

Background: Lipids are emerging as important constituents of the molecular machinery involved in the formation and release of exosomes.
Results: The ether lipid precursor hexadecylglycerol stimulates the release and changes the composition of exosomes.
Conclusion: Ether lipids are important modulators of exosome release.
Significance: By knowing the machinery involved in exosome release, it would be possible to modulate this process.
Keywords: Extracellular Vesicles, Lipid, lipid Ether, Membrane Lipid, Membrane Transport, Exosome Release, Exosomes, High Throughput Quantitative Lipidomics
Abstract
Exosomes are vesicles released by cells after fusion of multivesicular bodies with the plasma membrane. In this study, we have investigated whether ether lipids affect the release of exosomes in PC-3 cells. To increase the cellular levels of ether lipids, the ether lipid precursor hexadecylglycerol was added to cells. Lipidomic analysis showed that this compound was in fact able to double the cellular levels of ether lipids in these cells. Furthermore, increased levels of ether lipids were also found in exosomes released by cells containing high levels of these lipids. Interestingly, as measured by nanoparticle tracking analysis, cells containing high levels of ether lipids released more exosomes than control cells, and these exosomes were similar in size to control exosomes. Moreover, silver staining and Western blot analyses showed that the protein composition of exosomes released in the presence of hexadecylglycerol was changed; the levels of some proteins were increased, and the levels of others were reduced. In conclusion, this study clearly shows that an increase in cellular ether lipids is associated with changes in the release and composition of exosomes.
Introduction
Exosomes are vesicles released by cells to culture media (in vitro) or to physiological fluids (in vivo) after fusion of multivesicular bodies (MVBs)2 with the plasma membrane (1). Exosomes have been attributed to several important physiological and pathological functions, and today they are seen as a novel way for cell-to-cell communication (2, 3). The release of exosomes is the end point of a process that can be divided into three steps as follows: the biogenesis of MVBs, the transport of MVBs to the plasma membrane, and the fusion of the MVBs with the plasma membrane. It is not clear at the moment how and why some MVBs are transported to the plasma membrane to release exosomes, whereas others fuse with lysosomes to degrade their content. It is possible that several types of MVBs coexist (4–6). Importantly, the list of molecules that regulate the release of exosomes is growing. Exosome biogenesis starts at the limiting membrane of endosomes, which then buds and eventually pinches off into the endosomal lumen generating intraluminal vesicles (ILVs). Several endosomal sorting complexes required for transport proteins such as Hrs, TSG101, and Alix (7–9) have been shown to be implicated in this process. In addition, the sorting of molecules into ILVs and/or the formation of ILVs can occur independently of the endosomal sorting complex required for transport machinery. It has, for example, been reported that the tetraspanins CD9 and CD82 (10, 11) and neutral sphingomyelinase 2 (12) can be implicated in the process, although this is not always the case (13). The mechanism might very well be cell type-dependent. Upon maturation, MVBs can be transported and fuse with the plasma membrane to release ILVs, now named exosomes, to the extracellular environment. Several Rab proteins and synaptotagmin-7 have been implicated in these processes (14–17). Despite these interesting studies, there are still many unanswered questions concerning the mechanism of exosome release, and additional players will surely be identified in the coming years.
Here, we have investigated the possible role of ether phospholipids in exosome release. Ether lipids contain an ether-linked alkyl or alkenyl chain at the sn-1 position of the glycerol backbone and a polar headgroup, mainly ethanolamine or choline (18). Ether lipids are prevalent in humans and animals as structural components of membranes, although their distribution varies among tissues (19). These lipids are important for cell physiology, although their biological functions on the molecular levels are poorly understood. For example, in mice ether lipid deficiency leads to male infertility, defects in eye development, and abnormalities in the central nervous system (20). In addition, altered levels of ether lipids have been noted in several genetic disorders and pathologies such as Zellweger syndrome, Alzheimer disease, and diabetes (19–21). Furthermore, ether lipids may serve as a storage depot of polyunsaturated fatty acids or lipid mediators and function as antioxidative agents (21–25). In fact, a plasmalogen-selective phospholipase A2 that catalyzes the release of polyunsaturated fatty acid has been identified (26). Finally, ether lipids have been shown to be involved in signaling (27, 28), membrane fusion, and trafficking (29–33). Interestingly, the fusion rate of liposomes containing plasmenylethanolamine was found to be higher than the fusion rate of liposomes containing phosphatidylethanolamine in the presence of calcium (29). In addition, mutations in some of the genes involved in ether lipid biosynthesis were reported not only to change the morphology of caveolae, clathrin-coated pits, endoplasmic reticulum, and Golgi cisternae, but also to impair the uptake and recycling of transferrin in human skin fibroblasts (32). However, the uptake of transferrin is not affected in HEp-2 cells containing increased levels of ether lipids (34).
One way to change the lipid composition of cells is to grow cells in the presence of lipid precursors (35). Addition of the ether lipid precursor sn-1-O-hexadecylglycerol (HG) (Fig. 1), which enters the biosynthetic pathway after its phosphorylation by alkylglycerol kinase (18), has been shown to significantly increase the levels of ether phospholipids in a number of cell lines (34, 36–38). As control, we have used untreated cells or cells treated with dl-α-palmitin (DP), a control substance for HG that contains an acyl group instead of the ether group (Fig. 1). In this work, we treated human prostate cancer PC-3 cells with HG to investigate whether increased levels of ether lipids affect exosome release. Interestingly, our results show that cells containing higher levels of ether lipids release larger numbers of exosomes. Moreover, the protein profile of exosomes released from cells treated with HG is changed as compared with the exosomes released from control cells. It is, at the moment, unclear how HG alters exosome release, but this study demonstrates that lipids can be important modulators of this process.
FIGURE 1.
Chemical structure of HG and DP.
EXPERIMENTAL PROCEDURES
Materials
HG was from Santa Cruz Biotechnology. ProteoSilver Plus Silver Stain kit, DP, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and BSA were purchased from Sigma. EDTA-free protease inhibitor mixture was from Roche Applied Science. Bicinchoninic acid (BCA) protein assay kit was from Pierce (Thermo Scientific, Rockford, IL). Mini-protean TGX gels and Transfer-Blot Turbo Transfer Pack were from Bio-Rad. PVDF membranes and 0.02-μm Anotop 25 filters were from Millipore (Billerica, MA) and Whatman® (Dassel, Germany), respectively. The antibodies used for Western blotting were as follows: rabbit anti-caveolin-1, mouse anti-annexin A2, and mouse anti-TSG101 (all three from BD Biosciences); mouse anti-vinculin (Santa Cruz Biotechnology); rabbit anti-CD9 and rabbit anti-alkylglycerone phosphate synthase (AGPS) (Abcam, Cambridge, UK); and rabbit anti-Alix (Merck-Millipore). HRP-conjugated secondary antibodies were from Jackson ImmunoResearch (West Grove, PA), and Li-Cor infrared dye secondary antibodies were from Li-Cor Biosciences (Lincoln, NE). The antibodies used for immunoelectron microscopy were as follows: mouse anti-CD63 (H5C6) (Developmental Studies Hybridoma Bank, Iowa City, IA) and rabbit anti-mouse (Dako, Glostrup, Denmark). Protein A-gold conjugates (10 nm) were purchased from Cell Microscopy Center (Utrecht, Netherlands).
Cell Culture and Treatment
The human prostate cancer epithelial cell line PC-3 was obtained from ATCC (Boras, Sweden). PC-3 cells were cultured in DMEM/F-12 GlutaMAX-ITM (Invitrogen) supplemented with 7% FCS, 100 units/ml penicillin, and 100 μg/ml streptomycin, in a humidified 5% CO2 atmosphere at 37 °C. Cells were preincubated with 20 μm HG (dissolved in ethanol), 20 μm DP (dissolved in ethanol), or 0.1% (v/v) ethanol in complete medium for 24 h. At the end of the incubation, cells were washed twice and incubated again with the same concentration of the chemicals in serum-free medium for 17–19 h. For siRNA treatment, ON-TARGET plus siRNA oligos against AGPS or ON-target plus nontargeting siRNA (control) were purchased from Dharmacon RNAi Technologies (Lafayette, CO). Two different siRNA oligos against AGPS were used to down-regulate the level of AGPS (oligo1, GCGAAUUCCUGAUAUAGUU; oligo2, UCAAGGACCUCGUAUGUCA). siRNA oligos were delivered to the cells using LipofectamineTM RNAiMAX transfection reagent (Thermo Fisher Scientific, Waltham, MA) following the manufacturer's protocol. Two days after transfection, cells were washed twice and incubated for 17–19 h in serum-free medium to collect exosomes. Cells were lysed 2–3 days after transfection to measure knockdown efficiency. Knockdown efficiency after 2 days was measured to check that the levels of AGPS were reduced before serum-free medium was added to the cells for the collection of exosomes.
Cell Lysates
PC-3 cells were washed with cold PBS, and total cell lysates were prepared in lysis buffer (50 mm Tris-HCl, 300 mm NaCl, 1 mm EDTA, 0.5% Triton X-100, pH 7.4) in the presence of a protease inhibitor mixture. The suspension was incubated on ice for 20 min and centrifuged at 20,000 × g for 10 min at 4 °C. The supernatant was collected and stored at −20 °C.
