

Background: Oligomers of protein Tau are associated with neurodegenerative diseases.
Results: New antibodies were generated and validated that recognize different degrees of oligomerization of protein Tau.
Conclusion: Low order and higher order oligomers differ in C-terminal Tau phosphorylation and reflect consecutive stages in disease progression.
Significance: Antibodies recognizing Tau oligomers provide insight into disease etiology and are promising diagnostic tools.
Keywords: Alzheimer Disease, Antibody, Protein Folding, Tau Protein (Tau), Yeast, Protein Oligomer
Abstract
A link between Tau phosphorylation and aggregation has been shown in different models for Alzheimer disease, including yeast. We used human Tau purified from yeast models to generate new monoclonal antibodies, of which three were further characterized. The first antibody, ADx201, binds the Tau proline-rich region independently of the phosphorylation status, whereas the second, ADx215, detects an epitope formed by the Tau N terminus when Tau is not phosphorylated at Tyr18. For the third antibody, ADx210, the binding site could not be determined because its epitope is probably conformational. All three antibodies stained tangle-like structures in different brain sections of THY-Tau22 transgenic mice and Alzheimer patients, and ADx201 and ADx210 also detected neuritic plaques in the cortex of the patient brains. In hippocampal homogenates from THY-Tau22 mice and cortex homogenates obtained from Alzheimer patients, ADx215 consistently stained specific low order Tau oligomers in diseased brain, which in size correspond to Tau dimers. ADx201 and ADx210 additionally reacted to higher order Tau oligomers and presumed prefibrillar structures in the patient samples. Our data further suggest that formation of the low order Tau oligomers marks an early disease stage that is initiated by Tau phosphorylation at N-terminal sites. Formation of higher order oligomers appears to require additional phosphorylation in the C terminus of Tau. When used to assess Tau levels in human cerebrospinal fluid, the antibodies permitted us to discriminate patients with Alzheimer disease or other dementia like vascular dementia, indicative that these antibodies hold promising diagnostic potential.
Introduction
The microtubule-associated protein Tau is an “intrinsically disordered protein,” which undergoes posttranslational modifications like phosphorylation, glycosylation, truncation, oxidation, and acetylation (1–3). During Alzheimer disease (AD),5 a large pool of protein Tau becomes abnormally hyperphosphorylated, resulting in the loss of microtubule (MT) stabilization and the induction of conformational changes that allow the protein to oligomerize and aggregate into paired helical filaments (PHF) and neurofibrillary tangles (NFT) (4). The close correlation between the Tau pathology and the disease progression makes protein Tau a good biomarker for diagnosis. The protein is released from neurons into cerebrospinal fluid (CSF), and, indeed, several studies established that AD patients display CSF changes with elevated levels of total Tau and phosphorylated Tau, next to decreased levels of the amyloid β 42 peptide. Hence, the quantification of these parameters proved to be accurate for early diagnosis of AD (5, 6).
Data obtained from conditional Tau expression in transgenic mice point toward oligomeric Tau intermediates as toxic species and effectors in the etiology of AD (7). As such, recently developed oligomer-specific antibodies, like T22 (8) or TOC1 (9, 10), have been shown to preferentially label neurons at early stages of AD before the formation of NFT. Moreover, these antibodies also improved the discrimination between AD and control patients using total brain extracts, indicating their potential use as a pathophysiological biomarker (8–10). Most recent studies demonstrated that oligomers detecting antibodies can block Tau seeding activity and the propagation of Tau pathology in brains of transgenic mice when administered intracerebroventricularly, indicative that such antibodies have promising therapeutic potential (11, 12).
During the last decade, we and others reported on the use of Saccharomyces cerevisiae, or baker's yeast, as a valuable model organism to study protein aggregation disorders, such as Huntington and Parkinson diseases as well as AD (13). For the latter, this included not only toxicity models for the amyloid β peptides (14, 15) but also models based on expression of human wild-type Tau or its clinical mutants (16–19). These so-called humanized yeast models recapitulated robustly different important aspects of the Tau pathobiology associated with AD pathology, including Tau hyperphosphorylation, conformational change, and Tau self-aggregation (18, 19). Given the ease of genetic amenability of yeast, it was possible to modify major pathogenic phosphoepitopes on Tau, like the AD2 (Ser(P)396/Ser(P)404) and PG5 (Ser(P)409) epitopes, simply by deletion of the genes encoding for the protein kinases Mds1 and Pho85, the yeast orthologues of the two most important mammalian Tau kinases (i.e. GSK3-β and Cdk5, respectively). Importantly, whereas the lack of Mds1 coincides with reduced Tau phosphorylation at typical GSK3-β phosphoepitopes, the deletion of Pho85 triggers Tau hyperphosphorylation due to the secondary activation of different protein kinases, including Mds1/GSK3-β (18). Moreover, also in yeast, the phosphorylation status of protein Tau correlates with its immunoreactivity to the conformation-specific monoclonal antibody (mAb) MC1 and the amount of Sarkosyl-insoluble Tau, whereas an inverse correlation is found between Tau phosphorylation and its ability to bind and stabilize MT in vitro. In addition, purification of phosphorylated protein Tau from the yeast Δpho85 strain revealed spontaneous filament formation without the addition of anionic aggregation-inducing agents as well as the capacity of the hyperphosphorylated subfraction to drastically accelerate Tau aggregation in vitro (18). Notably, crude extracts and purified Tau preparations from humanized yeast contained higher molecular weight species, which, based on their apparent molecular weight, were tentatively identified as dimers and higher order oligomers (17, 19).
In this study, we used the hyperphosphorylated protein Tau isolated from the Δpho85 yeast strain to generate novel high affinity Tau mAbs. Their characterization and validation in different model systems and AD brains indicated that the mAbs stained tangle-like structures and neuritic plaques in brain sections and recognized either low order or higher order Tau oligomers and presumed prefibrillar structures besides different monomeric Tau isoforms in protein extracts from diseased brain. This revealed that Tau oligomerization occurs early in the disease process. Furthermore, the novel mAbs proved to be valuable diagnostic tools, allowing the discrimination of patients clinically diagnosed with AD or vascular dementia from control persons based on immunodetection of total Tau in CSF samples.
EXPERIMENTAL PROCEDURES
Yeast Strains, Culture Conditions, and Tau Purification
Yeast strains were obtained from the genome-wide yeast deletion collection and grown according to standard procedures on glucose-containing selective medium. Constructs and protocols for the expression of the longest human Tau isoform (Tau-2N/4R; 441 amino acids (aa)) were as described previously (17–19). The Y18E point mutation was introduced in Tau using the QuikChange II XL site-directed mutagenesis kit (Agilent, Diegem, Belgium) using the forward and reverse mutagenesis primers 5′-TCACGCTGGGACGGAGGGGTTGGGGGACA-3′ and 5′-TGTCCCCCAACCCCTCCGTCCCAGCGTGA-3′. Native protein Tau-2N/4R was purified from the Δpho85 yeast strain as reported earlier (18), concentrated using 50 kDa Centricon filters (Millipore, Overijse, Belgium), and dialyzed overnight against PBS buffer (137 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, and 2 mm KH2PO4, pH 7.0) at 4 °C. For immunizations, the purified Tau was activated with glutaraldehyde and coupled to keyhole limpet hemocyanin via a two-step enamine covalent coupling to generate keyhole limpet hemocyanin-coupled Tau. For the dephosphorylation studies, purified Tau extracted from the Δpho85 strain was treated with shrimp alkaline phosphatase (AP) (Roche Applied Science) according to the manufacturer's recommendations (17, 18).
Immunizations and Hybridoma Generation
After determination of the base titer via a preimmune tail bleeding, eight BALB/c mice were immunized with 5 μg of keyhole limpet hemocyanin-coupled Tau to start the immune response, followed by a monthly check-up and four booster immunizations using 5 μg of purified Tau in complete Freund's adjuvant, incomplete Freund's adjuvant (Lonza, Verviers, Belgium), and PBS, respectively. Titers were determined after the incomplete Freund's adjuvant immunization via tail bleeding. The spleen of the mouse with the highest titer was removed, washed in PBS + 0.5% BSA + 2.5 mm EDTA, and dissociated using the Gentle MACS dissociator T4171 until single cells were obtained. Cells were collected by centrifugation at 1000 rpm (228 × g) for 10 min at room temperature, and resuspended in ammonium/chloride/potassium lysis buffer (Lonza) for 5 min at room temperature in order to remove red blood cells. Hemolysis was stopped by adding PBS + 0.5% BSA + 2.5 mm EDTA. The remaining cells were collected by centrifugation at 1000 rpm (228 × g) for 10 min at room temperature and resuspended in PBS. Spleen cells were added to SP2/0 myeloma cells in a 1:1 ratio. The cell mixture was collected by centrifugation at 1000 rpm (228 × g) for 5 min at 4 °C. Cell fusion was performed using the HVJ viral fusion kit as described previously (20). The resulting cells were sown out over 36 96-well plates and selected using hypoxanthine-aminopterin-thymidine medium.
Antibody Screening
Antibodies were selected using a sandwich ELISA setup using purified Tau-2N/4R from yeast or bacterial recombinant Tau as coated antigen (0.1 μg/ml). Detection was performed using goat anti-mouse coupled to horseradish peroxidase (GAM-HRP; 0.05 μg/ml) (Jackson, Suffolk, UK), urea hydrogen peroxide (Sigma-Aldrich, Diegem, Belgium), and 3,3′,5,5′-tetramethylbenzidine (VWR, Haasrode, Belgium). Plate reading was done in a BioTek SL800 spectrophotometer (BioTek, Potton, UK) at 450 nm.
Biotinylation of Monoclonal Antibodies
For biotinylation, the antibodies were dialyzed overnight at 4 °C to boric acid buffer (113 mm, pH 8.6) in Slide-a-Lyzer cassettes with a molecular mass cut-off of 10 kDa (Thermo Fisher Scientific, Erembodegem, Belgium). After dialysis, all antibodies were diluted to a concentration of 0.5 mg/ml. Sulfo-NHS-LC-biotin (Pierce Thermo Fisher, Erembodegem, Belgium) was added to the antibodies at 32:1, 64:1, and 128:1 biotin/antibody ratios and incubated for 1 h, shaking (400 rpm) at room temperature. The reaction was stopped using an equal volume of 1 m glycine and again incubated for 1 h, shaking (400 rpm) at room temperature. The biotinylated antibodies were then dialyzed to PBS overnight at 4 °C.
