

Background: Oxidative stress promotes premature senescence in eukaryotic cells.
Results: Inhibition of Sirt1 by caveolin-1 is induced by free radicals and promotes cellular senescence and secretion of tumorigenic IL-6 in fibroblasts.
Conclusion: The caveolin-1/Sirt1/IL-6 signaling pathway contributes to explain the ability of senescent cells to stimulate cancer cell growth.
Significance: This novel signaling cascade brings new insights into how senescent cells regulate the tissue microenvironment.
Keywords: Caveolin, Oxidative Stress, p53, Senescence, Sirtuin 1 (SIRT1)
Abstract
Oxidative stress can induce premature cellular senescence. Senescent cells secrete various growth factors and cytokines, such as IL-6, that can signal to the tumor microenvironment and promote cancer cell growth. Sirtuin 1 (Sirt1) is a class III histone deacetylase that regulates a variety of physiological processes, including senescence. We found that caveolin-1, a structural protein component of caveolar membranes, is a direct binding partner of Sirt1, as shown by the binding of the scaffolding domain of caveolin-1 (amino acids 82–101) to the caveolin-binding domain of Sirt1 (amino acids 310–317). Our data show that oxidative stress promotes the sequestration of Sirt1 into caveolar membranes and the interaction of Sirt1 with caveolin-1, which lead to inhibition of Sirt1 activity. Reactive oxygen species stimulation promotes acetylation of p53 and premature senescence in wild-type but not caveolin-1 null mouse embryonic fibroblasts (MEFs). Either down-regulation of Sirt1 expression or re-expression of caveolin-1 in caveolin-1 null MEFs restores reactive oxygen species-induced acetylation of p53 and premature senescence. In addition, overexpression of caveolin-1 induces stress induced premature senescence in p53 wild-type but not p53 knockout MEFs. Phosphorylation of caveolin-1 on tyrosine 14 promotes the sequestration of Sirt1 into caveolar membranes and activates p53/senescence signaling. We also identified IL-6 as a caveolin-1-specific cytokine that is secreted by senescent fibroblasts following the caveolin-1-mediated inhibition of Sirt1. The caveolin-1-mediated secretion of IL-6 by senescent fibroblasts stimulates the growth of cancer cells. Therefore, by inhibiting Sirt1, caveolin-1 links free radicals to the activation of the p53/senescence pathway and the protumorigenic properties of IL-6.
Introduction
Most cells cannot divide indefinitely because of a process termed cellular senescence (1–4). Cellular senescence can be divided into two categories: replicative senescence and stress-induced premature senescence. Replicative senescence is dependent on the number of divisions the cell has completed. This type of senescence is spontaneously achieved by somatic cells. It is known that telomere shortening controls cell division counting, an unavoidable consequence of genome duplication (5–9). Senescence can be accelerated by a number of stressful stimuli, such as oncogene activation, DNA damage, cytotoxic drugs, and oxidative stress (10–14). This type of senescence is referred to as stress-induced premature senescence (SIPS).3 SIPS is independent of telomere status but shares many molecular and functional features with replicative senescence.
Although no single feature of the senescent phenotype is exclusively specific, hallmarks of cellular senescence include growth arrest, exit from the cell cycle, increased p53 activity, increased p21Waf1/Cip1 and p16 protein expression, hypophosphorylation of Retinoblastoma protein (pRb) and nuclear foci containing DNA damage response (DDR) proteins or heterochromatin (1–4). Senescent cells can also be experimentally identified by their enlarged and flattened morphology and by positive staining for β-galactosidase activity at pH 6 (2). Moreover, senescent cells acquire a senescence-associated secretory phenotype (SASP). They secrete a number of growth factors, cytokines, proteases, and other factors that possess strong autocrine and paracrine activities.
Cellular senescence is considered to be a powerful tumor suppressor mechanism because it prevents the propagation of cells with damaged DNA and of cells potentially carrying oncogenic mutations. However, senescent cells accumulate over time (2, 15–18), and they are believed to contribute to aging and age-related pathologies (19). In fact, the inability of senescent cells to proliferate contributes to reduced tissue function in aging organs. In addition, senescent cells secreting metalloproteinases and inflammatory cytokines (20–22) may also play a role in tissue aging by influencing the neighboring tissue microenvironment (19, 23). Cancer is an example of an age-related pathology that may be promoted by senescent cells. In fact, many SASP factors are known to stimulate phenotypes associated with aggressive cancer cells, and senescent cells have been shown to promote the growth of cells harboring preneoplastic or neoplastic mutations (24–27). Therefore, a delicate balance exists between the positive effects of cellular senescence on tumor suppression and the negative effects of cellular senescence on aging and age-related diseases. Because increased oxidative damage has been reported in aged animals and because oxidants can induce premature senescence in cells, understanding the molecular mechanisms that control the development of stress-induced premature senescence of eukaryotic cells and the acquisition of the SASP is therefore fundamental for gaining insight into aging and age-related diseases such as cancer.
The NAD+-dependent class III histone deacetylase Sirt1 plays a critical role in stress responses, cellular metabolism, and aging (28) by deacetylating a number of proteins, including p53 (29, 30). Deacetylation of p53 results in repression of p53 activity. Down-regulation of Sirt1 increases acetylation of p53 and premature senescence (31). In contrast, overexpression of Sirt1 prevents SIPS (31). Therefore, Sirt1 represses p53 activity and prevents p53-dependent senescence. However, the molecular mechanism that explains how the repression of p53 activity by Sirt1 is prevented under conditions of stress that lead to activation of p53 and premature senescence is not well understood.
Caveolae are flask-shaped invaginations of the plasma membrane. Caveolin-1 is a major structural protein component of caveolae in most cell types. Caveolin-1 acts as a scaffolding protein that concentrates and functionally regulates a variety of signaling molecules. Our laboratory was the first to show that caveolin-1 is a key mediator of the signaling events underlying oxidative stress-induced premature senescence (32–38). We demonstrated that disruption of caveolae inhibits SIPS both in cell culture models and in vivo (32–38). Here we investigated the molecular mechanisms through which caveolin-1 links free radicals to the protumorigenic properties of cellular senescence. We found that caveolin-1 is a novel endogenous inhibitor of Sirt1 and that the oxidant-induced and caveolin-1-mediated inhibition of Sirt1 promotes the acetylation/activation of p53 and the development of premature senescence in fibroblasts. Our findings also show that the inhibition of Sirt1 by caveolin-1 in senescent fibroblasts promotes the secretion of IL-6, which stimulates cancer cell growth. Together, our data provide novel mechanistic insights into the regulation of the tumor microenvironment by senescent cells.
EXPERIMENTAL PROCEDURES
Materials
Antibodies and their sources were as follows: anti-caveolin-1 IgG (N-20; pAb), anti-Sirt1 IgG (H-300; pAb), anti-p53 IgG (FL-393; pAb), anti-p21 IgG (pAb), and anti-β-actin (C4; mAb) were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-IL-6 (MAB406; mAb) was from R&D Systems (Minneapolis, MN). Anti-acetyl-p53 (K379; pAb) was from Cell Signaling Technology (Danvers, MA). Anti-FLAG IgG (M2; mAb) was from Sigma. Horseradish peroxidase-conjugated goat anti-mouse and anti-rabbit secondary antibodies were from Pierce. All other biochemicals used were of the highest purity available and were obtained from regular commercial sources.
Cell Culture and Oxidative Stress
Mouse embryonic fibroblasts (MEFs) were derived from wild-type and caveolin-1 null mice as described previously (32). MEFs and MDA-MB-231 cells were grown in DMEM supplemented with glutamine, antibiotics (penicillin and streptomycin), and 10% fetal bovine serum. NIH 3T3 cells were grown in DMEM supplemented with glutamine, antibiotics (penicillin and streptomycin), and 10% donor bovine calf serum. WI-38 cells were grown in Eagle's minimum essential medium supplemented with glutamine, antibiotics (penicillin and streptomycin), and 10% donor bovine calf serum. PC-3 human prostate cancer cells were grown in Ham's F-12 medium supplemented with glutamine, antibiotics (penicillin and streptomycin), and 10% fetal bovine serum. Oxidative stress was induced by subcytotoxic levels of hydrogen peroxide (150 μm for MEFs and 450 μm for WI-38 cells) for 2 h. Cells were then recovered in normal medium for different periods of time (see text for details).
