

Abstract
Nitrogenase, [FeFe]-hydrogenase, and [Fe]-hydrogenase enzymes perform catalysis at metal cofactors with biologically unusual non-protein ligands. The FeMo cofactor of nitrogenase has a MoFe7S9 cluster with a central carbon, whereas the H-cluster of [FeFe]-hydrogenase contains a 2Fe subcluster coordinated by cyanide and CO ligands as well as dithiomethylamine; the [Fe]-hydrogenase cofactor has CO and guanylylpyridinol ligands at a mononuclear iron site. Intriguingly, radical S-adenosyl-l-methionine enzymes are vital for the assembly of all three of these diverse cofactors. This minireview presents and discusses the current state of knowledge of the radical S-adenosylmethionine enzymes required for synthesis of these remarkable metal cofactors.
Keywords: Hydrogenase, Iron-Sulfur Protein, Nitrogenase, Radical, S-Adenosylmethionine (SAM)
Introduction
Despite the potential deleterious effects of radical reactions and unconstrained metal ions, the exquisitely controlled protein armature of metalloenzymes and the orchestration of their biosynthetic accessory proteins allow for the creation of complex metal centers that accomplish formidable catalysis. N2 reduction to ammonia occurs at the nitrogenase FeMo cofactor, which can be viewed as a complex bridged metal assembly consisting of a [4Fe-3S] cluster linked to a [Mo-3Fe-3S] cluster by three sulfides and a central carbon (Figs. 1A and 2A) (1, 2). Reversible reduction of protons to H2 occurs at the [FeFe]-hydrogenase H-cluster, a [4Fe-4S] cubane bridged by a cysteine to a 2Fe subcluster containing CO, CN−, and dithiomethylamine ligands (Figs. 1B and 2B) (3). The reversible conversion of methylenetetrahydromethanopterin to methenyltetrahydromethanopterin and H2 is catalyzed at a mononuclear iron active site, the ligands of which include two CO factors and one guanylylpyridinol cofactor (Figs. 1C and 2C) (4).
FIGURE 1.

Active-site metal centers within protein structures. A, nitrogenase with the FeMo cofactor magnified (oval) (PDB ID 1M1N). B, [FeFe]-hydrogenase with the H-cluster magnified (oval) (PDB ID 3C8Y). C, [Fe]-hydrogenase with the FeGP cofactor magnified (oval) (PDB ID 3F47). The additional cluster in nitrogenase is the P-cluster at the interface between NifD (green) and NifK (dark gray); the additional clusters in [FeFe]-hydrogenase are the F-clusters involved in electron transfer. Multiple protein subunits are distinguished by color: for nitrogenase, the MoFe protein tetramer is illustrated with one αβ-subunit including NifD (green) and NifK (dark gray), whereas the second αβ-subunit is light gray; for [Fe]-hydrogenase, one subunit of the dimer is pink, whereas the other is dark gray. In the cofactor magnifications, the protein-based ligands touch the outside of the oval. Blue, nitrogen; red, oxygen; orange, phosphorus; yellow-orange, sulfur; rust, iron; aqua, molybdenum; magenta, unknown; gray, carbon, unless part of a protein-based ligand, in which case, the carbons are the same color as the protein schematic. H4-MPT, tetrahydromethanopterin.
FIGURE 2.
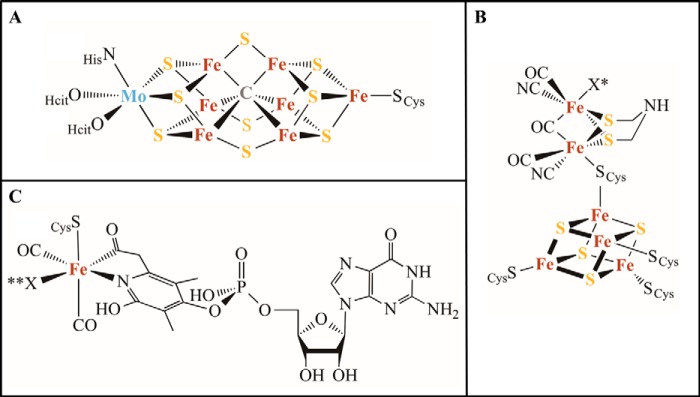
Structural drawings of the metallocofactors. A, FeMo cofactor of nitrogenase. B, H-cluster of [FeFe]-hydrogenase. C, FeGP cofactor of [Fe]-hydrogenase. X* and **X, locations of catalysis; Hcit, homocitrate).
Metal cofactor assembly begins with the synthesis of FeS clusters by either the housekeeping iron-sulfur cluster assembly machinery (5) in the case of [FeFe]-hydrogenase or the Nif (nitrogen fixation)-specific homologs in the case of nitrogenase. The biosynthetic steps necessary for preparation of [FeFe]-hydrogenase and nitrogenase active sites require additional dedicated proteins that include scaffolds for assembly of the nascent cluster, as well as radical S-adenosyl-l-methionine (SAM)2 enzymes that synthesize unique non-protein ligands. Whereas less is known about the assembly of the iron-guanylylpyridinol (FeGP) cofactor of the [Fe]-hydrogenase, it is clear that radical SAM chemistry is involved (6).
It is interesting to note that in all three enzymes discussed here, the metal cofactors are organometallic in nature and exist with only minimal protein coordination (Figs. 1 and 2). The FeMo cofactor of nitrogenase is covalently bound to the protein by only one histidine and one cysteine ligand; the 2Fe subcluster of the H-cluster of [FeFe]-hydrogenase and the FeGP cofactor of [Fe]-hydrogenase have only a single cysteine ligand.
Radical SAM chemistry is required for the synthesis of each of these inorganic cofactors, despite the fact that there is no single non-protein ligand shared by all three cofactors. The requirement for radical SAM chemistry to synthesize this diverse array of non-protein ligands reflects the wide variety of chemical transformations that can be initiated by hydrogen atom abstraction (7).
Nitrogenase FeMo Cofactor Biosynthesis
Found in a variety of bacteria and some methanogenic archaea, nitrogenase plays a critical role in the global nitrogen cycle by reducing N2 to NH3 (Fig. 1A). Cleavage of the triple bond in N2 requires considerable energy, as reflected in the high temperatures, high pressures, and catalysts required to attain practical rates of N2 reduction industrially (8). In contrast, nitrogenase employs ATP to drive the process at atmospheric pressure and ambient temperatures and accomplishes this through the activity of some of the most complex metal clusters observed in biology. Although alternative nitrogenases are known to contain vanadium or iron instead of molybdenum at the active-site cluster (9), the molybdenum nitrogenases are the most common and well characterized; thus, these will serve as the main source of examples for this discussion.