Collection of Cells for Lipidomics
Trypsinized PC-3 cells were pelleted, resuspended in cold PBS, and centrifuged again at 300 × g for 10 min at 4 °C. Finally, the supernatant was discarded, and the cell pellets were stored at −80 °C prior to analysis.
MTT Assay
A stock solution of MTT was prepared in PBS at 5 mg/ml. Cells were then treated with 0.5 mg/ml MTT for 3.5 h at 37 °C in a humidified 5% CO2 incubator. Following incubation, the supernatant was carefully removed, and dimethyl sulfoxide was added. The plate was read at 515 nm in a Synergy 2 instrument (BioTek Instruments Inc., Winooski, VT).
Exosome Isolation
Exosomes were isolated from the conditioned media of control and treated cells as described elsewhere (39). Briefly, the conditioned media were centrifuged at 300 × g for 10 min, 1,000 × g for 10 min, and 10,000 × g for 30 min, discarding the pellet at each step. The supernatants were then ultracentrifuged at 100,000 × g for 70 min. The exosome pellet was washed with PBS and then centrifuged again at 100,000× g for 70 min. The exosome pellets were resuspended in an equal volume of PBS and then used for further analyses. All centrifugation steps were carried out at 4 °C.
Electron Microscopy of Exosomes
Exosomes resuspended in PBS were fixed (4% formaldehyde, 0.2% glutaraldehyde) and deposited on Formvar/carbon-coated copper grids. For labeling, samples on grids were first blocked with 0.5% BSA and then successively incubated with mouse anti-CD63 followed by rabbit anti-mouse and then by 10-nm protein A-gold conjugates. Fixative, blocking solution, and antibody dilutions were prepared in PHEM buffer (60 mm PIPES, 25 mm HEPES, 10 mm EGTA, and 2 mm MgCl2, pH 6.9). Samples were then contrasted and embedded in a mixture of methylcellulose and uranyl acetate. Finally, exosomes were observed in a JEOL-JEM 1230 (JEOL Ltd., Tokyo, Japan) at 80 kV, and pictures were acquired using a Morada camera and iTEM software (Olympus, Münster, Germany).
Electron Microscopy of Cells
PC-3 cells were incubated with or without HG as described previously, and BSA-gold, 10 nm, was added to the cells during the last hour. Cells were then washed, fixed with 2% glutaraldehyde in 0.1 m cacodylate buffer, pH 7.2, for 10 min, scraped, and pelleted. Pellets were treated with 1% osmium tetroxide for 1 h, with 0.5% tannic acid for 30 min, and then with 1% Na2SO4 for 5 min. Finally, the samples were contrasted en bloc with 4% uranyl acetate, dehydrated with ethanol, and embedded in Epon. Cells were observed in a JEOL 1011 microscope. For quantitative analysis, two independent sets of sections were used, and ∼60 cell profiles were quantified (n = 10–20 cells). Only healthy mononucleate interphase cells with the nuclei sectioned were used. In these cells, all gold-labeled MVBs (one or more ILVs) were analyzed.
Nanoparticle Tracking Analysis (NTA)
NTA was used to determine the concentration and the size distribution of exosomes. Exosome pellets were resuspended in PBS filtered with a 0.02-μm Anotop 25 filter and vortexed for 1 min. Samples were diluted to be within the recommended range (2 × 108 to 1 × 109 particles per ml). The samples were then loaded into the NS500 instrument (NanoSight, Amesbury, UK) with a syringe pump. Five videos, each of 60 s, were acquired for every sample under the flow mode (infusion rate: 30, camera settings: shutter, 600; gain, 350–450). Videos were subsequently analyzed with the NTA 2.3 software, which identifies and tracks the center of each particle under Brownian motion to measure the average distance the particles move on a frame-by-frame basis.
Total Protein Measurements
The amount of total protein in exosomes and cells was determined using a BCA assay kit according to the manufacturer's instructions. BSA was used as standard protein.
SDS-PAGE and Silver Staining
Similar volumes of exosomal samples and similar volumes of cell lysates were mixed with loading buffer, and the samples were run on 4–20% polyacrylamide gels. The gels were stained using ProteoSilver Plus Silver Stain kit following the manufacturer's protocol.
SDS-PAGE and Immunoblotting
Similar volumes of exosomes and similar volumes of cell lysates were solubilized in loading buffer and run on 4–20% gradient TGX gels. In some experiments, specific amounts of cellular and exosomal proteins were loaded. The proteins were transferred to PVDF membranes using a Transfer-Blot Turbo Transfer Pack. Membranes were incubated with the specified primary and secondary antibodies. Blots were visualized with the Amersham Biosciences ECLTM Prime Western blot detection (GE Healthcare) on the Universal Hood II scanner (Bio-Rad). In some cases, the bands were detected using the Odyssey imaging system (Li-Cor Biosciences).
Annotations of Lipid Species
The lipid classes are abbreviated as follows: phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylglycerol (PG), phosphatidylinositol (PI), phosphatidylserine (PS), phosphatidic acid (PA), and diacylglycerol (DAG). Diacyl lipid species are listed with the two fatty acyl groups separated with an underscore, e.g. PC 16:0_18:1. The same annotation is applied for the ether-linked glycerophospholipids as follows: PC O (alkyl), PC P (alkenyl), PE O (alkyl), and PE P (alkenyl), e.g. PC O-16:0_18:1. In sphingomyelin (SM), ceramide (Cer), lactosylceramide (LacCer), and glucosylceramide (GlcCer), the N-amidated fatty acyl groups are shown after the slash, e.g. SM d18:1/18:1. Although the glycosphingolipids with only one hexose unit are called hexosylceramides, it is expected that almost all of these species are GlcCer. For lipids where isobaric structures cannot be discriminated, e.g. PE P-16:0_18:1 and PE O-16:1_18:1, both are shown. The abbreviation PL is used to indicate all phospholipids except SM. Cholesterol and cholesteryl esters are abbreviated as CHOL and CE, respectively.
Lipid Extraction
For lipidomic analysis, lipids were extracted from PC-3 cells-released vesicles and PC-3 cell lysates using a modified Folch lipid extraction procedure (40). To have sufficient exosomes to run the lipidomic analysis, exosomes collected on several days were pooled and analyzed together. The corresponding cells were also pooled before analysis. Known amounts of deuterium-labeled or heptadecanoyl-based synthetic internal standards were added to the samples for quantification of the endogenous lipid species (34). For quantification of free CHOL, an aliquot of each lipid extract was treated with acetyl chloride to derivatize CHOL to modified cholesteryl ester species according to Liebisch et al. (41). Lysates samples were analyzed in three replicates and exosomal samples in one replicate.
MS Analyses
Molecular glycerophospholipids, glycerolipids, sterols, and sphingomyelin were analyzed by shotgun analysis on a hybrid triple quadrupole/linear ion trap mass spectrometer (5500 QTRAP, AB SCIEX) equipped with a robotic nanoflow ion source (NanoMate HD, Advion Biosciences) (42). The analyses were performed in both positive and negative ion modes (e.g. [M + H]+ for PL, SM, Cer, GlcCer, and LacCer; [M + NH4]+ for CE and DAG; and [M − H]− for phospholipids with the exception of PC monitored as [M + CH3COO]−) using multiple precursor ion scanning and neutral loss-based methods (40, 43). Derivatized cholesterol was analyzed in positive ion mode (41). Neutral glycosphingolipids were analyzed by reverse phase ultra-high pressure liquid chromatography using an Acquity BEH C18, 2.1 × 50-mm column with a particle size of 1.7 μm (Waters, Milford, MA) coupled to a hybrid triple quadrupole/linear ion trap mass spectrometer (5500 QTRAP, AB SCIEX). A 25-min gradient using 10 mm ammonium acetate in water with 0.1% formic acid (mobile phase A) and 10 mm ammonium acetate in acetonitrile/2-propanol (4:3, v/v) containing 0.1% formic acid (mobile phase B) was used. Sphingolipids were monitored using multiple reactions monitoring (44).
Data Processing
The MS data files were processed using Lipid ProfilerTM and MultiQuantTM softwares for producing a list of lipid names and peak areas. Lipid identification was based on both parent mass and fragment ions and retention time where applicable. Masses and counts of detected peaks were converted into a list of corresponding lipid names. Lipids were normalized to their respective internal standard (34) and sample volume to retrieve their concentrations. The internal standards used were as follows: lysophosphatidylcholine (lyso-PC) 17:0; PC 17:0/17:0; PA 17:0/17:0; PE 17:0/17:0; PG 17:0/17:0; PS 17:0/17:0; DAG 17:0/17:0; D6-CE 18:0; D6-CHOL, SM d18:1/12:0; Cer d18:1/17:0; D3-GlcCer d18:1/16:0; D3-LacCer, d18:1/16:0; and Gb3 d18:1/17:0. The concentrations of molecular lipids are presented as nanomoles/mg protein and as mol % of total lipids. Quality control samples were utilized to monitor the overall quality of the lipid extraction and mass spectrometry analyses (45). The quality control samples were mainly used to remove technical outliers and lipid species that were detected below the lipid class lower limit of quantification. The lipid species PG, phosphatidylinositol, lyso-PC, and lyso-PE, constituting together ∼2% of the total lipid content, were not included in Fig. 3 due to their low amounts and larger variation between experiments. We show the lipid species from one such analysis (performed in triplicate) in which a pooled cell pellet consisting of cells collected on 10 different days was used. Similar results were obtained in an independent lipidomic analysis (also performed in triplicate) in which a pooled cell pellet consisting of cells collected on 3 different days was used. The concentration of lipid classes was calculated by adding the concentrations of each of the individual molecular species quantified within a specific lipid class.