GST Fusion Protein Pulldown Assay
The GST-caveolin-1 (GST-Cav-1) fusion protein constructs were transformed into Escherichia coli (BL21 strain, Novagen, Inc.). After induction of expression through addition of 5 mm isopropyl 1-thio-β-d-galactopyranoside (Sigma), GST-Cav-1 constructs were affinity-purified on glutathione-agarose beads using the detergent Sarcosyl for initial solubilization. GST-Cav-1 and GST alone (bound to glutathione-agarose beads) were washed three times with TNET buffer (50 mm Tris (pH 8.0), 150 mm NaCl, 5 mm EDTA, and 1% Triton X-100) containing protease inhibitors. SDS-PAGE followed by Coomassie staining was used to determine the concentration of GST-Cav-1 per 100 μl of packed bead volume. Precleared cell lysates were diluted in buffer A (10 mm Tris (pH 8.0) and 0.1% Tween 20) and added to ∼100 μl of equalized bead volume for overnight incubation at 4 °C. After binding, the beads were extensively washed with phosphate-buffered saline (six times). Finally, the beads were resuspended in 3× sample buffer and subjected to SDS-PAGE.
Immunoblotting
Cells were collected in boiling sample buffer. Cellular proteins were resolved by SDS-PAGE (12.5% acrylamide) and transferred to BA83 nitrocellulose membranes (Schleicher & Schüll). Blots were incubated for 2 h in TBST (10 mm Tris-HCl (pH 8.0), 150 mm NaCl, and 0.2% Tween 20) containing 2% powdered skim milk and 1% BSA. After three washes with TBST, membranes were incubated for 2 h with the primary antibody and for 1 h with horseradish peroxidase-conjugated goat anti-rabbit/mouse IgG. Bound antibodies were detected using an ECL detection kit (Pierce).
Preparation of Caveolae-enriched Membrane Fractions
Cells were scraped into 2 ml of Mes-buffered saline containing 1% (v/v) Triton X-100. Homogenization was carried out with 10 strokes of a loose-fitting Dounce homogenizer. The homogenate was adjusted to 40% sucrose by addition of 2 ml of 80% sucrose prepared in Mes-buffered saline and placed at the bottom of an ultracentrifuge tube. A 5–30% linear sucrose gradient was formed above the homogenate and centrifuged at 45,000 rpm for 16–20 h in a SW60 rotor (Beckman Coulter, Fullerton, CA). A light-scattering band confined to the 15–20% sucrose region was observed that contained endogenous caveolin-1 but excluded most of other cellular proteins. From the top of each gradient, 1-ml gradient fractions were collected to yield a total of 11 fractions. Fractions 4–6, representing caveolar membranes, and fractions 9–11, representing non-caveolar membranes, were pooled together. An equal amount of protein from each of the two groups was separated by SDS-PAGE and subjected to immunoblot analysis.
Coimmunoprecipitation
Cells were washed twice with PBS and lysed for 30 min at 4 °C in a buffer containing 10 mm Tris (pH 8.0), 0.15 m NaCl, 5 mm EDTA, 1% Triton X-100, and 60 mm octyl glucoside. Samples were precleared for 1 h at 4 °C using protein A-Sepharose (20-μl slurry, 1:1) and subjected to overnight immunoprecipitation at 4 °C using the intended antibody and protein A-Sepharose (30-μl slurry, 1:1). After three washes with the immunoprecipitation buffer, samples were separated by SDS-PAGE (12.5% acrylamide) and transferred to nitrocellulose. Then blots were probed with the intended antibody. Experiments were performed three times independently, and representative images are shown.
RNA Isolation and RT-PCR
Cells were collected, and total RNA was isolated using the RNeasy mini kit from Qiagen (Valencia, CA). Equal amounts of RNA were treated with RNase-free DNase and subjected to reverse transcription using the Advantage RT-for-PCR kit from Clontech (Mountain View, CA) according to the recommendations of the manufacturer. PCR was then performed in the exponential linear zone of amplification for each gene studied. The IL-6-specific primers used were as follows: forward, GAAGAACAACTTAAAAGATAA; reverse, GTCCTTAGCCACTCCTTCTGT. Sequences corresponding to either LR32 or GAPDH were also amplified as internal controls.
siRNA Treatment
Knockdown of Sirt1 expression was achieved by transfection of cells with siGENOME mouse Sirt1 siRNA SMARTpool from Thermo Scientific (catalog no. 93759) using Lipofectamine 2000 (Invitrogen).
Sirt1 Activity Assay
10 μg of either GST or GST-Cav-1 (82–101) were incubated with 1.5 μg of human recombinant Sirt1 at 4 °C for 3 h. Sirt1 activity was then measured using the Sirt1 assay kit from Sigma (catalog no. cs1040) according to the recommendation of the manufacturer.
IL-6 Promoter Reporter Assay
Cells were seeded in 60-mm dishes at 270,000 cells/dish. The following day, cells were treated with 150 μm H2O2 for 2 h, washed twice, and incubated in complete medium at 37 °C. After 24 h, cells were transiently transfected using a modified calcium phosphate precipitation method with 2 μg of a luciferase reporter construct carrying either wild-type or mutant IL-6 promoter sequences (provided by Dr. O. Eickelberg (39)) and 1 μg of a β-galactosidase-expressing construct. Forty-eight hours later, cells were lysed in 500 μl of extraction buffer. 200 μl was used to measure luciferase activity, and 150 μl was used to measure β-galactosidase activity. Three independent experiments were performed for each condition.
Acid β-Galactosidase Staining
Cells were subjected to acid β-galactosidase staining using the senescence-associated β-galactosidase staining kit (Cell Signaling Technology) according to the recommendations of the manufacturer. Briefly, cells were washed twice with PBS and fixed with fixative solution for 15 min. Then cells were washed twice with PBS and incubated overnight at 37 °C with the staining solution. Cells were then examined for the development of blue color. Cells were photographed at ×40 magnification using a BX50WI Olympus optical light microscope (Tokyo, Japan).
Measurement of Cytokine Production
Conditioned medium from untreated and hydrogen peroxide-treated WT and Cav-1 KO MEFs was collected, and proteins were concentrated using a centrifugal filter device (Amicon). Samples were then normalized by BCA protein assay, and the concentration of interleukins was determined using a multiplex bead-based Luminex assay (Bio-Rad) according to the instructions of the manufacturer.
Conditioned Medium Studies
Wild-type and caveolin-1 null MEFs were treated with 150 μm hydrogen peroxide for 2 h. After hydrogen peroxide treatment, cells were washed with PBS and cultured in complete medium for 4 days. MEFs were then serum-starved for 1 day. Conditioned medium was then collected and used to culture 37,500 serum-starved RasG12V-transformed NIH 3T3, PC-3, or MDA-MB-231 cells. Cells were grown for 5 days. Cancer cell growth was quantified by counting the number of nuclei, after DAPI staining, in 30 random fields; by counting the number of Ki67-positive cells in 30 random fields; and by crystal violet staining.
RESULTS
Caveolin-1 Binds Directly to Sirt1
The scaffolding domain of caveolin-1, which is represented by residues 82–101, mediates direct protein-protein interactions between caveolin-1 and a variety of signaling molecules carrying the caveolin binding domain (ΦXΦXXXXΦ, ΦXXXXΦXXΦ, or ΦXΦXXXXΦXXΦ, where Φ represents an aromatic amino acid and X represents any amino acid) (40–42). Analysis of the Sirt1 protein sequence indicates that Sirt1 has a putative caveolin-binding domain between amino acids 310 and 317 (Fig. 1A). Therefore, to investigate whether caveolin-1 is a binding partner of Sirt1, we performed pulldown assays using a series of caveolin-1 deletion mutants fused to GST (Fig. 1B). Fig. 1C shows that Sirt1 is a caveolin-1-binding protein and that the scaffolding domain of caveolin-1 was sufficient for binding to Sirt1. To determine whether Sirt1 binds directly to caveolin-1, we performed GST pulldown assays using recombinant Sirt1. We found that human recombinant Sirt1 bound to caveolin 1 (residues 82–101)-GST (Fig. 1D). From these data, we conclude that the scaffolding domain of caveolin-1 (amino acids 82–101) binds directly to Sirt1.
FIGURE 1.