ELISA on Biopeptides
For ELISA using biopeptides (Proteogenix, Strasbourg, France), Nunc 96-well plates (VWR, Haasrode, Belgium) were coated with streptavidin (Roche, Vilvoorde, Belgium) at a concentration of 5 μg/ml overnight at 4 °C. The plates were subsequently blocked with a PBS + 0.5% casein buffer solution. Biotinylated peptides were loaded onto the coated plates with a starting concentration of 100,000 pg/ml in PBS followed by a serial dilution of 1:4. The plate was incubated at 37 °C for 1 h. The wells were washed four times for 1 min with PBS + 0.05% Tween 20. The assay was then incubated with biotinylated antibodies ADx215 and ADx201 (0.125 μg/ml in PBS + 0.1% Tween 20) for 1 h at room temperature. Detection was performed using GAM-HRP, urea hydrogen peroxide and 3,3′,5,5′-tetramethylbenzidine. The plates were scanned using a BioTek SL800 spectrophotometer (BioTek) at 450 nm (and 630 nm for background). The peptide sequences are given in Tables 1 and 2.
TABLE 1.
Synthetic peptides used for epitope mapping and phosphodependence studies
| Specificity | Sequence |
|---|---|
| Tyr18 | MAEPRQEFEVMEDHAGTYGLGDRK |
| Tyr(P)18 | MAEPRQEFEVMEDHAGTpYGLGDRK |
| Y18E | MAEPRQEFEVMEDHAGTEGLGDRK |
| Y18F | MAEPRQEFEVMEDHAGTFGLGDRK |
| Thr217/Tyr18 | GSRSRTPSLPTPPTREPKKVAGEFEVMEDHAGTYGLGDRK |
| Thr(P)217/Tyr18 | GSRSRTPSLPpTPPTREPKKVAGEFEVMEDHAGTYGLGDRK |
TABLE 2.
Peptide scanning results for ADx215 epitope determination
| Start | Stop | Sequence | milli-A450 nm |
|---|---|---|---|
| 6 | 20 | QEFEVMEDHAGTYGL | 50 |
| 7 | 21 | EFEVMEDHAGTYGLG | 97 |
| 8 | 22 | FEVMEDHAGTYGLGD | 141 |
| 9 | 23 | EVMEDHAGTYGLGDR | 911 |
| 10 | 24 | VMEDHAGTYGLGDRK | 2055 |
| 11 | 25 | MEDHAGTYGLGDRKD | 2161 |
| 12 | 26 | EDHAGTYGLGDRKDQ | 2100 |
| 13 | 27 | DHAGTYGLGDRKDQG | 2082 |
| 14 | 28 | HAGTYGLGDRKDQGG | 2387 |
| 15 | 29 | AGTYGLGDRKDQGGY | 2434 |
| 16 | 30 | GTYGLGDRKDQGGYT | 2229 |
| 17 | 31 | TYGLGDRKDQGGYTM | 1364 |
| 18 | 32 | YGLGDRKDQGGYTMH | 171 |
| 19 | 33 | GLGDRKDQGGYTMHQ | 60 |
| 20 | 34 | LGDRKDQGGYTMHQD | 49 |
| 21 | 35 | GDRKDQGGYTMHQDQ | 49 |
NMR-based Epitope Mapping
15N-Labeled Tau-2N/4R was prepared as described previously (21). The protein was prepared without His tag; thus, purification after an initial heating step of the bacterial extract at 75 °C for 15 min involved one further step of cation exchange chromatography (using an SPFF column; GE Healthcare). The sampling was desalted on a G25 Superdex column in a 50 mm ammonium bicarbonate buffer before estimation of the concentration by absorbance at 280 nm and subsequent lyophilization of 1-mg aliquots. A sample of 30 μm Tau-2N/4R in 200 μl of buffer (100 mm phosphate, pH 6.6, 75 mm NaCl, 1 mm DTT, 5% D2O) was the reference, and equivalent samples of Tau-2N/4R with 30 μm ADx215 or ADx201 were prepared. The 1H,15N heteronuclear single-quantum correlation NMR spectrum was recorded on a 900-MHz Bruker Avance III spectrometer equipped with a cryogenic triple resonance probe head. The spectra were acquired with 4096 × 512 complex points in T2, T1 with 16 scans/increment. Total time for an experiment was 2 h 50 min. Spectra were zero-filled and transformed after multiplication with a squared sine bell apodization function. Signal intensities were determined with Bruker Topspin version 3.1 software.
Determination of the Antibody Affinity
A BIAcore 3000 instrument (GE Healthcare) was used at 25 °C with a BIAcore CM5 sensor mounted into the system. The sensor was preconditioned by a 1-min injection at 100 μl/min of 0.1% SDS, 50 mm NaOH, 10 mm HCl, and 100 mm H3PO4. As a running buffer, HBS-EP buffer was used (10 mm HEPES (pH 7.4), 150 mm NaCl, 1 mm EDTA, 0.05% (w/v) P20). The sample buffer was the system buffer supplemented with 1 mg/ml carboxymethyldextran (Sigma). An antibody capture system was established on the sensor surface. 6500 relative units of Fcγ-fragment rabbit anti-mouse IgG (GE Healthcare) were immobilized according to the manufacturer's instructions using EDC/NHS chemistry on all four flow cells. The sensor was deactivated using 1 m ethanolamine. Antibodies were captured at 35 nm concentration by a 1-min injection at 10 μl/min on the flow cells 2 and 3. Flow cell 1 served as a reference. The flow rate was set to 100 μl/min, and the 45-kDa Tau analyte in solution was injected for 2 min. The dissociation was monitored for 5 min. Analytes in solution were injected for 2 min in a concentration series of 0, 3, 11, 2 × 33, 90, and 180 nm. The dissociation was monitored for 5 min. The sensor was regenerated by three consecutive injections of 10 mm glycine buffer, pH 1.7, for 1 min at 100 μl/min. ADx201 showed a biphasic binding behavior for Tau-2N/4R. A four-parameter sigmoidal model (22) was used to describe an apparent affinity. ADx215 showed a single exponential sensorgram curvature and was evaluated according to a Langmuir fit. Because ADx215 showed a too high complex stability beyond the instrument limits of resolution, the dissociation rate, Kd, was set to 1.0E−05/s.
Immunoblotting and Immunohistochemistry
Samples for immunoblotting were prepared and processed as described previously (18, 23). Bacterial recombinant His-tagged human Tau-2N/4R (Merck Millipore (Nottingham, UK) and rPeptide (Bogart, GA)) was used as standard in the initial yeast-based assays. All antibodies used for detection of Tau or reference proteins are listed in Table 3. Dephosphorylation of Tau in yeast extracts was performed using alkaline phosphatase as described previously (17). For immunohistochemistry, we used free floating coronal sections of 40 μm for THY-Tau22 transgenic mice (23) and human hippocampal sections and paraffin-embedded sections of 7 μm in the case of human cortex. Human brain samples were obtained from the brain bank of University Hospital in Lille (Table 4). Protocols for tissue treatment, the preparation of the sections, and the immunostainings were described previously (23, 24). The phospho-dependent Tau antibody AT8 (Ser(P)202/Thr(P)205 Tau) (Pierce) served as a reference antibody for immunohistochemistry.
TABLE 3.
Antibodies used in these studies
| Antibody | Specificity | Source |
|---|---|---|
| Primary Abs | ||
| Actin (AC-18) | β-Actin | Sigma-Aldrich (Diegem, Belgium) |
| 9G3/P-Tyr18 | Tau; Tyr(P)18 | Medimabs (Montréal, Canada) |
| 12E8 | Tau; Ser(P)262/Ser(P)356 | Elan Pharmaceuticals (San Francisco, CA) |
| AD2 | Tau; Ser(P)396/Ser(P)404 | Sigma-Aldrich |
| AT8 | Tau; Ser(P)202/Thr(P)205 | Innogenetics (Ghent, Belgium) |
| AT180 | Tau; Thr(P)231 | Innogenetics |
| AT270 | Tau; Thr(P)181 | Innogenetics |
| P-S396 | Ser(P)396 | Merck-Millipore (Overijse, Belgium) |
| BT2 | Tau; aa 193–198 | Innogenetics |
| HT7 | Tau; aa 159–163 | Innogenetics |
| M19G | Tau; aa 1–19 | A. Delacourte (34) |
| MC1 | Tau; aa 5–15/312–322; conformation-dependent | Generous gift from P. Davies (59) |
| TauCter | Tau; aa 426–441 | A. Delacourte (34) |
| TAU1 | Tau; Ser198/Ser199/Ser202 | Chemicon (Temecula, CA) |
| TAU5 | Tau; aa 218–225 | BD Pharmingen (San Diego, CA) |
| TOC1 | Tau; aa 155–244/376–421; oligomer-specific | Generous gift from L. Binder (9) |
| ADx201 | Tau; aa 218–224 | Generated in this study |
| ADx201-bio | Tau; aa 218–224 | Generated in this study |
| ADx215 | Tau; aa 16–24 | Generated in this study |
| ADx215-bio | Tau; aa 16–24 | Generated in this study |
| ADx210 | Tau; conformational epitope | Generated in this study |
| ADx210-bio | Tau; conformational epitope | Generated in this study |
| Secondary Abs | ||
| GAM-HRP | Anti-mouse mAb | Thermo Fisher Scientific (Erembodegem, Belgium) |
| GAM-Alexa594 | Anti-mouse mAb | Abcam (Cambridge, UK) |
| RAMa-HRP | Anti-mouse mAb | Thermo Fisher Scientific |
a Rabbit anti-mouse.
TABLE 4.
Human samples used for validation of the mAbs
| Code | Age | Sex | PMIa | Pathology | Braak stage |
|---|---|---|---|---|---|
| years | h | ||||
| C49 | 49 | Male | 34 | None | |
| C52 | 52 | Female | 28 | None | |
| C53 | 53 | Male | 28 | None | |
| C61 | 61 | Male | 20 | None | |
| C75 | 75 | Male | 7 | None | |
| AD84 | 84 | Male | 18 | AD | 4 |
| AD91 | 91 | Female | 18 | AD | 4 |
| AD62 | 62 | Male | 10 | AD | 5 |
| AD73 | 73 | Male | 15 | AD | 5 |
| AD76 | 76 | Female | 3 | AD | 5 |
| AD77 | 77 | Male | 6 | AD | 5 |
| AD57 | 57 | Male | 23 | AD | 6 |
| AD59 | 59 | Female | 26 | AD | 6 |
| AD72 | 72 | Female | 5.5 | AD | 6 |
| AD73B | 73 | Female | 22 | AD | 6 |
| AD74 | 74 | Male | 4 | AD | 6 |
| AD76B | 76 | Male | 7 | AD | 6 |
| AD86 | 86 | Female | 10 | AD | 6 |
| AD86B | 86 | Male | 10 | AD | 6 |
a Post-mortem interval.