FIGURE 3.

Quantitative analysis of glycerophospholipids after HG or DP treatment. Cells were treated with HG (20 μm), DP (20 μm), or ethanol (0.1%, as control) for 24 h in complete medium and then for 17–19 h with similar substance concentrations in serum-free medium. The cellular lipidome was then analyzed by MS. This figure shows the concentration (in picomoles/μg protein) of the major species of PE O/PE P (a), PC O/PC P (b), PE (c), and PC (d). To make the figure easier to read, only species making up more than 0.04, 0.09, 0.07, and 0.3 pmol/μg protein, respectively, are shown. When the MS method was not able to distinguish between the two lipid species, one of the lipid species is in parentheses. The figure is based on analysis of three replicates of a pooled cell pellet consisting of cells collected on 10 different days. Similar results were obtained in an independent experiment.
To quantify the levels of proteins in Western blots Quantity One version 4.6.5 build 094 (Bio-Rad) or ImageJ (Fiji) were used. SigmaPlot, version 12.5 (Systat Software Inc., San Jose, CA), was used for statistical analyses. A paired t test was used to assess whether the changes in the amount of secreted exosomes from control and treated conditions were significant. The difference was considered significant for p value less than 0.05 if not specified otherwise in the text. All experiments were run independently in duplicate at least three times.
Profiling of the silver-stained gels was performed with GeneTools (version 4.03) (SynGene, Cambridge, UK). For comparison, the background was subtracted using a rolling disc of 50 pixels. Peak intensity threshold was held at 1% maximum peak height. For normalization, volumes were adjusted to control sample levels.
RESULTS
Ether Lipid Precursor HG Increases the Cellular Levels of Ether Lipids in PC-3 Cells
In this study, we aimed to investigate whether an increase in the cellular ether lipids affects exosome release. The ether lipid precursor HG has previously been shown to increase the cellular levels of ether lipids in several cell lines (34, 36–38). Therefore, we treated PC-3 cells with HG (20 μm), DP (20 μm), which is a control substance for HG that contains an acyl group instead of the ether group, or ethanol (0.1% as control) (Fig. 1). Treatment with HG or DP did not reveal any toxic effects in PC-3 cells as measured by the trypan blue assay and MTT assay, although HG reduced cell growth by 14% compared with control (data not shown).
To investigate whether HG changes the cellular levels of lipids in PC-3 cells, we performed lipidomic analyses of control, HG-, and DP-treated cells and quantified over 250 lipid species. As shown in Fig. 2, the levels of ether lipids (PE O/PE P and PC O/PC P),3 were ∼2-fold higher in cells treated with HG than in control and DP-treated cells. Furthermore, HG treatment also changed the levels of several other lipid classes, e.g. cholesterol was increased approximately by 25%, and PC, PE, and SM were reduced by 20–30%. These effects seem to be ether lipid-dependent because DP, the fatty acyl analog of HG, did not affect the levels of these lipid classes (Fig. 2). Also other lipid classes such as DAG and CE were shown to be reduced by ∼20% after HG treatment (Fig. 2). Moreover, higher Cer levels, almost similar GlcCer levels, and reduced LacCer levels were observed in HG-treated cells compared with control cells and DP-treated cells (Fig. 2).
FIGURE 2.
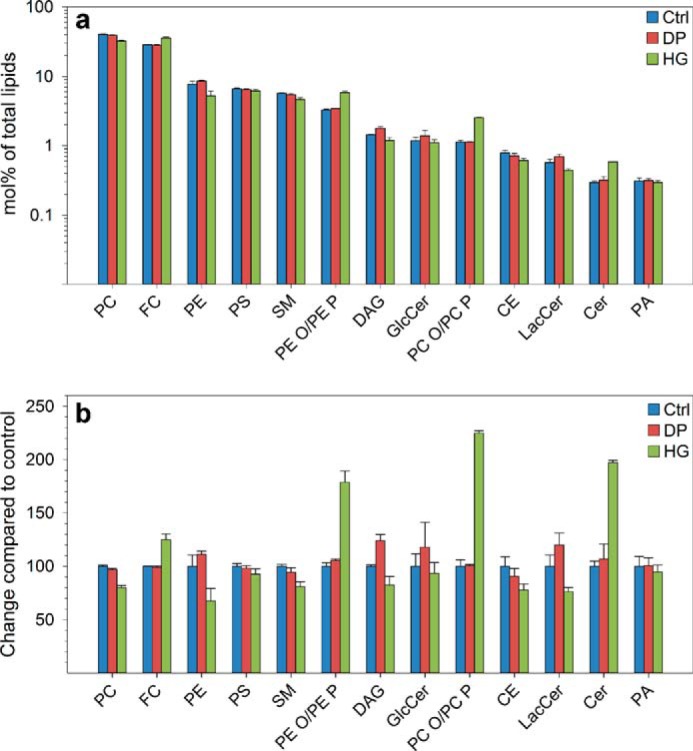
Changes in lipid classes after treatment of PC-3 cells with HG or DP. Cells were treated with HG (20 μm), DP (20 μm), or ethanol (0.1%, as control) for 24 h in complete medium and then for 17–19 h with similar substance concentrations in serum-free medium. The cellular lipidome was then analyzed by MS. The total amount of the different lipid classes is shown as mol % (note the logarithmic scale) (a) and in percentage of the control (b). The figure is based on analysis of three replicates of a pooled cell pellet consisting of cells collected on 10 different days. Similar results were obtained in an independent experiment.
Comparison of Lipid Species in HG-treated Cells, Control Cells, and DP-treated Cells
The quantification of the major PC and PE molecular species is shown in Fig. 3. All the PE O/PE P species with 16:0 in the sn-1 position were increased in cells treated with HG (Fig. 3a). Not surprisingly, the effect of HG is specific for PE ether lipid species with 16:0 in the sn-1 position because this ether lipid precursor has an alkyl group with 16 carbon atoms (Fig. 3a). Only minor differences between DP and control cells were observed (Fig. 3a). The PC ether lipid species are shown in Fig. 3b. All the PC ether lipids, with the exception of 18:0_16:0, were increased to some extent in cells treated with HG, although the effect of HG was larger in species with 16:0 in the sn-1 position (Fig. 3b). As for PE ether lipids, DP and control cells were quite similar with respect to the PC ether lipid composition (Fig. 3b). All PE species were decreased in HG-treated cells, and the effect of DP varied among the different PE lipid species (Fig. 3c). In particular, one of most abundant PE species, PE 16:0_18:1, was increased after DP treatment (Fig. 3c). HG-treated cells contained less of all PC species than control cells (Fig. 3d), including the most abundant PC species, PC 16:0_16:1 and 16:0_18:1. In general, DP and control cells gave similar results for the PC species (Fig. 3d).
It has recently been shown that HG treatment of HEp-2 cells can induce changes in the metabolism of glycosphingolipids (34). The major species of Cer, GlcCer, and LacCer quantified in this study are shown in Fig. 4. There were higher amounts of the most abundant Cer species in cells treated with HG (Fig. 4a), whereas GlcCer species (Fig. 4b) and to a larger extent LacCer species (Fig. 4c) were present in lower amounts in cells treated with HG. Surprisingly, the DP-treated cells showed increased levels of GlcCer and LacCer (Fig. 4), in contrast to our study in HEp-2 cells where control and DP-treated cells had very similar levels of these lipid classes (34). It should be noted that Gb3 species were not shown because they are present in very low amounts in PC-3 cells (∼1.0% of GlcCer levels), thus resulting in high uncertainty in Gb3 data.
FIGURE 4.
