

Background: Berberine inhibits PCSK9 transcription via the HNF1 binding site of the PCSK9 promoter, but the underlying mechanism is unclear.
Results: Berberine inhibits PCSK9 expression by inducing proteasomal degradation of hepatic HNF1α protein.
Conclusion: The ubiquitin-proteasomal system is intrinsically connected to the PCSK9-LDLR pathway through regulation of HNF1α protein stability.
Significance: We have uncovered a new aspect of PCSK9 regulation by ubiquitin-induced proteasomal degradation of HNF1α.
Keywords: Dyslipidemia, Lipid Metabolism, Nuclear Receptor, Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9), Proteasome, Ubiquitylation (Ubiquitination), HNF1a, LDL Receptor
Abstract
Our previous in vitro studies have identified hepatocyte nuclear factor 1α (HNF1α) as an obligated trans-activator for PCSK9 gene expression and demonstrated its functional involvement in the suppression of PCSK9 expression by berberine (BBR), a natural cholesterol-lowering compound. In this study, we investigated the mechanism underlying the inhibitory effect of BBR on HNF1α-mediated PCSK9 transcription. Administration of BBR to hyperlipidemic mice and hamsters lowered circulating PCSK9 concentrations and hepatic PCSK9 mRNA levels without affecting the gene expression of HNF1α. However, hepatic HNF1α protein levels were markedly reduced in BBR-treated animals as compared with the control. Using HepG2 cells as a model system, we obtained evidence that BBR treatment let to accelerated degradation of HNF1α protein. By applying inhibitors to selectively block the ubiquitin proteasome system (UPS) and autophagy-lysosomal pathway, we show that HNF1α protein content in HepG2 cells was not affected by bafilomycin A1 treatment, but it was dose-dependently increased by UPS inhibitors bortezomib and MG132. Bortezomib treatment elevated HNF1α and PCSK9 cellular levels with concomitant reductions of LDL receptor protein. Moreover, HNF1α protein displayed a multiubiquitination ladder pattern in cells treated with BBR or overexpressing ubiquitin. By expressing GFP-HNF1α fusion protein in cells, we observed that blocking UPS resulted in accumulation of GFP-HNF1α in cytoplasm. Importantly, we show that the BBR reducing effects on HNF1α protein and PCSK9 gene transcription can be eradicated by proteasome inhibitors. Altogether, our studies using BBR as a probe uncovered a new aspect of PCSK9 regulation by ubiquitin-induced proteasomal degradation of HNF1α.
Introduction
Mounting evidence has demonstrated that proprotein convertase subtilisin/kexin type 9 (PCSK9)2 is a critical player in LDL cholesterol (LDL-C) metabolism through its interaction with hepatic LDL receptor (LDLR) (1–4). PCSK9 is mainly synthesized in liver and is rapidly secreted into plasma after its maturation through a self-engaged autocatalytic cleavage in the endoplasmic reticulum (3). PCSK9 regulates plasma LDL-C levels by diverting cell surface LDLR of hepatocytes to lysosomes for degradation (5, 6). Thus, PCSK9 plasma levels directly influence the level of circulating LDL-C (7, 8). Recent successful demonstrations of neutralizing anti-PCSK9 antibodies that lowered serum LDL-C levels in dyslipidemic and hypercholesterolemic patients have provided strong validation to support the notion that lowering circulating PCSK9 levels to up-regulate hepatic LDLR is beneficial for reducing the risk of cardiovascular disease in humans (9).
In liver tissue, PCSK9 synthesis is largely controlled at the gene transcriptional level by two transcription factor families, sterol regulatory element-binding proteins (SREBPs) (10–12) and hepatocyte nuclear factor 1 (HNF1), a dimeric transcriptional activator containing homeodomain (13). PCSK9 gene expression is positively regulated by SREBP through an SRE motif of the proximal promoter in response to depletion of intracellular levels of sterols. Within the PCSK9 promoter, a highly conserved HNF1 binding site is located between the SRE and Sp1 site that functions as a tissue-specific cis-regulatory sequence of the PCSK9 promoter through the binding of the liver-enriched transcription factor HNF1α (14–16). We have previously reported that the interaction of HNF1α with HNF1 motif is not only requisite for the high level transcriptional activity of the PCSK9 promoter in hepatic cells; it is also a regulatory site to mediate the suppression of PCSK9 transcription by berberine (BBR), a natural cholesterol-lowering compound (17). In HepG2 cells, levels of PCSK9 mRNA and protein were substantially reduced after BBR treatment (14, 18). Mutation or deletion of the HNF1 binding site on the PCSK9 promoter resulted in the loss of BBR-mediated inhibition of PCSK9 promoter activity in HepG2 cells. Likewise, siRNA-mediated depletion of intracellular HNF1α protein attenuated the suppression of PCSK9 expression by BBR treatment (14).
Our subsequent in vivo study of dyslipidemic hamsters showed that BBR treatment of 100 mg/kg for 1 week lowered hepatic PCSK9 mRNA levels by 50% as compared with the PCSK9 mRNA levels in liver samples of control hamsters (15). However, the involvement of HNF1α in BBR-mediated reduction of PCSK9 mRNA in liver tissue was not examined in that hamster study. Thus, the in vivo evidence for a functional role of HNF1α in BBR-mediated inhibition of PCSK9 gene transcription is presently lacking. Furthermore, the underlying molecular mechanisms of how BBR inhibits PCSK9 gene expression via HNF1 site remain unclear. Because inhibition of PCSK9 transcription in liver tissue will directly reduce circulating PCSK9 levels and hence lower the risk for developing cardiovascular disease, it is important to conduct further investigations to elucidate the regulatory pathway that is elicited by BBR to constrain HNF1α-mediated transactivation of PCSK9 gene expression.
In this current study, by utilizing a hyperlipidemic mouse model, we demonstrate that BBR treatment reduced circulating PCSK9 concentrations and hepatic PCSK9 mRNA levels without affecting HNF1α gene expression. However, hepatic HNF1α protein content was greatly reduced in BBR-treated mice as compared with the control mice. Examination of liver tissues of BBR-treated hamsters further confirmed that BBR lowered HNF1α protein cellular abundance without inhibiting its gene expression. These in vivo observations from two different animal models suggest that BBR regulates HNF1α expression at translational levels. Through different lines of investigations conducted in cultured hepatic cells, we provide strong evidence to demonstrate that the ubiquitin proteasome system (UPS) is involved in BBR-mediated reduction of HNF1α protein cellular abundance, which negatively regulates PCSK9 gene transcription.
EXPERIMENTAL PROCEDURES
Cells and Reagents
The human hepatoma cell line HepG2 was obtained from American Type Culture Collection and cultured in Eagle's minimum essential medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin, streptomycin solution. HEK293 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS and 1% penicillin, streptomycin solution. FuGENE 6 transfection reagent (Roche Applied Science) was used to transfect plasmids into HepG2 cells or HEK293 cells according to the manufacturer's instructions. Anti-HNF1α, anti-Myc, and anti-HDAC1 antibodies were purchased from Santa Cruz Biotechnology, Inc. Anti-β-actin and anti-FLAG antibodies were purchased from Sigma-Aldrich. Anti-GAPDH antibody was obtained from Invitrogen. Anti-LDLR antibody was obtained from BioVision. Anti-hamster PCSK9 antibody was developed in our laboratory and was reported previously (19). Anti-human PCSK9 antibody was described previously (14). Anti-ubiquitin antibody (P4D1) was obtained from Cell Signaling. BBR, cycloheximide (CHX), bortezomib (BTZ), MG132, and bafilomycin A1 (BA1) were purchased from Sigma-Aldrich.
Animal Diet and BBR Treatment
2–3-month-old FVB mice expressing a luciferase reporter gene (20) were used in the BBR study. The expression of the luciferase in these mice is irrelevant to this study. Mice were housed (4 animals/cage) under controlled temperature (72 °F) and lighting (12-h light/dark cycle). Animals had free access to autoclaved water and food. Mice were fed a rodent high cholesterol diet containing 1.25% cholesterol (product D12108, Research Diet, Inc.) for 4 weeks. Mice were then divided into two groups (n = 10/group) and were given a daily dose of BBR at 200 mg/kg by oral gavage. The control group received vehicle (0.5% methyl cellulose). The drug treatment lasted 16 days. Serum samples were collected after a 4-h fasting before, during, and after the drug treatment. After the last dosing, all animals were euthanized for collection of serum and liver tissues. Sera and liver tissues were stored at −80 °C until analysis.
Male Syrian golden hamsters with body weights of 100–120 g were purchased from Harlan. Hamsters were fed a high fructose diet (60% fructose; Dyets, Inc., Bethlehem, PA) for 3 weeks to induce dyslipidemia (15). After 21 days, while continuously on the high fructose diet, 18 hamsters were randomly divided into vehicle control group or BBR group (n = 9/group). Hamsters were given a daily dose of BBR at 100 mg/kg by oral gavage. The control group received vehicle (10% 2-hydroxypropyl-β-cyclodextrin in autoclaved water) by oral gavage. The BBR treatment lasted 7 days. At the experimental termination, hamsters were fasted overnight before euthanization for serum and liver tissue collections. Sera and liver tissues were stored at −80 °C until analysis. Animal use and experimental procedures were approved by the Institutional Animal Care and Use Committee of the Veterans Affairs Palo Alto Health Care System.