The active molybdenum nitrogenase consists of two proteins: 1) an Fe protein (NifH) with a [4Fe-4S] cluster and 2) a MoFe protein (NifDK) with an [8Fe-7S] P-cluster and a [Mo-7Fe-9S-C-homocitrate] cluster, which is referred to as the FeMo cofactor. The Fe protein is a homodimer with a binding site for ATP hydrolysis in each subunit and a [4Fe-4S] cluster bridging the two subunits (Fig. 1A). The MoFe protein is a heterotetramer with the P-cluster sitting at each αβ-subunit interface, whereas the FeMo cofactors are located within each α-subunit (10–13). During catalysis, electrons are transferred from the [4Fe-4S] cluster in the Fe protein to the P-cluster in the MoFe protein and ultimately to the FeMo cofactor where N2 is reduced. However, to create this incredible protein assembly with the three critical clusters required for activity, a host of maturase enzymes is involved, with one of these being the radical SAM enzyme NifB.
The FeMo cofactor is synthesized (Fig. 3, panel I) by complex biosynthetic machinery, which includes the radical SAM enzyme NifB. FeMo cofactor assembly begins with the formation of [2Fe-2S] and [4Fe-4S] clusters, which are synthesized by the combined actions of NifS, a cysteine desulfurase, and NifU, a scaffolding protein (14, 15). Both NifS and NifU are homologs of IscS and IcsU, which have analogous roles in general FeS cluster assembly. After transfer of these [4Fe-4S] clusters to NifB, radical SAM chemistry is required to create a FeMo cofactor precursor cluster that has six to eight iron atoms, nine sulfur atoms, and one central carbon (16–18). This carbon-containing precursor has been referred to as the NifB-co (1) and is transferred to a scaffold protein, NifEN, which is structurally similar to the MoFe protein (16–22). Additional steps, including the addition of molybdenum (by NifQ) and homocitrate (by NifV), occur by the activity of additional accessory enzymes, not addressed here, en route to the synthesis of the FeMo cofactor (19, 20, 23–26). The final step of maturation (22) is the transfer and insertion of the FeMo cofactor to a cofactorless MoFe protein to produce an active nitrogenase (19, 20, 25, 27).
FIGURE 3.

Schematic illustration of the biosynthesis of the FeMo cofactor of nitrogenase (panel I) and the H-cluster of [FeFe]-hydrogenase (panel II). Similar steps in the two pathways are shown in the same shade of gray. From top to bottom, these steps are radical SAM-based ligand synthesis, assembly of the precursor on a scaffold protein, and transfer of a cluster precursor to the apoenzyme. Gray, carbon; blue, nitrogen; red, oxygen; orange, phosphorus; yellow-orange, sulfur; rust, iron; aqua, molybdenum; magenta, unknown. HC, homocitrate.
NifB contains the CX3CX2C motif characteristic of the radical SAM superfamily; however, until recently, the specific function of this enzyme in FeMo cofactor assembly was unclear. Due to a lack of solubility of NifB, Wiig et al. (17) investigated NifB via studies of a fusion protein, NifEN-B, overexpressed in Azotobacter vinelandii in the presence of all accessory proteins except the Fe protein (NifH) and the MoFe protein (NifDK). The goal was to halt cluster maturation at the NifB state until SAM was added; the addition of SAM then would allow modification of the cluster(s) on NifB and the subsequent transfer of the product cluster to NifEN. Initial EPR characterization demonstrated that, prior to the addition of SAM, a single axial EPR signal (g = 2.02, 1.95, and 1.90) existed (17); given that sequence annotation predicted that NifB contains both a radical SAM [4Fe-4S] cluster and two other [4Fe-4S] clusters, the observed paramagnetic signal could be explained by either all clusters having similar g values or by only one of these clusters being EPR-active. Upon SAM addition to NifEN-B, this axial signal disappeared, and an isotropic signal (g = 1.94) appeared and was assigned to the Fe8S9C cluster (17). Although the origin of these EPR spectral changes with SAM addition was not clear at the time, spectroscopic evidence for an interstitial carbon in the precursor and FeMo cofactor clusters (21, 22, 28–30) suggested that the NifB-catalyzed reaction served to install the central carbon atom.
NifB does not require substrate other than SAM to catalyze the transformation of two [4Fe-4S] clusters to the FeMo cofactor precursor FeS cluster with a central carbon. This result, together with the observation that both 5′-deoxyadenosine and S-adenosylhomocysteine (SAH) were formed as products, pointed to potential roles for SAM as both radical precursor and substrate. Reductive cleavage of SAM generally produces a 5′-deoxyadenosyl radical (dAdo•), which, upon hydrogen atom abstraction from substrate, is converted to 5′-deoxyadenosine. On the other hand, SAH is the expected product when SAM acts as a methyl donor (16). Use of methyl-d3-SAM in the NifB reaction yielded deuterated 5′-deoxyadenosine product, providing evidence that dAdo• abstracts a hydrogen atom from a methyl originating from another SAM (Fig. 4, upper panel) (16). Furthermore, incubation of [methyl-14C]SAM with the NifEN-B protein yielded 14C label incorporation into the cluster, but not the polypeptides of the protein, suggesting direct transfer of the carbon from SAM to the cluster without a protein-bound intermediate (16). Thus, multiple lines of evidence implicate the methyl group of SAM as the source of the central carbon in the cluster intermediate synthesized on NifB and ultimately in the FeMo cofactor on the mature MoFe protein.
FIGURE 4.

Schematic illustration of the reactions catalyzed by NifB (upper panel) and HydG (lower panel). For the NifB reaction, SN2 methylation of a cluster sulfide by SAM is followed by radical SAM chemistry, resulting in hydrogen atom abstraction from this cluster methyl and subsequent insertion of carbon to form a Fe6–8S9C cluster. For the HydG reaction, the dAdo• produced upon reductive cleavage of SAM abstracts an hydrogen atom from tyrosine, eventually generating a 4-hydroxybenzyl radical and DHG; further reaction of DHG leads to the formation of CO and CN− ligands ultimately found in the 2Fe subcluster of the H-cluster. SAH, S-adenosylhomocysteine.