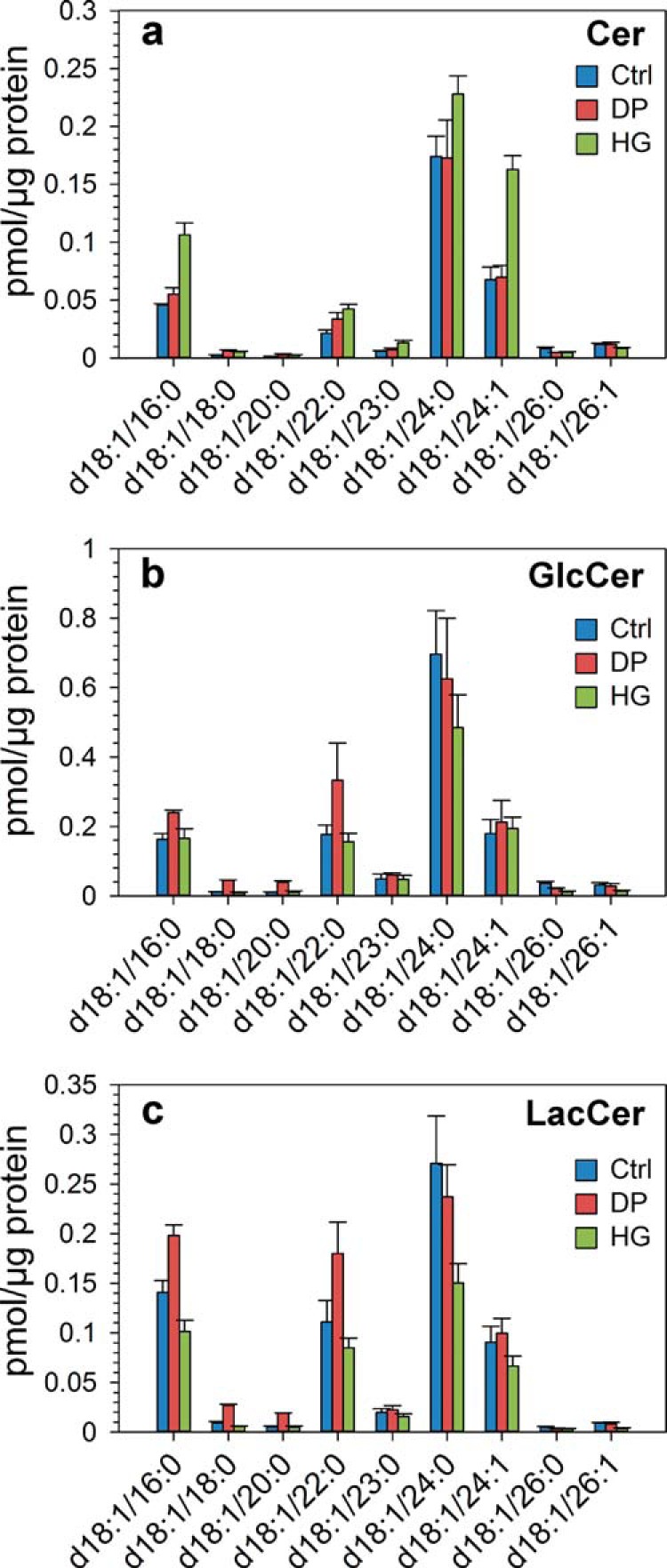
Quantitative analysis of Cer, GlcCer, and LacCer after HG or DP treatment. Cells were treated with HG (20 μm), DP (20 μm), or ethanol (0.1%, as control) for 24 h in complete medium and then for 17–19 h with similar substance concentrations in serum-free medium. The cellular lipidome was then analyzed by MS. This figure shows the concentration (in picomoles/μg protein) of the major species of Cer (a), GlcCer (b), and LacCer (c). The figure is based on analysis of three replicates of a pooled cell pellet consisting of cells collected on 10 different days. Similar results were obtained in an independent experiment.
Ether Lipid Precursor HG Increases the Exosomal Levels of Ether Lipids in PC-3 Cells
It has been shown that there is a specific sorting of lipid classes into exosomes (39, 46–48). Exosomes released by PC-3 cells are enriched in CHOL, SM, PS, and glycosphingolipids, although they contain less PC than the parent cells (39). Because the addition of the ether lipid precursor HG modifies the cellular lipidome (Figs. 2–4), we decided to investigate whether the lipid composition of exosomes released from PC-3 cells was changed after HG treatment. Exosomes released during a period of 17–19 h from control, HG-, and DP-treated cells were isolated from the cell culture medium by ultracentrifugation as described previously (39). As shown in Fig. 5, the morphology of the exosomes observed by electron microscopy was similar in control cells and HG-treated cells (Fig. 5, a and b). In addition, the exosomal marker CD63 was present in both exosomal samples, and no membrane fragments were observed in exosomes released by HG-treated cells (Fig. 5, a and b). Moreover, the size of the exosomes, measured with NTA, was not affected by HG or DP treatment (Fig. 5, c and d). Finally, we show that several typical exosomal markers were enriched in exosomes compared with cellular membranes both in control and HG exosomal samples (Fig. 5e). Altogether, these experiments indicate that the vesicles released by HG-treated cells are exosomes and not membrane fragments. Quantitative lipidomic analyses of exosomes released by control, HG-, and DP-treated were then performed, and the amounts of the different lipid classes were quantified in percent of the total amount of lipids in their respective exosome preparation (Fig. 6). These data revealed that, as expected from our previous study (39), exosomes released from control PC-3 cells are enriched in CHOL, SM, and glycosphingolipids and contained lower levels of PL than the parent cells (Fig. 6a, left and middle panels), and there is an ∼6-fold enrichment (6.2 ± 0.3; S.D., n = 3) of lipids/mg of protein in exosomes compared with the parent cells. Furthermore, exosomes isolated from DP-treated (Fig. 6b, left and middle panels) and HG-treated (Fig. 6c, left and middle panels) cells also had a similar lipid composition. Interestingly, a comparison of the different phospholipid classes (excluding SM) in exosomes released by control cells and HG- and DP-treated cells revealed that exosomes released by HG-treated cells contained a much higher percentage of ether lipids, especially ether-linked PE (Fig. 6, right panels). Interestingly, the decrease in PS in exosomes from HG-treated cells compared with control exosomes corresponds to the increase in ether-linked PE after HG treatment.
FIGURE 5.

HG treatment does not affect the morphology and size of exosomes. PC-3 cells were treated with HG (20 μm), DP (20 μm), or ethanol (0.1%, as control) for 24 h in complete medium, and exosomes released during a period of 17–19 h in serum-free medium in the presence of HG, DP, or ethanol were then isolated from the cell culture medium by ultracentrifugation. Exosomes (Exo) released from control (Ctrl) cells (a) and HG-treated cells (b) were inspected by electron microscopy. Exosomes were labeled with mouse anti-CD63 followed by rabbit anti-mouse and then by 10 nm protein A-gold conjugates. Vesicle size was measured by NTA. c, diameter of exosomes that were frequently detected (mode size). d, size distribution of whole exosome population. Error bars show S.E., n = 4. e, indicated amounts of exosomal or cellular proteins were loaded to gels, and typical exosomal markers were analyzed by Western blot.
FIGURE 6.

HG-treated cells release exosomes with high levels of ether lipids. PC-3 cells were treated with HG (20 μm), DP (20 μm), or ethanol (0.1%, as control) for 24 h in complete medium, and exosomes released during a period of 17–19 h in serum-free medium in the presence of HG, DP, or ethanol were then isolated from the cell culture medium by ultracentrifugation. Isolated exosomes were analyzed by MS. Lipid composition of exosomes released by control cells (a), DP-treated cells (b), and HG-treated cells (c) are shown. PL includes the sum of all phospholipid classes excluding SM. For cells and exosomes, the values represent percentages of total lipids in cells and exosomes, respectively. For the composition of the PL lipid classes in exosomes, 100% in the right panels corresponds to the percentage PL shown in the middle panels. The figure is based on the analysis of either a pooled cell pellet or a pooled exosome pellet of cells and exosomes, respectively, collected on 10 different days. Similar results were obtained in an independent experiment.
Treatment with the Ether Lipid Precursor HG Increases Exosome Release in PC-3 Cells
PC-3 cells were pretreated with HG, DP, or ethanol (0.1% as control) for 24 h, and exosomes released during a period of 17–19 h in the presence of the substances were then isolated from the cell culture medium by ultracentrifugation. Released exosomes were quantified by NTA, and their total protein content was measured using the BCA assay. Interestingly, treatment with HG increased the number of secreted vesicles per cell about 2-fold compared with control and DP treatment (Fig. 7a). In accordance with this, the total exosomal protein content was increased to the same extent after HG treatment compared with control and DP treatment (Fig. 7b). The total protein level in HG-treated cells was reduced by ∼20% (Fig. 7c), in agreement with the observed reduction in cell growth (data not shown). Finally, to investigate whether lower levels of ether lipids had the opposite effect on exosome release than HG, the cellular levels of AGPS, a peroxisomal enzyme involved in the rate-limiting step of the biosynthetic pathway of ether lipids, were depleted with siRNA. In particular, this enzyme catalyzes the exchange of an acyl group for a long-chain alkyl group and forms the ether bond. The two siRNA oligos that were used reduced the cellular levels of the enzyme by ∼70%. Based on the reported half-life of this enzyme (23 ± 12 h) (50) and on the half-life of peroxisomes (∼2 days) (51), we cannot expect to achieve a much larger knockdown. Under these conditions, the amount of exosomes released per cell, as measured with nanoparticle tracking analysis, was reduced by ∼15% (Fig. 8).
FIGURE 7.