Serum Isolation and Cholesterol Determination
Fasting blood samples (0.1 ml) were collected from the retro-orbital plexus using heparinized capillary tubes under anesthesia (2–3% isoflurane and 1–2 liters/min oxygen), and serum was isolated at room temperature and stored at −80 °C. Standard enzymatic methods were used to determine TC and LDL-C with commercially available kits purchased from Stanbio Laboratory (Boerne, TX). Each sample was assayed in duplicate.
Detection of Mouse PCSK9 in Serum
Secreted PCSK9 in mouse serum samples were measured using a mouse PCSK9 ELISA kit obtained from R&D Systems according to the manufacturer's instructions.
Detection of Hamster PCSK9 in Serum
Hamster serum PCSK9 was directly measured using the mouse PCSK9 ELISA kit to obtain relative PCSK9 levels. Additionally, PCSK9 in hamster serum samples was detected by immunoprecipitation (IP), followed by Western blotting using anti-hamster PCSK9 antibody as we described previously (19, 22).
RNA Isolation, cDNA Generation, and Real-time Quantitative PCR (qPCR)
Total RNA isolation, generation of cDNA, and real-time PCR were conducted as previously reported (23). Each cDNA sample was run in duplicate for liver samples and triplicate for HepG2 cells. Target mRNA expression in each sample was normalized to the housekeeping gene GAPDH. The 2−ΔΔCt method was used to calculate relative mRNA expression levels. Primer sequences of mouse, hamster, and human genes used in real-time PCR are listed in Table 1.
TABLE 1.
Real-time PCR primer sequences
| Forward | Reverse | |
|---|---|---|
| Mouse | ||
| Gapdh | AACTTTGGCATTGTGGAAGG | GGATGCAGGGATGATGTTCT |
| Hmgcr | CTTTCAGAAACGAACTGTAGCTCAC | CTAGTGGAAGATGAATGGACATGAT |
| Hnf1α | GCACCAGAGACCCACGTGCC | GGCTTCCCCTCAGCTCCCGA |
| Ldlr | ACCTGCCGACCTGATGAATTC | GCAGTCATGTTCACGGTCACA |
| Pcsk9 | TTGCAGCAGCTGGGAACTT | CCGACTGTGATGACCTCTGGA |
| Srebp1 | CAAGGCCATCGACTACATCCG | CACCACTTCGGGTTTCATGC |
| Srebp2 | CCAAAGAAGGAGAGAGGCGG | CGCCAGACTTGTGCATCTTG |
| Hamster | ||
| Gapdh | AACTTTGGCATTGTGGAAGG | GGATGCAGGGATGATGTTCT |
| Hnf1α | GAGGTGGCTCAGCAATTCAC | CACTCCTCCACCAAGGTCTC |
| Pcsk9 | TGCTCCAGAGGTCATCACAG | GTCCCACTCTGTGACATGAAG |
| Srebp1 | GCACTTTTTGACACGTTTCTTC | CTGTACAGGCTCTCCTGTGG |
| Srebp2 | GAGAGCTGTGAATTTTCCAGTG | CTACAGATGATATCCGGACCAA |
| Human | ||
| GAPDH | ATGGGGAAGGTGAAGGTCG | GGGGTCATTGATGGCAACAATA |
| HNF1α | TGGCGCAGCAGTTCACCCAT | TGAAACGGTTCCTCCGCCCC |
| PCSK9 | AGGGGAGGACATCATTGGTG | CAGGTTGGGGGTCAGTACC |
Western Blot Detection of HNF1α Protein in Liver Tissues of Mouse and Hamster and in HepG2 Cells
For tissue samples, ∼50 mg of frozen liver tissue was homogenized in radioimmune precipitation assay buffer containing 1 mm PMSF and protease inhibitor mixture (Roche Applied Science). For HepG2 cells, total cell lysates and cytoplasmic and nuclear fractions were isolated as described previously (24, 25). Protein concentration was determined using BCA protein assay reagent (Pierce). Protein samples were resolved by SDS-PAGE and transferred onto a nitrocellulose membrane. Immunoreactive bands were visualized using Super Signal West Substrate (Thermo Scientific) and quantified with the Alpha View Software.
Plasmid Constructions
For construction of FLAG-tagged HNF1α expression vector, human HNF1α coding sequence was amplified with the following primers containing an SgfI site (underlined) at the 5′-end and MluI site (boldface type) at the 3′-end: 5′-gccgcgatcgccatggtttctaaactgagccagc-3′ and 5′-gtacgcgtctgggaggaagaggccatc-3′. The PCR product was then inserted into pCMV6-entry vector (Origen), at the SgfI and MluI sites of the cut vector to express HNF1α with a FLAG tag at the C terminus. The final sequence of the vector pCMV6-HNF1α and the gene product HNF1α-FLAG were validated by sequencing and Western blotting, respectively.
For construction of GFP-HNF1α fusion protein expression vector, human HNF1α coding sequence was amplified with the following primers containing BglII site (underlined) at the 5′-end and EcoRI site (boldface type) at the 3′-end: 5′-CTCAGATCTATGGTTTCTAAACTGAGCCA-3′ and 5′-GCAGAATTCTTACTGGGAGGAAGAGGCCA-3′. The PCR product was then inserted into pAcGFP1-C vector (Clontech) at the BglII and EcoRI sites of the cut vector to express GFP-HNF1α. The final sequence of the vector pAcGFP-HNF1α and the gene product GFP-HNF1α were validated by sequencing and Western blotting, respectively.
Transient Transfection of PCSK9 Promoter Reporter Construct
PCSK9 promoter reporter pGL3-PCSK9-D4 (14) was cotransfected with pCMV-β-galactosidase control vector. One day post-transfection, the medium was changed to 0.5% FBS in Eagle's minimum essential medium overnight, and BBR at a dose of 40 μm was added for 24 h in the absence or presence of increasing concentrations of UPS inhibitors. Cells were lysed in 100 μl of reporter lysis buffer per well, of which 50 μl of cell lysate were used to measure β-galactosidase activity by using the β-galactosidase enzyme assay system (Promega). The remaining 50 μl of lysate were used to measure the firefly luciferase activity by using the luciferase assay system (Promega). Absolute luciferase activity was normalized against β-galactosidase activity to correct for transfection efficiency. Triplicate wells were assayed for each transfection condition.
Detection of Ubiquitinated HNF1α by IP
HEK293 cells were cotransfected with plasmid pCMV6-HNF1α and plasmid pCW7 expressing an N-terminal Myc-tagged ubiquitin (Myc-Ub). Mock transfections with empty vectors (pCMV6-entry or pCMV-Myc) were performed in parallel as a control. At 48 h post-transfection, cells were lysed in lysis buffer containing 250 mm NaCl, 25 mm Tris/HCl, pH 7.4, 1 mm EDTA, 1% Nonidet P-40, and protease inhibitors. Anti-FLAG M2-agarose (Sigma-Aldrich) or Protein A-agarose (Millipore) and anti-Myc antibody were individually mixed with 200 μg of cell lysates at 4 °C overnight. After incubation, the agarose beads were washed three times with lysis buffer. All proteins were released from agarose beads by boiling in 20 μl of 1× Laemmli sample buffer and then subjected to SDS-PAGE and Western blotting with the indicated antibodies.
Detection of Endogenously Ubiquitinated HNF1α in HepG2 Cells
HepG2 cells were treated for 24 h with BBR or control. During the last 8 h of BBR treatment, proteasome inhibitor MG132 (20 μm) or DMSO was added to cells, and cells were lysed by the addition of modified radioimmune precipitation assay buffer (50 mm Tris, pH 7.4, 1% Nonidet P-40, 0.25% sodium deoxycholate, 150 mm NaCl, 1 mm EDTA). Protein A-agarose (Millipore) beads were mixed with 1 μg of anti-HNF1α antibody or a control antibody (goat IgG) at 4 °C for 1 h. 0.5 ml of cell lysates containing 500 μg of protein was then added to the mixture and incubated at 4 °C overnight under agitation. The beads were collected by centrifugation and washed three times by modified radioimmune precipitation assay buffer. All proteins were released from agarose beads by boiling in 20 μl of 1× Laemmli sample buffer and then subjected to SDS-PAGE and Western blotting using anti-Ub or anti-HNF1α antibodies.