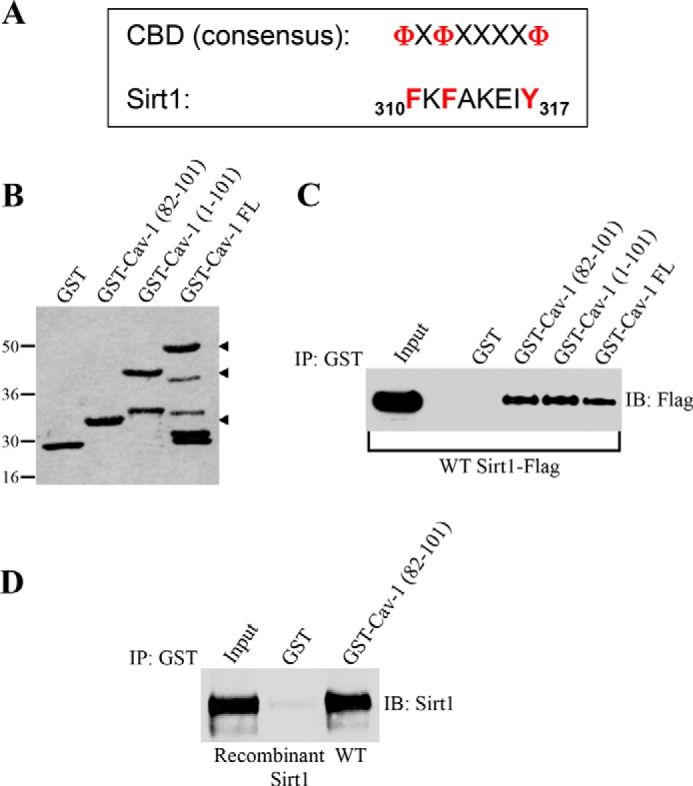
Sirt1 directly interacts with caveolin-1 in vitro. A, schematic showing the consensus caveolin-binding domain (CBD) and the caveolin-binding domain of Sirt1 (amino acids 310–317). Φ represents an aromatic amino acid, and X represents any amino acid. B, Ponceau S staining of GST alone and caveolin-1-GST fusion proteins. FL, full-length. C, caveolin-1-GST fusion protein pulldown assays were performed using cell lysates from NIH 3T3 cells transiently transfected with Sirt1-FLAG. IP, immunoprecipitation; IB, immunoblot. D, pulldown assays between recombinant Sirt1 and affinity-purified GST alone or caveolin-1 (residues 82–101)-GST fusion protein.
Oxidative Stress Promotes the Interaction of Caveolin-1 with Sirt1
Because we observed a direct interaction between caveolin-1 and Sirt1 in in vitro studies, we asked whether Sirt1 can interact with caveolin-1 in cells. Coimmunoprecipitation studies in WI-38 human diploid fibroblasts showed that Sirt1 and caveolin-1 did not interact under resting conditions (Fig. 2, A and B). However, Sirt1 strongly interacted with caveolin-1 72 h after stimulation of WI-38 cells with sublethal doses of hydrogen peroxide, which, as we have shown previously, induces premature senescence (32–38) (Fig. 2, A and B). Consistent with the data of Fig. 1, only the scaffolding domain of caveolin-1 (82–101), but not deletion mutants of caveolin-1 lacking the caveolin-1 scaffolding domain, fused to GFP interacted with Sirt1 72 h after oxidative stress (Fig. 2C). Because caveolin-1 is a structural component of caveolae, these data were confirmed in Fig. 2, D and E, by showing that Sirt1 was excluded from caveolar membranes before oxidative stress and moved to caveolae 72 h after oxidant stimulation in both WI-38 cells and MEFs. Importantly, the Sirt1/caveolin-1 interaction was not specific to hydrogen peroxide stimulation, as shown by the ability of UV-C light to promote binding of caveolin-1 to Sirt1 (Fig. 2F).
FIGURE 2.
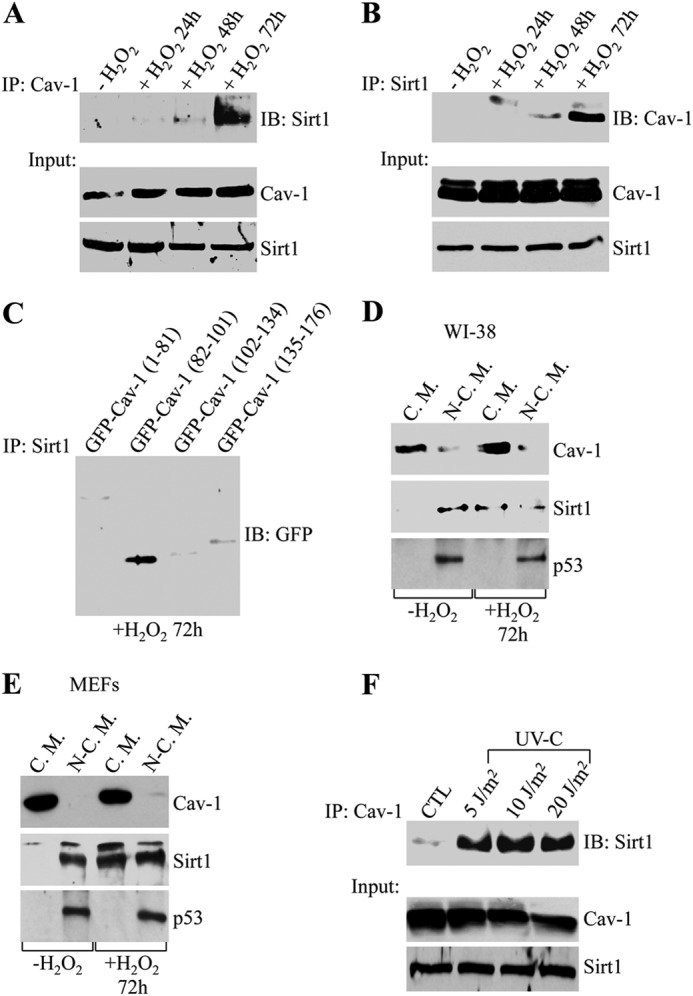
Sirt1 interacts with caveolin-1 in vivo. WI-38 human diploid fibroblasts were treated with sublethal doses of hydrogen peroxide (450 μm) for 2 h. Cells were then recovered in complete medium for different periods of time (24, 48, and 72 h). Untreated cells (−H2O2) were used as a control. A, cell lysates were immunoprecipitated (IP) using an antibody probe specific for caveolin-1 (Cav-1), and immunoprecipitates were subjected to immunoblot (IB) analysis with anti-Sirt1 IgGs. Total expression of Sirt1 and caveolin-1 is shown in the bottom panels. B, cell lysates were immunoprecipitated using an antibody probe specific for Sirt1, and immunoprecipitates were subjected to immunoblot analysis with anti-caveolin-1 IgGs. Total expression of Sirt1 and caveolin-1 is shown in the bottom panels. C, caveolin-1-GFP fusion proteins were expressed in WI-38 cells at comparable levels (data not shown). Cells were treated with 450 μm H2O2 for 2 h and recovered in complete medium for 72 h. Cell lysates were immunoprecipitated using an antibody probe specific for Sirt1, and immunoprecipitates were subjected to immunoblot analysis with anti-GFP IgGs. D and E, cellular fractions containing caveolar membranes (C.M.) were isolated from cellular fractions containing the rest of cellular proteins (N-C.M.) by sucrose gradient centrifugation in WI-38 cells (D) and MEFs (E). Expression of caveolin-1, Sirt1, and p53 into caveolar and non-caveolar fractions was determined by immunoblot analysis using antibody probes specific for caveolin-1, Sirt1, and p53, respectively. F, WI-38 cells were exposed to sublethal doses of UV-C light (5, 10, and 20 J/m2). Cells were allowed to recover in complete medium for 72 h. Untreated cells (CTL) were used as a control. Cell lysates were immunoprecipitated using an antibody probe specific for caveolin-1, and immunoprecipitates were subjected to immunoblot analysis with anti-Sirt1 IgGs. Total expression of Sirt1 and caveolin-1 is shown in the bottom panels.
Oxidant-induced Binding of Sirt1 to Caveolin-1 Inhibits Sirt1 Activity, Promotes Acetylation of p53, and Induces Premature Senescence
What is the functional consequence of the binding of Sirt1 to caveolin-1 after oxidative stress? To directly answer this question, we first assessed Sirt1 activity in vitro in the presence or absence of the scaffolding domain of caveolin-1 fused to GST (GST-Cav-1 (82–101)). We show in Fig. 3A that the incubation with the purified GST-Cav-1 (82–101) peptide inhibited Sirt1 activity by ∼45% compared to GST alone. These data suggest that the recruitment of Sirt1 into caveolar membranes, after oxidative stress, inhibits Sirt1 activity. If this were the case, one would expect increased acetylation of Sirt1 targets after oxidative stress in caveolin-1-expressing but not caveolin-1-lacking cells. Since (i) activation of the tumor suppressor p53 is associated with the development of cellular senescence, (ii) acetylation activates p53, (iii) p53 is a Sirt1 target protein, and (iv) we have shown previously that overexpression of caveolin-1 activates p53 (37), we examined the acetylation state of p53 in wild-type and caveolin-1 null mouse embryonic fibroblasts under resting conditions and under conditions of oxidative stress that lead to premature senescence. We found that sublethal doses of hydrogen peroxide promoted acetylation of p53 48–72 h after oxidative stress in wild-type MEFs (Fig. 3B). In contrast, free radical-induced acetylation of p53 was significantly inhibited in cells lacking caveolin-1 (Fig. 3B). Interestingly, down-regulation of Sirt1 by siRNA enhanced oxidant-induced acetylation of p53 in caveolin-1 null MEFs to levels seen in wild-type MEFs (Fig. 3E). Because Sirt1 expression is similarly up-regulated in both wild-type and caveolin-1 null MEFs after oxidative stress (Fig. 3, C and D), the reduced acetylation of p53 in caveolin-1 null fibroblasts is the consequence of Sirt1 not having been sequestered in caveolar membranes and having been inhibited by caveolin-1. Therefore, sequestration/inhibition of Sirt1 into caveolar membranes, after oxidative stress, prevents Sirt1-mediated deacetylation of p53. In support of these data, p53 is excluded from caveolar membranes before and after oxidant stimulation (Fig. 2, D and E). Importantly, down-regulation of Sirt1 restored the ability of caveolin-1 null MEFs to undergo oxidative stress-induced premature senescence (Fig. 3F). Moreover, re-expression of caveolin-1 in caveolin-1 null MEFs rescued oxidative stress-induced acetylation of p53 (Fig. 4A) and premature senescence (Fig. 4, B and C). Together, these data indicate that oxidative stress promotes the acetylation/activation of p53 and the development of premature senescence through the caveolin-1-mediated inhibition of Sirt1.
