

Background: The known mammalian glycerophosphodiester phosphodiesterases hydrolyze glycerophosphodiesters.
Results: New members of the glycerophosphodiester phosphodiesterase family, GDE4 and GDE7, cannot hydrolyze glycerophosphodiesters but show lysophospholipase D activity.
Conclusion: GDE4 and GDE7 can hydrolyze 1-acyl-lyso-PC and lyso-PAF to produce 1-acyl-lysophosphatidic acid (LPA) and alkyl-LPA, respectively.
Significance: The mammalian glycerophosphodiester phosphodiesterase family may have a new function in LPA signaling.
Keywords: Enzyme, Enzyme Kinetics, Gene Amplification, Glycerophospholipid, Lysophospholipid, Phosphatidylcholine, Phospholipase D, Phospholipid, Phospholipid Metabolism
Abstract
The known mammalian glycerophosphodiester phosphodiesterases (GP-PDEs) hydrolyze glycerophosphodiesters. In this study, two novel members of the mammalian GP-PDE family, GDE4 and GDE7, were isolated, and the molecular basis of mammalian GP-PDEs was further explored. The GDE4 and GDE7 sequences are highly homologous and evolutionarily close. GDE4 is expressed in intestinal epithelial cells, spermatids, and macrophages, whereas GDE7 is particularly expressed in gastro-esophageal epithelial cells. Unlike other mammalian GP-PDEs, GDE4 and GDE7 cannot hydrolyze either glycerophosphoinositol or glycerophosphocholine. Unexpectedly, both GDE4 and GDE7 show a lysophospholipase D activity toward lysophosphatidylcholine (lyso-PC). We purified the recombinant GDE4 and GDE7 proteins and show that these enzymes can hydrolyze lyso-PC to produce lysophosphatidic acid (LPA). Further characterization of purified recombinant GDE4 showed that it can also convert lyso-platelet-activating factor (1-O-alkyl-sn-glycero-3-phosphocholine; lyso-PAF) to alkyl-LPA. These data contribute to our current understanding of mammalian GP-PDEs and of their physiological roles via the control of lyso-PC and lyso-PAF metabolism in gastrointestinal epithelial cells and macrophages.
Introduction
Glycerophosphodiesters (GPs),3 such as glycerophosphocholine (GroPCho), glycerophosphoinositol (GroPIns), glycerophosphoserine (GroPSer), and glycerophosphoethanolamine (GroPEth), are water-soluble metabolites of the glycerophospholipids. GPs are produced via phospholipase A1 and phospholipase A2 activities, and they are degraded by GP phosphodiesterases (GP-PDEs) (1–4). Six mammalian GP-PDEs were previously isolated, and investigations have been carried out to explore their physiological significance (5, 6). In renal cells, GDE2 contributes to osmotic regulation as a GroPCho phosphodiesterase, which is supported by increasing evidence that GroPCho acts as an organic osmolyte (7–9). Moreover, our recent study demonstrated that GDE5 is a unique cytosolic protein that can regulate intracellular GroPCho concentration and myogenic differentiation (10). Previous work showed that both GDE1 and GDE3 can hydrolyze GroPIns (11, 12) and that the biological function of GDE3 in osteoblast proliferation and differentiation appears to be mediated by GroPIns. Indeed, GroPIns has been shown to regulate cell growth in thyroid cells (4), and induction of its hydrolysis through GDE3 expression results in reduced osteoblast proliferation and the appearance of markers of osteoblast differentiation (12). Thus, mammalian GP-PDEs have an intriguing feature; they show restricted substrate specificities, which prompted us to consider the possibility that mammalian GP-PDEs modulate GroPCho and/or GroPIns concentrations, because these intracellular GPs are increasingly recognized as bioactive molecules that are involved in a variety of cellular events (6). Recently, Simon and Cravatt (13, 14) reported that GDE1 is involved in the production of anandamide from glycerophospho-N-arachidonoylethanolamine in the nervous system. Moreover, a very recent study by Park et al. (15) demonstrated that GDE2 can cleave glycosylphosphatidylinositol anchors to induce spinal motor neuron differentiation. These studies inspired us to further consider that there might be additional physiological functions of mammalian GP-PDEs that are independent of their regulation of the levels of GPs, such as GroPCho and GroPIns.
In the present study, we explored novel mammalian GP-PDE cDNAs using sections of the catalytic sequence of the GDE domain to locate expressed sequence tags. We isolated two novel members of the GP-PDE family, GDE4 and GDE7. We explored their enzymatic activities to understand their distinct biological relevance and found that both GDE4 and GDE7 do not show GP-PDE activity toward GPs, such as GroPCho and GroPIns. Unexpectedly, these two new GP-PDEs have a lysophospholipase D activity, because they can convert lysophosphatidylcholine (lyso-PC) and 1-O-alkyl-sn-glycero-3-phosphocholine (lyso-PAF) to acyl-lysophosphatidic acid (LPA) and alkyl-LPA, respectively. This study identifies a novel pathway leading to the formation of LPA and alkyl-LPA that is based on the activity of GDE4 and GDE7. These mammalian GP-PDEs might therefore play roles in the regulation of LPA and alkyl-LPA biological activities.