Transient Transfection of GFP-HNF1α Construct and Fluorescence Microscopy
HEK293 or HepG2 cells seeded in 6-well plates were transiently transfected with plasmid pAc-GFP-HNF1α or the control vector pAc-GFP. At 48 h after transfection, cells were treated with BTZ for 24 h. Three wells were used in each condition. The cellular localizations of GFP and GFP-HNF1α fusion protein were examined with a fluorescence microscope. To obtain the percentage of cells with GFP-HNF1α localization in cytoplasm, pictures were taken for 10–15 fields of view in each well, and the number of GFP-positive cells with nuclear localization or with cytoplasmic and nuclear localization were separately counted and recorded.
Proteasome Activity Assays
HepG2 cells cultured in Eagle's minimum essential medium containing 0.5% FBS were treated with BBR (40 μm), BTZ (100 nm), or MG132 (1 μm) for 4 or 24 h. At the indicated treatment time, cells were incubated for 10 min with the Promega Proteasome-Glo cell-based assay reagent (Proteasome-GloTM 3-substrate cell-based assay system). The chymotrypsin-like, trypsin-like activity or caspase-like proteasome activities were detected as the relative light units generated from the cleaved substrate in the reaction mixture. Luminescence generated from each reaction condition was measured by a 96-well plate reader (SpectraMax® L microplate luminometer, Molecular Devices).
Statistical Analysis
Values are presented as means ± S.E. Significant differences between treatment groups were assessed by two-tailed Student's t test (nonparametric Mann-Whitney test). Statistical significance is displayed as p < 0.05 (*), p < 0.01 (**), or p < 0.001 (***).
RESULTS
Reduction of Circulating PCSK9 and Hepatic HNF1α Protein Levels in Hyperlipidemic Mice and Hamsters Treated with BBR
To obtain in vivo evidence for BBR-mediated suppression of Pcsk9 gene expression via HNF1α, we employed a dyslipidemic mouse model. Mice fed a high cholesterol diet for 4 weeks were given BBR at a daily dose of 200 mg/kg (n = 10) or the vehicle (n = 10) for 16 days. BBR treatment led to a time-dependent reduction of serum TC (Fig. 1A) and LDL-C (Fig. 1B), whereas TC and LDL-C levels in control mice remained stable during the dosing period. At the end of treatment, serum PCSK9 levels were measured that showed a ∼50% reduction in BBR-treated mice as compared with that in the control mice (Fig. 1C). qRT-PCR measurement of mRNA levels of PCSK9, HNF1α, and four other SREBP target genes including LDLR in all liver samples revealed that mRNA levels of PCSK9 were reduced 46% (p < 0.05) by BBR treatment, whereas mRNA levels of LDLR and other SREBP2 target genes were unchanged (Fig. 1D). Importantly, HNF1α mRNA levels were not reduced by BBR treatment.
FIGURE 1.

BBR treatment lowered serum PCSK9 and cholesterol levels with reduced HNF1α protein content in liver of mice fed a high cholesterol diet. Male mice fed a high cholesterol diet for 4 weeks were orally dosed with 200 mg/kg/day BBR (n = 10) or an equal volume of vehicle (0.5% methyl cellulose) as the control group (n = 10) for 16 days. Mouse serum TC (A) and LDL-C (B) were measured before, during, and after BBR treatment. C, mouse serum PCSK9 levels after BBR treatment were quantified by ELISA. D, real-time PCR analysis of mouse liver mRNA levels of the indicated genes after BBR treatment. E, two mouse liver samples of the same treatment group were pooled together. For each group, 50 μg of homogenate proteins of five pooled samples were resolved by SDS-PAGE. HNF1α, LDLR, and β-actin were detected by immunoblotting. The protein abundance of HNF1α and LDLR was quantified with the Alpha View software with normalization by signals of β-actin. The values are presented as mean ± S.E. (error bars). *, p < 0.05; **, p < 0.01; ***, p < 0.001 as compared with the vehicle group.
We analyzed protein levels of HNF1α and LDLR in individual mouse liver homogenates by Western blotting, and the signals were quantified (Fig. 1E). In contrast to the steady HNF1α mRNA levels, amounts of HNF1α protein were 42% lower (p < 0.05), and LDLR protein levels were ∼67% higher (p < 0.05), respectively, in the BBR-treated group as compared with the vehicle group. Taken together, these results demonstrate that BBR treatment reduced HNF1α protein levels that attenuated Pcsk9 gene expression. As a consequence, serum PCSK9 levels were decreased in BBR-treated mice that led to an up-regulation of hepatic LDLR protein and an increased removal of circulating LDL-C.
We questioned next whether BBR could reduce the expression of HNF1α protein in liver tissue of other animal models. To this end, we examined the hepatic expression of HNF1α protein and mRNA levels from dyslipidemic hamsters (15) that were treated with BBR at a daily dose of 100 mg/kg (n = 9) or the vehicle (n = 9) for 7 days. Similar to the mice study results, BBR treatment reduced hepatic PCSK9 mRNA and HNF1α protein levels without any effect on HNF1α and SREBP2 mRNA expression (Fig. 2, A and B). Because an ELISA kit for hamster PCSK9 is not available, we used the mouse ELISA kit to estimate serum PCSK9 levels in all hamster serum samples (Fig. 2C). Serum PCSK9 concentration was 30% (p < 0.01) lower in the BBR group as compared with the control group. To validate the ELISA results, utilizing a highly specific anti-hamster PCSK9 antibody, we performed IP of pooled serum samples obtained from BBR and vehicle groups, and the presence of PCSK9 in immunoprecipitates was detected by Western blotting. Fig. 2D shows that serum PCSK9 levels in the BBR group were significantly lower as compared with the vehicle, which agreed with the ELISA results. Taken together, these in vivo data showed that BBR inhibited HNF1α-mediated Pcsk9 transcription by reducing hepatic HNF1α protein content without affecting its mRNA levels.
FIGURE 2.
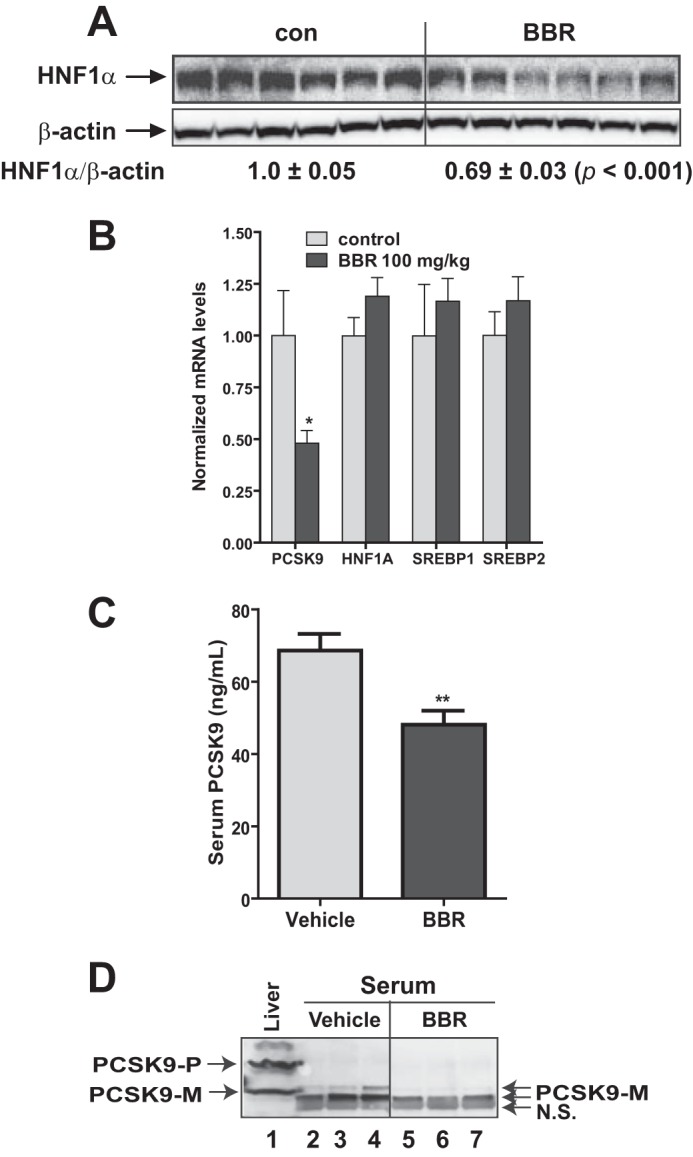
Reduction of serum PCSK9 and hepatic HNF1α protein in dyslipidemic hamsters by BBR treatment. Male hamsters were fed a high fructose diet for 3 weeks, which elevated serum TC and LDL-C (15). Hamsters on the high fructose diet were orally dosed with 100 mg/kg/day BBR (n = 9) or an equal volume of vehicle (10% 2-hydroxypropyl-β-cyclodextrin) as the control group (n = 9) for 7 days. A, 50 μg of homogenate proteins of six representative hamster liver samples from each group were resolved by SDS-PAGE. HNF1α and β-actin were detected by immunoblotting. The protein abundance of HNF1α was quantified with the Alpha View software with normalization by signals of β-actin. B, qPCR analysis of hamster liver mRNA levels of the indicated genes after BBR treatment. C, hamster serum PCSK9 levels after BBR treatment were determined using a mouse PCSK9 ELISA kit. D, three hamster serum samples of the same treatment group were pooled together. For each group, 20 μl of pooled serum of three pooled samples were used for conducting PCSK9 IP and Western blotting. Hamster liver homogenate protein was loaded in lane 1 as a positive control for anti-PCSK9 antibody Western blotting. The values are presented as mean ± S.E. (error bars). *, p < 0.05; **, p < 0.01; ***, p < 0.001 as compared with the vehicle group.