One mechanism proposed for the carbon atom insertion process (Fig. 4, upper panel) involves the SN2 transfer of the methyl on SAM to a sulfide of a [4Fe-4S] cluster on NifB. Reductive cleavage of the second equivalent of SAM then produces a dAdo• that abstracts a hydrogen atom from the methyl just added to the [4Fe-4S] cluster. This mechanism has chemical precedent in radical SAM enzymes, as it is similar to that proposed for the RNA methyltransferase radical SAM enzymes RlmN and Cfr (31, 32). An alternative mechanism (not shown) requires that the first equivalent of SAM is reductively cleaved to form a methyl radical, which is then transferred to an iron of a [4Fe-4S] cluster on NifB. The reductive cleavage of the second equivalent of SAM forms dAdo•, which then abstracts a hydrogen atom from this methyl group. This second scheme seems less likely because it requires two distinct radical-forming reactions at a single radical SAM active site; however, further studies should decipher this. After these initial steps, both mechanisms are expected to converge and involve proton loss from the intermediate species in conjunction with the further incorporation of the carbon to form the Fe6–8S9C cluster. The carbon atom insertion is concomitant with incorporation of a sulfide of unknown origin that bridges the two clusters, as well as rearrangement of the two clusters, although the details of the mechanism by which this occurs have yet to be elucidated.
[FeFe]-Hydrogenase H-cluster Biosynthesis
[FeFe]-Hydrogenases are distinguished by the organometallic H-cluster at the active site and are found in numerous species of anaerobic prokaryotes (such as sulfate reducers and Firmicutes) lower eukaryotes, including green alga and protists (33, 34). The H-cluster (Fig. 1B) consists of a [4Fe-4S] cluster linked via the thiolate of a cysteine residue to a 2Fe subcluster coordinated by three CO and two CN− ligands and a bridging dithiomethylamine (Fig. 2B). Only three accessory proteins are required for generation of this active-site cluster; these include the radical SAM enzymes HydE and HydG and the GTPase HydF (35, 36). Evidence suggests that these three accessory proteins are specifically directed toward the synthesis of only the 2Fe subcluster of the H-cluster (Fig. 3, panel II) (36–40). When these three maturation proteins are expressed together in Escherichia coli and then HydF is purified from the cells, this HydFEG is able to generate an active hydrogenase in the absence of other proteins or exogenous small molecules, indicating that HydF serves as a scaffold or carrier during H-cluster assembly (37, 38). Observation of vibrational bands associated with Fe-CO and Fe-CN and Fe-CO-Fe species (41, 42) on HydFEG, but not on HydF expressed without HydE and HydG (HydFΔEG) (41), further supports a scaffolding role for HydF. Most recently, synthetic models of the 2Fe subcluster of the H-cluster have been inserted into HydFΔEG, and this semisynthetic holo-HydF has been shown to be competent for hydrogenase maturation, providing yet further evidence that HydF acts as a scaffold/carrier for the 2Fe subcluster precursor (43).
HydG Catalyzes CO and CN− Biosynthesis
HydG (44) exhibits significant sequence homology to ThiH, a radical SAM enzyme that catalyzes the Cα–Cβ bond cleavage of tyrosine to produce p-cresol and dehydroglycine (DHG), the latter of which is a precursor in thiamine biosynthesis (45). This sequence homology ultimately led to testing of tyrosine as a substrate for HydG, and p-cresol was observed as a product (46). At the time, it was proposed that the presumed DHG product of HydG served as a precursor for the dithiomethylamine of the H-cluster; however, the source of this ligand remains unknown. Driesener et al. (47) demonstrated that CN− was formed in the HydG-catalyzed reaction at an ∼1:1:1 stoichiometric ratio with p-cresol and 5′-deoxyadenosine. Subsequent HydG assays in which deoxyhemoglobin was included in the assay mixture demonstrated the conversion of deoxyhemoglobin to carboxyhemoglobin, resulting from the HydG-catalyzed production of CO (48). In both the CN− and CO assays, use of isotopically labeled tyrosine coupled with mass spectral or FTIR analysis demonstrated that the diatomic products are derived from tyrosine (47, 48). Interestingly, similar rate constants for formation of both CO (kcat = 11 × 10−4 s−1 at 30 °C for carboxyhemoglobin) (48) and CN− (kcat = 20 × 10−4 s−1 at 37 °C for the CN− adduct) (47) in separate experiments provided support that these species arose from the same intermediate, presumably DHG. Furthermore, isotopic labeling has revealed that all of the diatomic ligands of the H-cluster are derived from tyrosine (49), implying that HydG provides all of the diatomic ligands of the H-cluster. Intriguingly, the reaction catalyzed by HydG as shown in Fig. 4 should provide CO and CN− in a 1:1 stoichiometric ratio, and yet the H-cluster contains these ligands in a 3:2 ratio. Therefore, assembly of the H-cluster may require three turnovers of HydG, producing three CN− and three CO molecules. As only two CN− molecules are incorporated into the H-cluster, the rate of the third CN− is undefined at this time.
The mechanism by which HydG catalyzes this intriguing reaction is currently under active investigation (Fig. 4). HydG binds two [4Fe-4S] clusters (48, 50), both the radical SAM cluster at the N-terminal CX3CX2C motif and a second site-differentiated cluster in the C-terminal domain at a CX2CX22C motif. Both clusters are essential for synthesis of the diatomic products and for achieving hydrogenase maturation (36, 48, 50). A recent study has shown that the N-terminal cluster alone is involved in the reductive cleavage of SAM and the Cα–Cβ bond cleavage of tyrosine to produce p-cresol (50). However, the C-terminal domain is essential for the subsequent production of the diatomic products from DHG (50–52). These two site-differentiated [4Fe-4S] clusters are expected to be bound at opposite ends of a (βα)8 TIM (triose-phosphate isomerase) barrel based on comparison with PylB and HydE, structurally characterized radical SAM enzymes with considerable sequence similarity (3, 53).
To separately analyze the SAM and C-terminal clusters, variant proteins were created: 1) a C-terminal deletion (ΔCTD) variant, in which the entire C-terminal domain was eliminated, and 2) a C96A/C100A/C103A variant, in which the cysteines of the SAM-binding cluster were mutated to alanines. EPR analysis of purified, reconstituted, and photoreduced HydGΔCTD demonstrated an axial signal akin to wild-type HydG that was perturbed by addition of SAM (50); in contrast, the C96A/C100A/C103A variant lacking the N-terminal SAM-binding cluster showed no perturbation of the wild-type axial EPR signal with addition of SAM (50). Furthermore, the ΔCTD variant catalyzed reductive cleavage of SAM and tyrosine to generate p-cresol, but could produce neither CO nor CN− (50–52). Together, these results suggest that the N-terminal cluster alone is responsible for the radical SAM chemistry resulting in the initial tyrosine reaction to produce p-cresol, whereas diatomic ligand production requires elements in the C-terminal domain and the accessory cluster itself.