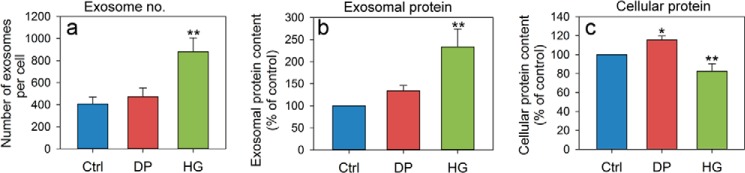
Exosome release is stimulated in HG-treated PC-3 cells. Cells were treated with HG (20 μm), DP (20 μm), or ethanol (0.1%, as control) for 24 h in complete medium, and exosomes released during a period of 17–19 h in serum-free medium in the presence of HG, DP, or ethanol were then isolated from the cell culture medium by ultracentrifugation. a, exosome concentration was measured by NTA. The total exosomal (b) and cellular (c) protein content was measured using the BCA assay. In the figure, error bars show S.E. for n = 4. *, p value < 0.05 for DP versus control (Ctrl); **, p value < 0.05 for both HG versus control and HG versus DP.
FIGURE 8.

Role of AGPS on exosome release. PC-3 cells were transfected with control siRNA or with two different siRNA oligonucleotides (50 nm) against APGS (APGS #1 and APGS #2). After 2 days, serum-free medium was added, and exosomes were collected after 17–19 h. a, representative Western blots showing the levels of APGS and the internal control GAPDH in the different conditions 3 days after transfection. b, quantification graphs showing values (% of control) normalized to GAPDH. c, exosome concentration as measured by nanoparticle tracking analysis. The error bars show S.E. for three independent experiments performed in duplicate. The reduction was not statistically significant (n.s.) (p value >0.05). Ctrl, control.
Protein Profile of Released Exosomes Is Changed by HG Treatment of PC-3 Cells
Because HG treatment increases the number of released exosomes (Fig. 7), we decided to characterize the protein composition of exosomes by performing silver staining of exosomal proteins in polyacrylamide gels (Fig. 9a). The GeneTools quantification software was used to analyze the protein profile and total intensity of the silver-stained proteins in exosomes released from control and HG- and DP-treated cells. The protein amount of exosomes released by HG-treated cells increased to ∼190% compared with exosomes released by control cells. This is in agreement with the results obtained by measuring the exosomal protein amount with the BCA assay (Fig. 7b). The protein content of exosomes released by DP-treated cells was ∼90% that in control exosomes. The positions of the protein bands in the profiles were highly reproducible between the samples (Fig. 9). To detect changes in protein composition, the profiles were normalized to the same protein level, i.e. to the same area under the curve. As shown in Fig. 9b, the normalized profiles revealed some peaks, as the peak at 50 kDa, that were increased in exosomes released from HG-treated cells compared with control cells. Conversely, a significant decrease for the protein peak at 160 kDa and for several peaks below 30 kDa in exosomes released from HG-treated cells was observed. A more detailed analysis (intensity values, ratio of signals for DP-treated and control cells, and for HG-treated and control cells) of the 12 main peaks can be found in Table 1 and Fig. 9c. As shown in Table 1, not all the peaks were significantly changed (p < 0.05) compared with the control.
FIGURE 9.
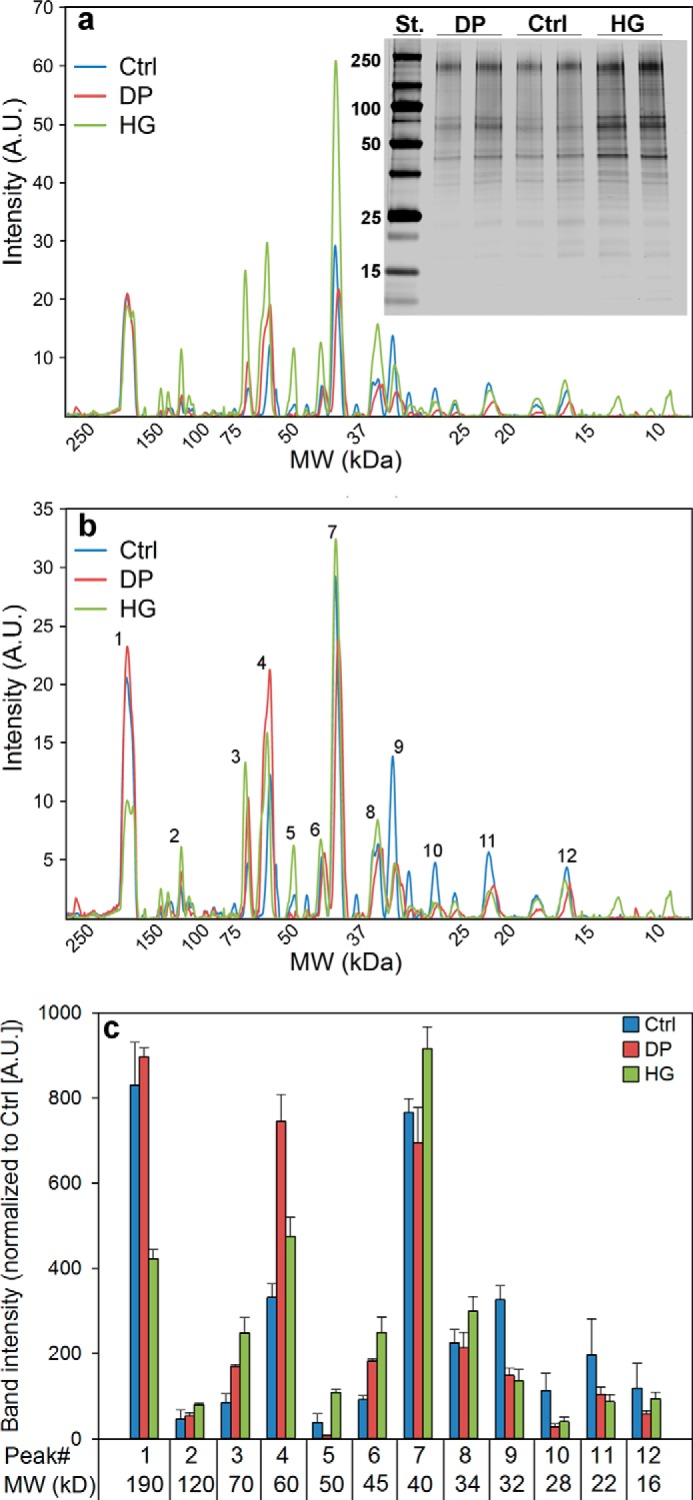
Protein profile of exosomes after treatment of PC-3 cells with HG or DP. Cells were treated with HG (20 μm), DP (20 μm), or ethanol (0.1%, as control) for 24 h in complete medium. Exosomes released during a period of 17–19 h in serum-free medium in the presence of HG, DP, or ethanol were then isolated from the cell culture medium by ultracentrifugation. The exosomal fraction was run on SDS-PAGE, and the proteins were stained by silver staining. a, silver-stained gel and protein profile in exosomes released by cells treated in the three conditions. Lane St., standard. b, protein profile after normalization to the same area under the curve. c, analyses of the 12 peaks specified in b (see also Table 1). The values are normalized to control. The panel shows the S.E. of four replicates. A.U., arbitrary units. MW, molecular weight. The figure shows the results from four replicates.
TABLE 1.
Intensities (areas under the curve, in arbitrary units) of the 12 main peaks in silver-stained protein SDS-PAGE of exosomes released from control (ctrl), DP-treated cells, and HG-treated cells
The values are normalized to control. Ratios were calculated, and significance was evaluated with a paired t test (p < 0.05). Four replicates were used in the calculations. Boldface cells show significant differences.
| Mass | Peak no. | Ctrl | DP | HG | Ratio HG/Ctrl | p value HG/Ctrl | Ratio DP/Ctrl | p value DP/Ctrl |
|---|---|---|---|---|---|---|---|---|
| kDa | ||||||||
| 190 | 1 | 830.0 | 896.9 | 422.0 | 0.51 | 0.004 | 1.08 | 0.159 |
| 120 | 2 | 46.3 | 54.5 | 80.0 | 1.73 | 0.159 | 1.18 | 0.088 |
| 70 | 3 | 84.9 | 169.7 | 248.8 | 2.93 | 0.006 | 2.00 | 0.063 |
| 60 | 4 | 331.6 | 745.3 | 475.5 | 1.43 | 0.011 | 2.25 | 0.000 |
| 50 | 5 | 38.7 | 7.1 | 108.6 | 2.80 | 0.044 | 0.18 | 0.328 |
| 45 | 6 | 92.5 | 182.6 | 249.3 | 2.69 | 0.108 | 1.97 | 0.037 |
| 40 | 7 | 766.0 | 694.9 | 916.2 | 1.20 | 0.002 | 0.91 | 0.183 |
| 34 | 8 | 225.3 | 213.3 | 299.1 | 1.33 | 0.003 | 0.95 | 0.153 |
| 32 | 9 | 326.5 | 148.9 | 136.1 | 0.42 | 0.000 | 0.46 | 0.480 |
| 28 | 10 | 113.3 | 28.4 | 40.4 | 0.36 | 0.037 | 0.25 | 0.780 |
| 22 | 11 | 197.2 | 104.4 | 87.2 | 0.44 | 0.103 | 0.53 | 0.607 |
| 16 | 12 | 118.68 | 58.81 | 94.25 | 0.79 | 0.102 | 0.50 | 0.278 |
Furthermore, Western blotting was performed to investigate changes in the amount of several exosome-associated proteins. Some of these proteins are typically found in exosomes, such as Alix and TSG101, and others were chosen due to their presence in PC-3-released exosomes (13). The exosomal samples were dissolved in similar volumes of sample buffer and applied to the gels. Because there are more exosomes released in the presence of HG, the HG exosomal samples will then contain approximately double the amount of protein compared with control exosomes (Fig. 7). This implies that one should expect to find approximately the double amount of a specific protein in HG exosomes if the amount of this protein per exosome was unchanged by HG treatment. As shown in Fig. 10a, none of the tested proteins showed a 2-fold increase in exosomes released by HG-treated cells compared with control cells. This indicates that exosomes isolated in the presence of HG have lower amounts of these proteins compared with control cells. The levels of the same proteins in exosomes released by DP-treated cells were in general similar to control exosomes (Fig. 10a). Finally, HG treatment did not change the cellular levels of the five tested proteins (Fig. 10b). With the exception of Alix, which showed a large variation between experiments, the levels of the other proteins in DP-treated cells were also quite similar to control cells (Fig. 10b).