BBR Down-regulates Hepatic HNF1α Expression by Accelerating Its Protein Degradation in Cultured Hepatic Cells
We examined the time-dependent effects of BBR on the mRNA and protein expressions of PCSK9 and HNF1α in HepG2 cells. Reductions of both PCSK9 and HNF1α protein levels in total cell lysates were clearly observed after 16 and 24 h of BBR treatment (Fig. 3A). In contrast to the concomitant reductions of both proteins, qRT-PCR analysis showed that HNF1α mRNA levels remained constant during the 24-h treatment course, whereas PCSK9 mRNA levels decreased with similar kinetics as PCSK9 protein in BBR-treated cells (Fig. 3B). These results are consistent with the in vivo results described above and suggest that HepG2 cells are a suitable in vitro model system to further decipher the molecular mechanism by which BBR down-regulates HNF1α protein cellular abundance.
FIGURE 3.

BBR down-regulates HNF1α expression by accelerating its protein degradation in HepG2 cells. A, HepG2 cells were treated with 40 μm BBR for the indicated times. Total cell lysates were isolated from cells and analyzed for PCSK9 and HNF1α protein levels by Western blotting. The results shown are representative of three separate experiments with similar results. B, HepG2 cells were treated with 40 μm BBR for the indicated times. Total RNA was isolated, and PCSK9 and HNF1α mRNA levels were assessed by qRT-PCR using human-specific PCR primers, and triplicate measurements were conducted for each cDNA sample. The results shown are representative of three separate experiments with similar results. C, HepG2 cells were treated with CHX at a 5 μg/ml concentration at the indicated times in the absence or the presence of 40 μm BBR. Total cell lysates were subjected to Western blotting, and bands were visualized with antibody against HNF1α or β-actin. The data shown are representative of two separate experiments with similar results. D, after normalization to β-actin, the HNF1α signal intensity was plotted against the CHX treatment time to calculate the t½ of HNF1α protein. Half-life was calculated from each of the two separate experiments. Error bars, S.E.
The effect of BBR on HNF1α protein could be a result of accelerated protein degradation or the reduction of new protein synthesis. Because the protein half-life of HNF1α has not been reported previously, initially we decided to use CHX to block new protein synthesis and assess the HNF1α protein turnover rate in control and BBR-treated cells. HepG2 cells were treated for 0, 1, 2, 4, and 8 h with CHX in the absence or the presence of BBR, and total lysates were isolated for analyzing HNF1α protein levels by Western blotting. Two separate experiments with identical conditions were performed. Fig. 3C is a representative Western blot analysis. Fig. 3D shows the protein decay curves of both experiments. BBR treatment caused an accelerated degradation of HNF1α protein with approximate t½ = 8.3 h as compared with t½ = 16.2 h calculated from DMSO-treated control cells (Fig. 3D, left). A similar 1.8-fold reduction of HNF1α t½ by BBR was obtained from the second assay (t½ = 7.3 h in BBR versus t½ = 12.9 h in DMSO control).
Evaluation of Autophagy-Lysosomal Pathway and Ubiquitin Proteasome System in HNF1α Protein Degradation in Hepatic Cells
The reduction in HNF1α protein half-life in BBR-treated cells suggested that HNF1α is subject to an enhanced degradation process upon BBR treatment. The autophagy-lysosomal pathway and the UPS are the two major cellular proteolytic systems for intracellular protein degradation in eukaryotic cells. It was recently reported that HCV infection of human hepatoma-derived Huh-7.5 cells resulted in HNF1α protein degradation in lysosomes (26). Matsui et al. (26) further demonstrated that the HCV NS5A protein is responsible for the HCV-induced degradation of HNF1α protein. Thus, we first assessed whether lysosomes are involved in the normal clearance of HNF1α in HepG2 cells without HCV infection. We treated HepG2 cells with autophagy/lysosome inhibitor BA1 at an effective dose of 50 nm or DMSO as the vehicle control for 24 h, and total cell lysates were isolated for Western blotting. Fig. 4A shows that cellular levels of LDLR and PCSK9 were markedly increased by BA1 treatment, which was in line with literature reports for lysosome-mediated degradations of PCSK9 and LDLR (5, 6). In contrast, the cellular abundance of HNF1α was not affected by the lysosomal inhibitor, suggesting that HNF1α is not degraded by the autophagy-lysosomal pathway in HepG2 cells.
FIGURE 4.
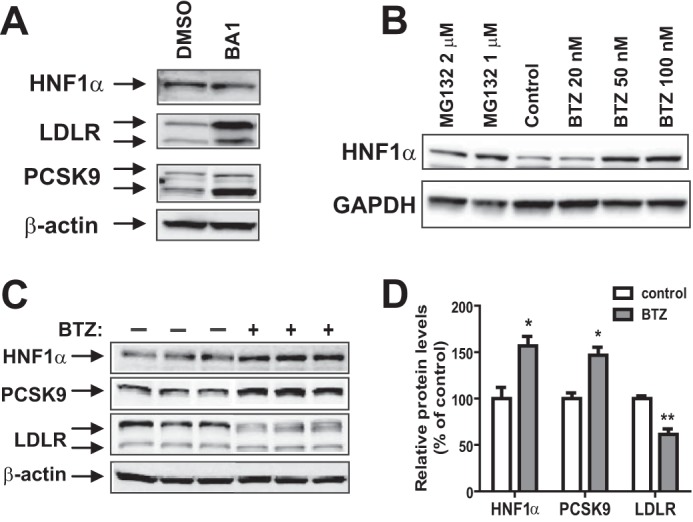
Evaluation of the effect of lysosome inhibitor BA1 and proteasome inhibitors BTZ and MG132 on HNF1α protein stability in HepG2 cells. A, HepG2 cells were incubated with 50 nm BA1 or DMSO for 24 h. Total cell lysates were harvested; 30 μg of total proteins were resolved by SDS-PAGE; and HNF1α, LDLR, and PCSK9 were detected by Western blotting. The data shown are representative of three separate experiments with similar results. B, HepG2 cells were treated with the indicated concentrations of BTZ or MG132 for 24 h. The data shown are representative of three separate experiments with similar results. C, HepG2 cells in triplicate dishes were treated with BTZ at 80 nm or DMSO for 24 h, and total cell lysates were analyzed by Western blotting. The data shown are representative of three separate experiments with similar results. D, the protein abundance of HNF1α, PCSK9, and LDLR in C was quantified with the Alpha View software with normalization by signals of β-actin. Values are mean ± S.E. (error bars) of 3 samples/group. *, p < 0.05; **, p < 0.01 compared with DMSO.
We next utilized the proteasome inhibitors BTZ and MG132 to examine the role of UPS in HNF1α degradation in HepG2 cells. Cells were treated with different concentrations of BTZ or MG132 for 24 h, and total cell lysates were analyzed for HNF1α protein abundance. Fig. 4B shows that proteasome inhibitors dose-dependently increased the protein level of HNF1α in HepG2 cells. In parallel experiments, we examined the cell viability in control and treated cells, and we did not detect significant changes induced by BBR or by the proteasome inhibitors.
To assess the impact of blocking proteasome-mediated HNF1α degradation on cellular levels of PCSK9 and LDLR, HepG2 cells seeded in triplicate dishes were treated with either 100 nm BTZ or DMSO for 24 h, and cell lysates were harvested. Fig. 4C shows the results of Western blotting of HNF1α, PCSK9, and LDLR. The signal intensity of each protein was quantified and normalized to β-actin. The quantitative data are presented in Fig. 4D. BTZ treatment increased HNF1α abundance 60% (p < 0.05) over control, which was accompanied by a 40% increase in PCSK9 protein amount (p < 0.05) and a 30% reduction of LDLR protein (p < 0.01). These results demonstrate that inhibition of proteasome inversely affected PCSK9 and LDLR protein levels through HNF1α protein stabilization.