FIGURE 3.

Inhibition of Sirt1 by caveolin-1 promotes oxidative stress-induced acetylation of p53 and premature senescence. A, 10 μg of either purified GST alone or caveolin-1 (residues 82–101)-GST was incubated with 1.5 μg of human recombinant Sirt1 for 3 h at 4 °C. Sirt1 activity was then measured using the Sirt1 assay kit (Sigma-Aldrich). Values represent mean ± S.E. *, p < 0.005. B–D, WT and caveolin-1 null (Cav-1 KO) mouse embryonic fibroblasts were treated with sublethal doses of hydrogen peroxide (150 μm) for 2 h. Cells were then recovered in complete medium for different periods of time (24, 48, and 72 h). Untreated cells (−H2O2) were used as a control. Expression of total p53 (B), acetylated p53 (B), and Sirt1 (C) was measured by immunoblot analysis using specific antibody probes. Quantification of Sirt1 expression is shown in (D). E, WT and Cav-1 KO MEFs were transfected with either scrambled (Sc.) siRNA or Sirt1 siRNA. After 24 h, cells were treated with sublethal doses of hydrogen peroxide (150 μm) for 2 h. Cells were then recovered in complete medium for 72 h. Total expression of p53 and Sirt1 and that of acetylated p53 (Ac-p53) were measured by immunoblotting analysis using specific antibody probes. F, WT and Cav-1 KO MEFs were transfected with either scrambled siRNA or Sirt1 siRNA as indicated. After 24 h, cells were treated with sublethal doses of hydrogen peroxide (150 μm) for 2 h. Cells were then recovered in complete medium for 7 days and subjected to senescence-associated β-galactosidase (SA-βgal) activity assays. Values represent mean ± S.E. *, p < 0.001.
FIGURE 4.

Re-expression of caveolin-1 in caveolin-1 null MEFs rescues oxidative stress-induced acetylation of p53 and premature senescence. Overexpression of caveolin-1 promotes premature senescence in wild-type but not p53 null MEFs. A–C, caveolin-1 was re-expressed in caveolin-1 null MEFs using a viral vector (pLVX-Cav1) at levels comparable with that of wild-type cells (data not shown). Infection of wild-type and caveolin-1 null MEFs with vector alone (pLVX) was used as a control. After 24 h, cells were treated with hydrogen peroxide (150 μm) for 2 h and recovered in complete medium for either 72 h (A) or 7 days (B and C). A, cells were collected, and cell lysates were subjected to immunoblot analysis using antibody probes specific for total p53 and acetylated p53 (Ac-p53). B and C, cells were subjected to senescence-associated β-galactosidase (SA-βgal) activity assays. Quantification of the assays is shown in B. Representative images are shown in C. Values in B represent mean ± S.E. *, p < 0.001. D–F, WT and p53 KO MEFs were infected with either vector alone (pLVX) or pLVX-Cav-1. After 24 h, cells were subjected to sublethal doses of hydrogen peroxide (150 μm) for 2 h and allowed to recover for 4 days. Overexpression of caveolin-1 was confirmed by immunoblot analysis (data not shown). D and E, cells were subjected to senescence-associated β-galactosidase activity assays. Quantification of the assays is shown in D. Representative images are shown in E. Values in D represent mean ± S.E. *, p < 0.001. F, cell lysates were subjected to immunoblot analysis with antibody probes specific for p53 and p21. Immunoblotting with anti-β-actin IgGs was performed as an internal control.
Caveolin-1-mediated Premature Senescence Requires p53
We have shown previously that overexpression of caveolin-1 promotes premature senescence (37). To independently confirm the data of Figs. 1–3 showing that it is the activation of p53 that mediates caveolin-1-induced premature senescence, we asked whether caveolin-1 fails to induce premature senescence in a null p53 background. To this end, we took advantage of p53 null mouse embryonic fibroblasts. Wild-type and p53 null MEFs were infected with a retroviral vector expressing caveolin-1. Cells were then subjected to oxidative stress to induce premature senescence. We found that wild-type MEFs expressing vector alone underwent premature senescence 7 days after oxidative stress (data not shown), similarly to non-infected WT MEFs (Fig. 3F). In contrast, wild-type MEFs overexpressing caveolin-1 developed a senescent phenotype 4 days after oxidative stress, as determined by senescent-associated β-galactosidase staining (Fig. 4, D and E) and p21 up-regulation (Fig. 4F). Importantly, overexpression of caveolin-1 failed to accelerate the development of SIPS in MEFs lacking p53 expression (Figs. 4, D–F). Together, these data indicate that activation of p53 is a major molecular mechanism through which caveolin-1 promotes SIPS.
Phosphorylation of Caveolin-1 Mediates Sirt1/p53/Senescence Signaling
What is the upstream molecular signal that promotes the localization of Sirt1 in caveolar membranes and the consequent activation of the p53/senescence pathway? To directly address this question, we determined whether posttranslational modifications of caveolin-1 could play a role. More specifically, we focused on phosphorylation of caveolin-1 on tyrosine 14, which we have shown previously to occur in response to cellular stress (43). WI-38 cells were subjected to oxidative stress, and phosphorylation of caveolin-1 was determined by immunoblot analysis. We show in Fig. 5A that oxidant stimulation promoted phosphorylation of caveolin-1 on tyrosine 14. Under the same conditions of oxidative stress, p38 MAPK was activated (Fig. 5A). Caveolin-1 phosphorylation was inhibited by the p38 MAPK-specific inhibitor SB203580 (Fig. 5A). These results suggest that p38 MAPK links oxidative stress to phosphorylation of caveolin-1. Interestingly, inhibition of p38 MAPK also inhibited the sequestration of Sirt1 into caveolar membranes (Fig. 5B) and SIPS (Fig. 5C). Because our data show that sequestration of Sirt1 into caveolar membranes plays an important role in SIPS, that oxidative stress promotes the activation of p38 MAPK and the phosphorylation of caveolin-1, and that inhibition of p38 MAPK inhibits phosphorylation of caveolin-1 and sequestration of Sirt1 into caveolar membranes and SIPS, we tested the hypothesis that phosphorylation of caveolin-1 on tyrosine 14 is involved in the regulation of caveolin-1-mediated SIPS. To directly test this hypothesis, we used a tyrosine-to-alanine mutant form of caveolin-1 that is unable to undergo tyrosine phosphorylation at residue 14 (Y14A-Cav-1 (44)) and stably expressed Y14A-Cav-1 in WI-38 cells. We found that oxidative stress failed to promote the localization of Sirt1 into caveolar membranes in cells expressing Y14A-Cav-1 (Fig. 5D) in which oxidant-induced acetylation of p53 was inhibited (Fig. 5E). Consistent with these data, oxidant-induced premature senescence was inhibited in Y14A-Cav-1-expressing cells (Fig. 5, F and G). Together, these data suggest that Y14A-Cav-1 acts in a dominant negative manner and that phosphorylation of caveolin-1 on tyrosine 14 is a posttranslational modification that promotes the oxidant-induced localization of Sirt1 in caveolae and the subsequent activation of the p53/senescence pathway in fibroblasts.
FIGURE 5.

