.
Supplemental Table S6. Abbreviations and nomenclature of metabolites.
Supplemental Table S7. Modeling constraints used in growth simulations with FBA.
Supplemental Table S8. Net fluxes with ses determined by INST-MFA.
Supplemental Text S1. Autotrophic model of the C. protothecoides metabolic network presented in SBML format.
Supplemental Text S2. Heterotrophic model of the C. protothecoides metabolic network presented in SBML format.
Acknowledgments
We thank Jamey D. Young (Vanderbilt University) for providing software and technical assistance on 13C metabolic modeling, Dr. Yi Ding and Yu Tian (Center of Biomedical Analysis of Tsinghua University) for valuable advice and discussions in compound determination using LC-MS, and Daniel Brune and David J. Menn (Arizona State University) for helpful comments and English improvement in preparing the article.
Glossary
- FBA
flux balance analysis
- FBA1
flux balance analysis model 1
- FBA2
flux balance analysis model 2
- GC
gas chromatography
- INST-MFA
isotopically nonstationary metabolic flux analysis
- LC
liquid chromatography
- MFA
metabolic flux analysis
- MS
mass spectrometry
- PhPP
phenotype-phase plane
- PP
pentose phosphate
Footnotes
This work was supported by National Natural Science Foundation of China (project nos. 31370282 and 41030210), Chinese Ministry of Science and Technology (project nos. 2011BAD14B05 and 2014AA02200 to Q.W.), the Tsinghua University Initiative Scientific Research Program (grant no. 2012Z08128 to J.D.), and the National Renewable Energy Laboratory Director's Postdoctorate Fellowship (to W.X.).
Articles can be viewed without a subscription.
References
- Annesley TM. (2003) Ion suppression in mass spectrometry. Clin Chem 49: 1041–1044 [DOI] [PubMed] [Google Scholar]
- Antoniewicz MR, Kelleher JK, Stephanopoulos G (2007) Elementary metabolite units (EMU): a novel framework for modeling isotopic distributions. Metab Eng 9: 68–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker SA, Bassham JA, Calvin M, Quarck UC (1956) Intermediates in the photosynthetic cycle. Biochim Biophys Acta 21: 376–377 [DOI] [PubMed] [Google Scholar]
- Bassham JA, Benson AA, Calvin M (1950) The path of carbon in photosynthesis. J Biol Chem 185: 781–787 [PubMed] [Google Scholar]
- Bennett BD, Yuan J, Kimball EH, Rabinowitz JD (2008) Absolute quantitation of intracellular metabolite concentrations by an isotope ratio-based approach. Nat Protoc 3: 1299–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benthin S, Nielsen J, Villadsen J (1991) A simple and reliable method for the determination of cellular RNA content. Biotechnol Tech 5: 39–42 [Google Scholar]
- Boyle NR, Morgan JA (2009) Flux balance analysis of primary metabolism in Chlamydomonas reinhardtii. BMC Syst Biol 3: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvin M. (1956) The photosynthetic cycle. Bull Soc Chim Biol (Paris) 38: 1233–1244 [PubMed] [Google Scholar]
- Chang RL, Ghamsari L, Manichaikul A, Hom EF, Balaji S, Fu W, Shen Y, Hao T, Palsson BO, Salehi-Ashtiani K, et al. (2011) Metabolic network reconstruction of Chlamydomonas offers insight into light-driven algal metabolism. Mol Syst Biol 7: 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Alonso AP, Allen DK, Reed JL, Shachar-Hill Y (2011) Synergy between (13)C-metabolic flux analysis and flux balance analysis for understanding metabolic adaptation to anaerobiosis in E. coli. Metab Eng 13: 38–48 [DOI] [PubMed] [Google Scholar]
- Chou YC, Prakash E, Huang CF, Lien TW, Chen X, Su IJ, Chao YS, Hsieh HP, Hsu JT (2008) Bioassay-guided purification and identification of PPARalpha/gamma agonists from Chlorella sorokiniana. Phytother Res 22: 605–613 [DOI] [PubMed] [Google Scholar]
- Codd GA, Schmid GH, Kowallik W (1972) Enzymic evidence for peroxisomes in a mutant of Chlorella vulgaris. Arch Mikrobiol 81: 264–272 [DOI] [PubMed] [Google Scholar]
- Cogne G, Gros JB, Dussap CG (2003) Identification of a metabolic network structure representative of Arthrospira (spirulina) platensis metabolism. Biotechnol Bioeng 84: 667–676 [DOI] [PubMed] [Google Scholar]
- Coleman JS, Bhattacharjee JK (1975) Regulation of citrate synthase activity of Saccharomyces cerevisiae. Antonie van Leeuwenhoek 41: 249–256 [DOI] [PubMed] [Google Scholar]
- Dal’Molin CG, Quek LE, Palfreyman RW, Nielsen LK (2011) AlgaGEM—a genome-scale metabolic reconstruction of algae based on the Chlamydomonas reinhardtii genome. BMC Genomics (Suppl 4) 12: S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JS, Covert M, Palsson B (2002) Metabolic modelling of microbes: the flux-balance approach. Environ Microbiol 4: 133–140 [DOI] [PubMed] [Google Scholar]