Blocking UPS Abolished Reducing Effects of BBR on HNF1α Protein and PCSK9 mRNA and Protein Expressions
We utilized BTZ to examine the involvement of UPS in BBR-mediated down-regulation of HNF1α and PCSK9 protein levels in HepG2 cells. Cells in triplicate wells were treated with vehicle control DMSO, BBR, BTZ, and a combination of BBR and BTZ. Fig. 5A shows results of Western blotting of HNF1α, PCSK9, and LDLR. The signal intensity of each protein was quantified and normalized to β-actin. The quantitative data are presented in Fig. 5B. In the absence of proteasome inhibitor BTZ, BBR treatment significantly lowered HNF1α and PCSK9 while increasing LDLR protein levels. In the presence of BTZ, however, these effects of BBR were largely lost. BTZ treatment led to a marked elevation of cellular abundances of HNF1α and PCSK9 and reversals of BBR-induced decrease in HNF1α and PCSK9 protein contents (Fig. 5B).
FIGURE 5.

Proteasomal pathway participates in BBR-induced degradation of HNF1α protein. A, HepG2 cells were treated with 40 μm BBR in the absence or the presence of BTZ (1 μm) for 16 h, and cell lysates were utilized for immunoblotting using anti-HNF1α, anti-PCSK9, and anti-LDLR antibodies. Triplicate wells were used for each treatment. The data shown are representative of three separate experiments with similar results. B, the protein abundances of HNF1α, PCSK9, and LDLR in A were quantified with the Alpha View software with normalization by signals of β-actin. The values are presented as means ± S.E. (error bars). *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with DMSO. C, HepG2 cells were treated with 40 μm BBR in the absence or the presence of BTZ for 16 h, and total RNA was isolated for qPCR analysis of mRNA levels of HNF1α, PCSK9, and LDLR. Duplicate wells were used for each treatment, and individual cDNA samples were measured in triplicates. The values are presented as mean ± S.E. The data shown are representative of two separate experiments with similar results. D, the PCSK9 promoter reporter construct pGL3-PCSK9-D4 was transiently cotransfected with pSV-β-gal vector into HepG2 cells. One day post transfection, cells were incubated in 0.5% FBS medium overnight, followed by a 24-h treatment of 40 μm BBR in the absence or the presence of BTZ at the indicated concentrations. Cells were harvested, and the luciferase and β-galactosidase activities were measured as described under “Experimental Procedures.” Normalized luciferase activity of untreated cells is expressed as 100%. The data shown are representative of three separate experiments with similar results. E, plasmids encoding FLAG-tagged HNF1α and Myc-tagged ubiquitin were cotransfected into HEK293 cells. The empty vectors of pCMV-entry and pCMV-Myc were transfected as a mock control. Cell lysates were harvested after 2 days of transfection and immunoprecipitated with anti-Myc or anti-FLAG antibodies. Ubiquitinated HNF1α was detected by immunoblotting (WB) using anti-Myc or anti-FLAG antibodies, respectively. The data shown are representative of three separate experiments with similar results. F, HepG2 cells were treated for 24 h with BBR or DMSO as a control. During the last 8 h of BBR treatment, MG132 (20 μm) or DMSO was added to cells. Total cell lysates were prepared, and 500 μg of proteins from total lysates were subjected to IP with anti-HNF1α antibody or control antibody goat IgG. IP complexes were analyzed for ubiquitinated HNF1α with anti-HNF1α antibody and anti-ubiquitin antibody. The data shown are representative of two separate experiments with similar results.
In parallel experiments, we examined the effects of BTZ on mRNA expressions of HNF1α, PCSK9, and LDLR (Fig. 5C) by qPCR. As we expected, whereas HNF1α mRNA levels were barely changed by any of the treatments, PCSK9 mRNA expression levels were reduced by BBR and increased by BTZ without or with BBR, which was consistent with the results of Western blot analysis. It is worthy to note that our previous studies have demonstrated that BBR increases LDLR mRNA levels by mRNA stabilization mediated through the LDLR mRNA 3′-untranslated region (17, 20), and this BBR effect was not affected by the proteasome inhibitor. BTZ treatment did not alter LDLR mRNA levels in uninduced control cells or in BBR-stimulated cells.
To provide a direct link between proteasome blockage and BBR-mediated suppression of PCSK9 transcription, we analyzed PCSK9 promoter activity in HepG2 cells that were treated with BBR in the absence or the presence of BTZ at concentrations of 50 and 100 nm. A reporter assay confirmed that the inhibitory effect of BBR on PCSK9 promoter activity was largely attenuated by co-treatment with BTZ (Fig. 5D).
The UPS recognizes cellular proteins that are tagged by ubiqintin (27–29). Thus, it is important to obtain direct evidence linking the UPS to BBR-regulated HNF1α degradation. To this end, first, we overexpressed a C-terminal FLAG-tagged HNF1α and Myc-Ub in HEK293 cells. In parallel, cells were transfected with respective empty vectors as negative controls. Whole cell lysates were subjected to IP with either anti-FLAG- or anti-Myc-conjugated agarose beads. We blotted anti-Myc precipitates with anti-HNF1α antibody. A multiubiquitination ladder pattern was readily observed in cells expressing HNF1α (Fig. 5E, lane 1) but not in cells transfected with the empty vector (Fig. 5E, lane 2). Likewise, blotting anti-FLAG precipitates with anti-ubiquitin antibody detected the polyubiquitinated HNF1α in cells that were cotransfected with FLAG-HNF1α and Myc-Ub (Fig. 5E, lane 3). Without overexpression of Myc-Ub, a single band possibly representing the monoubiquitinated HNF1α was detected by anti-ubiquitin antibody in cells transfected with FLAG-HNF1α (Fig. 5E, lane 4).
Next, we examined the ubiquitination status of endogenous HNF1α in HepG2 cells without and with BBR treatment. HepG2 cells were treated for 24 h with BBR or DMSO as a control. During the last 8 h of BBR treatment, proteasome inhibitor MG132 (20 μm) or DMSO was added to cells. Equal amounts of whole cell lysates were subjected to IP with anti-HNF1α antibody or a control antibody (goat IgG), followed by Western blotting using anti-HNF1α or anti-ubiquitin antibody. Detection of HNF1α and ubiquitin in IP samples showed that the amount of pulled down unubiquitinated HNF1α, as shown by an anti-HNF1α Western blot, was lower in the precipitates of the BBR-treated sample than in those of the control sample (Fig. 5F, compare lane 8 with lane 7); however, the BBR-treated sample clearly had a higher amount of polyubiquitinated HNF1α than untreated control. In contrast with anti-HNF1α IP, Western blotting with anti-HNF1α or anti-ubiquitin antibodies did not detect specific bands in control IgG immunoprecipitates (Fig. 5F, lanes 1–4). These data provide direct evidence that BBR exposure led to enhanced HNF1α ubiquitination and its proteasomal degradation.
Blocking the Ubiquitin Proteasome System Affects HNF1α Subcellular Localization
We were interested in learning whether HNF1α subcellular localization could be affected by BBR or proteasome inhibitor. We isolated cytosol and nuclear fractions of HepG2 cells that were treated with BBR without or with BTZ. Detections of HNF1α by Western blotting were conducted using different exposure times with a 10-s exposure for nuclear fraction and 60 s for cytosolic fraction (Fig. 6A). HNF1α was largely present in the nuclear fraction. The reduction of HNF1α by BBR was observed in both nuclear and cytosol fractions. Interestingly, blocking proteasome by BTZ nearly abolished the BBR reducing effect on HNF1α level in the cytosolic fraction, but it only partially blunted the BBR effect on nuclear HNF1α.
FIGURE 6.