FIGURE 10.

Analysis of specific proteins in exosomes by Western blot. PC-3 cells were incubated with HG (20 μm), DP (20 μm), or ethanol (0.1%, as control) for 24 h in complete medium, and then exosomes released during a period of 17–19 h in serum-free medium in the presence of HG, DP, or ethanol were isolated from the cell culture medium by ultracentrifugation. Exosomes and cellular fractions were run on SDS-PAGE, and Western blotting was used to analyze changes of specific proteins. a, representative Western blots and protein quantification of exosome samples in several experiments. b, representative Western blots and protein quantification of cellular samples in several experiments. Error bars indicate S.E., n = 3–4.
Quantitative Electron Microscopy Analysis Reveals a Lower Number of MVBs in HG Cells
As mentioned in the Introduction, the release of exosomes is the end point of a process that can be divided into three main steps as follows: the biogenesis of MVBs, the transport of MVBs to the plasma membrane, and the fusion of the MVBs with the plasma membrane. The experiments that we have performed so far show that HG treatment increases the release of exosomes in PC-3 cells. However, these experiments do not allow us to establish at what stage HG affects exosome release. In an attempt to obtain more information about the effect of HG, we performed a quantitative analysis by electron microscopy. Electron micrographs show that the morphology and size of MVBs and ILVs are quite similar in control (Fig. 11a) and in HG-treated cells (Fig. 11b). However, quantification of MVBs and ILVs showed that the numbers of MVBs per cell profile is lower in HG-treated cells and that these cells also contain less ILVs per MVB (Fig. 11c). In agreement with these results, the total number of ILVs per cell profile is also reduced in HG-treated cells (Fig. 11c). Finally, it should be mentioned that the number of gold particles per MVB was quite similar in control and HG-treated cells (Fig. 11c).
FIGURE 11.
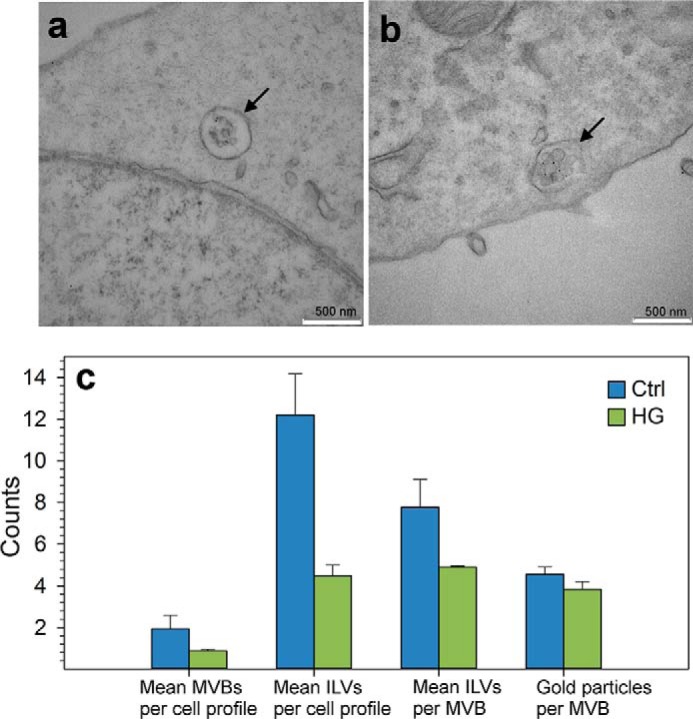
Electron microscopy analysis of control cells and HG-treated cells. One hour before the collection of exosomes was stopped, cells were incubated with BSA-gold 10 nm. Cells were then fixed, and Epon sections were prepared and visualized by electron microscopy. The figure shows an electron micrograph of control cells (a) and HG-treated cells (b) containing MVB (arrow). c, quantitative analysis of MVBs and ILVs in micrographs of control and HG-treated cells. Two independent sets of sections were used, and ∼60 cell profiles were quantified (n = 10–20 cells). Only healthy mononucleate interphase cells with the nucleus sectioned were used. In these cells, all gold-labeled MVBs (one or more ILVs) were analyzed.
DISCUSSION
Ether lipids have not obtained much attention even though they constitute a considerable proportion of membrane lipids, and their absence causes several diseases. Notably, these lipids play a role in membrane trafficking and fusion (29–33). In this study, we have investigated how increased levels of ether lipids affect the release of exosomes from the human prostate cancer cell line PC-3. The ether lipid precursor HG was used to increase the cellular levels of ether lipids. Interestingly, HG treatment of cells induces the release of higher levels of exosomes to the extracellular environment. Moreover, these exosomes contain much higher levels of ether lipids, especially ether-linked PE, and their protein composition is altered compared with exosomes released from control cells.
Although an increased level of ether lipids leads to increased exosome release, one cannot assume that a reduction will lead to the opposite. There is no reason to expect a linear relationship. However, to investigate whether a reduction in the cellular levels of ether lipids interferes with exosome release, the process was measured in cells with reduced levels of AGPS, a peroxisomal enzyme essential for the biosynthesis of ether lipids. In these cells, exosome release was slightly reduced. However, we have to consider that the levels of this enzyme were only depleted by ∼70%, probably due to the long half-life of peroxisomes and of this enzyme (50, 51).
The release of exosomes is the end point of a process that can be divided into several steps. At the moment there are not many experimental approaches that allow a clear differentiation between these stages. In this study, we have used quantitative electron microscopy in an attempt to get more information about the stage of exosome release that is affected by HG. The lower number of MVBs per cell profile observed in HG-treated cells is in agreement with the idea that more MVBs have fused with the plasma membrane in HG-treated cells and more ILVs have been released. Considering that ether lipids have previously been suggested to be implicated in membrane fusion (29), it is possible that the addition of HG increases the fusion of MVBs with the plasma membrane. Moreover, lower numbers of ILVs per MVB are found in HG-treated cells. This could indicate the possibility that less mature MVBs with fewer ILVs are left after increased fusion of mature MVBs with the plasma membrane. It is important to mention that even if these experiments have provided us with some clues about the stage of exosome release that may be affected in HG-treated cells, further experiments are required to confirm our hypothesis.
Our results suggest that HG is able to affect protein sorting at the level of MVBs and thereby the composition of the ILVs that later are released as exosomes. We do not know at the moment how HG exerts this effect. A possibility is that the ether lipid content of the limiting membrane of MVBs is higher in HG-treated cells and that this affects the physical properties of this membrane. In particular, it is possible that mainly the raft domains at the limiting membrane of MVBs are affected by HG treatment, because it has been reported that some ether lipids are enriched in lipid rafts (52). This is interesting because raft domains have been implicated in exosomal protein sorting (46). It should also be mentioned that not all the vesicular pathways are changed after HG treatment. We have previously found that although HG treatment of HEp-2 cells inhibited retrograde transport of Shiga toxin through the Golgi apparatus (33), there was no effect on the transport of the plant toxin ricin all the way from the cell surface, through the Golgi apparatus, and to the cytosol (34). Furthermore, we have recently reported that HG treatment of PC-3 cells does not affect the cAMP production caused by cholera toxin, thus suggesting that the endocytosis and retrograde trafficking of this toxin, which binds the ganglioside GM1, is unaltered (33).
Our lipidomic analyses clearly show that HG increases the levels of ether lipids in PC-3 cells compared with control cells or cells treated with DP, a substance that contains an acyl group instead of an ether group. However, HG treatment also induced changes in other lipid classes suggesting that there is a metabolic link between ether-linked phospholipids and other lipid classes. We recently showed that the levels of glycosphingolipids were reduced in HEp-2 cells after HG treatment (34), and we observed a similar effect in this study. However, in PC-3 cells, HG treatment resulted in higher levels of Cer, the substrate for GlcCer synthesis. This might explain why in PC-3 cells we observed only small differences between GlcCer in HG-treated cells and control cells, whereas the LacCer level was much lower in HG-treated cells. The mechanism behind the effect of HG in these lipids is not understood at the moment.