In Situ Immunodetection in Yeast Cells
Exponentially growing pho85Δ yeast cells expressing human Tau 2N/4R were fixed by adding formaldehyde at a final concentration of 4% and incubated for 15 min at room temperature. Cells were then collected and suspended in 0.1 m phosphate buffer, pH 6.5. Formaldehyde was added a second time to a final concentration of 4%. The suspension was incubated at 30 °C for 2 h. Cells were then washed in phosphate buffer and then in PBSorb (phosphate buffer containing 1.2 m sorbitol). For spheroplasting, cells were immersed in PBsorb containing β-mercaptoethanol and lyticase (Sigma-Aldrich), incubated for 40 min at 30 °C, and then collected by centrifugation at low speed (200 × g). Next, spheroplasts were added onto a polylysine slide (Menzel-Gläser, Braunschweig, Germany) and incubated for 10 min at room temperature in a humidified chamber. The liquid was then drained, and the slides were left to dry. The samples were subsequently submerged in methanol and acetone (each for 6 min 30 s at −20 °C) and blocked with PBSB (phosphate buffer containing BSA at 10 mg/ml) for 30 min. After washing, solutions of PBSB containing antibodies TAU5, TOC1, and MC1 at a concentration of 1:200 were added, and the slides were incubated overnight at 4 °C. The day after, the samples were washed with PBSB and dried at room temperature in the dark. Finally, a solution of PBSB containing the secondary antibody GAM-Alexa594 (Molecular Probes, Inc., concentration 1:200) was added, and the slides were incubated for an additional 2 h in the dark. In the last step, samples were washed, and mounting medium was added. At any critical step, quality of the spheroplast was checked by microscopy. Analysis was performed with a fluorescence microscope (model DFC420C, Leica).
CSF Samples and ELISA Detection Total Tau
Cerebrospinal fluid (CSF) from patients recruited in the Memory Clinic and the Department of Neurology of Hospital Network Antwerp (ZNA) were selected from the Biobank of the Institute Born-Bunge (Antwerp, Belgium). Patients with AD (n = 20), vascular dementia (VAD; n = 20), and controls (n = 20) were included (Table 5). Patients with dementia due to AD were diagnosed according to the NIA-Alzheimer's Association criteria (25). VAD was diagnosed according to the NINDS-AIREN criteria (26). The inclusion criteria for the control group were: 1) no neurological or psychiatric antecedents and 2) no organic disease involving the central nervous system following extensive clinical examination. The study was approved by the local ethics committee. CSF was obtained by lumbar puncture (lateral posterior nucleus of the thalamus) at the L3/L4 or L4/L5 interspace. CSF samples were collected in polypropylene vials (Nalgene® catalog no. 5000-1020), immediately frozen in liquid nitrogen, and subsequently stored at −80 °C until analysis.
TABLE 5.
Summary of the demographic, clinical, and biomarker data of the population used for CSF-based ELISA
| AD | VAD | Control | |
|---|---|---|---|
| n (female/male) | 20 (7/13) | 20 (11/9) | 20 (11/9) |
| Average age (years) (range) | 81 (62–97) | 77 (56–87) | 48 (25–85) |
| MMSEa score (range) | 13.5 (4–27) | 15 (0–26) | NAb |
| Age at onset (years) (range) | 79 (60–95) | 75 (56–85) | NA |
| Disease duration (years) (range) | 2 (1–7) | 2 (0–9) | NA |
| Median CSF-Tau (pg/ml) (quartiles)c | 596 (457–881)*** | 391 (305–629)* | 271 (178–355) |
a Minimal mental state examination (score of 30).
b NA, not available.
c CSF-Tau data are presented as median values with 25th and 75th quartiles because these data are not normally distributed. A Kruskall-Wallis test was used to compare groups separately.
*, p < 0.05 versus control;
***, p < 0.001 versus control.
For the CSF total Tau ELISA assay we used a peptide comparable with the peptide Thr217/Tyr18 (Table 1) as calibrator and mixed 25 μl of CSF sample or calibrator with 100 μl of sample diluent (0.1% casein in PBS, 0.2% Triton X-705 (Sigma T70570)) containing the biotinylated detector antibody, ADx215bio. ADx201 was coated as a capture antibody on the plate. Plates were washed five times with 400 μl/well wash buffer (0.05% Tween (Merck S6299684) in PBS) after the 3-h sample incubation at room temperature. Detection of the biotinylated antibody was done with a ready-to-use streptavidine-peroxidase complex (SA-PolyHRP80, SDT GmbH SP80C), and after another five washes, color development was done at room temperature for 30 min. The enzymatic reaction was stopped with 100 μl of stop solution (0.5 n H2SO4), and optical density was measured at 450 and 630 nm for background. Conversion of OD values to pg/ml was done using a four-parameter logistic curve fitting using the values assigned to the calibrator.
Lipid Raft Isolation
The procedure for lipid raft isolation has been described previously (27). Briefly, cortex homogenates were prepared in TBS containing 1% Triton X-100, phosphatase inhibitors (1 mm Na3VO4, 0.1 μg/ml okadaic acid), and a protease inhibitor mix (Roche Applied Science) and then incubated on ice for 30 min. The homogenates were centrifuged at 1000 × g for 10 min, and supernatants were collected. The total protein concentrations were then determined by using the BCA protein assay kit (Pierce). A total amount of 1 mg of protein extract was taken for fractionation and mixed with a sucrose solution to reach 40% concentration (m/v) and then overlaid with 6 ml of 30% sucrose (m/v) and subsequently with 1.3 ml of 5% sucrose (m/v). The samples were centrifuged at 100,000 × g for 18 h at 4 °C in SW60 rotor (Beckman Coulter, Brea, CA). Eight equal fractions were taken, and every fraction was precipitated by adding 100% trichloroacetic acid. Samples were centrifuged and protein pellets were suspended in 2× SDS-PAGE buffer. Equal volumes of particular fractions were analyzed by SDS-PAGE and Western blotting.
RESULTS
Generation and Characterization of the Novel mAbs
The expression of human Tau (longest isoform, 2N/4R; 441 aa) in yeast has been characterized extensively before, thereby showing robust traits found in AD (17–19). In this study, we focused mainly on expression of Tau in the pho85Δ strain, which lacks the yeast orthologue of Cdk5 and displays extensive hyperphosphorylation of protein Tau and higher levels of Sarkosyl-insoluble Tau as compared with the BY4741 wild type strain (18). Using in situ immunodetection with the pan-antibody TAU5, we now show that human Tau is not evenly distributed throughout the cytoplasm in pho85Δ cells as the protein forms diffuse foci. With the mAbs MC1 and TOC1, we confirm that also in yeast protein, Tau does adopt an aggregation-prone conformation and forms prefibrillar oligomers (Fig. 1A). For the generation of novel mAbs, human Tau was purified from the Δpho85 strain using anion exchange chromatography.
FIGURE 1.

In situ immunodetection of Tau and characterization of novel monoclonal antibodies in yeast. A, in situ immunodetection of total Tau (TAU5), the pathogenic Tau conformation (MC1), and oligomeric Tau (TOC1) in spheroplasts of the BY4741 wild-type strain expressing human Tau-2N/4R. Scale bar, 2 μm. B, Western blot analysis of protein extracts made from the BY4741 wild-type strain with the empty vector (lane 1), the BY4741 wild type strain expressing Tau-2N/4R (lane 2), the Δmds1 strain expressing 2N/4R Tau (lane 3), and the Δpho85 strain expressing human Tau-2N/4R (lane 4). TAU5 was used as a control antibody. Exposure times were 10 s for ADx215, 2 min for ADx201 and ADx210, and 1 min for TAU5. The open arrows indicate oligomeric Tau as detected by ADx210 and ADx215. The inset for ADx210 shows that this mAb also recognizes monomeric Tau at longer exposure times. C, Western blot analysis of AP-treated samples. Lane 1, extracts of the Δpho85 strain transformed with the empty vector. Lane 2, untreated extracts of the Δpho85 strain expressing Tau; lanes 3–6, the same extract but treated with SAP for 30, 60, 90, and 120 min, respectively.
After ELISA-based selection of the hybridomas, the newly generated Tau-mAbs were further characterized using different methods. As evidenced by SDS-PAGE and Western blot analysis of total protein extracts from the humanized BY4741 wild type yeast strain and the isogenic Δmds1 and Δpho85 deletion mutants (Fig. 1B), two of the mAbs (i.e. ADx201 and ADx215) recognized monomeric Tau phosphoisoforms in all yeast extracts and provided a banding pattern in the 65–75 kDa molecular mass region that was comparable with that obtained with TAU5, which was used as a control. Both antibodies also recognized non-phosphorylated bacterial recombinant His-tagged human Tau. Interestingly, ADx215 reacted as well to Tau oligomers found at 100 kDa and above (Fig. 1B). These oligomers were observed at similar levels in extracts of the wild type strain and the Δmds1 or Δpho85 kinase deletion mutants, indicative that their formation is not dependent on the hyperphosphorylation of Tau. Another mAb (i.e. ADx210) had weaker affinity for monomeric Tau isoforms in the extracts of wild type yeast and the Δmds1 mutant, where its detection required overexposure (Fig. 1B). In contrast, in the extract of the Δpho85 strain, ADx210 displayed strong reactivity toward monomeric hyperphosphorylated Tau as well as Tau oligomers, thereby suggesting that the formation of these oligomers would require Tau hyperphosphorylation. In line with this, ADx210 did not react to bacterial recombinant Tau, which is not phosphorylated.
To confirm differences in phosphorylation dependence of the generated mAbs, we treated the protein extracts of the Δpho85 strain with AP for different time intervals and again checked the immunoreactivity but with lower exposure times in order to be able to distinguish the different monomeric phosphoisoforms. As shown for ADx201 and ADx215 (Fig. 1C), the AP treatment resulted in the disappearance of the highest monomeric Tau phosphoisoforms, seen on Western blot at an approximate molecular mass around 75 kDa and in an increase of less phosphorylated isoforms running at lower molecular mass. Similar to the pan-Tau antibody TAU5, the overall immunoreactivity of ADx201 or ADx215 remained equal in all samples examined. In contrast, an increased immunoreactivity was observed after prolonged AP treatment with the commercially available antibody BT2, specifically recognizing non-phosphorylated protein Tau (28), whereas a gradually decreased immunoreactivity was seen with the Thr181 phosphospecific antibody AT270 (29). Hence, these data suggest that ADx201 and ADx215 do not have phosphodependence.