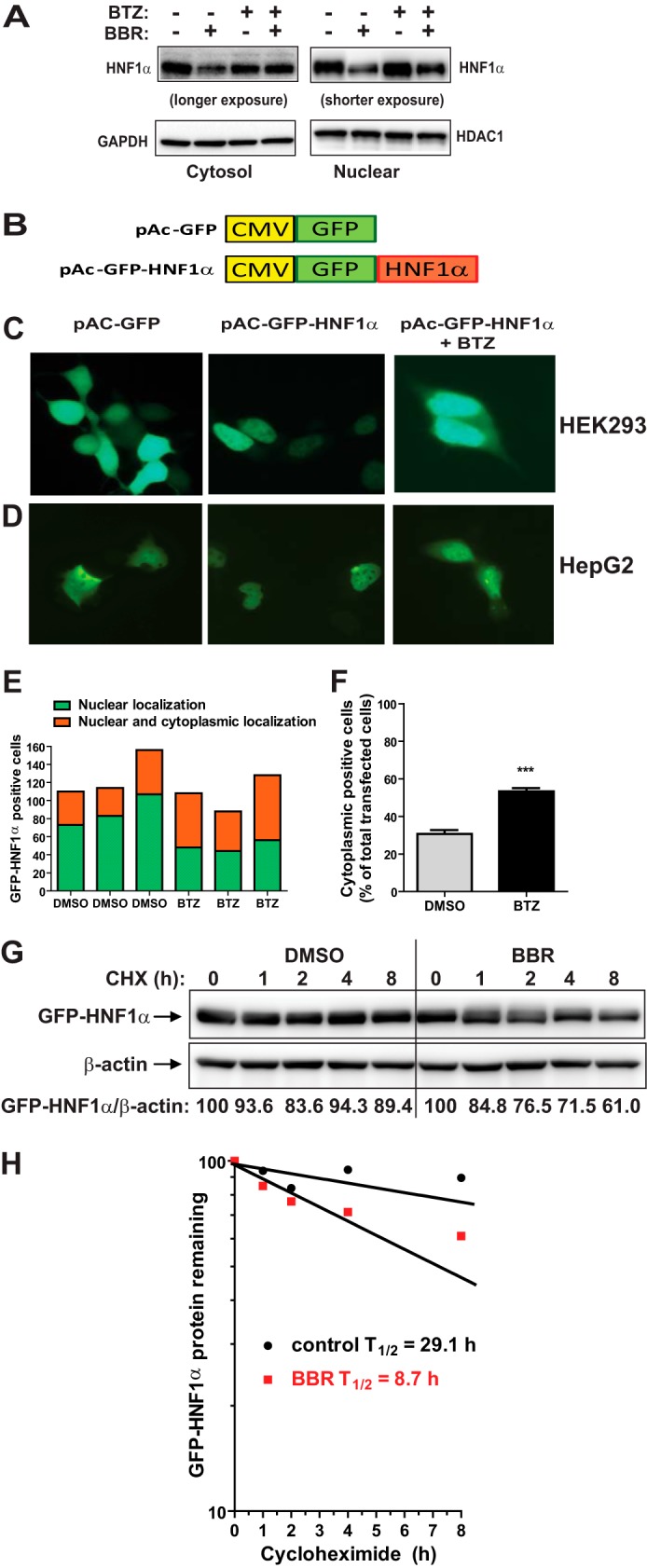
Accumulation of HNF1α in cytoplasm by proteasome inhibition. A, HepG2 cells were treated with BBR without and with 100 nm BTZ for 24 h. Nuclear fraction and cytoplasmic fraction of total cell lysates from each sample were prepared and analyzed for HNF1α protein levels by Western blotting. The membranes were reprobed with anti-HDAC1 antibody as a control of equal nuclear protein loading or GAPDH as a control of equal cytoplasmic protein loading. The data shown are representative of two separate experiments with similar results. B, diagram of GFP and GFP-HNF1α fusion constructs. Human HNF1α coding sequence was subcloned into pAcGFP-C1 vector at the BglII and EcoRI sites to produce the N-terminal GFP-tagged fusion protein. C and D, HEK293 cells or HepG2 cells were transfected with pAc-GFP or pAc-GFP-HNF1α. Two days after transfection, cells were treated with either BTZ or DMSO for 24 h. The subcellular localization of GFP-HNF1α fusion protein was examined with a fluorescence microscope. The pictures shown are representative of three separate experiments with similar results. E, from each well, ∼80–160 GFP-HNF1α-positive HepG2 cells were examined for their subcellular localization and graphed. F, the percentage of transfected HepG2 cells with GFP-HNF1α in cytoplasm was calculated. The values are presented as mean ± S.E. (error bars) of triplicate wells per condition. G, HepG2 cells transfected with pAc-GFP-HNF1α were treated with CHX at 5 μg/ml concentration at the indicated times in the absence or the presence of 40 μm BBR. Total cell lysates were subjected to Western blotting and bands were visualized with antibody against GFP or β-actin. The data shown are representative of two separate experiments with similar results. H, after normalization to β-actin, the GFP-HNF1α signal intensity in G was plotted against the CHX treatment time to calculate t½ of GFP-HNF1α fusion protein.
To further examine the impact of blocking the activity of UPS on HNF1α subcellular localization, we constructed a plasmid, pAc-GFP-HNF1α, to express a GFP-tagged HNF1α fusion protein (Fig. 6B). HEK293 cells were transfected with pAc-GFP-HNF1α or the GFP control vector, and the transfected cells were subjected to BTZ treatment. Fluorescent microscopy revealed that GFP was present in both the cytoplasm and nucleus (Fig. 6C, left) which was in contrast to the GFP-HNF1α that was primarily localized in the nucleus (Fig. 6C, middle). Interestingly, upon BTZ treatment, GFP-HNF1α displayed both a nuclear and cytoplasmic localization pattern in some transfected cells (Fig. 6C, right).
We repeated this transfection experiment in HepG2 cells and consistently observed the enhanced cytoplasmic localization of GFP-HNF1α protein upon BTZ treatment (Fig. 6D). As shown in Fig. 6, E and F, there was a significant increase in GFP detection in the cytoplasm upon BTZ treatment.
We could not apply fluorescent microscopy to detect BBR-induced cytoplasmic accumulation of GFP-HNF1α due to the autofluorescent property of BBR, which caused a high fluorescent background in BBR-treated cells and confounded the GFP signal detection. Thus, we examined the effect of BBR on the protein stability of GFP-HNF1α after transient transfection of pAc-GFP-HNF1α plasmid into HepG2 cells. Fig. 6G is a representative Western blot analysis. Fig. 6H shows the GFP-HNF1α protein decay curves of untreated and BBR-treated samples. Similar to the endogenous HNF1α, BBR treatment caused an accelerated degradation of GFP-HNF1α protein.
BBR Treatment Did Not Globally Affect Proteasome Activities in HepG2 Cells
The 26 S proteasome is a 2.5-MDa multiprotein complex found in both the nucleus and cytosol of all eukaryotic cells and is composed of a single 20 S core particle and 19 S regulatory particles (30). Three major proteolytic activities described as chymotrypsin-like, trypsin-like, and caspase-like are contained within the 20 S core. Together, these three activities are responsible for much of the protein degradation mediated by proteasomes. We considered the possibility that BBR treatment might directly activate the proteasome activity in HepG2 cells, which could be a causal factor for the enhanced turnover rate of HNF1α in BBR-treated cells. To examine this possibility, we treated HepG2 cells with BBR for 4 and 24 h, and the proteasome activities in untreated and BBR-treated cells were assessed by employing cell-based proteasome assays that individually measure the chymotrypsin-like, trypsin-like, or caspase-like protease activity. In parallel, we treated HepG2 cells with proteasome inhibitors BTZ and MG132 as the assay control. Fig. 7 shows that BBR treatments of 4 and 24 h did not affect any of the protease activities. In contrast, the proteasome inhibitors effectively blocked all protease activities. These data suggest that BBR might trigger some signal transduction pathways that lead to the selective degradation of HNF1α protein by proteasome. This possibility requires further investigation.
FIGURE 7.
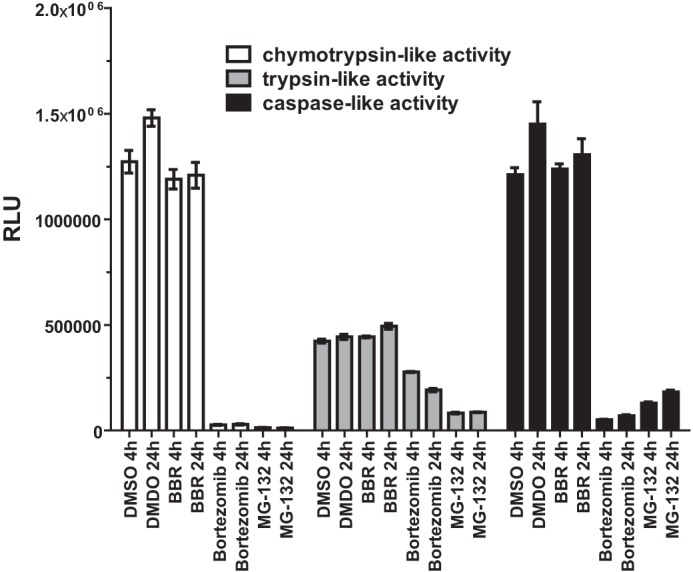
BBR treatment is not associated with altered cellular proteasomal activity. HepG2 cells were treated with BBR (40 μm), BTZ (100 nm), and MG132 (1 μm) for 4 or 24 h. At the indicated treatment time, cells were incubated for 10 min with the Promega Proteasome-Glo cell-based assay reagent (Proteasome-GloTM 3-substrate cell-based assay system). The chymotrypsin-like, trypsin-like activity or caspase-like proteasome activities were detected as the relative light units (RLU) generated from the cleaved substrate in the reaction mixture. The values are presented as mean ± S.E. (error bars). The data shown are representative of two separate experiments with similar results.