The specific mechanistic steps involved in the synthesis and delivery of the diatomic products of HydG remain a subject of current inquiry. Although tyrosine was proposed to coordinate to the C-terminal cluster (54) in a manner similar to the coordination of GTP to the C-terminal cluster of the radical SAM enzyme MoaA (55), recent results demonstrate that the C-terminal cluster is not involved in or required for tyrosine cleavage, making tyrosine binding to this cluster unlikely (3, 44, 50). However, spectroscopic evidence does suggest that a fragment of tyrosine may bind to this cluster (56), consistent with a role for this cluster in breaking down DHG to CO and CN−. It was originally proposed that HydG synthesized and delivered only the diatomic products; however, recent studies provide evidence that CO and CN− produced by HydG coordinate to an iron on the protein, possibly the unique iron of the C-terminal cluster (56). Furthermore, use of 57Fe-enriched HydG has provided evidence that iron from HydG becomes incorporated into the H-cluster (56). Although some specifics of the diatomic ligand production and delivery by HydG are debated, the steps illustrated in Fig. 4 provide a reasonable framework consistent with current data. In the initial step, SAM bound to the N-terminal [4Fe-4S] cluster undergoes reductive cleavage to generate dAdo•. This dAdo• abstracts a hydrogen from tyrosine to produce a radical that undergoes heterolytic Cα–Cβ bond cleavage to give a 4-hydroxybenzyl radical and DHG (44, 50, 54). Presumably, DHG decomposition occurs in the C-terminal domain and is key for the generation of CO and CN−. The breakdown of DHG to these diatomics is unprecedented and novel chemistry, and it seems most likely to occur via a radical mechanism, possibly initiated by hydrogen atom abstraction from DHG by the 4-hydroxybenzyl radical.
Role of HydE in H-cluster Biosynthesis
In contrast to HydG, several crystal structures of HydE have been solved (Protein Data Bank (PDB) ID 3IIZ, 3IIX, 3CIX, and 3CIW) (57–59). These structures illustrate a complete (βα)8 TIM barrel with an internal electropositive channel that extends from the SAM-binding cluster at the top of the barrel to the bottom of the barrel. The channel appears to include three specific anion-binding locations, including one near the bottom of the barrel, which was found to bind thiocyanate in soaking experiments (58). Thus, at the top of the barrel, radical SAM chemistry could act on substrate to form intermediates that travel the length of the barrel before being transferred to another protein such as HydF (58, 60). Although previously annotated as having sequence similarity to the radical SAM enzyme biotin synthase (BioB) (58), a greater similarity exists with the methylornithine synthase PylB (61).
Because HydG is known to synthesize the diatomic ligands of the H-cluster, HydE is presumed to synthesize the dithiomethylamine ligand (62, 63); however, the substrate for HydE has yet to be identified. It has been suggested that HydE plays no essential role in hydrogenase maturation (64), although this seems unlikely given the in vivo and in vitro evidence that HydE is essential to the assembly process and conservation of hydE across species with hydF and hydG (35, 36, 38).
Although the substrate and product of HydE have yet to be identified, the interactions between HydE and the other maturase enzymes have been investigated. Both HydG and HydE have been observed to stimulate the rate of GTP hydrolysis in HydF (38), possibly implying a connection between GTP hydrolysis on HydF and the interactions with the other two maturases. Surface plasmon resonance experiments corroborated this result, as the presence of GTP increased the rate of dissociation of HydG and HydE from HydF (65). Furthermore, although surface plasmon resonance experiments revealed that neither HydG nor HydE could displace each other from HydF, a stronger interaction between HydE and HydF compared with that between HydG and HydF was observed (65). The strength of this interaction is perhaps related to the observation that HydE and HydF are fused in some organisms (36), as the radical SAM enzyme NifB is fused to the NifEN scaffold in Clostridium acetobutylicum (66), and could be interpreted to emphasize the independent role of both HydE and HydG in hydrogenase maturation. As depicted in Fig. 3 (panel II), the radical SAM enzymes are expected to deliver the CO and CN− (HydG) and dithiomethylamine-bridging (HydE) ligands to form the 2Fe subcluster on HydF, which is essential for the formation of the complete H-cluster and activation of hydrogenase.
[Fe]-Hydrogenase (Hmd) Cofactor Biosynthesis
[Fe]-hydrogenase, or H2-forming methylenetetrahydromethanopterin dehydrogenase (Hmd) (Fig. 1C), is an enzyme in the pathway that produces methane from CO2 (4). The enzyme utilizes a novel cofactor, FeGP (Fig. 2C) (67), to catalyze the reversible reduction of methenyltetrahydromethanopterin+ with H2 to yield methylenetetrahydromethanopterin and a proton (68–70). The FeGP cofactor contains a low-spin iron(II) (71, 72) that is octahedrally coordinated to 1) the thiol of the cysteine ligand (73, 74), 2) two CO ligands (75), and 3) nitrogen and carbon from the guanylylpyridinol. The sixth ligand remains undefined but presumably is the site of the methenyltetrahydromethanopterin+ reduction (Fig. 2C) (67, 76, 77). The nine carbons in the pyridinol moiety of the FeGP cofactor are derived from acetate, methionine, pyruvate, and CO2 (78).
[Fe]-Hydrogenases are found in hydrogenotrophic methanogenic archaea and are the only other hydrogenases in these organisms aside from various [NiFe]-hydrogenases; thus, when the nickel content is below a certain threshold, [Fe]-hydrogenase is up-regulated as compensation (79). Although the hmd gene codes for [Fe]-hydrogenase, the accessory proteins are coded for by the hcgA-hcgG suite of genes, and all seven are required for Hmd activation (80). Although the specific roles of each of these genes have yet to be determined, annotation has provided some clues: hcgA is annotated as a radical SAM enzyme; hcgB appears to have a phosphate-binding domain that shares structural similarities with pyrophosphatases (PDB ID 3BRC) and catalyzes the guanylyltransferase reaction to form the guanylylpyridinol part of the cofactor (80, 81); hcgC has a putative NAD(P)-binding Rossmann-like domain; hcgD is proposed to be an iron chaperone for the FeGP cofactor (82); and hcgG may have a SAM-binding domain but is annotated as a fibrillarin-like protein (80). The detailed roles of each of these accessory proteins remain undefined.
The accessory gene hcgA codes for a protein that harbors a motif (CX5CX2C) similar to the canonical radical SAM CX3CX2C motif. This CX5CX2C motif found in HcgA is expected to coordinate three irons in a [4Fe-4S] cluster, leaving a site-differentiated iron that is coordinated by SAM (6). Although the cluster-binding motif is slightly different from the canonical motif, other radical SAM proteins such as Elp3 and ThiC (83, 84) have also been shown to have altered motifs. Although the substrate of HcgA is not known, the purified protein has been shown to catalyze the uncoupled reductive cleavage of SAM (6). Upon reduction, the purified protein gives rise to an EPR signal consistent with the presence of a [4Fe-4S]+ cluster, which is altered upon addition of SAM (6). Due to sequence similarity with HydG, the role of HcgA was tentatively assigned to be generation of the CO ligands for the FeGP cofactor; however, in vivo labeling studies illustrated that the CO ligands of the FeGP cofactor come from CO2 (78). Furthermore, transfer of the methyl group on l-[methyl-2H3]methionine to the pyridinol ring implied that a potential SAM-dependent methyltransferase reaction might occur (78). However, without further evidence, the role of HcgA in the synthesis of the FeGP cofactor remains the subject of current inquiry.