Phosphorylation of caveolin-1 on tyrosine 14 promotes the oxidant-induced sequestration of Sirt1 in caveolae and the activation of the p53/senescence pathway. A–C, WI-38 cells were subjected to oxidative stress (450 μm H2O2 for 2 h) and recovered in complete medium for 72 h (A and B) or 7 days (C) in the presence or absence of the p38 MAPK inhibitor SB203580 (SB, 5 μm). DMSO treatment was performed as a control. A, expression levels of phosphorylated caveolin-1 (P-Cav-1), total caveolin-1, phosphorylated p38 MAPK (P-p38 MAPK), and total p38 MAPK was quantified by immunoblot analysis using specific antibody probes. Immunoblotting with anti-β-actin IgGs was performed to show equal loading. B, cellular fractions containing caveolar membranes (C.M.) were isolated from cellular fractions containing the rest of cellular proteins (N-C.M.) by sucrose gradient centrifugation. Expression of Sirt1 into caveolar and non-caveolar fractions was determined by immunoblot analysis using anti-Sirt1 IgGs. C, cells were subjected to senescence-associated β-galactosidase (SA-βgal) activity assays. Quantification of the assays is shown. Values represent mean ± S.E. *, p < 0.001. D–G, WI-38 cells were infected with a viral vector (pLVX) expressing a mutant form of caveolin-1 in which tyrosine 14 was substituted with alanine (Y14A Cav-1-Myc). Expression of wild-type caveolin-1 (WT Cav-1-Myc) was used as a control. After 24 h, cells were subjected to oxidative stress (450 μm H2O2) for 2 h. Cells were recovered in complete medium for 72 h (D and E) or 7 days (F and G). D, cellular fractions containing caveolar membranes were isolated from cellular fractions containing the rest of cellular proteins by sucrose gradient centrifugation. Levels of exogenously expressed WT and Y14A caveolin-1 and that of Sirt1 into caveolar and non-caveolar fractions were determined by immunoblot analysis using antibody probes specific for Myc and Sirt1, respectively. E, expression levels of p16, acetylated p53 (Ac-p53), total p53, and Sirt1 were quantified by immunoblot analysis using specific antibody probes. Immunoblotting with anti-β-actin IgGs was performed to show equal loading. F and G, cells were subjected to senescence-associated β-galactosidase activity assays. d, days. Quantification of the assays is shown in F. Representative images are shown in G. Values in F represent mean ± S.E. *, p < 0.001.
IL-6 Is Released by Senescent Fibroblasts in a Caveolin-1-specific Manner, and Inhibition of Sirt1 by Caveolin-1 Stimulates IL-6 Gene Transcription
Because senescent cells can secrete factors that stimulate the growth of cancer cells, we decided to test the hypothesis that caveolin-1-specific, protumorigenic mediators are released by senescent fibroblasts following inhibition of Sirt1 by caveolin-1. To this end, wild-type and caveolin-1 null MEFs were subjected to oxidative stress to induce premature senescence. Untreated cells were used as controls. Seven days after oxidative stress, when mouse embryonic fibroblasts displayed a senescent phenotype (Fig. 3F and as we described previously (32–34, 36–38)), conditioned medium from actively proliferating and senescent MEFs was collected, and the expression level of a panel of interleukins was determined by Luminex analysis. We show in Fig. 6A that oxidative stress promoted the expression/secretion of IL-6 in a caveolin-1-dependent manner. This finding was confirmed by immunoblot analysis in which we showed that H2O2 induced the accumulation of IL-6 into the conditioned medium of wild-type but not caveolin-1 null MEFs (Fig. 6B). The accumulation of IL-6 into the conditioned medium of wild-type MEFs is the consequence of increased transcription of the IL-6 gene, as shown by increased IL-6 mRNA levels in wild-type but not caveolin-1 null MEFs following oxidative stress (Fig. 6C). Consistent with these results, down-regulation of caveolin-1 expression by siRNA in fibroblasts inhibited oxidant-induced activation of the IL-6 promoter (Fig. 6D). The lack of up-regulation of IL-6 gene transcription following oxidative stress in caveolin-1-lacking fibroblasts is due to the loss of caveolin-1-mediated inhibition of Sirt1, as shown by the restored free radical-induced up-regulation of IL-6 mRNA (Fig. 6C) and activation of the IL-6 promoter (Fig. 6D) in caveolin-1-lacking fibroblasts in which Sirt1 expression was down-regulated by siRNA. Because inhibition of Sirt1 by caveolin-1 leads to the activation of p53, one would expect reduced transcriptional activation of IL-6 by oxidative stress in cells lacking p53 expression. As predicted, we show in Fig. 6, E and F, that oxidative stress activated the IL-6 promoter and up-regulated IL-6 mRNA levels, respectively, in p53 WT but not in p53 knockout MEFs. In addition, we find that re-expression of caveolin-1 in caveolin-1 null MEFs rescued oxidant-induced up-regulation of IL-6 mRNA (Fig. 6G). Finally, we demonstrate that oxidant-induced up-regulation of IL-6 expression was inhibited in Cav-1-Y14A-expressing cells (Fig. 6H). Therefore, IL-6 is a caveolin-1-specific component of the senescent associated secretory phenotype whose expression by senescent fibroblasts occurs through the caveolin-1-mediated inhibition of Sirt1.
FIGURE 6.

IL-6 is a caveolin-1-specific senescence-associated cytokine. A and B, WT and caveolin-1 null (Cav-1 KO) MEFs were treated with sublethal doses (150 μm) of hydrogen peroxide for 2 h. Untreated cells (−H2O2) were used as a control. A, cells were recovered in complete medium for 6 days. MEFs were then serum-starved for 24 h, and their conditioned medium was subjected to multiplex bead-based Luminex assays. Values represent mean ± S.E. *, p < 0.001. B, cells were recovered in complete medium for different periods of time (1 day, 4 days, and 6 days) and serum-starved for 24 h. Conditioned medium was then subjected to immunoblotting analysis using an antibody probe specific for IL-6. Immunoblotting with anti-β-actin IgGs was performed as an internal control. C, wild-type and caveolin-1 null MEFs were transfected with either scrambled (Sc.) or Sirt1 siRNA as indicated. After 24 h, cells were left untreated (−H2O2) or treated with 150 μm hydrogen peroxide for 2 h and recovered in complete medium for 72 h. IL-6 mRNA levels were determined by RT-PCR using primers specific for IL-6. RT-PCR using LR32-specific primers was performed as a control. D, NIH 3T3 fibroblasts were treated with sublethal doses of hydrogen peroxide (150 μm) for 2 h and recovered in complete medium for 24 h. Cells were then transfected with either pGL3 or p-IL-6-Luc651 in the presence of either scrambled or Sirt1 siRNA as indicated. pIL-6-Luc651 is a previously characterized luciferase-based reporter in which the IL-6 promoter was cloned upstream of the luciferase gene (39). After 48 h, cells were collected, and luciferase activity was measured. Untreated cells (−H2O2) were used as a control. Values represent mean ± S.E. * and #, p < 0.001. E, wild-type and p53 KO MEFs were treated with 150 μm hydrogen peroxide for 2 h and recovered in complete medium for 24 h. Cells were then transfected with pIL-6-Luc651. After 48 h, cells were collected, and the luciferase activity was measured. Untreated cells (−H2O2) were used as a control. Values represent mean ± S.E. *, p < 0.001. F, wild-type and p53 KO MEFs were treated with 150 μm hydrogen peroxide for 2 h and recovered in complete medium for 72 h. IL-6 mRNA levels were determined by RT-PCR using primers specific for IL-6. RT-PCR using LR32-specific primers was performed as a control. G, caveolin-1 was re-expressed in caveolin-1 null MEFs using a viral vector (pLVX-Cav1) at levels comparable to that of wild-type cells (data not shown). Infection of wild-type and caveolin-1 null MEFs with vector alone (pLVX) was used as a control. After 24 h, cells were treated with hydrogen peroxide (150 μm) for 2 h and recovered in complete medium for 72 h. IL-6 mRNA levels were determined by RT-PCR using primers specific for IL-6. RT-PCR using LR32-specific primers was performed as a control. H, WI-38 cells were infected with a viral vector (pLVX) expressing Y14A Cav-1-Myc. Expression of WT Cav-1-Myc was used as a control. After 24 h, cells were subjected to oxidative stress (450 μm H2O2) for 2 h. Cells were recovered in complete medium for 72 h. IL-6 mRNA levels were determined by RT-PCR using primers specific for IL-6. RT-PCR using GAPDH-specific primers was performed as a control.