Exosomes have a characteristic lipid composition (39, 48, 53). In a previous study of the lipidome of exosomes released from PC-3 cells, we observed an enrichment of PS 18:0_18:1 in exosomes compared with cells (39). This enrichment was similar to that of CHOL and long-chain sphingolipids (N-amidated C22 and C24 species). Furthermore, assuming a diameter of 70 nm for exosomes, PS 18:0_18:1 in the inner leaflet could theoretically occupy ∼80% of the membrane area occupied by long-chain sphingolipids in the outer leaflet of exosomes, and we speculated that this was due to specific interactions between the two membrane leaflets (39). Such interactions might be required for exosome formation and for signaling across the membrane like that observed following binding of Shiga toxin to its lipid receptor on the outer leaflet of cells (54). Interestingly, in this study control exosomes and exosomes released in the presence of DP or HG showed ratios of PS 18:0_18:1 to long-chain sphingolipids (N-amidated C24 and C26 species) fitting with PS 18:0_18:1 and being able to occupy ∼95, 79, and 71%, respectively, in the inner leaflet of the area covered by these long-chain sphingolipids in the outer leaflet. It should be noted that the experimental conditions used in the two studies were not completely similar, and this may explain why long-chain glycosphingolipids contributed only 6% to the total long-chain sphingolipids in exosomes in the previous study (39) but 32–37% in the three exosomal preparations analyzed in this study. Despite this, we observed similar ratios of PS/sphingolipids in the two studies, and exosomes released by HG- and DP-treated cells showed similar ratios as in the control exosomes. Thus, these data provide further evidence for our previous theory that there may be “handshaking” between PS 18:0_18:1 on the inner leaflet and long-chain sphingolipids in the outer leaflet (39).
In conclusion, our studies reveal that ether lipids are able to modulate the release of exosomes as well as to change the composition of exosomes and highlight the importance of lipids in exosome release.
Acknowledgments
We thank Anne Engen for excellent assistance with cell culturing; Sirpa Sutela-Tuominen for assistance with the lipidomic analyses; the electron microscopy core facility at the Institute for Cancer Research; and Charles Ferguson for electron microscopy analyses. The facilities and scientific and technical assistance from the staff at the Australian Microscopy and Microanalysis Facility, Centre for Microscopy and Microanalysis, University of Queensland, are acknowledged.
This work was supported by the Norwegian Cancer Society, the South-Eastern Norway Regional Health Authority, The Research Council of Norway through its Centres of Excellence funding scheme, Project Number 179571, and by Grant 1045092 and Fellowship 1058565 (to R. G. P.) from the National Health and Medical Research Council of Australia.
For lipid abbreviations, see “Annotations of Lipid Species” under “Experimental Procedures.”
- MVB
- multivesicular body
- ILV
- intraluminal vesicle
- HG
- sn-1-O-hexadecylglycerol
- DP
- dl-α-palmitin
- MTT
- [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]
- BCA
- bicinchoninic acid
- NTA
- nanoparticle tracking analysis
- AGPS
- alkylglycerone phosphate synthase
- PC
- phosphatidylcholine
- PE
- phosphatidylethanolamine
- PG
- phosphatidylglycerol
- PS
- phosphatidylserine
- DAG
- diacylglycerol
- CE
- cholesteryl ester
- CHOL
- cholesterol
- SM
- sphingomyelin
- Cer
- ceramide
- LacCer
- lactosylceramide
- PL
- phospholipid
- oligo
- oligonucleotide.
REFERENCES
- 1. Raposo G., Stoorvogel W. (2013) Extracellular vesicles: exosomes, microvesicles, and friends. J. Cell Biol. 200, 373–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Record M., Carayon K., Poirot M., Silvente-Poirot S. (2014) Exosomes as new vesicular lipid transporters involved in cell-cell communication and various pathophysiologies. Biochim. Biophys. Acta 1841, 108–120 [DOI] [PubMed] [Google Scholar]
- 3. Robbins P. D., Morelli A. E. (2014) Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 14, 195–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Edgar J. R., Eden E. R., Futter C. E. (2014) Hrs- and CD63-dependent competing mechanisms make different sized endosomal intraluminal vesicles. Traffic 15, 197–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Möbius W., Ohno-Iwashita Y., van Donselaar E. G., Oorschot V. M., Shimada Y., Fujimoto T., Heijnen H. F., Geuze H. J., Slot J. W. (2002) Immunoelectron microscopic localization of cholesterol using biotinylated and non-cytolytic perfringolysin O. J. Histochem Cytochem 50, 43–55 [DOI] [PubMed] [Google Scholar]
- 6. White I. J., Bailey L. M., Aghakhani M. R., Moss S. E., Futter C. E. (2006) EGF stimulates annexin 1-dependent inward vesiculation in a multivesicular endosome subpopulation. EMBO J. 25, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baietti M. F., Zhang Z., Mortier E., Melchior A., Degeest G., Geeraerts A., Ivarsson Y., Depoortere F., Coomans C., Vermeiren E., Zimmermann P., David G. (2012) Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 14, 677–685 [DOI] [PubMed] [Google Scholar]
- 8. Colombo M., Moita C., van Niel G., Kowal J., Vigneron J., Benaroch P., Manel N., Moita L. F., Théry C., Raposo G. (2013) Analysis of ESCRT functions in exosome biogenesis, composition, and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 126, 5553–5565 [DOI] [PubMed] [Google Scholar]
- 9. Tamai K., Tanaka N., Nakano T., Kakazu E., Kondo Y., Inoue J., Shiina M., Fukushima K., Hoshino T., Sano K., Ueno Y., Shimosegawa T., Sugamura K. (2010) Exosome secretion of dendritic cells is regulated by Hrs, an ESCRT-0 protein. Biochem. Biophys. Res. Commun. 399, 384–390 [DOI] [PubMed] [Google Scholar]
- 10. Chairoungdua A., Smith D. L., Pochard P., Hull M., Caplan M. J. (2010) Exosome release of β-catenin: a novel mechanism that antagonizes Wnt signaling. J. Cell Biol. 190, 1079–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Buschow S. I., Nolte-'t Hoen E. N., van Niel G., Pols M. S., ten Broeke T., Lauwen M., Ossendorp F., Melief C. J., Raposo G., Wubbolts R., Wauben M. H., Stoorvogel W. (2009) MHC II in dendritic cells is targeted to lysosomes or T cell-induced exosomes via distinct multivesicular body pathways. Traffic 10, 1528–1542 [DOI] [PubMed] [Google Scholar]
- 12. Trajkovic K., Hsu C., Chiantia S., Rajendran L., Wenzel D., Wieland F., Schwille P., Brügger B., Simons M. (2008) Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 319, 1244–1247 [DOI] [PubMed] [Google Scholar]
- 13. Phuyal S., Hessvik N. P., Skotland T., Sandvig K., Llorente A. (2014) Regulation of exosome release by glycosphingolipids and flotillins. FEBS J. 281, 2214–2227 [DOI] [PubMed] [Google Scholar]
- 14. Hoshino D., Kirkbride K. C., Costello K., Clark E. S., Sinha S., Grega-Larson N., Tyska M. J., Weaver A. M. (2013) Exosome secretion is enhanced by invadopodia and drives invasive behavior. Cell Rep. 5, 1159–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hsu C., Morohashi Y., Yoshimura S., Manrique-Hoyos N., Jung S., Lauterbach M. A., Bakhti M., Grønborg M., Möbius W., Rhee J., Barr F. A., Simons M. (2010) Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A-C. J. Cell Biol. 189, 223–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ostrowski M., Carmo N. B., Krumeich S., Fanget I., Raposo G., Savina A., Moita C. F., Schauer K., Hume A. N., Freitas R. P., Goud B., Benaroch P., Hacohen N., Fukuda M., Desnos C., Seabra M. C., Darchen F., Amigorena S., Moita L. F., Thery C. (2010) Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 12, 19–30 [DOI] [PubMed] [Google Scholar]
- 17. Savina A., Fader C. M., Damiani M. T., Colombo M. I. (2005) Rab11 promotes docking and fusion of multivesicular bodies in a calcium-dependent manner. Traffic 6, 131–143 [DOI] [PubMed] [Google Scholar]
- 18. Nagan N., Zoeller R. A. (2001) Plasmalogens: biosynthesis and functions. Prog. Lipid Res. 40, 199–229 [DOI] [PubMed] [Google Scholar]
- 19. Braverman N. E., Moser A. B. (2012) Functions of plasmalogen lipids in health and disease. Biochim. Biophys. Acta 1822, 1442–1452 [DOI] [PubMed] [Google Scholar]