Concluding Remarks
Despite significant progress, important aspects of the overall metallocofactor assembly pathways and the specific roles of the radical SAM enzymes remain unresolved. With regard to FeMo cofactor biosynthesis, aspects of the mechanism of carbon insertion, sulfide incorporation, and cluster rearrangement have yet to be elucidated. For H-cluster biogenesis, the mechanism of formation of CO and CN− from tyrosine, the identity of the substrate of HydE, and the detailed steps involved in assembly and delivery of the 2Fe subcluster of the H-cluster will be the focus of future work. For the FeGP cofactor maturation in [Fe]-hydrogenase, the role of the radical SAM enzyme HcgA is currently unresolved. The discovery of the involvement of radical SAM chemistry in the synthesis of organometallic cofactors in biology adds fascinating new examples of the remarkable chemistry that can be catalyzed by this diverse superfamily, using the simple combination of an iron-sulfur cluster and SAM.
This work was supported by United States Department of Energy Grant DE-FG02-10ER16194 (to J. B. B., J. W. P., and E. M. S.). This is the fourth article in the Thematic Minireview Series on Radical SAM Enzymes.
- SAM
- S-adenosylmethionine
- SAH
- S-adenosylhomocysteine
- FeGP
- iron-guanylylpyridinol
- dAdo•
- 5′-deoxyadenosyl radical
- DHG
- dehydroglycine
- ΔCTD
- C-terminal deletion
- PDB
- Protein Data Bank.
REFERENCES
- 1. Rubio L. M., Ludden P. W. (2008) Biosynthesis of the iron-molybdenum cofactor of nitrogenase. Annu. Rev. Microbiol. 62, 93–111 [DOI] [PubMed] [Google Scholar]
- 2. Ribbe M. W., Hu Y., Hodgson K. O., Hedman B. (2014) Biosynthesis of nitrogenase metalloclusters. Chem. Rev. 114, 4063–4080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shepard E. M., Mus F., Betz J. N., Byer A. S., Duffus B. R., Peters J. W., Broderick J. B. (2014) [FeFe]-Hydrogenase maturation. Biochemistry 53, 4090–4104 [DOI] [PubMed] [Google Scholar]
- 4. Thauer R. K., Kaster A.-K., Goenrich M., Schick M., Hiromoto T., Shima S. (2010) Hydrogenases from methanogenic archaea, nickel, a novel cofactor, and H2 storage. Annu. Rev. Biochem. 79, 507–536 [DOI] [PubMed] [Google Scholar]
- 5. Bandyopadhyay S., Chandramouli K., Johnson M. K. (2008) Iron-sulfur cluster biosynthesis. Biochem. Soc. Trans. 36, 1112–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McGlynn S. E., Boyd E. S., Shepard E. M., Lange R. K., Gerlach R., Broderick J. B., Peters J. W. (2010) Identification and characterization of a novel member of the radical AdoMet enzyme superfamily and implications for the biosynthesis of the Hmd hydrogenase active site cofactor. J. Bacteriol. 192, 595–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Broderick J. B., Duffus B. R., Duschene K. S., Shepard E. M. (2014) Radical S-adenosylmethionine enzymes. Chem. Rev. 114, 4229–4317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schlögl R. (2003) Catalytic synthesis of ammonia–a “never-ending story”? Angew. Chem. Int. Ed. Engl. 42, 2004–2008 [DOI] [PubMed] [Google Scholar]
- 9. Eady R. R. (1996) Structure-function relationships of alternative nitrogenases. Chem. Rev. 96, 3013–3030 [DOI] [PubMed] [Google Scholar]
- 10. Kim J., Rees D. C. (1994) Nitrogenase and biological nitrogen fixation. Biochemistry 33, 389–397 [DOI] [PubMed] [Google Scholar]
- 11. Einsle O., Tezcan F. A., Andrade S. L., Schmid B., Yoshida M., Howard J. B., Rees D. C. (2002) Nitrogenase MoFe-protein at 1.16 Å resolution: a central ligand in the FeMo-cofactor. Science 297, 1696–1700 [DOI] [PubMed] [Google Scholar]
- 12. Spatzal T., Aksoyoglu M., Zhang L., Andrade S. L., Schleicher E., Weber S., Rees D. C., Einsle O. (2011) Evidence for interstitial carbon in nitrogenase FeMo cofactor. Science 334, 940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lancaster K. M., Roemelt M., Ettenhuber P., Hu Y., Ribbe M. W., Neese F., Bergmann U., DeBeer S. (2011) X-ray emission spectroscopy evidences a central carbon in the nitrogenase iron-molybdenum cofactor. Science 334, 974–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dos Santos P. C., Johnson D. C., Ragle B. E., Unciuleac M.-C., Dean D. R. (2007) Controlled expression of nif and isc iron-sulfur protein maturation components reveals target specificity and limited functional replacement between the two systems. J. Bacteriol. 189, 2854–2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smith A. D., Jameson G. N., Dos Santos P. C., Agar J. N., Naik S., Krebs C., Frazzon J., Dean D. R., Huynh B. H., Johnson M. K. (2005) NifS-mediated assembly of [4Fe-4S] clusters in the N- and C-terminal domains of the NifU scaffold protein. Biochemistry 44, 12955–12969 [DOI] [PubMed] [Google Scholar]
- 16. Wiig J. A., Hu Y., Lee C. C., Ribbe M. W. (2012) Radical SAM-dependent carbon insertion into the nitrogenase M-cluster. Science 337, 1672–1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wiig J. A., Hu Y., Ribbe M. W. (2011) NifEN-B complex of Azotobacter vinelandii is fully functional in nitrogenase FeMo cofactor assembly. Proc. Natl. Acad. Sci. U.S.A. 108, 8623–8627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boal A. K., Rosenzweig A. C. (2012) Biochemistry. A radical route for nitrogenase carbide insertion. Science 337, 1617–1618 [DOI] [PubMed] [Google Scholar]