The Caveolin-1-dependent Secretion of IL-6 by Senescent Fibroblasts Stimulates Cancer Cell Growth
What is the functional consequence of the caveolin-1-mediated secretion of IL-6 by senescent fibroblasts? Evidence shows that senescent stromal cells, through the development of a senescent-associated secretory phenotype, can stimulate the hyperproliferation and malignant progression of preneoplastic and neoplastic epithelial cells (24–27). Because IL-6 is a major component of the senescence-associated secretory phenotype that promotes the proliferation of a variety of epithelial cancer cells, we asked whether caveolin-1-expressing senescent fibroblasts stimulate the proliferation of cancer cells through the secretion of IL-6. To this end, wild-type and caveolin-1 null mouse embryonic fibroblasts were subjected to sublethal doses of oxidative stress to induce premature senescence. Untreated cells were used as a control. Then conditioned medium from either wild-type or caveolin-1 null MEFs was used to grow PC3 prostate cancer epithelial cells. We chose prostate cancer cells because the accumulation of senescent cells has been described at sites of preneoplastic prostate cancer lesions (45, 46). We show in Fig. 7A that conditioned medium from free radical-treated wild-type MEFs stimulated the growth of PC3 cells compared with PC3 cells grown with unconditioned medium, as determined by Ki67 staining. In contrast, conditioned medium from H2O2-treated caveolin-1 null MEFs only marginally enhanced the growth of PC3 cells (Fig. 7A). The ability of senescent fibroblasts to stimulate the proliferation of PC3 cells was mostly due to the caveolin-1-dependent secretion of IL-6, as shown by a 60% reduction of PC-3 cell proliferation when PC-3 cells were grown in the presence of conditioned medium from wild-type fibroblasts containing a well characterized IL-6-specific neutralizing antibody (Fig. 7, B and C). To determine whether the enhancement of proliferation by the caveolin-1-mediated secretion of IL-6 was not limited to prostate cancer cells, the proliferation of MDA-MB-231 breast cancer and H-Ras (G12V)-transformed NIH-3T3 cells was tested using conditioned medium from senescent wild-type MEFs in the presence or absence of neutralizing IL-6 IgGs. We found that the neutralization of IL-6 inhibited the enhancement of H-Ras (G12V)-transformed cell proliferation that was induced by conditioned medium from senescent caveolin-1-expressing MEFs, as determined by both Ki67 (Fig. 7D) and DAPI (Fig. 7E) staining. Similarly, IL-6 IgGs inhibited the ability of conditioned medium from wild-type MEFs to stimulate the growth of MDA-MB-231 breast cancer cells (Fig. 7F). We conclude that the prostimulatory properties of IL-6 that is secreted by senescent fibroblasts are caveolin-1-dependent.
FIGURE 7.

Stimulation of cancer cell growth by the caveolin-1-mediated release of IL-6 from senescent fibroblasts. A, wild-type and caveolin-1 null MEFs were treated with sublethal doses of hydrogen peroxide (150 μm) for 2 h. Cells were recovered in complete medium for 6 days. Cells were then serum-starved for 24 h, and their conditioned medium was used to culture serum-starved PC3 cells for 5 days. The number of proliferating cells was quantified by Ki67 staining. PC3 cells grown with 0% serum were used as a control. Values represent mean ± S.E. * and #, p < 0.001. B and C, conditioned medium from wild-type MEFs was derived as described in A and used to culture serum-starved PC3 cells in the presence of either control (CTL) IgGs or anti-IL-6 IgGs. B, cell proliferation was quantified by Ki67 staining. Values represent mean ± S.E. * and #, p < 0.001. C, cell proliferation was measured by crystal violet staining. D and E, conditioned medium from wild-type MEFs was derived as described in A and used to culture serum-starved Ras-transformed NIH 3T3 cells in the presence of either control IgGs or anti-IL-6 IgGs. D, cell proliferation was quantified by Ki67 staining. Values represent mean ± S.E. * and #, p < 0.001. E, cell number was measured by DAPI staining. Values represent mean ± S.E. * and #, p < 0.001. F, conditioned medium from wild-type MEFs was derived as described in A and used to culture serum-starved MDA-MB-231 cells in the presence of either control IgGs or anti-IL-6 IgGs. Cell proliferation was quantified by crystal violet staining.
DISCUSSION
Our understanding of the molecular mechanisms through which oxidative stress promotes premature senescence remains to be fully explored. Activation of the p53/p21Waf1/Cip1 pathway plays a critical role in the development of SIPS. Acetylation is a posttranslational modification that can activate p53 functions. Oxidative stress promotes acetylation of p53 by removing the inhibitory effect of Sirt1 on p53. How prosenescent levels of oxidative stress prevent Sirt-1-mediated deacetylation/inactivation of p53 remains unknown. Our findings directly fill this gap of knowledge. We demonstrate that caveolin-1 is a novel endogenous inhibitor of Sirt1. Although caveolin-1 and Sirt1 do not interact under resting conditions, oxidative stress promotes the sequestration of Sirt1 into caveolar membranes, where it interacts with caveolin-1. Because we found that p53 is excluded from caveolae before and after oxidative stress, targeting of Sirt1 in caveolae following oxidant stimulation frees p53 from the inhibitory action of Sirt1. As a result, acetylation of p53 is increased dramatically. Acetylation of p53 leads to activation of downstream targets, such as p21, and development of premature senescence. When the oxidant-induced sequestration of Sirt1 into caveolar membranes is prevented in caveolin-1 null cells, acetylation/activation of p53 and development of SIPS are inhibited. Importantly, when Sirt1 expression is down-regulated in caveolin-1 null cells, SIPS is improved significantly, providing direct evidence that sustained deacetylation of p53 by Sirt1 contributes to reduced SIPS in caveolin-1 null fibroblasts. Our results also suggest that caveolin-1, in addition to indirectly inhibiting deacetylation of Sirt1 targets by keeping Sirt1 in caveolae after oxidative stress, may directly inhibit Sirt1 activity. In fact, our in vitro studies show that purified Sirt1 is inhibited by recombinant caveolin-1. Consistent with this possibility, the caveolin-binding domain of Sirt1 (amino acids 310–317) is within the catalytic core domain of Sirt1 (amino acids 244–498). More specifically, it overlaps with the catalytic core small domain of Sirt1, which is represented by amino acids 272–318. Therefore, we provide evidence that the caveolin-1-mediated inhibition of Sirt1 plays a pivotal role in the signaling linking oxidative stress to the development of premature senescence.
We found that stress-induced premature senescence is inhibited in caveolin-1 null MEFs. We also show that, although down-regulation of Sirt1 expression in caveolin-1 null MEFs significantly improves SIPS, it does not totally rescue it. These data suggest that inhibition of Sirt1 by caveolin-1 is only one of the mechanisms through which caveolin-1 promotes SIPS. This is consistent with our previous findings that caveolin-1 activates p53 by inhibiting Mdm2 (32), a negative regulator of p53, and activating Ataxia Telangiectasia Mutated (ATM) (36), a positive regulator of p53. Therefore, caveolin-1 acts as a master regulator of p53 activation by directly regulating independent signaling cascades that converge to activate the p53/p21Waf1/Cip1/senescence pathway. It remains to be determined whether, similar to inhibition of Sirt1, both inhibition of Mdm2 and activation of ATM contribute to the caveolin-1-mediated secretion of IL-6.
Tumor growth is dependent on the molecular changes that occur within cancer cells. However, the tumor microenvironment also plays a pivotal role in tumor progression. Growing evidence suggests that senescent cells, through the release of SASP factors, can modulate the tissue microenvironment and, therefore, contribute to the development of age-associated diseases, including cancer. IL-6 is a well established SASP factor that has been linked to the pathogenesis of a variety of cancers. Therefore, the identification of the molecular mechanisms mediating IL-6 expression and secretion by senescent cells represents a critical step toward the development of novel therapeutic interventions aimed at limiting and/or preventing age-associated phenotypes. Our findings show that IL-6 is a caveolin-1-dependent senescence-associated factor. Although IL-6 is released by senescent wild-type fibroblasts, IL-6 expression and secretion are inhibited in oxidative stress-treated caveolin-1 null MEFs. Caveolin-1-lacking fibroblasts fail to up-regulate IL-6 expression because Sirt1 function cannot be inhibited by caveolin-1 after oxidative stress. In fact, IL-6 expression is rescued in caveolin-1 null cells following down-regulation of Sirt1. Therefore, inhibition of Sirt1 function, through either interaction with caveolin-1 or down-regulation of Sirt1 itself by siRNA, leads to IL-6 expression. Activation of p53 by the caveolin-1-mediated inhibition of Sirt1 contributes to oxidant-induced transcriptional regulation of IL-6, as shown by reduced activation of the IL-6 promoter following oxidative stress in cells lacking p53 expression. These data are in contrast to previous studies showing that secretion of senescent-associated factors, including IL-6, occurs independently of p53 in HCA2, WI-38, and IMR90 fibroblasts following replicative senescence or premature senescence induced by high doses of ionizing radiation and oncogenic Ras (47, 48). How can we explain this apparent discrepancy? One possibility is the different cell type used in our studies, i.e. MEFs. More importantly, the p53 dependence of IL-6 transcription in our system may be explained by the different stimulus used to induce cellular senescence, hydrogen peroxide. Although, at high levels, H2O2 is toxic to living organisms, at moderate/low concentrations it acts as a second messenger and regulates a variety of signaling processes. Sublethal concentrations of hydrogen peroxide reach eukaryotic cells from extracellular sources (for example, cigarette smoke) or are produced endogenously. One of the effects that subcytotoxic hydrogen peroxide has on a variety of cell types, including fibroblasts and epithelial cells, is the induction of premature senescence. It is possible that, although both ionizing radiation and hydrogen peroxide induce premature senescence in a p53-dependent manner, the signaling that controls IL-6 expression, and possibly that of other cytokines, may rely on the activation of p53 only upon hydrogen peroxide stimulation. High doses of ionizing radiation promote cytokine secretion through a persistent DNA damage response in a p53-independent manner. However, hydrogen peroxide-initiated signaling may initiate a cascade that is qualitatively different from that produced by ionizing radiation and results in the caveolin-1/p53-mediated secretion of cytokines. In addition, because we observe a caveolin-1-dependent secretion of IL-6 48–72 h after hydrogen peroxide stimulation but a senescent phenotype 7 days after oxidative stress, release of IL-6 through caveolin-1-mediated signaling occurs independently of the development of premature senescence. Moreover, because cytokine signaling can reinforce senescence (49–51), caveolin-1-dependent cytokines may actually reinforce the acquisition of a senescent phenotype of cells that are near senescence.