Although AP treatment resulted in the appearance of intermediate bands running between 75 and 85 kDa, most of the Tau oligomers detected with ADx215 were resistant to AP treatment and persisted even after 2 h of treatment at 37 °C. In contrast, the Tau oligomers detected with ADx210 steadily diminished upon prolonged AP treatment, and, interestingly, this coincided with the appearance of monomeric Tau isoforms. The observation of a gradual decrease of ADx210-positive oligomeric Tau upon dephosphorylation corroborates a link between Tau phosphorylation and Tau self-assembly. Moreover, these data nicely illustrate that oligomers detected by ADx210 have different properties than those detected by ADx215. Note, however, that there is no inverse correlation with the disappearance of oligomers and the appearance of the monomeric forms, as detected by ADx210, whereas such correlation is found for the appearance of non-phospho-Tau based on staining with BT2. This suggests that monomeric Tau detected by ADx210 still has some degree of phosphorylation and thus that the ADx210 epitope may be destroyed upon further dephosphorylation. This could explain in part the lower affinity of ADx210 for Tau monomers.
Determination of the mAb Epitopes
Different strategies were applied to determine the epitopes of ADx201 and ADx215. First, for the phospho-independent mAbs, the use of recombinant Tau fragments, covering different regions of the Tau protein (30), allowed us to roughly position the epitope for ADx201 within the proline-rich region of Tau and that of ADx215 closer to the N terminus. Next, different sandwich ELISA formats were performed, whereby ADx201 and ADx215 competed with previously characterized commercial and non-commercial antibodies for binding of the recombinant protein Tau fragments. This approach successfully determined the approximate epitope for ADx201 because it turned out that the AT120 antibody (30) inhibited ADx201 reactivity. This was confirmed by using synthetic biotinylated peptides that contained the epitope 218PPTREPK224 (Fig. 2A and Table 1). In contrast to ADx201, no significant competition was found when ADx215 was used in the sandwich ELISA assays, indicative that this mAb recognized a novel epitope. Therefore, the epitope was mapped by scanning the N terminus of Tau using a series of small overlapping peptides (Table 2). This approach eventually narrowed the binding site of ADx215 to 16GTYGLGDRK24 (Fig. 2A), an epitope that is specific for primates and not present in Tau of bovine or rodents.
FIGURE 2.

Epitope mapping and phosphorylation dependence. A, schematic representation of human Tau-2N/4R with the shaded regions corresponding to the two N-terminal inserts and the four MTBR as indicated. Shown are the epitope of ADx215 (aa 16–24), which is specific for primates, and the epitope of ADx201 (aa 218–224), which is found in different mammalian Tau sequences. B and C, ELISA using synthetic biotinylated peptides to analyze phosphorylation dependence of ADx201 and ADx215 binding. The peptides used in B combined the epitopes of ADx201 and ADx215 and differed by the presence (closed symbols) or absence (open symbols) of a phospho-Thr, corresponding to position 217 in human Tau-2N/4R. Circles, data for ADx201; squares, data for ADx215. The peptides used in C all contained the epitope for ADx215 but differed by the presence (closed triangles) or absence (open diamonds) of a phospho-Tyr18 or contained the substitution Tyr → Phe18 (open squares) or the phosphomimic substitution Tyr → Glu18 (closed circles). The sequences of the peptides are given in Table 1. D, Western blot analysis of protein extracts made from the BY4741 wild-type strain and mutant strains lacking the dual specificity kinase Yak1, Swe1, Kns1, or Mck1 as indicated. All strains express human Tau-2N/4R, of which various phosphoisoforms were detected with TAU5, 9G3, or ADx215. Error bars, S.E.
The ADx201 epitope is just adjacent to the Thr217 residue, a known phosphosite of Tau associated with AD that is targeted by different kinases (4). To test whether the phosphorylation of Thr217 influences the binding of ADx201, we performed an ELISA using synthesized peptides, one of which contained the phosphorylated Thr217 (Table 1). As shown, phosphorylation of the latter had only a minimal effect on ADx201 binding (Fig. 2B), thereby confirming the results obtained with Tau dephosphorylation. Because the peptides used also contained the ADx215 epitope, the relative affinities of both mAbs could be compared. As expected, the affinity of ADx215 was better than that of ADx201 with EC50 values for the non-phosphorylated peptide of 7.99 ± 1.79 and 23.26 ± 2.58 pg/ml, respectively. To document this in more detail, the apparent KD values were also calculated based on Biacore biomolecular interaction assays using full-length recombinant bacterial Tau-2N/4R. This revealed apparent KD values of 92 pm for ADx201 and 14 pm for ADx215.
In another ELISA, we tested the phosphorylation dependence of ADx215 using peptides modified on Tyr18 (Table 1). This residue is a known phosphosite targeted by the Src non-receptor tyrosine kinase family member Fyn that increases during disease progression (31). However, consistent with the Tau dephosphorylation data, ADx215 shows a preference for non-phosphorylated Tyr18 (Fig. 2C). Furthermore, in contrast to a peptide containing a Tyr → Phe18 substitution, ADx215 did not recognize a peptide containing a Tyr → Glu18 substitution or the equivalent full-length phosphomimic Tyr → Glu18 Tau protein when this was expressed in yeast (Fig. 2C) (data not shown). In this context, it is interesting to note that whereas the yeast genome does not encode for bona fide tyrosine kinases, comparative Western blot analysis with the phospho-Tyr18 antibody 9G3 (31) revealed Tyr18 to be targeted by the dual specificity kinase Yak1, the yeast orthologue of human Dyrk1 (Fig. 2D). Consistently, immunodetection with ADx215 yielded different Tau phosphoisoform profiles in wild type cells and cells lacking Yak1.
We could not use competitive sandwich ELISA or peptide scanning to determine the exact epitope of ADx210 because this mAb has a higher affinity for oligomeric Tau than for monomeric Tau and thus apparently recognizes a non-linear epitope. Nonetheless, some of the data described below suggest that the ADx210 epitope most likely includes exposed sequences of the proline-rich region.
Finally, we used a recently developed NMR assay to directly map the epitope on full-length Tau (32). Hereby, the 1H,15N heteronuclear single-quantum correlation spectrum of Tau-2N/4R when isolated in solution is compared with its spectrum when in a 1:1 complex with the antibody. Immobilization of the epitope at the surface of the large antibody thereby changes the NMR properties of the amide functions involved, leading to line broadening and concomitant disappearance of the epitope from the spectrum. For the ADx201 epitope, the relative intensity profile shows a pronounced intensity dip between Ser214 and Ser235 (Fig. 3A), whereas the addition of the ADx215 antibody causes signal disappearance in the very N-terminal part of the protein, with detectable intensity only starting from Asp25 (Fig. 3B). We cannot exclude the possibility that a couple of residues have their signal broadened because of the steric hindrance due to the antibody surface. Nonetheless, the NMR results confirm in a direct manner the epitopes obtained from the previous experiments.
FIGURE 3.

Confirmation of the ADx215 and ADx201 epitopes by NMR spectroscopy. NMR analysis of the ADx201 (A) and ADx215 (B) epitopes is shown. The 1H,15N heteronuclear single-quantum correlation NMR spectra at 900 MHz of a 1:1 Tau antigen to antibody sample (red) and a pure antigen sample (black) were recorded. Some relevant regions of the spectra showing peak disappearance are depicted at the top. The ratio of the relative intensities per amino acid is shown at the bottom. A ratio close to zero indicates that this particular amino acid is in the potential epitope of the antibody.
Validation of the mAbs in Samples of Transgenic Rodents and AD Brains
To further validate the generated mAbs in the mammalian brain, we performed immunoblotting using hippocampal homogenates from 3-month-old wild type mice and 3- or 12-month-old THY-Tau22 transgenic mice (Fig. 4) that showed, respectively, weak and severe Tau pathology (23). ADx201 and ADx210 recognized a 55-kDa endogenous mouse Tau isoform in samples of non-transgenic mice. ADx215 failed to detect this isoform, which is consistent with the finding that this mAb recognized a primate-specific epitope. In protein extracts of the THY-Tau22 mice, all mAbs reacted well to monomeric human Tau isoforms running at ∼60–62 kDa as well as some breakdown products. In addition, ADx215 easily detected Tau oligomers with an approximate mass of 115 kDa, which may correspond to Tau dimers. ADx215 also recognized a number of discrete intermediate bands around 75 and 85 kDa. These oligomers and intermediate bands were hardly recognized by ADx201 and ADx210. Although there was a slight decrease in the overall intensity of ADx215-positive oligomers and monomeric Tau isoform as the transgenic mice grew older, their ratio did not appear to change significantly with increasing age. This is interesting because previous characterization of the THY-Tau22 transgenic mice clearly documented Tau hyperphosphorylation increasing dramatically with age (23). Hence, it appears that these ADx215-positive oligomers may be formed prior to severe Tau hyperphosphorylation and thus that there is no strict correlation between Tau hyperphosphorylation and the formation of these low order oligomers.
FIGURE 4.
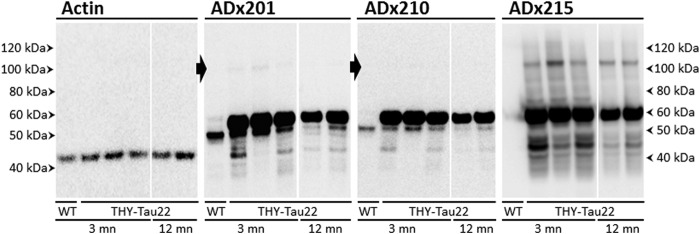
Biochemical validation in brain of transgenic rodents. Western blot analysis is shown of hippocampal brain extracts obtained from wild type control mice (WT) or the transgenic THY-Tau22 mouse model at the age of 3 or 12 months (mn) using the mAb ADx201, ADx210, or ADx215. The closed arrows beside the pictures obtained with ADx201 or ADx210 mark the position where Tau oligomers are faintly detected in the THY-Tau22 samples. The filter probed with ADx201 was stripped and used to detect actin, which served as loading control.
Next, we analyzed homogenates of the frontal and parietal cortex obtained from post-mortem biopsies of healthy persons and AD patients at Braak stages 4, 5, and 6 (Table 4). Besides recognizing various monomeric Tau isoforms in the different samples tested, the three mAbs stained Tau oligomers specifically in the homogenates of AD diseased brains (Fig. 5, A and B). Indeed, ADx215 recognized mainly low order Tau oligomers of about 130 kDa and lower intermediate bands. ADx201 and ADx210, on the other hand, displayed a quite different pattern because they had the capacity to visualize additional folding intermediates, higher order oligomers, and presumed prefibrillary structures of Tau. Notably, Tau oligomers were already present at Braak stage 4 (Fig. 5B), suggesting that they mark an earlier stage in AD pathology. This corroborates previously reported data (8, 9).
FIGURE 5.