- 19. Hu Y., Corbett M. C., Fay A. W., Webber J. A., Hodgson K. O., Hedman B., Ribbe M. W. (2006) FeMo cofactor maturation on NifEN. Proc. Natl. Acad. Sci. U.S.A. 103, 17119–17124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hu Y., Corbett M. C., Fay A. W., Webber J. A., Hodgson K. O., Hedman B., Ribbe M. W. (2006) Nitrogenase Fe protein: a molybdate/homocitrate insertase. Proc. Natl. Acad. Sci. U.S.A. 103, 17125–17130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Corbett M. C., Hu Y., Fay A. W., Ribbe M. W., Hedman B., Hodgson K. O. (2006) Structural insights into a protein-bound iron-molybdenum cofactor precursor. Proc. Natl. Acad. Sci. U.S.A. 103, 1238–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kaiser J. T., Hu Y., Wiig J. A., Rees D. C., Ribbe M. W. (2011) Structure of a precursor-bound NifEN: a nitrogenase FeMo cofactor maturase/insertase. Science 331, 91–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hu Y., Corbett M. C., Fay A. W., Webber J. A., Hedman B., Hodgson K. O., Ribbe M. W. (2005) Nitrogenase reactivity with P-cluster variants. Proc. Natl. Acad. Sci. U.S.A. 102, 13825–13830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zheng L., White R. H., Dean D. R. (1997) Purification of the Azotobacter vinelandii nifV-encoded homocitrate synthase. J. Bacteriol. 179, 5963–5966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yoshizawa J. M., Blank M. A., Fay A. W., Lee C. C., Wiig J. A., Hu Y., Hodgson K. O., Hedman B., Ribbe M. W. (2009) Optimization of FeMoco maturation on NifEN. J. Am. Chem. Soc. 131, 9321–9325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hernandez J. A., Curatti L., Aznar C. P., Perova Z., Britt R. D., Rubio L. M. (2008) Metal trafficking for nitrogen fixation: NifQ donates molybdenum to NifEN/NifH for the biosynthesis of the nitrogenase FeMo-cofactor. Proc. Natl. Acad. Sci. U.S.A. 105, 11679–11684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fay A. W., Blank M. A., Yoshizawa J. M., Lee C. C., Wiig J. A., Hu Y., Hodgson K. O., Hedman B., Ribbe M. W. (2010) Formation of a homocitrate-free iron-molybdenum cluster on NifEN: implications for the role of homocitrate in nitrogenase assembly. Dalton Trans. 39, 3124–3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fay A. W., Blank M. A., Lee C. C., Hu Y., Hodgson K. O., Hedman B., Ribbe M. W. (2011) Spectroscopic characterization of the isolated iron-molybdenum cofactor (FeMoco) precursor from the protein NifEN. Angew. Chem. Int. Ed. Engl. 50, 7787–7790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lancaster K. M., Hu Y., Bergmann U., Ribbe M. W., DeBeer S. (2013) X-ray spectroscopic observation of an interstitial carbide in NifEN-bound FeMoco precursor. J. Am. Chem. Soc. 135, 610–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. George S. J., Igarashi R. Y., Xiao Y., Hernandez J. A., Demuez M., Zhao D., Yoda Y., Ludden P. W., Rubio L. M., Cramer S. P. (2008) Extended x-ray absorption fine structure and nuclear resonance vibrational spectroscopy reveal that NifB-co, a FeMo-co precursor, comprises a 6Fe core with an interstitial light atom. J. Am. Chem. Soc. 130, 5673–5680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boal A. K., Grove T. L., McLaughlin M. I., Yennawar N. H., Booker S. J., Rosenzweig A. C. (2011) Structural basis for methyl transfer by a radical SAM enzyme. Science 332, 1089–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grove T. L., Benner J. S., Radle M. I., Ahlum J. H., Landgraf B. J., Krebs C., Booker S. J. (2011) A radically different mechanism for S-adenosylmethionine-dependent methyltransferases. Science 332, 604–607 [DOI] [PubMed] [Google Scholar]
- 33. Vignais P. M., Billoud B. (2007) Occurrence, classification, and biological function of hydrogenases: an overview. Chem. Rev. 107, 4206–4272 [DOI] [PubMed] [Google Scholar]
- 34. Vignais P. M., Billoud B., Meyer J. (2001) Classification and phylogeny of hydrogenases. FEMS Microbiol. Rev. 25, 455–501 [DOI] [PubMed] [Google Scholar]
- 35. Posewitz M. C., King P. W., Smolinski S. L., Zhang L., Seibert M., Ghirardi M. L. (2004) Discovery of two novel radical S-adenosylmethionine proteins required for the assembly of an active [Fe] hydrogenase. J. Biol. Chem. 279, 25711–25720 [DOI] [PubMed] [Google Scholar]
- 36. King P. W., Posewitz M. C., Ghirardi M. L., Seibert M. (2006) Functional studies of [FeFe] hydrogenase maturation in an Escherichia coli biosynthetic system. J. Bacteriol. 188, 2163–2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McGlynn S. E., Ruebush S. S., Naumov A., Nagy L. E., Dubini A., King P. W., Broderick J. B., Posewitz M. C., Peters J. W. (2007) In vitro activation of [FeFe] hydrogenase: new insights into hydrogenase maturation. J. Biol. Inorg. Chem. 12, 443–447 [DOI] [PubMed] [Google Scholar]
- 38. McGlynn S. E., Shepard E. M., Winslow M. A., Naumov A. V., Duschene K. S., Posewitz M. C., Broderick W. E., Broderick J. B., Peters J. W. (2008) HydF as a scaffold protein in [FeFe] hydrogenase H-cluster biosynthesis. FEBS Lett. 582, 2183–2187 [DOI] [PubMed] [Google Scholar]
- 39. Mulder D. W., Boyd E. S., Sarma R., Lange R. K., Endrizzi J. A., Broderick J. B., Peters J. W. (2010) Stepwise [FeFe]-hydrogenase H-cluster assembly revealed in the structure of HydAΔEFG. Nature 465, 248–251 [DOI] [PubMed] [Google Scholar]
- 40. Mulder D. W., Ortillo D. O., Gardenghi D. J., Naumov A. V., Ruebush S. S., Szilagyi R. K., Huynh B., Broderick J. B., Peters J. W. (2009) Activation of HydAΔEFG requires a preformed [4Fe-4S] cluster. Biochemistry 48, 6240–6248 [DOI] [PubMed] [Google Scholar]
- 41. Shepard E. M., McGlynn S. E., Bueling A. L., Grady-Smith C. S., George S. J., Winslow M. A., Cramer S. P., Peters J. W., Broderick J. B. (2010) Synthesis of the 2Fe-subcluster of the [FeFe]-hydrogenase H-cluster on the HydF scaffold. Proc. Natl. Acad. Sci. U.S.A. 107, 10448–10453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Czech I., Silakov A., Lubitz W., Happe T. (2010) The [FeFe]-hydrogenase maturase HydF from Clostridium acetobutylicum contains a CO and CN− ligated cluster. FEBS Lett. 584, 638–642 [DOI] [PubMed] [Google Scholar]