DISCUSSION
One important aspect of the PCSK9-LDLR pathway in mediating LDL clearance is that their transcription is coordinately regulated by sterols through a common SRE motif embedded in their gene promoters and is co-induced by current cholesterol lowering drugs, such as statins, through activation of SREBPs (10–12). Statin treatment increases the transcription of both LDLR and PCSK9. The undesirable inducing effect of statins on PCSK9 transcription is increasingly recognized as a major limitation to statin therapeutic efficacy in further lowering plasma LDL-C (31–33). Because the HNF1 binding site is unique to the PCSK9 promoter and is not present in the LDLR promoter, modulations of PCSK9 transcriptions through HNF1 sequence will not affect LDLR gene expression. Thus, the HNF1 binding site represents a divergent point to disconnect the coregulation of PCSK9 with LDLR and other SREBP target genes. Indeed, in this study, we have shown that the natural cholesterol-lowering compound BBR suppressed hepatic Pcsk9 gene expression and reduced serum PCSK9 concentrations in mice without affecting mRNA levels of LDLR and other SREBP target genes. Importantly, in the absence of an increase in gene expression, liver LDLR protein levels in BBR-treated mice were significantly higher than that of control mice, which was convoyed by a 30% reduction of serum LDL-C levels. Analysis of HNF1α mRNA and protein levels in all liver samples clearly demonstrated that BBR attenuated Pcsk9 transcription by reducing the liver content of HNF1α protein without lowering its mRNA expression. These results provided the first in vivo example of down-regulation of HNF1α alone by a lipid-lowering compound and the beneficial impact on plasma LDL-C metabolism through the PCSK9-LDLR pathway. By analyzing liver samples of control and BBR-treated hamsters, we observed essentially the same phenomenon, that BBR lowered Pcsk9 expression by reducing HNF1α protein levels. We further confirmed these in vivo findings by experiments conducted in HepG2 cells, a model human hepatic cell line. Hence, it is likely that a similar cellular mechanism is utilized by BBR to suppress PCSK9 gene expression in liver tissue in vivo and in cultured hepatic cells.
HNF1α is a homeodomain-containing transcription factor (13, 34) that is important for diverse metabolic functions of liver, pancreatic islets, kidney, and intestine (35–37). Although the regulatory network that controls HNF1α gene expression has been well studied (38–40), less is known about the regulation of HNF1α at the protein level. Searching the literature, we found two reports that described the regulation of HNF1α protein. An early study conducted by Park et al. (41) reported that ceramide treatment of H4IIE, a rat hepatocyte-derived cell line, repressed HNF1α protein via the ubiquitin proteasome system, whereas a recent study reported that HCV infection of human hepatoma-derived Huh-7.5 cells resulted in HNF1α protein degradation in lysosomes (26). To determine which protein degradation pathway might be operated by BBR to induce HNF1α protein degradation in HepG2 cells, initially we applied BA1 and BTZ to separately shut down the cellular protein clearance machineries. The cellular content of HNF1α was not affected by BA1 but was markedly elevated by proteasome inhibitors BTZ and MG132. It is noteworthy that the elevated HNF1α protein levels were accompanied by increased PCSK9 and reduced LDLR abundance in BTZ-treated cells. Our results provided the first evidence that the UPS-mediated degradation of HNF1α is directly linked to the PCSK9-LDLR pathway.
BBR treatment consistently lowered HNF1α protein and PCSK9 mRNA and protein levels in HepG2 cells; however, this effect was not observed in the presence of UPS inhibitors. Both MG132 and BTZ showed antagonism to BBR-mediated repression of HNF1α protein. Utilizing BTZ, we further showed that BBR-mediated suppression of PCSK9 promoter activity was largely abolished. By co-transfection of FLAG-tagged HNF1α and Myc-tagged ubiquitin in HEK293 cells, we detected a multiubiquitination ladder pattern of HNF1α, which provided additional evidence suggesting that HNF1α is targeted by the ubiquitin-induced proteasomal degradation for its intracellular clearance. Importantly, by detection of ubiquitinated endogenous HNF1α in HepG2 cells, our study provides strong evidence showing that BBR treatment resulted in an enhanced HNF1α ubiquitination and its proteasomal degradation, which negatively impacted PCSK9 gene transcription and protein abundance in hepatic cells. Possibly, these BBR effects, at least in part, account for the LDL-C-lowering effect of BBR in hypercholesterolemic patients.
The precise mechanism by which BBR induces HNF1α degradation via the UPS is presently unknown. Assessment of proteasome activities in HepG2 cells indicated that BBR treatment did not affect any of the three major proteolytic activities that are contained within the proteasome 20 S core. By analysis of HNF1α subcellular localization, we observed that HNF1α is accumulated in the cytoplasm upon BTZ treatment. Additionally, BTZ treatment totally reversed the reducing effect of BBR on HNF1α in the cytoplasmic fraction, but it only partially reversed the BBR effect on nuclear HNF1α. It is tempting to speculate that BBR might increase HNF1α nuclear export activity, resulting in enhanced degradation in cytoplasm. Verification of this point awaits further experimental examination.
In summary, we have demonstrated that BBR inhibits HNF1α-mediated transactivation of PCSK9 gene expression by down-regulation of hepatic HNF1α protein content in animal models and in cultured cells where BBR treatment accelerated HNF1α protein degradation. This effect can be blocked by proteasome inhibitors that increase intracellular content of HNF1α in untreated cells. Importantly, we demonstrate that blocking the ubiquitin-proteasome pathway in HepG2 cells unfavorably affected the PCSK9-LDLR pathway by increasing PCSK9 and lowering LDLR. Considering that BTZ is currently used in the clinic as an anticancer drug (21, 42) and new proteasome inhibitors are under development for treating patients with certain types of cancer, our findings in this study warrant further investigations in animal models to determine whether proteasome inhibitors would increase HNF1α protein levels in liver tissue and elevate circulating PCSK9 and LDL-C levels.
This work was supported, in whole or in part, by National Institutes of Health, NCCIH, Grants 1R01 AT002543-01A1 and 1R01AT006336-01A1. This work was also supported by the Department of Veterans Affairs (Office of Research and Development, Medical Research Service).
- PCSK9
- proprotein convertase subtilisin/kexin type 9
- BA1
- bafilomycin A1
- BBR
- berberine
- BTZ
- bortezomib
- HNF1α
- hepatocyte nuclear factor 1α
- LDLR
- LDL receptor
- SRE
- sterol regulatory element
- SREBP
- sterol regulatory element-binding protein
- UPS
- ubiquitin proteasome system
- LDL-C
- low density lipoprotein cholesterol
- CHX
- cycloheximide
- IP
- immunoprecipitation
- qPCR and qRT-PCR
- quantitative PCR and RT-PCR, respectively
- Ub
- ubiquitin
- DMSO
- dimethyl sulfoxide.
REFERENCES
- 1. Peterson A. S., Fong L. G., Young S. G. (2008) PCSK9 function and physiology. J. Lipid Res. 49, 1595–1599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Qian Y.-W., Schmidt R. J., Zhang Y., Chu S., Lin A., Wang H., Wang X., Beyer T. P., Bensch W. R., Bensch W. R., Li W., Ehsani M. E., Lu D., Konrad R. J., Eacho P. I., Moller D. E., Karathanasis S. K., Cao G. (2007) Secreted PCSK9 downregulates low density lipoprotein receptor through receptor-mediated endocytosis. J. Lipid Res. 48, 1488–1498 [DOI] [PubMed] [Google Scholar]
- 3. Seidah N. G., Benjannet S., Wickham L., Marcinkiewicz J., Jasmin S. B., Stifani S., Basak A., Prat A., Chretien M. (2003) The secretory proprotein convertase neutal apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. U.S.A. 100, 928–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Seidah N. G. (2009) PCSK9 as a therapeutic target of dyslipidemia. Expert Opin. Ther. Targets 13, 19–28 [DOI] [PubMed] [Google Scholar]
- 5. Zhang D. W., Lagace T. A., Garuti R., Zhao Z., McDonald M., Horton J. D., Cohen J. C., Hobbs H. H. (2007) Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J. Biol. Chem. 282, 18602–18612 [DOI] [PubMed] [Google Scholar]
- 6. McNutt M. C., Kwon H. J., Chen C., Chen J. R., Horton J. D., Lagace T. A. (2009) Antagonism of secreted PCSK9 increases low density lipoprotein receptor expression in HepG2 cells. J. Biol. Chem. 284, 10561–10570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lambert G., Ancellin N., Charlton F., Comas D., Pilot J., Keech A., Patel S., Sullivan D. R., Cohn J. S., Cohn J. S., Rye K. A., Barter P. J. (2008) Plasma PCSK9 concentration correlate with LDL and total cholesterol in diabetic patients and are decreased by fenofibrate treatment. Clin. Chem. 54, 1038–1045 [DOI] [PubMed] [Google Scholar]