Biochemical validation in diseased human brain. A, Western blot analysis of frontal cortex brain extracts obtained from healthy persons (C numbers) and AD patients (AD numbers) at Braak stages 5 and 6 (BR5 and BR6, respectively) using the mAb ADx201, ADx210, or ADx215. The open arrow beside the picture obtained with ADx215 indicates Tau oligomers with a molecular mass below 100 kDa in brain samples of healthy controls. B, Western blot analysis of parietal cortex brain extracts obtained from AD patients (AD numbers) at Braak stages 4 (BR4), 5 (BR3), and 6 (BR6) using the mAbs ADx201, ADx210, and ADx215, as indicated. The molecular masses are given on each picture. Filters probed with ADx201 were stripped and used to detect actin, which served as loading control in A and B. Details on the samples can be found in Table 4.
Tau Dimers Are Phosphorylated in the N-terminal Half
Our data on analysis of transgenic mice and human brain samples suggested that the formation of oligomers may precede severe Tau hyperphosphorylation, meaning that oligomerization would already start when Tau is aberrantly phosphorylated at only a few specific sites and that further hyperphosphorylation of Tau would subsequently drive low order oligomers to grow into higher order oligomeric structures. To test this scenario in more detail, we performed Western blot analysis with frontal cortex homogenates prepared from healthy brain and AD patient brain at Braak stage 5. We used different antibodies to determine which of those would preferentially detect the 130-kDa oligomers or the higher order oligomers. As shown in Fig. 6A, TAU1, a mAb specific for dephosphorylated Tau in the region between amino acids 189 and 207, failed to detect Tau oligomers, indicative that oligomers are indeed phosphorylated in this region. In contrast, the mAb HT7, which recognizes an epitope formed by the amino acids 159–163 independently of the phosphorylation status, readily visualized 130-kDa oligomers in the AD brain sample and thereby displayed a profile similar to that obtained by ADx215. Also, AT180 and 12E8 clearly visualized these 130-kDa oligomers in the AD sample, and because these mAbs recognize the phosphoepitopes Ser(P)231/Thr(P)235 and Ser(P)262/Ser(P)356, respectively, this observation confirms that the 130-kDa oligomers contain phosphorylated Tau. As mentioned above, ADx215 shows a preference for non-phosphorylated Tyr18; therefore, we also included immunodetection with the phospho-Tyr18-specific mAb 9G3. As shown, 9G3 reasonably stained Tau monomeric isoforms and oligomers of an approximate molecular mass of 100 and 150 kDa in both the healthy and AD brain sample, but it did not stain the 130-kDa oligomers. When using mAbs that recognize phosphoepitopes C-terminal of the microtubule-binding repeat (MTBR) region, such as AD2 (Ser(P)396/Ser(P)404; Fig. 6A) or P-S396 (Ser(P)396; Fig. 6B), a completely different pattern was obtained. This pattern more closely resembles that obtained with ADx201/ADx210 (see Fig. 5, A and B) and is characterized by the detection of additional folding intermediates and higher order oligomeric structures in the AD sample. Note that, similar to 9G3, AD2 also recognized oligomers of ∼100 and 150 kDa in the healthy control sample. Combined, these data suggest that the ADx215-positive 130-kDa oligomers are typified by exposure of different phosphoepitopes N-terminal of and in the MTBR region, whereas the higher order oligomeric structures additionally expose phosphoepitopes in the Tau C terminus.
FIGURE 6.

Phosphoepitope mapping in Tau oligomers and Tau-lipid raft interaction. A, Western blot analysis of frontal cortex brain extracts obtained from a healthy person (C75) and an AD patient (AD62) at Braak stage 5 using different phosphorylation-specific mAbs as indicated. B, comparison of ADx201 detection and Tau Ser396 phosphorylation using the mAb P-S396 in frontal cortex brain extracts obtained from a healthy person (C49), an AD patient at Braak stage 5 (AD62), and the transgenic THY-Tau22 mouse model at the age of 12 months. Exposure times (min) for the two mAbs are indicated above the pictures. C, detection of Tau in membrane fractions obtained from post-mortem human frontal cortex samples of an AD patient at Braak stage 5 (AD62) using the mAb ADx215 and the polyclonal antibodies M19G and TauCter. Lipid rafts were identified using the mammalian lipid raft marker flotillin-1 and were enriched in fractions 3 and 4 (underlined). The black arrow beside the pictures indicates the 85-kDa Tau folding variant detected by the polyclonal antibodies. Details on the samples can be found in Table 4.
To evaluate whether differences in Tau C-terminal phosphorylation may explain, at least in part, why we were unable to detect higher order Tau oligomeric structures in the mice samples (Fig. 4), as opposed to the human AD samples (Fig. 5, A and B), we compared the degree of Ser396 Tau phosphorylation in cortex homogenates of a healthy person, an AD patient at Braak stage 5, and a THY-Tau22 transgenic mouse sacrificed at 12 months of age. As shown, P-S396 reacted very strongly with Tau monomers and higher order Tau oligomeric structures in the sample of the AD patient but only faintly detected Tau monomers and no oligomers in the sample of the transgenic mice, this despite the fact that the levels of the Tau monomers in the mouse sample were significantly higher, as evidenced by immunodetection with ADx201 (Fig. 6B). The latter was expected because the THY-Tau22 mice expresses 10 copies of the human Tau transgene (23). Thus, in comparison with human AD brain, Tau is significantly less phosphorylated in its C terminus in the brain of the THY-Tau22 transgenic mice.
Finally, because Tau oligomers are suspected to have an important role in disease propagation (11) and because previous studies indicated that phosphorylated Tau accumulated in detergent-resistant membrane domains or lipid rafts during disease progression (33), we evaluated whether the 130-kDa oligomers would preferentially interact with these lipid domains. To this end, we separated the insoluble membranes of AD frontal cortex homogenates at Braak stage 6 and performed fractionations by flotation on a sucrose gradient. Besides ADx215 and an antibody raised against the lipid raft marker flotillin-1, we also used polyclonal antibodies raised against the very N terminus of protein Tau (i.e. M19G) or the C terminus of protein Tau (i.e. TauCter) (34). As shown, the study clearly confirmed the accumulation of Tau monomeric isoforms and the 130-kDa oligomers in the lipid raft enriched fractions (Fig. 6C). Interestingly, the two polyclonal antibodies TauCter and M19G additionally detected an 85-kDa Tau folding variant, which is mainly recovered in the Triton-soluble fractions.
Performance of the mAbs for Immunohistochemistry
The performance of the mAbs for immunohistochemistry was tested on sections of the hippocampal region of 12-month-old THY-Tau22 transgenic mice brains as well as on hippocampal and cortical sections of advanced AD patients (Braak stage 6). Here, we used both the native mAbs ADx201, ADx210, and ADx215 and biotinylated counterparts, whereas the phospho-dependent antibody AT8 served as control. As shown for mouse brain (Fig. 7), all mAbs robustly stained tangle-like structures that appeared as a dense perinuclear rim in neurons of the CA1 pyramidal layer, the CA3 field, and the dentate gyrus. With ADx215, positive neurons were also clearly observed in the lateral posterior nucleus of the thalamus. In the human brain, the different mAbs densely stained neurons with tangle-like structures in both the hippocampus (Fig. 8). In the cortex, tangle-like structures were only detected with ADx201 and ADx210 (Fig. 8) and not with ADx215 because this mAb gave a very high background as compared with the labeling obtained with the other mAbs (data not shown). Interestingly, ADx201 and ADx210 also labeled neuritic plaques in the human cortex.
FIGURE 7.

Immunohistochemical validation in transgenic rodents. Shown are representative pictures obtained with immunohistochemical analysis using the mAbs ADx201, ADx210, and ADx215 on different regions in the hippocampus of THY-Tau22 mouse brain. AT8 was used as a reference antibody. The brain regions analyzed are indicated in the boxes in the left lower resolution pictures and correspond to the higher resolution pictures shown on the right. Scale bar, 200 μm. CA, Ammon's horn; DG, dentate gyrus.
FIGURE 8.

Immunohistochemical validation in brain of AD patients. Immunohistochemical analysis with ADx201, ADx210, and ADx215 of the cortex and hippocampus in the brain of an AD patient at Braak stage 6. AT8 was used as a reference antibody. The boxes in the left pictures correspond to the higher resolution pictures shown in the right panels. Insets in the higher resolution pictures of the cortex sections show neuritic plaques. Scale bar, 200 μm.
Diagnostic Potential of the mAbs
Having validated the generated mAbs in vivo, we next completed a pilot study to evaluate their potential use for the quantitative detection of Tau in CSF and the ability to discriminate between healthy persons and patients suffering from AD and VAD (Table 5). To this end, we performed ELISA using ADx201 as capturing antibody and biotinylated ADx215 as detector antibody. For optimal comparison, we also included a calibrator peptide, allowing us to convert OD values into concentration values (pg/ml). Consistent with data reported previously (35–37), the levels of total Tau were significantly increased in samples of VAD and AD patients as compared with those of the healthy controls (Fig. 9). Using receiver operating characteristic analysis, the area under the curve for AD versus controls is 0.87 (with a confidence interval set at 95%, ranges were 0.75–0.98), and at a cut-off level of 404 pg/ml, the sensitivity and specificity were 80%. 13 of the 20 VAD patients had CSF Tau levels below this cut-off level, meaning 65% specificity as compared with AD.
FIGURE 9.
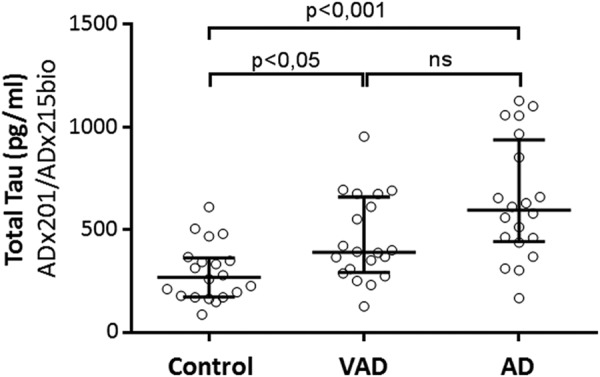
Diagnostic performance of the novel mAbs in CSF. Shown are the scatter dot blot of levels of total Tau in CSF obtained from healthy persons (n = 20) and patients suffering from AD (n = 20) or VAD (n = 20). The line presented in each scatter dot blot is the median level, and bars show the interquartile range. ns, non-significant. Details on the samples can be found in Table 5. *, p < 0.05; ***, p < 0.001.