- 43. Berggren G., Adamska A., Lambertz C., Simmons T. R., Esselborn J., Atta M., Gambarelli S., Mouesca J. M., Reijerse E., Lubitz W., Happe T., Artero V., Fontecave M. (2013) Biomimetic assembly and activation of [FeFe]-hydrogenases. Nature 499, 66–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nicolet Y., Pagnier A., Zeppieri L., Martin L., Amara P., Fontecilla-Camps J. C. (2014) Crystal structure of HydG from Carboxydothermus hydrogenoformans: a trifunctional [FeFe]-hydrogenase maturase. ChemBioChem. 10.1002/cbic.201402661 [DOI] [PubMed] [Google Scholar]
- 45. Kriek M., Martins F., Leonardi R., Fairhurst S. A., Lowe D. J., Roach P. L. (2007) Thiazole synthase from Escherichia coli. An investigation of the substrates and purified proteins required for activity in vitro. J. Biol. Chem. 282, 17413–17423 [DOI] [PubMed] [Google Scholar]
- 46. Pilet E., Nicolet Y., Mathevon C., Douki T., Fontecilla-Camps J. C., Fontecave M. (2009) The role of the maturase HydG in [FeFe]-hydrogenase active site synthesis and assembly. FEBS Lett. 583, 506–511 [DOI] [PubMed] [Google Scholar]
- 47. Driesener R. C., Challand M. R., McGlynn S. E., Shepard E. M., Boyd E. S., Broderick J. B., Peters J. W., Roach P. L. (2010) [FeFe]-Hydrogenase cyanide ligands derived from S-adenosylmethionine-dependent cleavage of tyrosine. Angew. Chem. Int. Ed. Engl. 49, 1687–1690 [DOI] [PubMed] [Google Scholar]
- 48. Shepard E. M., Duffus B. R., George S. J., McGlynn S. E., Challand M. R., Swanson K. D., Roach P. L., Cramer S. P., Peters J. W., Broderick J. B. (2010) [FeFe]-Hydrogenase maturation: HydG-catalyzed synthesis of carbon monoxide. J. Am. Chem. Soc. 132, 9247–9249 [DOI] [PubMed] [Google Scholar]
- 49. Kuchenreuther J. M., George S. J., Grady-Smith C. S., Cramer S. P., Swartz J. R. (2011) Cell-free H-cluster synthesis and [FeFe] hydrogenase activation: all five CO and CN− ligands derive from tyrosine. PLoS ONE 6, e20346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Driesener R. C., Duffus B. R., Shepard E. M., Bruzas I. R., Duschene K. S., Coleman N. J., Marrison A. P., Salvadori E., Kay C. W., Peters J. W., Broderick J. B., Roach P. L. (2013) Biochemical and kinetic characterization of radical S-adenosylmethionine enzyme HydG. Biochemistry 52, 8696–8707 [DOI] [PubMed] [Google Scholar]
- 51. Nicolet Y., Martin L., Tron C., Fontecilla-Camps J. C. (2010) A glycyl free radical as the precursor in the synthesis of carbon monoxide and cyanide by the [FeFe]-hydrogenase maturase HydG. FEBS Lett. 584, 4197–4202 [DOI] [PubMed] [Google Scholar]
- 52. Tron C., Cherrier M. V., Amara P., Martin L., Fauth F., Fraga E., Correard M., Fontecave M., Nicolet Y., Fontecilla-Camps J. C. (2011) Further characterization of the [FeFe]-hydrogenase maturase HydG. Eur. J. Inorg. Chem. 2011, 1121–1127 [Google Scholar]
- 53. Vey J. L., Drennan C. L. (2011) Structural insights into radical generation by the radical SAM superfamily. Chem. Rev. 111, 2487–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kuchenreuther J. M., Myers W. K., Stich T. A., George S. J., Nejatyjahromy Y., Swartz J. R., Britt R. D. (2013) A radical intermediate in tyrosine scission to the CO and CN− ligands of FeFe hydrogenase. Science 342, 472–475 [DOI] [PubMed] [Google Scholar]
- 55. Hänzelmann P., Schindelin H. (2006) Binding of 5′-GTP to the C-terminal FeS cluster of the radical S-adenosylmethionine enzyme MoaA provides insights into its mechanism. Proc. Natl. Acad. Sci. U.S.A. 103, 6829–6834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kuchenreuther J. M., Myers W. K., Suess D. L., Stich T. A., Pelmenschikov V., Shiigi S. A., Cramer S. P., Swartz J. R., Britt R. D., George S. J. (2014) The HydG enzyme generates an Fe(CO)2(CN) synthon in assembly of the FeFe hydrogenase H-cluster. Science 343, 424–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nicolet Y., Rohac R., Martin L., Fontecilla-Camps J. C. (2013) X-ray snapshots of possible intermediates in the time course of synthesis and degradation of protein-bound [4Fe-4S] clusters. Proc. Natl. Acad. Sci. U.S.A. 110, 7188–7192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nicolet Y., Rubach J. K., Posewitz M. C., Amara P., Mathevon C., Atta M., Fontecave M., Fontecilla-Camps J. C. (2008) X-ray structure of the [FeFe]-HydE from Thermotoga maritima. J. Biol. Chem. 283, 18861–18872 [DOI] [PubMed] [Google Scholar]
- 59. Nicolet Y., Amara P., Mouesca J. M., Fontecilla-Camps J. C. (2009) Unexpected electron transfer mechanism upon AdoMet cleavage in radical SAM proteins. Proc. Natl. Acad. Sci. U.S.A. 106, 14867–14871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nicolet Y., Fontecilla-Camps J. C. (2012) Structure-function relationships in [FeFe]-hydrogenase active site maturation. J. Biol. Chem. 287, 13532–13540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Quitterer F., List A., Eisenreich W., Bacher A., Groll M. (2012) Crystal structure of methylornithine synthase (PylB): insights into the pyrrolysine biosynthesis. Angew. Chem. Int. Ed. Engl. 51, 1339–1342 [DOI] [PubMed] [Google Scholar]
- 62. Peters J. W., Broderick J. B. (2012) Emerging paradigms for complex iron-sulfur cofactor assembly and insertion. Annu. Rev. Biochem. 81, 429–450 [DOI] [PubMed] [Google Scholar]
- 63. Shepard E. M., Boyd E. S., Broderick J. B., Peters J. W. (2011) Biosynthesis of complex iron-sulfur enzymes. Curr. Opin. Chem. Biol. 15, 319–327 [DOI] [PubMed] [Google Scholar]