- Edwards JS, Ibarra RU, Palsson BO (2001) In silico predictions of Escherichia coli metabolic capabilities are consistent with experimental data. Nat Biotechnol 19: 125–130 [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D (1998) Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA 95: 14863–14868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu PC. (2009) Genome-scale modeling of Synechocystis sp. PCC 6803 and prediction of pathway insertion. J Chem Technol Biotechnol 84: 473–483 [Google Scholar]
- Gao C, Wang Y, Shen Y, Yan D, He X, Dai J, Wu Q (2014) Oil accumulation mechanisms of the oleaginous microalga Chlorella protothecoides revealed through its genome, transcriptomes, and proteomes. BMC Genomics 15: 582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendimenico GJ, Bouquin PL, Tramposch KM (1988) Diphenylamine-colorimetric method for DNA assay: a shortened procedure by incubating samples at 50 degrees C. Anal Biochem 173: 45–48 [DOI] [PubMed] [Google Scholar]
- Gouveia L, Oliveira AC (2009) Microalgae as a raw material for biofuels production. J Ind Microbiol Biotechnol 36: 269–274 [DOI] [PubMed] [Google Scholar]
- Grant NG, Hommersand MH (1974) The respiratory chain of Chlorella protothecoides: I. Inhibitor responses and cytochrome components of whole cells. Plant Physiol 54: 50–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hädicke O, Grammel H, Klamt S (2011) Metabolic network modeling of redox balancing and biohydrogen production in purple nonsulfur bacteria. BMC Syst Biol 5: 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Salt IP, Hawley SA, Davies SP (1999) AMP-activated protein kinase: an ultrasensitive system for monitoring cellular energy charge. Biochem J 338: 717–722 [PMC free article] [PubMed] [Google Scholar]
- Holdsworth JE, Veenhuis M, Ratledge C (1988) Enzyme activities in oleaginous yeasts accumulating and utilizing exogenous or endogenous lipids. J Gen Microbiol 134: 2907–2915 [DOI] [PubMed] [Google Scholar]
- Huege J, Goetze J, Schwarz D, Bauwe H, Hagemann M, Kopka J (2011) Modulation of the major paths of carbon in photorespiratory mutants of synechocystis. PLoS ONE 6: e16278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28: 27–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher JK, Masterson TM (1992) Model equations for condensation biosynthesis using stable isotopes and radioisotopes. Am J Physiol 262: E118–E125 [DOI] [PubMed] [Google Scholar]
- Klamt S, Saez-Rodriguez J, Gilles ED (2007) Structural and functional analysis of cellular networks with CellNetAnalyzer. BMC Syst Biol 1: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klanchui A, Khannapho C, Phodee A, Cheevadhanarak S, Meechai A (2012) iAK692: a genome-scale metabolic model of Spirulina platensis C1. BMC Syst Biol 6: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoop H, Zilliges Y, Lockau W, Steuer R (2010) The metabolic network of Synechocystis sp. PCC 6803: systemic properties of autotrophic growth. Plant Physiol 154: 410–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Gianchandani EP, Papin JA (2006) Flux balance analysis in the era of metabolomics. Brief Bioinform 7: 140–150 [DOI] [PubMed] [Google Scholar]
- Lee YK. (2001) Microalgal mass culture systems and methods: their limitation and potential. J Appl Phycol 13: 307–315 [Google Scholar]
- Manichaikul A, Ghamsari L, Hom EF, Lin C, Murray RR, Chang RL, Balaji S, Hao T, Shen Y, Chavali AK, et al. (2009) Metabolic network analysis integrated with transcript verification for sequenced genomes. Nat Methods 6: 589–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matuszewski BK, Constanzer ML, Chavez-Eng CM (1998) Matrix effect in quantitative LC/MS/MS analyses of biological fluids: a method for determination of finasteride in human plasma at picogram per milliliter concentrations. Anal Chem 70: 882–889 [DOI] [PubMed] [Google Scholar]
- Miao X, Wu Q (2006) Biodiesel production from heterotrophic microalgal oil. Bioresour Technol 97: 841–846 [DOI] [PubMed] [Google Scholar]
- Miller GL. (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31: 426–428 [Google Scholar]