DISCUSSION
It is well established that the deposition of protein Tau into NFT correlates with neuronal cytopathology in AD. This self-assembly of the protein is believed to be triggered by its hyperphosphorylation and concomitant conformational changes (4). The latter is supported by several studies, including CSF-based studies indicating a correlation between phospho-Tau and cognitive decline in patients with mild cognitive impairment as well as the neocortical neurofibrillary pathology in AD (38, 39). Despite these correlations, it is still a matter of debate whether or not NFT should be considered as toxic agents in AD and other tauopathies, especially because more and more data suggest NFT to be the inert or even protective end stage deposits in a cascade where soluble oligomeric and prefibrillar forms of Tau may represent the true pathogenic entities (see Refs. 40 and 41 and references therein). The interest in these oligomeric and prefibrillar forms of Tau has gained further momentum by the data obtained with Tau oligomer-specific antibodies and the presumed role of prefibrillar Tau oligomers in the propagation of Tau pathology during disease progression (8, 9, 11, 12). Similarly to amyloid β oligomers, Tau oligomers were suggested to act as seeds that are able to recruit and drive further oligomerization of monomeric Tau isoforms. In addition, the formation of the seeding oligomers was suggested to be influenced by post-translational modifications, including aberrant Tau phosphorylation (42).
The generation of monoclonal antibodies against different characteristics of Tau pathology has long been based on the extraction of NFT from AD brains and on recombinant Tau purified from Escherichia coli or synthetic peptides (8, 10, 12). Studies on oligomerization relied on chemically treated recombinant Tau or required preformed aggregates as seeding material (43, 44). Such studies generate Tau dimers and trimers of various size ranging from an apparent 110 to 180 kDa (8, 9, 12, 45). The humanized yeast model discussed here offers an alternative in cellulo system for the production of post-translational modified mono- and oligomeric Tau, without the need of additional cross-linkers or seeding materials. In line with our previous studies, the purification of human Tau from the yeast model generates relatively pure Tau bearing many of the post-translational modifications typifying the protein in AD (17–19). Based on this, we generated an array of new high affinity mAbs with differential phospho- and oligospecificity. Indeed, besides monomeric Tau, two of the mAbs (i.e. ADx201 and ADx210) have affinity toward higher order oligomeric Tau structures, whereas the third (i.e. ADx215) recognizes low order Tau oligomers with an apparent size of 130 kDa that may represent Tau dimers. It has been suggested that Tau oligomers consist of an antiparallel intermolecular interaction of the MTBR domains (9, 46). Also, the oxidation of cysteine residues and the formation of intramolecular or intermolecular disulfide bridges was proposed as a determining factor for oligomer formation and PHF assembly, whereby intramolecular bonds would lead to compacted monomers that do not further assemble, whereas intermolecular bonds would give rise to stabilized dimers and thereby accelerate further oligomerization (47). Interestingly, our previous studies in the yeast already revealed the presence of oligomers with an apparent molecular mass of 130 kDa in yeast extracts, which then could only be detected under non-reducing SDS-PAGE conditions (19). Hence, it remains to be clarified whether those oligomers correspond to the ADx215-positive oligomers detected in the current studies because these are visualized under standard SDS-PAGE conditions with sampling buffer containing DTT as a reducing agent.
In general, the data produced with ADx215 correspond well to those obtained in recent studies that used the oligomer-specific mAbs T22 and TOC1. Similar to ADx215, T22 and TOC1 also revealed significantly increased levels of low order Tau oligomers in samples of AD patients as compared with healthy controls. Co-localization of T22- and TOC1-positive inclusions with early markers of NFT evolution demonstrated increased prefibrillar Tau oligomerization to mark a very early disease stage (8, 9). Furthermore, although we found the ADx215-positive oligomers to be phosphorylated mainly in the N-terminal part and MTBR domain of Tau, the study with T22 nicely documented oligomerization to start once Tau is phosphorylated at Thr231 (8). Phosphorylation of Thr231 by Gsk-3β is known to prevent Tau from binding microtubules (4) and to relieve the inhibitory activity of the N terminus over the C terminus of Tau so as to allow kinases such as Gsk-3β to access and subsequently phosphorylate Tau at other epitopes (48). Indeed, monomeric Tau is thought to adopt a so-called “paperclip” conformation when in solution, where the C-terminal end of Tau folds over the MTBR domain and the N terminus folds back to come in close proximity to the C terminus (49, 50). It is conceivable that the opening of this paperclip conformation is a first essential step for Tau oligomerization. However, it remains to be studied in more detail whether this relies solely on phosphorylation of Thr231 or requires the additional phosphorylation of some of the other sites we identified by phosphoepitope mapping in the ADx215-positive oligomers. In our opinion, these additional phosphorylation events may help to stabilize the oligomers while being formed. This idea comes from the observation that the ADx215-positive oligomers are only weakly detected and of lower molecular weight in healthy brain as compared with AD brain, and this indeed suggests differences in the phosphorylation status and stability of the Tau oligomers between both conditions.
Besides its supposed importance to initiate the formation of Tau oligomers, the opening of the paperclip and exposure of the Tau N terminus has additional consequences. Recent studies have shown that the N terminus harbors a phosphatase-activating domain (PAD) between the residues Ala2 and Tyr18 that, when exposed in Tau filaments, activates protein phosphatase 1 and GSK3 and results in the phosphorylation of kinesin light chains, the dissociation of kinesin from its cargo, and the disruption of fast axonal transport (FAT) (51, 52). PAD overlaps in part with the minimal epitope of ADx215, and thus it appears that PAD might be exposed in the ADx215-positive oligomers detected in our present study. Moreover, a follow-up study recently showed that phosphorylation or pseudophosphorylation of Tyr18 and, to a lesser extent, of Tyr29 abrogates the inhibitory effect of PAD on FAT (53), whereas we found that Tyr18 phosphorylation disrupts Tau detection by ADx215. Of particular importance are the immunohistochemical studies that used the anti-phospho-Tyr18 mAb 9G3 and the PAD-specific mAb TNT1 to classify increased PAD exposure as an early pathogenic event occurring before and more frequently than Tyr18 phosphorylation in the path of NFT formation in AD (53). This led the authors to suggest that Tyr18 phosphorylation is part of a compensatory mechanism aiming to reduce the toxicity associated with exposure of the Tau N terminus. Whether exposure of PAD in the ADx215-positive oligomers already affects FAT and thus defines these oligomers as soluble toxic entities needs to be examined in more detail because an inhibitory effect of PAD on FAT has so far only been observed with Tau filaments (51, 53).
Concerning the Tau C terminus, we observed that exposure of phosphoepitopes in this region comes along with the appearance of higher order Tau oligomers and prefibrillary structures. There are several observations pointing toward an important role of Tau C-terminal phosphorylation in determining the Tau polymerization propensity and Tau toxicity. Similarly to the N terminus, pseudophosphorylation of Ser396 and Ser404 in the AD2/PHF1 epitope of Tau was shown to move the C-terminal domain away from the MTBR region and thus to open the paperclip configuration. However, the combined pseudophosphorylation in N- and C-terminal epitopes was found to result in compaction of the paperclip, such that the N terminus approaches the MTBR region to form the conformation-dependent MC1/Alz50 epitope (49). The latter defines a pathogenic conformation of Tau that enhances its assembly into paired helical filaments (54). Consistently, we previously reported on a strong correlation between phosphorylation of Tau at the AD2/PHF1 and PG5 (Ser(P)409) epitopes, the immunoreactivity with the MC1 antibody, and the level of Sarkosyl-insoluble Tau in Tau-expressing yeast cells and demonstrated the capacity of this MC1-positive fraction to seed spontaneous Tau polymerization in vitro (18, 19). Similar correlations have been reported based on studies in transgenic mice (55). In the human brain, the AD2/PHF1 and PG5 phosphoepitopes and the MC1/Alz50 conformational epitope are established markers for tauopathy, as seen in AD (56–59), and several studies confirmed the appearance of the MC1/Alz50 conformation to precede the formation of PHF and NFT (60–62). It is therefore tempting to speculate that C-terminal phosphorylation of Tau and the establishment of the MC1/Alz50 epitope define the transition between Tau low order and higher order oligomeric structures. Another important aspect is disease propagation by the secretion and uptake of Tau oligomers that act as seeding templates for Tau misfolding and toxicity (63). In this context, we recently developed a lentivirus-mediated rat model that allowed us to monitor the spreading of Tau pathology from the CA1 region of the hippocampus to other brain areas, including the most distant ones (64, 65). We then already used ADx215 as marker to demonstrate that the propagating Tau entity was not phosphorylated at Tyr18 (65). Because ADx215 does not recognize higher order oligomers, the data suggest that Tau monomers or the 130-kDa low order oligomers are propagating Tau entities. In line with this are the data from a study that tested the ability of anti-Tau mAbs to block the trans-cellular seeding activity when infused into the lateral ventricle of P301S transgenic mice (11). Here the most potent mAb was HJ8.5, which recognizes an N-terminal epitope between Asp25 and Thr30. Because this epitope is just adjacent to the minimal epitope identified for ADx215, it indeed suggests that exposure of the N terminus may be crucial for seeding. Hence, it would be useful to confirm the ability of ADx215 to block the Tau seeding and propagation activity because this would make it a good candidate for immune therapy.
Finally, we tested the use of our novel high affinity mAbs for diagnostics and documented that the combined use of ADx201 and ADx215 as capturing and detecting antibodies, respectively, has the potential to discriminate patients clinically diagnosed with VAD or AD from healthy persons based on increased levels in CSF of total Tau. This is in agreement with previously published studies (35–37). Recently, improved characterization of CSF Tau using reverse-phase high performance liquid chromatography led to the evaluation of five total Tau assays side by side, and the two assays containing N-terminal sequences exhibited the most significant differences between AD and control CSF (66). This suggests that the use of mAbs such as ADx215, recognizing the N terminus of Tau, may indeed improve diagnostic accuracy. That ADx215 might also recognize a presumed propagating Tau seeding conformation only adds relevance for its discriminative capacity between the healthy and diseased persons.
In conclusion, we expressed human Tau-2N/4R in pho85Δ yeast cells and used the purified protein as antigen to generate novel mAbs. These mAbs recognize specific Tau oligomeric species in diseased brain besides monomeric Tau upon Western blot analysis, and they have capacity to stain tangle-like structures and neuritic plaques in diseased brain sections upon immunohistochemical analysis. Moreover, the mAbs proved to be ideal tools to discriminate patients suffering from AD or VAD from healthy controls using CSF-based ELISA. As such, our studies unequivocally demonstrate that Tau must adopt pathologically relevant conformations when expressed in yeast and illustrate how the use of this simple eukaryotic model organism can contribute in the quest to understand the etiology of tauopathies.
Acknowledgments
We thank the CNRS Large Scale Facility NMR THC Fr3050 for support; M. Michels, A. Chauderlier, and M-E. Grosjean for excellent technical assistance; Dr. F.-X. Cantrelle for maintenance of the 900-MHz NMR spectrometer; and Prof. Dr. F. Van Leuven for advice and additional validation in mice models. We also thank Dr. M. Schraeml (Roche Applied Science) and Dr. T. Bittner (Roche Applied Science) for providing the Biacore affinity data and Dr. Lester (Skip) Binder for providing the TOC1 antibody.