- 64. Kuchenreuther J. M., Britt R. D., Swartz J. R. (2012) New insights into [FeFe] hydrogenase activation and maturase function. PLoS ONE 7, e45850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vallese F., Berto P., Ruzzene M., Cendron L., Sarno S., De Rosa E., Giacometti G. M., Costantini P. (2012) Biochemical analysis of the interactions between the proteins involved in the [FeFe]-hydrogenase maturation process. J. Biol. Chem. 287, 36544–36555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Boyd E. S., Anbar A. D., Miller S., Hamilton T. L., Lavin M., Peters J. W. (2011) A late methanogen origin for molybdenum-dependent nitrogenase. Geobiology 9, 221–232 [DOI] [PubMed] [Google Scholar]
- 67. Shima S., Pilak O., Vogt S., Schick M., Stagni M. S., Meyer-Klaucke W., Warkentin E., Thauer R. K., Ermler U. (2008) The crystal structure of [Fe]-hydrogenase reveals the geometry of the active site. Science 321, 572–575 [DOI] [PubMed] [Google Scholar]
- 68. Thauer R. K., Klein A. R., Hartmann G. C. (1996) Reactions with molecular hydrogen in microorganisms: evidence for a purely organic hydrogenation catalyst. Chem. Rev. 96, 3031–3042 [DOI] [PubMed] [Google Scholar]
- 69. Buurman G., Shima S., Thauer R. K. (2000) The metal-free hydrogenase from methanogenic archaea: evidence for a bound cofactor. FEBS Lett. 485, 200–204 [DOI] [PubMed] [Google Scholar]
- 70. Vogt S., Lyon E. J., Shima S., Thauer R. K. (2008) The exchange activities of [Fe] hydrogenase (iron-sulfur-cluster-free hydrogenase) from methanogenic archaea in comparison with the exchange activities of [FeFe] and [NiFe] hydrogenases. J. Biol. Inorg. Chem. 13, 97–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wang X., Li Z., Zeng X., Luo Q., Evans D. J., Pickett C. J., Liu X. (2008) The iron centre of the cluster-free hydrogenase (Hmd): low-spin Fe(II) or low-spin Fe(0)? Chem. Commun. 30, 3555–3557 [DOI] [PubMed] [Google Scholar]
- 72. Salomone-Stagni M., Stellato F., Whaley C. M., Vogt S., Morante S., Shima S., Rauchfuss T. B., Meyer-Klaucke W. (2010) The iron-site structure of [Fe]-hydrogenase and model systems: an x-ray absorption near edge spectroscopy study. Dalton Trans. 39, 3057–3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Korbas M., Vogt S., Meyer-Klaucke W., Bill E., Lyon E. J., Thauer R. K., Shima S. (2006) The iron-sulfur cluster-free hydrogenase (Hmd) is a metalloenzyme with a novel iron binding motif. J. Biol. Chem. 281, 30804–30813 [DOI] [PubMed] [Google Scholar]
- 74. Hiromoto T., Ataka K., Pilak O., Vogt S., Stagni M. S., Meyer-Klaucke W., Warkentin E., Thauer R. K., Shima S., Ermler U. (2009) The crystal structure of C176A mutated [Fe]-hydrogenase suggests an acyl-iron ligation in the active site iron complex. FEBS Lett. 583, 585–590 [DOI] [PubMed] [Google Scholar]
- 75. Lyon E. J., Shima S., Boecher R., Thauer R. K., Grevels F. W., Bill E., Roseboom W., Albracht S. P. (2004) Carbon monoxide as an intrinsic ligand to iron in the active site of the iron-sulfur-cluster-free hydrogenase H2-forming methylenetetrahydromethanopterin dehydrogenase as revealed by infrared spectroscopy. J. Am. Chem. Soc. 126, 14239–14248 [DOI] [PubMed] [Google Scholar]
- 76. Shima S., Lyon E. J., Sordel-Klippert M., Kauss M., Kahnt J., Thauer R. K., Steinbach K., Xie X., Verdier L., Griesinger C. (2004) The cofactor of the iron-sulfur-cluster-free hydrogenase Hmd: structure of the light-inactivation product. Angew. Chem. Int. Ed. Engl. 43, 2547–2551 [DOI] [PubMed] [Google Scholar]
- 77. Pilak O., Mamat B., Vogt S., Hagemeier C. H., Thauer R. K., Shima S., Vonrhein C., Warkentin E., Ermler U. (2006) The crystal structure of the apoenzyme of the iron-sulphur cluster-free hydrogenase. J. Mol. Biol. 358, 798–809 [DOI] [PubMed] [Google Scholar]
- 78. Schick M., Xie X., Ataka K., Kahnt J., Linne U., Shima S. (2012) Biosynthesis of the iron-guanylylpyridinol cofactor of [Fe]-hydrogenase in methanogenic archaea as elucidated by stable-isotope labeling. J. Am. Chem. Soc. 134, 3271–3280 [DOI] [PubMed] [Google Scholar]
- 79. Afting C., Kremmer E., Brucker C., Hochheimer A., Thauer R. K. (2000) Regulation of the synthesis of H2-forming methylenetetrahydromethanopterin dehydrogenase (Hmd) and of HmdII and HmdIII in Methanothermobacter marburgensis. Arch. Microbiol. 174, 225–232 [DOI] [PubMed] [Google Scholar]
- 80. Lie T. J., Costa K. C., Pak D., Sakesan V., Leigh J. A. (2013) Phenotypic evidence that the function of the [Fe]-hydrogenase Hmd in Methanococcus maripaludis requires seven hcg (hmd co-occurring genes) but not hmdII. FEMS Microbiol. Lett. 343, 156–160 [DOI] [PubMed] [Google Scholar]
- 81. Fujishiro T., Tamura H., Schick M., Kahnt J., Xie X., Ermler U., Shima S. (2013) Identification of the HcgB enzyme in [Fe]-hydrogenase-cofactor biosynthesis. Angew. Chem. Int. Ed. Engl. 52, 12555–12558 [DOI] [PubMed] [Google Scholar]
- 82. Fujishiro T., Ermler U., Shima S. (2014) A possible iron delivery function of the dinuclear iron center of HcgD in [Fe]-hydrogenase cofactor biosynthesis. FEBS Lett. 588, 2789–2793 [DOI] [PubMed] [Google Scholar]
- 83. Martinez-Gomez N. C., Downs D. M. (2008) ThiC is an [Fe-S] cluster protein that requires AdoMet to generate the 4-amino-5-hydroxymethyl-2-methylpyrimidine moiety in thiamin synthesis. Biochemistry 47, 9054–9056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Paraskevopoulou C., Fairhurst S. A., Lowe D. J., Brick P., Onesti S. (2006) The elongator subunit Elp3 contains a Fe4S4 cluster and binds S-adenosylmethionine. Mol. Microbiol. 59, 795–806 [DOI] [PubMed] [Google Scholar]