- Montagud A, Navarro E, Fernández de Córdoba P, Urchueguía JF, Patil KR (2010) Reconstruction and analysis of genome-scale metabolic model of a photosynthetic bacterium. BMC Syst Biol 4: 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger J, Bennett BD, Parikh A, Feng XJ, McArdle J, Rabitz HA, Shenk T, Rabinowitz JD (2008) Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat Biotechnol 26: 1179–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales J, Gudmundsson S, Knight EM, Palsson BO, Thiele I (2012) Detailing the optimality of photosynthesis in cyanobacteria through systems biology analysis. Proc Natl Acad Sci USA 109: 2678–2683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberhardt MA, Palsson BO, Papin JA (2009) Applications of genome-scale metabolic reconstructions. Mol Syst Biol 5: 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang Z, Wen X, Geng Y, Mei H, Hu H, Zhang G, Li Y (2010) The effects of light intensities, temperatures, pH and salinities on photosynthesis of Chlorella. J Wuhan Bot Res 28: 49–55 [Google Scholar]
- Pan X, Li J, Hao Y (2001) A improved method for extraction of microalga chlorophyll using DMF. Biotechnology 11: 39–41 [Google Scholar]
- Pearce J, Leach CK, Carr NG (1969) The incomplete tricarboxylic acid cycle in the blue-green alga Anabaena variabilis. J Gen Microbiol 55: 371–378 [DOI] [PubMed] [Google Scholar]
- Pulz O, Gross W (2004) Valuable products from biotechnology of microalgae. Appl Microbiol Biotechnol 65: 635–648 [DOI] [PubMed] [Google Scholar]
- Satoh A, Kurano N, Miyachi S (2001) Inhibition of photosynthesis by intracellular carbonic anhydrase in microalgae under excess concentrations of CO(2). Photosynth Res 68: 215–224 [DOI] [PubMed] [Google Scholar]
- Schwarz D, Orf I, Kopka J, Hagemann M (2013) Recent applications of metabolomics toward cyanobacteria. Metabolites 3: 72–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifar RM, Zhao Z, van Dam J, van Winden W, van Gulik W, Heijnen JJ (2008) Quantitative analysis of metabolites in complex biological samples using ion-pair reversed-phase liquid chromatography-isotope dilution tandem mass spectrometry. J Chromatogr A 1187: 103–110 [DOI] [PubMed] [Google Scholar]
- Shastri AA, Morgan JA (2005) Flux balance analysis of photoautotrophic metabolism. Biotechnol Prog 21: 1617–1626 [DOI] [PubMed] [Google Scholar]
- Sorokin C, Krauss RW (1959) Maximum growth rates of chlorella in steady-state and in synchronized cultures. Proc Natl Acad Sci USA 45: 1740–1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhauser D, Fernie AR, Araújo WL (2012) Unusual cyanobacterial TCA cycles: not broken just different. Trends Plant Sci 17: 503–509 [DOI] [PubMed] [Google Scholar]
- Tardif M, Atteia A, Specht M, Cogne G, Rolland N, Brugière S, Hippler M, Ferro M, Bruley C, Peltier G, et al. (2012) PredAlgo: a new subcellular localization prediction tool dedicated to green algae. Mol Biol Evol 29: 3625–3639 [DOI] [PubMed] [Google Scholar]
- Taylor BF. (1973) Fine control of citrate synthase activity in blue-green algae. Arch Mikrobiol 92: 245–249 [DOI] [PubMed] [Google Scholar]
- Underwood SA, Buszko ML, Shanmugam KT, Ingram LO (2002) Flux through citrate synthase limits the growth of ethanologenic Escherichia coli KO11 during xylose fermentation. Appl Environ Microbiol 68: 1071–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, Gao C, Yan D, Wu C, Wu Q (2010a) Double CO(2) fixation in photosynthesis-fermentation model enhances algal lipid synthesis for biodiesel production. Bioresour Technol 101: 2287–2293 [DOI] [PubMed] [Google Scholar]
- Xiong W, Li X, Xiang J, Wu Q (2008) High-density fermentation of microalga Chlorella protothecoides in bioreactor for microbio-diesel production. Appl Microbiol Biotechnol 78: 29–36 [DOI] [PubMed] [Google Scholar]
- Xiong W, Liu L, Wu C, Yang C, Wu Q (2010b) 13C-tracer and gas chromatography-mass spectrometry analyses reveal metabolic flux distribution in the oleaginous microalga Chlorella protothecoides. Plant Physiol 154: 1001–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Miao X, Wu Q (2006) High quality biodiesel production from a microalga Chlorella protothecoides by heterotrophic growth in fermenters. J Biotechnol 126: 499–507 [DOI] [PubMed] [Google Scholar]
- Yang Y, Gao K (2003) Effects of CO2 concentrations on the freshwater microalgae, Chlamydomonas reinhardtii, Chlorella pyrenoidosa and Scenedesmus obliquus (Chlorophyta). J Appl Phycol 15: 379–389 [Google Scholar]
- Young JD. (2014) INCA: a computational platform for isotopically non-stationary metabolic flux analysis. Bioinformatics 30: 1333–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JD, Shastri AA, Stephanopoulos G, Morgan JA (2011) Mapping photoautotrophic metabolism with isotopically nonstationary 13C flux analysis. Metab Eng 13: 656–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JD, Walther JL, Antoniewicz MR, Yoo H, Stephanopoulos G (2008) An elementary metabolite unit (EMU) based method of isotopically nonstationary flux analysis. Biotechnol Bioeng 99: 686–699 [DOI] [PubMed] [Google Scholar]