This work was supported by grants from the Research Foundation Flanders (FWO-Vlaanderen) (to J. W. and S. E.), KU Leuven Research Fund (KU Leuven-BOF; KU Leuven-IOF), KU Leuven R&D (to J. W.), and the Agency for Innovation by Science and Technology (IWT) (to J. W., E. V., and S. E.). This work was also supported by an IWT Ph.D. fellowship (to J. V. d. B.) and FWO-Vlaanderen postdoctoral fellowships (to M. C. and E. S.). This work was also supported through the LabEx DISTALZ (Excellent Laboratory-Development of Innovative Strategies for a Transdisciplinary Approach to Alzheimer's Disease), the CNRS Large Scale Facility NMR THC Fr3050, INSERM, CNRS, University of Lille 2, Lille Métropole Communauté Urbaine (LMCU), Région Nord/Pas-de-Calais, FEDER, the University of Antwerp Research Fund, the Alzheimer Research Foundation (SAO-FRA), the central Biobank facility of the Institute Born-Bunge/University Antwerp, the Belgian Science Policy Office Interuniversity Attraction Poles (IAP) program, and the Flemish Government initiated Methusalem excellence grant. J. W. and E. V. are both co-founders of ADx NeuroSciences and co-inventors on the patent entitled Antibodies to Phosphorylated Tau Aggregates (WO2013007839 (A1)).
This paper is dedicated to Skip Binder as a token of our appreciation for his generosity and his scientific legacy, which includes numerous and significant contributions to the Tau field.
- AD
- Alzheimer disease
- aa
- amino acid(s)
- AP
- alkaline phosphatase
- CSF
- cerebrospinal fluid
- FAT
- fast axonal transport
- GAM
- goat anti-mouse
- MT
- microtubule(s)
- MTBR
- microtubule-binding repeat
- NFT
- neurofibrillary tangle(s)
- PAD
- phosphatase-activating domain
- PHF
- paired helical filament(s)
- VAD
- vascular dementia.
REFERENCES
- 1. Buée L., Bussière T., Buée-Scherrer V., Delacourte A., Hof P. R. (2000) Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 33, 95–130 [DOI] [PubMed] [Google Scholar]
- 2. Irwin D. J., Cohen T. J., Grossman M., Arnold S. E., Xie S. X., Lee V. M., Trojanowski J. Q. (2012) Acetylated tau, a novel pathological signature in Alzheimer's disease and other tauopathies. Brain 135, 807–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang Y., Garg S., Mandelkow E. M., Mandelkow E. (2010) Proteolytic processing of tau. Biochem. Soc. Trans. 38, 955–961 [DOI] [PubMed] [Google Scholar]
- 4. Sergeant N., Bretteville A., Hamdane M., Caillet-Boudin M. L., Grognet P., Bombois S., Blum D., Delacourte A., Pasquier F., Vanmechelen E., Schraen-Maschke S., Buée L. (2008) Biochemistry of Tau in Alzheimer's disease and related neurological disorders. Expert Rev. Proteomics 5, 207–224 [DOI] [PubMed] [Google Scholar]
- 5. Irwin D. J., McMillan C. T., Toledo J. B., Arnold S. E., Shaw L. M., Wang L. S., Van Deerlin V., Lee V. M., Trojanowski J. Q., Grossman M. (2012) Comparison of cerebrospinal fluid levels of tau and Aβ 1–42 in Alzheimer disease and frontotemporal degeneration using 2 analytical platforms. Arch. Neurol. 69, 1018–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Le Bastard N., Coart E., Vanderstichele H., Vanmechelen E., Martin J. J., Engelborghs S. (2013) Comparison of two analytical platforms for the clinical qualification of Alzheimer's disease biomarkers in pathologically-confirmed dementia. J. Alzheimers Dis. 33, 117–131 [DOI] [PubMed] [Google Scholar]
- 7. Berger Z., Roder H., Hanna A., Carlson A., Rangachari V., Yue M., Wszolek Z., Ashe K., Knight J., Dickson D., Andorfer C., Rosenberry T. L., Lewis J., Hutton M., Janus C. (2007) Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J. Neurosci. 27, 3650–3662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lasagna-Reeves C. A., Castillo-Carranza D. L., Sengupta U., Sarmiento J., Troncoso J., Jackson G. R., Kayed R. (2012) Identification of oligomers at early stages of tau aggregation in Alzheimer's disease. FASEB J. 26, 1946–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Patterson K. R., Remmers C., Fu Y., Brooker S., Kanaan N. M., Vana L., Ward S., Reyes J. F., Philibert K., Glucksman M. J., Binder L. I. (2011) Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J. Biol. Chem. 286, 23063–23076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ward S. M., Himmelstein D. S., Lancia J. K., Fu Y., Patterson K. R., Binder L. I. (2013) TOC1: characterization of a selective oligomeric Tau antibody. J. Alzheimers Dis. 37, 593–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yanamandra K., Kfoury N., Jiang H., Mahan T. E., Ma S., Maloney S. E., Wozniak D. F., Diamond M. I., Holtzman D. M. (2013) Anti-Tau antibodies that block Tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 80, 402–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Castillo-Carranza D. L., Gerson J. E., Sengupta U., Guerrero-Muñoz M. J., Lasagna-Reeves C. A., Kayed R. (2014) Specific targeting of Tau oligomers in Htau mice prevents cognitive impairment and Tau toxicity following injection with brain-derived Tau oligomeric seeds. J. Alzheimers Dis. 40, S97–S111 [DOI] [PubMed] [Google Scholar]
- 13. Winderickx J., Delay C., De Vos A., Klinger H., Pellens K., Vanhelmont T., Van Leuven F., Zabrocki P. (2008) Protein folding diseases and neurodegeneration: lessons learned from yeast. Biochim. Biophys. Acta 1783, 1381–1395 [DOI] [PubMed] [Google Scholar]
- 14. D'Angelo F., Vignaud H., Di Martino J., Salin B., Devin A., Cullin C., Marchal C. (2013) A yeast model for amyloid-β aggregation exemplifies the role of membrane trafficking and PICALM in cytotoxicity. Dis. Model. Mech. 6, 206–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Treusch S., Hamamichi S., Goodman J. L., Matlack K. E., Chung C. Y., Baru V., Shulman J. M., Parrado A., Bevis B. J., Valastyan J. S., Han H., Lindhagen-Persson M., Reiman E. M., Evans D. A., Bennett D. A., Olofsson A., DeJager P. L., Tanzi R. E., Caldwell K. A., Caldwell G. A., Lindquist S. (2011) Functional links between Abeta toxicity, endocytic trafficking, and Alzheimer's disease risk factors in yeast. Science 334, 1241–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. De Vos A., Anandhakumar J., Van den Brande J., Verduyckt M., Franssens V., Winderickx J., Swinnen E. (2011) Yeast as a model system to study tau biology. Int. J. Alzheimers Dis. 2011, 428970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vandebroek T., Terwel D., Vanhelmont T., Gysemans M., Van Haesendonck C., Engelborghs Y., Winderickx J., Van Leuven F. (2006) Microtubule binding and clustering of human Tau-4R and Tau-P301L proteins isolated from yeast deficient in orthologues of glycogen synthase kinase-3β or Cdk5. J. Biol. Chem. 281, 25388–25397 [DOI] [PubMed] [Google Scholar]
- 18. Vandebroek T., Vanhelmont T., Terwel D., Borghgraef P., Lemaire K., Snauwaert J., Wera S., Van Leuven F., Winderickx J. (2005) Identification and isolation of a hyperphosphorylated, conformationally changed intermediate of human protein tau expressed in yeast. Biochemistry 44, 11466–11475 [DOI] [PubMed] [Google Scholar]
- 19. Vanhelmont T., Vandebroek T., De Vos A., Terwel D., Lemaire K., Anandhakumar J., Franssens V., Swinnen E., Van Leuven F., Winderickx J. (2010) Serine-409 phosphorylation and oxidative damage define aggregation of human protein tau in yeast. FEMS Yeast Res. 10, 992–1005 [DOI] [PubMed] [Google Scholar]
- 20. Kato Y., Jin G., Kuan C. T., McLendon R. E., Yan H., Bigner D. D. (2009) A monoclonal antibody IMab-1 specifically recognizes IDH1R132H, the most common glioma-derived mutation. Biochem. Biophys. Res. Commun. 390, 547–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lippens G., Wieruszeski J. M., Leroy A., Smet C., Sillen A., Buée L., Landrieu I. (2004) Proline-directed random-coil chemical shift values as a tool for the NMR assignment of the tau phosphorylation sites. Chembiochem 5, 73–78 [DOI] [PubMed] [Google Scholar]
- 22. Gottschalk P. G., Dunn J. R. (2005) The five-parameter logistic: a characterization and comparison with the four-parameter logistic. Anal. Biochem. 343, 54–65 [DOI] [PubMed] [Google Scholar]
- 23. Schindowski K., Bretteville A., Leroy K., Bégard S., Brion J. P., Hamdane M., Buée L. (2006) Alzheimer's disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am. J. Pathol. 169, 599–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Delacourte A., Defossez A. (1986) Alzheimer's disease: Tau proteins, the promoting factors of microtubule assembly, are major components of paired helical filaments. J. Neurol. Sci. 76, 173–186 [DOI] [PubMed] [Google Scholar]
- 25. McKhann G. M., Knopman D. S., Chertkow H., Hyman B. T., Jack C. R., Jr., Kawas C. H., Klunk W. E., Koroshetz W. J., Manly J. J., Mayeux R., Mohs R. C., Morris J. C., Rossor M. N., Scheltens P., Carrillo M. C., Thies B., Weintraub S., Phelps C. H. (2011) The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 7, 263–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Román G. C., Tatemichi T. K., Erkinjuntti T., Cummings J. L., Masdeu J. C., Garcia J. H., Amaducci L., Orgogozo J. M., Brun A., Hofman A. (1993) Vascular dementia: diagnostic criteria for research studies: report of the NINDS-AIREN International Workshop. Neurology 43, 250–260 [DOI] [PubMed] [Google Scholar]
- 27. Büttner S., Delay C., Franssens V., Bammens T., Ruli D., Zaunschirm S., de Oliveira R. M., Outeiro T. F., Madeo F., Buée L., Galas M. C., Winderickx J. (2010) Synphilin-1 enhances α-synuclein aggregation in yeast and contributes to cellular stress and cell death in a Sir2-dependent manner. PLoS One 5, e13700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rösner H., Rebhan M., Vacun G., Vanmechelen E. (1994) Expression of a paired helical filament tau epitope in embryonic chicken central nervous system. Neuroreport 5, 1164–1166 [DOI] [PubMed] [Google Scholar]