- 8. Grefhorst A., McNutt M. C., Lagace T. A., Horton J. D. (2008) Plasma PCSK9 preferentially reduces liver LDL receptors in Mice. J. Lipid Res. 49, 1303–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ling H., Burns T. L., Hilleman D. E. (2014) An update on the clinical development of proprotein convertase subtilisin kexin 9 inhibitors, novel therapeutic agents for lowering low-density lipoprotein cholesterol. Cardiovasc. Ther. 32, 82–88 [DOI] [PubMed] [Google Scholar]
- 10. Horton J. D., Shah N. A., Warrington J. A., Anderson N. N., Park S. W., Brown M. S., Goldstein J. L. (2003) Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc. Natl. Acad. Sci. U.S.A. 100, 12027–12032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dubuc G., Chamberland A., Wassef H., Davignon J., Seidah N. G., Bernier L., Prat A. (2004) Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 24, 1454–1459 [DOI] [PubMed] [Google Scholar]
- 12. Jeong H. J., Lee H.-S., Kim K.-S., Kim Y.-K., Yoon D., Park S. W. (2008) Sterol-dependent regulation of proprotein convertase subtilisin/kexin type 9 expression by sterol-regulatroy element binding protein-2. J. Lipid Res. 49, 399–409 [DOI] [PubMed] [Google Scholar]
- 13. Mendel D. B., Crabtree G. R. (1991) HNF-1, a member of a novel class of dimerizing homeodomain proteins. J. Biol. Chem. 266, 677–680 [PubMed] [Google Scholar]
- 14. Li H., Dong B., Park S. W., Lee H. S., Chen W., Liu J. (2009) Hepatocyte nuclear factor 1α plays a critical role in PCSK9 gene transcription and regulation by the natural hypocholesterolemic compound berberine. J. Biol. Chem. 284, 28885–28895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dong B., Wu M., Li H., Kraemer F. B., Adeli K., Seidah N. G., Park S. W., Liu J. (2010) Strong induction of PCSK9 gene expression through HNF1α and SREBP2: mechanism for the resistance to LDL-cholesterol lowering effect of statins in dyslipidemic hamsters. J. Lipid Res. 51, 1486–1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li H., Liu J. (2012) The novel function of HINFP as a co-activator in sterol-regulated transcription of PCSK9 in HepG2 cells. Biochem. J. 443, 757–768 [DOI] [PubMed] [Google Scholar]
- 17. Kong W., Wei J., Abidi P., Lin M., Inaba S., Li C., Wang Y., Wang Z., Si S., Pan H., Wang S., Wu J., Wang Y., Li Z., Liu J., Jiang J. D. (2004) Berberine is a promising novel cholesterol-lowering drug working through a unique mechanism distinct from statins. Nat. Med. 10, 1344–1351 [DOI] [PubMed] [Google Scholar]
- 18. Cameron J., Ranheim T., Kulseth M. A., Leren T. P., Berge K. E. (2008) Berberine decreases PCSK9 expression in HepG2 cells. Atherosclerosis 201, 266–273 [DOI] [PubMed] [Google Scholar]
- 19. Cao A., Wu M., Li H., Liu J. (2011) Janus kinase activation by cytokine oncostatin M decreases PCSK9 expression in liver cells. J. Lipid Res. 52, 518–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Singh A. B., Kan C. F., Shende V., Dong B., Liu J. (2014) A novel posttranscriptional mechanism for dietary cholesterol-mediated suppression of liver LDL receptor expression. J. Lipid Res. 55, 1397–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thompson J. L. (2013) Carfilzomib: a second-generation proteasome inhibitor for the treatment of relapsed and refractory multiple myeloma. Ann. Pharmacother. 47, 56–62 [DOI] [PubMed] [Google Scholar]
- 22. Wu M., Dong B., Cao A., Li H., Liu J. (2012) Delineation of molecular pathways that regulate hepatic PCSK9 and LDL receptor expression during fasting in normolipidemic hamsters. Atherosclerosis 224, 401–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Astudillo A. M., Pérez-Chacon G., Meana C., Balgoma D., Pol A., Del Pozo M. A., Balboa M. A., Balsinde J. (2011) Altered arachidonate distribution in macrophages from caveolin-1 null mice leading to reduced eicosanoid synthesis. J. Biol. Chem. 286, 35299–35307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dong B., Kan C. F., Singh A. B., Liu J. (2013) High-fructose diet downregulates long-chain acyl-CoA synthetase 3 expression in liver of hamsters via impairing LXR/RXR signaling pathway. J. Lipid Res. 54, 1241–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dong B., Singh A. B., Fung C., Kan K., Liu J. (2014) CETP inhibitors downregulate hepatic LDL receptor and PCSK9 expression in vitro and in vivo through a SREBP2 dependent mechanism. Atherosclerosis 235, 449–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Matsui C., Shoji I., Kaneda S., Sianipar I. R., Deng L., Hotta H. (2012) Hepatitis C virus infection suppresses GLUT2 gene expression via downregulation of hepatocyte nuclear factor 1α. J. Virol. 86, 12903–12911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hershko A. (2005) The ubiquitin system for protein degradation and some of its roles in the control of the cell-division cycle (Nobel lecture). Angew. Chem. Int. Ed. Engl. 44, 5932–5943 [DOI] [PubMed] [Google Scholar]
- 28. Hershko A., Ciechanover A., Varshavsky A. (2000) Basic Medical Research Award. The ubiquitin system. Nat. Med. 6, 1073–1081 [DOI] [PubMed] [Google Scholar]
- 29. Huang L., Chen C. H. (2009) Proteasome regulators: activators and inhibitors. Curr. Med. Chem. 16, 931–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Careskey H. E., Davis R. A., Alborn W. E., Troutt J. S., Cao G., Konrad R. J. (2008) Atovastatin increases human serum levels of proprotein convertase subtilisin/kexin type 9. J. Lipid Res. 49, 394–398 [DOI] [PubMed] [Google Scholar]
- 31. Raal F., Panz V., Immelman A., Pilcher G. (2013) Elevated PCSK9 levels in untreated patients with heterozygous or homozygous familial hypercholesterolemia and the response to high-dose statin therapy. J. Am. Heart Assoc. 2, e000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Costet P., Hoffmann M. M., Cariou B., Guyomarc'h Delasalle B., Konrad T., Winkler K. (2010) Plasma PCSK9 is increased by fenofibrate and atorvastatin in a non-additive fashion in diabetic patients. Atherosclerosis 212, 246–251 [DOI] [PubMed] [Google Scholar]
- 33. Welder G., Zineh I., Pacanowski M. A., Troutt J. S., Cao G., Konrad R. J. (2010) High-dose atorvastatin causes a rapid sustained increase in human serum PCSK9 and disrupts its correlation with LDL cholesterol. J. Lipid Res. 51, 2714–2721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cereghini S. (1996) Liver-enriched transcription factors and hepatocyte differentiation. FASEB J. 10, 267–282 [PubMed] [Google Scholar]
- 35. Shih D. Q., Bussen M., Sehayek E., Ananthanarayanan M., Shneider B. L., Suchy F. J., Shefer S., Bollileni J. S., Gonzalez F. J., Breslow J. L., Stoffel M. (2001) Hepatocyte nuclear factor-1α is an essential regulator of bile acid and plasma cholesterol metabolism. Nat. Genet. 27, 375–382 [DOI] [PubMed] [Google Scholar]
- 36. Pontoglio M., Barra J., Hadchouel M., Doyen A., Kress C., Bach J. P., Babinet C., Yaniv M. (1996) Hepatocyte nuclear factor 1 inactivation results in hepatic dysfunction, phenylketonuria, and renal Fanconi syndrome. Cell 84, 575–585 [DOI] [PubMed] [Google Scholar]
- 37. Shih D. Q., Screenan S., Munoz K. N., Philipson L., Pontoglio M., Yaniv M., Polonsky K. S., Stoffel M. (2001) Loss of HNF-1α function in mice leads to abnormal expression of genes involved in pancreatic islet development and metabolism. Diabetes 50, 2472–2480 [DOI] [PubMed] [Google Scholar]
- 38. Costa R. H., Kalinichenko V. V., Holterman A. X., Wang X. (2003) Transcription factors in liver development, differentiation, and regeneration. Hepatology 38, 1331–1347 [DOI] [PubMed] [Google Scholar]
- 39. Armendariz A. D., Krauss R. M. (2009) Hepatic nuclear factor 1-α: inflammation, genetics, and atherosclerosis. Curr. Opin. Lipidol. 20, 106–111 [DOI] [PubMed] [Google Scholar]
- 40. Pontoglio M., Pausa M., Doyen A., Viollet B., Yaniv M., Tedesco F. (2001) Hepatocyte nuclear factor 1α controls the expression of terminal complement genes. J. Exp. Med. 194, 1683–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Park I. N., Cho I. J., Kim S. G. (2004) Ceramide negatively regulates glutathione S-transferase gene transactivation via repression of hepatic nuclear factor-1 that is degraded by the ubiquitin proteasome system. Mol. Pharmacol. 65, 1475–1484 [DOI] [PubMed] [Google Scholar]
- 42. Yoshizawa K., Mukai H. Y., Miyazawa M., Miyao M., Ogawa Y., Ohyashiki K., Katoh T., Kusumoto M., Gemma A., Sakai F., Sugiyama Y., Hatake K., Fukuda Y., Kudoh S. (2014) Bortezomib therapy-related lung disease in Japanese patients with multiple myeloma: incidence, mortality and clinical characterization. Cancer Sci. 105, 195–201 [DOI] [PMC free article] [PubMed] [Google Scholar]