Although our data strongly suggest that IL-6 is a prostimulatory factor that is released by senescent fibroblasts in a caveolin-1-dependent manner, it may not be the only one. In fact, although the enhancement of cancer cell proliferation by conditioned medium derived from oxidative stress-treated MEFs is inhibited by ∼70% when caveolin-1 expression is genetically ablated in the fibroblasts, it is only inhibited by ∼50% when the conditioned medium from wild-type MEFs is incubated in the presence of neutralizing IL-6 antibodies. Therefore, there may be additional factors that contribute, together with IL-6, to define a caveolin-1-specific secretory phenotype. Given the role of cellular senescence in the development of age-associated phenotypes, it will be important to fully identify the composition of the caveolin-1-specific secretory phenotype and to determine which detrimental effects senescent cells have on the surrounding microenvironment occur through the caveolin-1-specific secretory phenotype.
Pulmonary emphysema is a cigarette smoke-induced and age-related disease. Chronic lung inflammation caused by cigarette smoking promotes alveolar destruction, which leads to air space enlargement with a reduction of the alveolar capillary exchange area. Emerging evidence indicates that senescent cells accumulate in the lungs of emphysema patients. Because cigarette smoke can induce premature senescence and because senescent cells secrete a variety of mediators of inflammation, including interleukins, senescence of lung cells has been proposed as a potential novel contributor to the pathogenesis of emphysema. We have demonstrated previously that the cigarette smoke-induced accumulation of senescent fibroblasts in the lungs of mice is caveolin-1-dependent and that caveolin-1 null mice are protected against cigarette smoke-induced emphysema (36). Interestingly, Sirt1 expression is reduced in emphysema patients (52, 53), and both the overexpression of Sirt1 and the pharmacological activation of Sirt1 inhibit stress-induced premature senescence and protect against emphysema following cigarette smoke exposure (54). In addition, ablation of Sirt1 expression in the airway epithelium exacerbates emphysematous phenotypes (54). Therefore, it is possible to speculate that the protection against cigarette smoke-induced emphysema in caveolin-1 null mice may be due, at least in part, to the lack of inhibition of Sirt1 by caveolin-1. The absence of caveolin-1-mediated inhibition of Sirt1 may limit the secretion of IL-6 and other caveolin-1-specific factors by senescent cells, which would then reduce the deleterious inflammatory response that promotes lung damages in emphysema patients.
This work was supported, in whole or in part, by NIA/National Institutes of Health Grant R01-AG030636 and NHLBI/National Institutes of Health Grant R56-HL124747. This work was also supported by American Heart Association Grant 13GRNT16560012 and the University of Pittsburgh Cancer Institute (UPCI) Specialized Program of Research Excellence (SPORE) in Lung Cancer (to F. G.) and by American Heart Association Grant 12SDG8800012 (to D. V.).
- SIPS
- stress-induced premature senescence
- SASP
- senescence-associated secretory phenotype
- pAb
- polyclonal antibody
- MEF
- mouse embryonic fibroblast.
REFERENCES
- 1. Lundberg A. S., Hahn W. C., Gupta P., Weinberg R. A. (2000) Genes involved in senescence and immortalization. Curr. Opin. Cell Biol. 12, 705–709 [DOI] [PubMed] [Google Scholar]
- 2. Dimri G. P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E. E., Linskens M., Rubelj I., Pereira-Smith O. (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. U.S.A. 92, 9363–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sherr C. J., DePinho R. A. (2000) Cellular senescence: mitotic clock or culture shock? Cell 102, 407–410 [DOI] [PubMed] [Google Scholar]
- 4. Wynford-Thomas D. (1999) Cellular senescence and cancer. J. Pathol. 187, 100–111 [DOI] [PubMed] [Google Scholar]
- 5. Bodnar A. G., Ouellette M., Frolkis M., Holt S. E., Chiu C. P., Morin G. B., Harley C. B., Shay J. W., Lichtsteiner S., Wright W. E. (1998) Extension of life-span by introduction of telomerase into normal human cells. Science 279, 349–352 [DOI] [PubMed] [Google Scholar]
- 6. Harley C. B., Futcher A. B., Greider C. W. (1990) Telomeres shorten during ageing of human fibroblasts. Nature 345, 458–460 [DOI] [PubMed] [Google Scholar]
- 7. Kim N. W., Piatyszek M. A., Prowse K. R., Harley C. B., West M. D., Ho P. L., Coviello G. M., Wright W. E., Weinrich S. L., Shay J. W. (1994) Specific association of human telomerase activity with immortal cells and cancer. Science 266, 2011–2015 [DOI] [PubMed] [Google Scholar]
- 8. Lansdorp P. M. (2000) Repair of telomeric DNA prior to replicative senescence. Mech. Ageing Dev. 118, 23–34 [DOI] [PubMed] [Google Scholar]
- 9. Martens U. M., Chavez E. A., Poon S. S., Schmoor C., Lansdorp P. M. (2000) Accumulation of short telomeres in human fibroblasts prior to replicative senescence. Exp. Cell Res. 256, 291–299 [DOI] [PubMed] [Google Scholar]
- 10. Chen Q., Fischer A., Reagan J. D., Yan L. J., Ames B. N. (1995) Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc. Natl. Acad. Sci. U.S.A. 92, 4337–4341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Frippiat C., Chen Q. M., Zdanov S., Magalhaes J. P., Remacle J., Toussaint O. (2001) Subcytotoxic H2O2 stress triggers a release of transforming growth factor-β 1, which induces biomarkers of cellular senescence of human diploid fibroblasts. J. Biol. Chem. 276, 2531–2537 [DOI] [PubMed] [Google Scholar]
- 12. Robles S. J., Adami G. R. (1998) Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene 16, 1113–1123 [DOI] [PubMed] [Google Scholar]
- 13. Serrano M., Lin A. W., McCurrach M. E., Beach D., Lowe S. W. (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602 [DOI] [PubMed] [Google Scholar]
- 14. Zhu J., Woods D., McMahon M., Bishop J. M. (1998) Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 12, 2997–3007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Herbig U., Ferreira M., Condel L., Carey D., Sedivy J. M. (2006) Cellular senescence in aging primates. Science 311, 1257. [DOI] [PubMed] [Google Scholar]
- 16. Jeyapalan J. C., Ferreira M., Sedivy J. M., Herbig U. (2007) Accumulation of senescent cells in mitotic tissue of aging primates. Mech. Ageing Dev. 128, 36–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kishi S. (2004) Functional aging and gradual senescence in zebrafish. Ann. N.Y. Acad. Sci. 1019, 521–526 [DOI] [PubMed] [Google Scholar]
- 18. Melk A., Kittikowit W., Sandhu I., Halloran K. M., Grimm P., Schmidt B. M., Halloran P. F. (2003) Cell senescence in rat kidneys in vivo increases with growth and age despite lack of telomere shortening. Kidney Int. 63, 2134–2143 [DOI] [PubMed] [Google Scholar]
- 19. Campisi J. (1997) The biology of replicative senescence. Eur. J. Cancer 33, 703–709 [DOI] [PubMed] [Google Scholar]
- 20. Linskens M. H., Feng J., Andrews W. H., Enlow B. E., Saati S. M., Tonkin L. A., Funk W. D., Villeponteau B. (1995) Cataloging altered gene expression in young and senescent cells using enhanced differential display. Nucleic Acids Res. 23, 3244–3251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Millis A. J., Hoyle M., McCue H. M., Martini H. (1992) Differential expression of metalloproteinase and tissue inhibitor of metalloproteinase genes in aged human fibroblasts. Exp. Cell Res. 201, 373–379 [DOI] [PubMed] [Google Scholar]
- 22. West M. D., Pereira-Smith O. M., Smith J. R. (1989) Replicative senescence of human skin fibroblasts correlates with a loss of regulation and overexpression of collagenase activity. Exp. Cell Res. 184, 138–147 [DOI] [PubMed] [Google Scholar]
- 23. Campisi J. (2005) Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 120, 513–522 [DOI] [PubMed] [Google Scholar]