- 20. Gorgas K., Teigler A., Komljenovic D., Just W. W. (2006) The ether lipid-deficient mouse: tracking down plasmalogen functions. Biochim. Biophys. Acta 1763, 1511–1526 [DOI] [PubMed] [Google Scholar]
- 21. Wallner S., Schmitz G. (2011) Plasmalogens, the neglected regulatory and scavenging lipid species. Chem. Phys. Lipids 164, 573–589 [DOI] [PubMed] [Google Scholar]
- 22. Broniec A., Klosinski R., Pawlak A., Wrona-Krol M., Thompson D., Sarna T. (2011) Interactions of plasmalogens and their diacyl analogs with singlet oxygen in selected model systems. Free Radic. Biol. Med. 50, 892–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hahnel D., Beyer K., Engelmann B. (1999) Inhibition of peroxyl radical-mediated lipid oxidation by plasmalogen phospholipids and α-tocopherol. Free Radic. Biol. Med. 27, 1087–1094 [DOI] [PubMed] [Google Scholar]
- 24. Kuczynski B., Reo N. V. (2006) Evidence that plasmalogen is protective against oxidative stress in the rat brain. Neurochem. Res. 31, 639–656 [DOI] [PubMed] [Google Scholar]
- 25. Morand O. H., Zoeller R. A., Raetz C. R. (1988) Disappearance of plasmalogens from membranes of animal cells subjected to photosensitized oxidation. J. Biol. Chem. 263, 11597–11606 [PubMed] [Google Scholar]
- 26. Farooqui A. A. (2010) Studies on plasmalogen-selective phospholipase A2 in brain. Mol. Neurobiol. 41, 267–273 [DOI] [PubMed] [Google Scholar]
- 27. Hossain M. S., Ifuku M., Take S., Kawamura J., Miake K., Katafuchi T. (2013) Plasmalogens rescue neuronal cell death through an activation of AKT and ERK survival signaling. PLoS One 8, 10.1371/journal.pone.0083508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhan Y., Wang L., Liu J., Ma K., Liu C., Zhang Y., Zou W. (2013) Choline plasmalogens isolated from swine liver inhibit hepatoma cell proliferation associated with caveolin-1/Akt signaling. PLoS One 8, 10.1371/journal.pone.0077387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Glaser P. E., Gross R. W. (1994) Plasmenylethanolamine facilitates rapid membrane fusion: a stopped-flow kinetic investigation correlating the propensity of a major plasma membrane constituent to adopt an HII phase with its ability to promote membrane fusion. Biochemistry 33, 5805–5812 [DOI] [PubMed] [Google Scholar]
- 30. Lohner K., Balgavy P., Hermetter A., Paltauf F., Laggner P. (1991) Stabilization of non-bilayer structures by the etherlipid ethanolamine plasmalogen. Biochim. Biophys. Acta 1061, 132–140 [DOI] [PubMed] [Google Scholar]
- 31. Munn N. J., Arnio E., Liu D., Zoeller R. A., Liscum L. (2003) Deficiency in ethanolamine plasmalogen leads to altered cholesterol transport. J. Lipid Res. 44, 182–192 [DOI] [PubMed] [Google Scholar]
- 32. Thai T. P., Rodemer C., Jauch A., Hunziker A., Moser A., Gorgas K., Just W. W. (2001) Impaired membrane traffic in defective ether lipid biosynthesis. Hum. Mol. Genet. 10, 127–136 [DOI] [PubMed] [Google Scholar]
- 33. Bergan J., Skotland T., Lingelem A. B., Simm R., Spilsberg B., Lindback T., Sylvanne T., Simolin H., Ekroos K., Sandvig K. (2014) The ether lipid precursor hexadecylglycerol protects against Shiga toxins. Cell. Mol. Life Sci. 71, 4285–4300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bergan J., Skotland T., Sylvanne T., Simolin H., Ekroos K., Sandvig K. (2013) The ether lipid precursor hexadecylglycerol causes major changes in the lipidome of HEp-2 cells. PLoS One 8, 10.1371/journal.pone.0075904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Spector A. A., Yorek M. A. (1985) Membrane lipid composition and cellular function. J. Lipid Res. 26, 1015–1035 [PubMed] [Google Scholar]
- 36. Brites P., Ferreira A. S., da Silva T. F., Sousa V. F., Malheiro A. R., Duran M., Waterham H. R., Baes M., Wanders R. J. (2011) Alkyl-glycerol rescues plasmalogen levels and pathology of ether-phospholipid deficient mice. PLoS One 6, e28539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zoeller R. A., Grazia T. J., LaCamera P., Park J., Gaposchkin D. P., Farber H. W. (2002) Increasing plasmalogen levels protects human endothelial cells during hypoxia. Am. J. Physiol. Heart Circ. Physiol. 283, H671–H679 [DOI] [PubMed] [Google Scholar]
- 38. Zoeller R. A., Lake A. C., Nagan N., Gaposchkin D. P., Legner M. A., Lieberthal W. (1999) Plasmalogens as endogenous antioxidants: somatic cell mutants reveal the importance of the vinyl ether. Biochem. J. 338, 769–776 [PMC free article] [PubMed] [Google Scholar]
- 39. Llorente A., Skotland T., Sylvänne T., Kauhanen D., Róg T., Orl̸owski A., Vattulainen I., Ekroos K., Sandvig K. (2013) Molecular lipidomics of exosomes released by PC-3 prostate cancer cells. Biochim. Biophys. Acta 1831, 1302–1309 [DOI] [PubMed] [Google Scholar]
- 40. Ekroos K., Chernushevich I. V., Simons K., Shevchenko A. (2002) Quantitative profiling of phospholipids by multiple precursor ion scanning on a hybrid quadrupole time-of-flight mass spectrometer. Anal. Chem. 74, 941–949 [DOI] [PubMed] [Google Scholar]
- 41. Liebisch G., Binder M., Schifferer R., Langmann T., Schulz B., Schmitz G. (2006) High throughput quantification of cholesterol and cholesteryl ester by electrospray ionization tandem mass spectrometry (ESI-MS/MS). Biochim. Biophys. Acta 1761, 121–128 [DOI] [PubMed] [Google Scholar]
- 42. Ståhlman M., Ejsing C. S., Tarasov K., Perman J., Borén J., Ekroos K. (2009) High-throughput shotgun lipidomics by quadrupole time-of-flight mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 877, 2664–2672 [DOI] [PubMed] [Google Scholar]
- 43. Ekroos K., Ejsing C. S., Bahr U., Karas M., Simons K., Shevchenko A. (2003) Charting molecular composition of phosphatidylcholines by fatty acid scanning and ion trap MS3 fragmentation. J. Lipid Res. 44, 2181–2192 [DOI] [PubMed] [Google Scholar]
- 44. Merrill A. H., Jr., Sullards M. C., Allegood J. C., Kelly S., Wang E. (2005) Sphingolipidomics: high-throughput, structure-specific, and quantitative analysis of sphingolipids by liquid chromatography tandem mass spectrometry. Methods 36, 207–224 [DOI] [PubMed] [Google Scholar]
- 45. Jung H. R., Sylvänne T., Koistinen K. M., Tarasov K., Kauhanen D., Ekroos K. (2011) High throughput quantitative molecular lipidomics. Biochim. Biophys. Acta 1811, 925–934 [DOI] [PubMed] [Google Scholar]
- 46. de Gassart A., Geminard C., Fevrier B., Raposo G., Vidal M. (2003) Lipid raft-associated protein sorting in exosomes. Blood 102, 4336–4344 [DOI] [PubMed] [Google Scholar]
- 47. Subra C., Laulagnier K., Perret B., Record M. (2007) Exosome lipidomics unravels lipid sorting at the level of multivesicular bodies. Biochimie 89, 205–212 [DOI] [PubMed] [Google Scholar]
- 48. Wubbolts R., Leckie R. S., Veenhuizen P. T., Schwarzmann G., Möbius W., Hoernschemeyer J., Slot J. W., Geuze H. J., Stoorvogel W. (2003) Proteomic and biochemical analyses of human B cell-derived exosomes. Potential implications for their function and multivesicular body formation. J. Biol. Chem. 278, 10963–10972 [DOI] [PubMed] [Google Scholar]
- 49. Deleted in proof. [Google Scholar]
- 50. Biermann J., Gootjes J., Wanders R. J., van den Bosch H. (1999) Stability of alkyl-dihydroxyacetonephosphate synthase in human control and peroxisomal disorder fibroblasts. IUBMB Life 48, 635–639 [DOI] [PubMed] [Google Scholar]
- 51. Huybrechts S. J., Van Veldhoven P. P., Brees C., Mannaerts G. P., Los G. V., Fransen M. (2009) Peroxisome dynamics in cultured mammalian cells. Traffic 10, 1722–1733 [DOI] [PubMed] [Google Scholar]
- 52. Pike L. J., Han X., Chung K. N., Gross R. W. (2002) Lipid rafts are enriched in arachidonic acid and plasmenylethanolamine and their composition is independent of caveolin-1 expression: a quantitative electrospray ionization/mass spectrometric analysis. Biochemistry 41, 2075–2088 [DOI] [PubMed] [Google Scholar]
- 53. Laulagnier K., Motta C., Hamdi S., Roy S., Fauvelle F., Pageaux J. F., Kobayashi T., Salles J. P., Perret B., Bonnerot C., Record M. (2004) Mast cell- and dendritic cell-derived exosomes display a specific lipid composition and an unusual membrane organization. Biochem. J. 380, 161–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sandvig K., Bergan J., Kavaliauskiene S., Skotland T. (2014) Lipid requirements for entry of protein toxins into cells. Prog Lipid Res. 54, 1–13 [DOI] [PubMed] [Google Scholar]