- 29. Goedert M., Jakes R., Crowther R. A., Cohen P., Vanmechelen E., Vandermeeren M., Cras P. (1994) Epitope mapping of monoclonal antibodies to the paired helical filaments of Alzheimer's disease: identification of phosphorylation sites in tau protein. Biochem. J. 301, 871–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vanmechelen E., Vanderstichele H., Davidsson P., Van Kerschaver E., Van Der Perre B., Sjögren M., Andreasen N., Blennow K. (2000) Quantification of tau phosphorylated at threonine 181 in human cerebrospinal fluid: a sandwich ELISA with a synthetic phosphopeptide for standardization. Neurosci. Lett. 285, 49–52 [DOI] [PubMed] [Google Scholar]
- 31. Bhaskar K., Hobbs G. A., Yen S. H., Lee G. (2010) Tyrosine phosphorylation of tau accompanies disease progression in transgenic mouse models of tauopathy. Neuropathol. Appl. Neurobiol. 36, 462–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Amniai L., Lippens G., Landrieu I. (2011) Characterization of the AT180 epitope of phosphorylated Tau protein by a combined nuclear magnetic resonance and fluorescence spectroscopy approach. Biochem. Biophys. Res. Commun. 412, 743–746 [DOI] [PubMed] [Google Scholar]
- 33. Kawarabayashi T., Shoji M., Younkin L. H., Wen-Lang L., Dickson D. W., Murakami T., Matsubara E., Abe K., Ashe K. H., Younkin S. G. (2004) Dimeric amyloid β protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer's disease. J. Neurosci. 24, 3801–3809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sergeant N., Sablonnière B., Schraen-Maschke S., Ghestem A., Maurage C. A., Wattez A., Vermersch P., Delacourte A. (2001) Dysregulation of human brain microtubule-associated tau mRNA maturation in myotonic dystrophy type 1. Hum. Mol. Genet. 10, 2143–2155 [DOI] [PubMed] [Google Scholar]
- 35. Olsson A., Vanderstichele H., Andreasen N., De Meyer G., Wallin A., Holmberg B., Rosengren L., Vanmechelen E., Blennow K. (2005) Simultaneous measurement of β-amyloid(1–42), total tau, and phosphorylated tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clin. Chem. 51, 336–345 [DOI] [PubMed] [Google Scholar]
- 36. Sjögren M., Davidsson P., Gottfries J., Vanderstichele H., Edman A., Vanmechelen E., Wallin A., Blennow K. (2001) The cerebrospinal fluid levels of tau, growth-associated protein-43 and soluble amyloid precursor protein correlate in Alzheimer's disease, reflecting a common pathophysiological process. Dement. Geriatr. Cogn. Disord. 12, 257–264 [DOI] [PubMed] [Google Scholar]
- 37. Hertze J., Minthon L., Zetterberg H., Vanmechelen E., Blennow K., Hansson O. (2010) Evaluation of CSF biomarkers as predictors of Alzheimer's disease: a clinical follow-up study of 4.7 years. J. Alzheimers Dis. 21, 1119–1128 [DOI] [PubMed] [Google Scholar]
- 38. Buerger K., Ewers M., Andreasen N., Zinkowski R., Ishiguro K., Vanmechelen E., Teipel S. J., Graz C., Blennow K., Hampel H. (2005) Phosphorylated tau predicts rate of cognitive decline in MCI subjects: a comparative CSF study. Neurology 65, 1502–1503 [DOI] [PubMed] [Google Scholar]
- 39. Buerger K., Ewers M., Pirttilä T., Zinkowski R., Alafuzoff I., Teipel S. J., DeBernardis J., Kerkman D., McCulloch C., Soininen H., Hampel H. (2006) CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer's disease. Brain 129, 3035–3041 [DOI] [PubMed] [Google Scholar]
- 40. Cowan C. M., Mudher A. (2013) Are tau aggregates toxic or protective in tauopathies? Front. Neurol. 4, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Iqbal K., Gong C. X., Liu F. (2013) Hyperphosphorylation-induced tau oligomers. Front. Neurol. 4, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gerson J. E., Kayed R. (2013) Formation and propagation of tau oligomeric seeds. Front. Neurol. 4, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dinkel P. D., Siddiqua A., Huynh H., Shah M., Margittai M. (2011) Variations in filament conformation dictate seeding barrier between three- and four-repeat tau. Biochemistry 50, 4330–4336 [DOI] [PubMed] [Google Scholar]
- 44. Guo J. L., Lee V. M. (2011) Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 286, 15317–15331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lasagna-Reeves C. A., Castillo-Carranza D. L., Guerrero-Muoz M. J., Jackson G. R., Kayed R. (2010) Preparation and characterization of neurotoxic tau oligomers. Biochemistry 49, 10039–10041 [DOI] [PubMed] [Google Scholar]
- 46. Wille H., Drewes G., Biernat J., Mandelkow E. M., Mandelkow E. (1992) Alzheimer-like paired helical filaments and antiparallel dimers formed from microtubule-associated protein tau in vitro. J. Cell Biol. 118, 573–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Barghorn S., Mandelkow E. (2002) Toward a unified scheme for the aggregation of tau into Alzheimer paired helical filaments. Biochemistry 41, 14885–14896 [DOI] [PubMed] [Google Scholar]
- 48. Lin Y. T., Cheng J. T., Liang L. C., Ko C. Y., Lo Y. K., Lu P. J. (2007) The binding and phosphorylation of Thr231 is critical for Tau's hyperphosphorylation and functional regulation by glycogen synthase kinase 3β. J. Neurochem. 103, 802–813 [DOI] [PubMed] [Google Scholar]
- 49. Jeganathan S., Hascher A., Chinnathambi S., Biernat J., Mandelkow E. M., Mandelkow E. (2008) Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J. Biol. Chem. 283, 32066–32076 [DOI] [PubMed] [Google Scholar]
- 50. Jeganathan S., von Bergen M., Brutlach H., Steinhoff H. J., Mandelkow E. (2006) Global hairpin folding of tau in solution. Biochemistry 45, 2283–2293 [DOI] [PubMed] [Google Scholar]
- 51. Kanaan N. M., Morfini G. A., LaPointe N. E., Pigino G. F., Patterson K. R., Song Y., Andreadis A., Fu Y., Brady S. T., Binder L. I. (2011) Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J. Neurosci. 31, 9858–9868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. LaPointe N. E., Morfini G., Pigino G., Gaisina I. N., Kozikowski A. P., Binder L. I., Brady S. T. (2009) The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J. Neurosci. Res. 87, 440–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kanaan N. M., Morfini G., Pigino G., LaPointe N. E., Andreadis A., Song Y., Leitman E., Binder L. I., Brady S. T. (2012) Phosphorylation in the amino terminus of tau prevents inhibition of anterograde axonal transport. Neurobiol. Aging 10.1016/j.neurobiolaging.2011.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gamblin T. C., Berry R. W., Binder L. I. (2003) Tau polymerization: role of the amino terminus. Biochemistry 42, 2252–2257 [DOI] [PubMed] [Google Scholar]
- 55. Sahara N., Lewis J., DeTure M., McGowan E., Dickson D. W., Hutton M., Yen S. H. (2002) Assembly of tau in transgenic animals expressing P301L tau: alteration of phosphorylation and solubility. J. Neurochem. 83, 1498–1508 [DOI] [PubMed] [Google Scholar]
- 56. Galván M., David J. P., Delacourte A., Luna J., Mena R. (2001) Sequence of neurofibrillary changes in aging and Alzheimer's disease: a confocal study with phospho-tau antibody, AD2. J. Alzheimers Dis. 3, 417–425 [DOI] [PubMed] [Google Scholar]
- 57. Kimura T., Ono T., Takamatsu J., Yamamoto H., Ikegami K., Kondo A., Hasegawa M., Ihara Y., Miyamoto E., Miyakawa T. (1996) Sequential changes of tau-site-specific phosphorylation during development of paired helical filaments. Dementia 7, 177–181 [DOI] [PubMed] [Google Scholar]
- 58. Mondragón-Rodríguez S., Perry G., Luna-Muñoz J., Acevedo-Aquino M. C., Williams S. (2014) Phosphorylation of tau protein at sites Ser 396–404 is one of the earliest events in Alzheimer's disease and Down syndrome. Neuropathol. Appl. Neurobiol. 40, 121–135 [DOI] [PubMed] [Google Scholar]
- 59. Jicha G. A., Lane E., Vincent I., Otvos L., Jr., Hoffmann R., Davies P. (1997) A conformation- and phosphorylation-dependent antibody recognizing the paired helical filaments of Alzheimer's disease. J. Neurochem. 69, 2087–2095 [DOI] [PubMed] [Google Scholar]
- 60. Haroutunian V., Davies P., Vianna C., Buxbaum J. D., Purohit D. P. (2007) Tau protein abnormalities associated with the progression of alzheimer disease type dementia. Neurobiol. Aging 28, 1–7 [DOI] [PubMed] [Google Scholar]
- 61. Luna-Muñoz J., Chávez-Macías L., García-Sierra F., Mena R. (2007) Earliest stages of tau conformational changes are related to the appearance of a sequence of specific phospho-dependent tau epitopes in Alzheimer's disease. J. Alzheimers Dis. 12, 365–375 [DOI] [PubMed] [Google Scholar]
- 62. Weaver C. L., Espinoza M., Kress Y., Davies P. (2000) Conformational change as one of the earliest alterations of tau in Alzheimer's disease. Neurobiol. Aging 21, 719–727 [DOI] [PubMed] [Google Scholar]
- 63. Gendreau K. L., Hall G. F. (2013) Tangles, toxicity, and Tau secretion in AD: new approaches to a vexing problem. Front. Neurol. 4, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Caillierez R., Bégard S., Lécolle K., Deramecourt V., Zommer N., Dujardin S., Loyens A., Dufour N., Aurégan G., Winderickx J., Hantraye P., Déglon N., Buée L., Colin M. (2013) Lentiviral delivery of the human wild-type tau protein mediates a slow and progressive neurodegenerative tau pathology in the rat brain. Mol. Ther. 21, 1358–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dujardin S., Lécolle K., Caillierez R., Bégard S., Zommer N., Lachaud C., Carrier S., Dufour N., Aurégan G., Winderickx J., Hantraye P., Déglon N., Colin M., Buée L. (2014) Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: relevance to sporadic tauopathies. Acta Neuropathol. Commun. 2, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Meredith J. E., Jr., Sankaranarayanan S., Guss V., Lanzetti A. J., Berisha F., Neely R. J., Slemmon J. R., Portelius E., Zetterberg H., Blennow K., Soares H., Ahlijanian M., Albright C. F. (2013) Characterization of novel CSF Tau and ptau biomarkers for Alzheimer's disease. PLoS One 8, e76523. [DOI] [PMC free article] [PubMed] [Google Scholar]