- 24. Bavik C., Coleman I., Dean J. P., Knudsen B., Plymate S., Nelson P. S. (2006) The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res. 66, 794–802 [DOI] [PubMed] [Google Scholar]
- 25. Krtolica A., Parrinello S., Lockett S., Desprez P. Y., Campisi J. (2001) Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc. Natl. Acad. Sci. U.S.A. 98, 12072–12077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu D., Hornsby P. J. (2007) Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 67, 3117–3126 [DOI] [PubMed] [Google Scholar]
- 27. Parrinello S., Coppe J. P., Krtolica A., Campisi J. (2005) Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J. Cell Sci. 118, 485–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bordone L., Guarente L. (2005) Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat. Rev. Mol. Cell Biol. 6, 298–305 [DOI] [PubMed] [Google Scholar]
- 29. Langley E., Pearson M., Faretta M., Bauer U. M., Frye R. A., Minucci S., Pelicci P. G., Kouzarides T. (2002) Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 21, 2383–2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vaziri H., Dessain S. K., Ng Eaton E., Imai S. I., Frye R. A., Pandita T. K., Guarente L., Weinberg R. A. (2001) hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107, 149–159 [DOI] [PubMed] [Google Scholar]
- 31. Ota H., Akishita M., Eto M., Iijima K., Kaneki M., Ouchi Y. (2007) Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J. Mol. Cell Cardiol. 43, 571–579 [DOI] [PubMed] [Google Scholar]
- 32. Bartholomew J. N., Volonte D., Galbiati F. (2009) Caveolin-1 regulates the antagonistic pleiotropic properties of cellular senescence through a novel Mdm2/p53-mediated pathway. Cancer Res. 69, 2878–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dasari A., Bartholomew J. N., Volonte D., Galbiati F. (2006) Oxidative stress induces premature senescence by stimulating caveolin-1 gene transcription through p38 mitogen-activated protein kinase/Sp1-mediated activation of two GC-rich promoter elements. Cancer Res. 66, 10805–10814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Volonte D., Galbiati F. (2009) Inhibition of thioredoxin reductase 1 by caveolin 1 promotes stress-induced premature senescence. EMBO Rep. 10, 1334–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Volonte D., Galbiati F. (2011) Polymerase I and transcript release factor (PTRF)/cavin-1 is a novel regulator of stress-induced premature senescence. J. Biol. Chem. 286, 28657–28661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Volonte D., Kahkonen B., Shapiro S., Di Y., Galbiati F. (2009) Caveolin-1 expression is required for the development of pulmonary emphysema through activation of the ATM-p53-p21 pathway. J. Biol. Chem. 284, 5462–5466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Volonte D., Zhang K., Lisanti M. P., Galbiati F. (2002) Expression of caveolin-1 induces premature cellular senescence in primary cultures of murine fibroblasts. Mol. Biol. Cell 13, 2502–2517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Volonte D., Liu Z., Musille P. M., Stoppani E., Wakabayashi N., Di Y. P., Lisanti M. P., Kensler T. W., Galbiati F. (2013) Inhibition of nuclear factor-erythroid 2-related factor (Nrf2) by caveolin-1 promotes stress-induced premature senescence. Mol. Biol. Cell 24, 1852–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eickelberg O., Pansky A., Mussmann R., Bihl M., Tamm M., Hildebrand P., Perruchoud A. P., Roth M. (1999) Transforming growth factor-β1 induces interleukin-6 expression via activating protein-1 consisting of JunD homodimers in primary human lung fibroblasts. J. Biol. Chem. 274, 12933–12938 [DOI] [PubMed] [Google Scholar]
- 40. Couet J., Li S., Okamoto T., Ikezu T., Lisanti M. P. (1997) Identification of peptide and protein ligands for the caveolin-scaffolding domain: implications for the interaction of caveolin with caveolae-associated proteins. J. Biol. Chem. 272, 6525–6533 [DOI] [PubMed] [Google Scholar]
- 41. Jagannadham M. V., Sharadadevi A., Nagaraj R. (2002) Effects of deleting a tripeptide sequence observed in muscular dystrophy patients on the conformation of synthetic peptides corresponding to the scaffolding domain of caveolin-3. Biochem. Biophys. Res. Commun. 298, 203–206 [DOI] [PubMed] [Google Scholar]
- 42. Song K. S., Tang Z., Li S., Lisanti M. P. (1997) Mutational analysis of the properties of caveolin-1: a novel role for the C-terminal domain in mediating homotypic caveolin-caveolin interactions. J. Biol. Chem. 272, 4398–4403 [DOI] [PubMed] [Google Scholar]
- 43. Volonté D., Galbiati F., Pestell R. G., Lisanti M. P. (2001) Cellular stress induces the tyrosine phosphorylation of caveolin-1 (Tyr(14)) via activation of p38 mitogen-activated protein kinase and c-Src kinase: evidence for caveolae, the actin cytoskeleton, and focal adhesions as mechanical sensors of osmotic stress. J. Biol. Chem. 276, 8094–8103 [DOI] [PubMed] [Google Scholar]
- 44. Lee H., Volonte D., Galbiati F., Iyengar P., Lublin D. M., Bregman D. B., Wilson M. T., Campos-Gonzalez R., Bouzahzah B., Pestell R. G., Scherer P. E., Lisanti M. P. (2000) Constitutive and growth factor-regulated phosphorylation of caveolin-1 occurs at the same site (Tyr-14) in vivo: identification of a c-Src/Cav-1/Grb7 signaling cassette. Mol. Endocrinol. 14, 1750–1775 [DOI] [PubMed] [Google Scholar]
- 45. Dean J. P., Nelson P. S. (2008) Profiling influences of senescent and aged fibroblasts on prostate carcinogenesis. Br. J. Cancer 98, 245–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ewald J. A., Desotelle J. A., Church D. R., Yang B., Huang W., Laurila T. A., Jarrard D. F. (2013) Androgen deprivation induces senescence characteristics in prostate cancer cells in vitro and in vivo. Prostate 73, 337–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Coppé J. P., Patil C. K., Rodier F., Sun Y., Muñoz D. P., Goldstein J., Nelson P. S., Desprez P. Y., Campisi J. (2008) Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, 2853–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rodier F., Coppé J. P., Patil C. K., Hoeijmakers W. A., Muñoz D. P., Raza S. R., Freund A., Campeau E., Davalos A. R., Campisi J. (2009) Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 11, 973–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Acosta J. C., O'Loghlen A., Banito A., Guijarro M. V., Augert A., Raguz S., Fumagalli M., Da Costa M., Brown C., Popov N., Takatsu Y., Melamed J., d'Adda di Fagagna F., Bernard D., Hernando E., Gil J. (2008) Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133, 1006–1018 [DOI] [PubMed] [Google Scholar]
- 50. Kortlever R. M., Higgins P. J., Bernards R. (2006) Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat. Cell Biol. 8, 877–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kuilman T., Michaloglou C., Vredeveld L. C., Douma S., van Doorn R., Desmet C. J., Aarden L. A., Mooi W. J., Peeper D. S. (2008) Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133, 1019–1031 [DOI] [PubMed] [Google Scholar]
- 52. Nakamaru Y., Vuppusetty C., Wada H., Milne J. C., Ito M., Rossios C., Elliot M., Hogg J., Kharitonov S., Goto H., Bemis J. E., Elliott P., Barnes P. J., Ito K. (2009) A protein deacetylase SIRT1 is a negative regulator of metalloproteinase-9. FASEB J. 23, 2810–2819 [DOI] [PubMed] [Google Scholar]
- 53. Rajendrasozhan S., Yang S. R., Kinnula V. L., Rahman I. (2008) SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 177, 861–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yao H., Chung S., Hwang J. W., Rajendrasozhan S., Sundar I. K., Dean D. A., McBurney M. W., Guarente L., Gu W., Rönty M., Kinnula V. L., Rahman I. (2012) SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J. Clin. Invest. 122, 2032–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]