EXPERIMENTAL PROCEDURES
Materials
Restriction endonucleases and DNA-modifying enzymes were from TaKaRa Bio (Kyoto, Japan) and TOYOBO (Osaka, Japan). l-α-Lyso-PC from egg yolk, l-α-phosphatidylcholine from egg yolk, 1-palmityl-sn-glycero-3-phosphocholine, oleoyl-l-α-LPA, sphingomyelin, and 1,2-dibutyryl-sn-glycero-3-phosphatidylcholine were from Sigma. 1-Hexadecanoyl-sn-glycero-3-phosphocholine, lysophosphatidylinositol (lyso-PI; soybean), lysophosphatidylserine (lyso-PS), and lysophosphatidylethanolamine (lyso-PE; egg yolk) were from Avanti Polar Lipids (Alabaster, AL). 1-Hexadecyl-2-hydroxy-sn-glycero-3-phosphate, 1-hexadecy-2-acetyl-sn-glycero-3-phosphocholine, and autotaxin were from Cayman (Ann Arbor, MI). GroPCho, GroPIns, GroPSer, and GroPEth were prepared as described previously (16).
Database Search for Novel Members of the Mammalian GP-PDE Family
An amino acid sequence containing a putative GP-PDE domain of mouse GDE3 (residues 500–875) (17) was used as the query to search the GenBankTM database with BLAST (18).
Cell Culture, Expression, and Measurement of Enzymatic Activity
HEK293T, COS7, 3T3-L1, and RAW264.7 cells were cultured in maintenance medium (10% fetal calf serum, 100 units/ml penicillin, 100 μg/ml streptomycin in DMEM) at 37 °C in 5% CO2, 95% humidified air. The full-length GDE7 cDNA was subcloned into the expression vector pCMV-EGFPN1. HEK293T cells were transfected with full-length GDE1, GDE3, GDE4, or GDE7 cDNA in pCMV-EGFPN1 using Lipofectamine 2000 (Invitrogen). Twenty-four h after transfection, the cells were washed twice with ice-cold phosphate-buffered saline (PBS), harvested in the homogenizing buffer (10 mm Tris-HCl, pH 7.5, 5 μg/ml leupeptin, 0.5 mm phenylmethylsulfonyl fluoride, 1 mm dithiothreitol), and centrifuged at 100,000 × g for 30 min. The pellet was homogenized in the assay buffer (10). HEK293T cells were also transfected with pEF-GDE5, washed with ice-cold PBS, and scraped in the homogenizing buffer. The homogenates were centrifuged at 10,000 × g for 10 min. These protein samples were subjected to the enzymatic assays. The GP-PDE activity for GroPIns was examined using an enzyme-coupled spectrophotometric assay as described previously (10). The GP-PDE activity for GroPCho and lysophospholipase D activity toward lyso-PC were measured by the released choline. The choline was measured by a commercial kit (BioVision, Mountain View, CA). pGAL4-PPARγ chimera receptor and the reporter plasmid 6xUAS-luc plasmid were provided by Dr. Junko Mizukami (Tanabe Seiyaku Co., Osaka, Japan). HEK293T cells were transfected with pCMV-EGFPN1 or pCMV-EGFPN1-GDE4 and pGAL4-PPARγ chimera receptor plasmid, 6xUAS-luc plasmid, and pCMV-β-gal (Invitrogen). After 24 h, the cells were scraped and subjected to a luciferase assay. O-Nitrophenyl-β-d-galactropyranoside was used as a substrate for β-galactosidase activity.
Expression and Purification of Recombinant Mouse GDE4 and GDE7 in Insect Cells
The mouse cDNAs encoding GDE4 and GDE7 were obtained from FANTOM2 clones (clone ID 3000003A09 and 1110015E22, respectively) (19). The GDE4 and GDE7 cDNAs were inserted into BaculoDirect C-Term Linear DNA, which added the V5 peptide and His6 tag at the C terminus. Sf9 cells were transfected by the DNA to produce the baculovirus. Sf9 cells were cultured in Sf-900II SFM (Invitrogen) containing 10% Grace insect medium (Invitrogen), 1% fetal calf serum, 1 μg/ml gentamicin (Invitrogen), 0.25 μg/ml amphotericin B, and 0.2% pluronic (Sigma). The cells were collected at 48 h after transfection and disrupted by sonication in 20 mm Tris-HCl, pH 8.0, 500 mm NaCl. After centrifugation at 800 × g for 10 min, the supernatant was further centrifuged at 105,000 × g for 60 min at 4 °C to obtain the membrane fraction of Sf9 cells. This was solubilized in 2% dodecyl-β-d-maltoside (DDM), 50 mm Tris-HCl, pH 8.0, 10 mg/ml membrane protein, 500 mm NaCl, and 5 mm 2-mercaptoethanol at 4 °C for 1 h. After centrifugation at 105,000 × g for 1 h, the supernatant was subjected to a Ni2+-bound HiTrap chelating column (GE Healthcare) equilibrated with 20 mm Tris-HCl buffer (pH 8.0), containing 500 mm NaCl, 0.1% DDM, and 50 mm imidazole. The protein was eluted with the same buffer, except for imidazole (500 mm). The eluate was then applied to a HiLoad 16/60 Superdex 200 column (GE Healthcare) equilibrated with 20 mm Tris-HCl, pH 8.0, 200 mm NaCl, and 0.02% DDM.
Measurement of GP-PDE and Lysophospholipase D Activities of the Recombinant Mouse GDE4 and GDE7 Proteins
GP-PDE activity for GroPCho, GroPIns, GroPSer, and GroPEth was examined as described previously (10). Lysophospholipase D activity of the recombinant GDE4 and GDE7 proteins toward lyso-PC, PC, and sphingomyelin was measured by the released choline. Lysophospholipase D activities of GDE4 and GDE7 toward lyso-PC, lyso-PAF, lyso-PE, lyso-PI, and lyso-PS were also examined by measuring the acyl-LPA or alkyl-LPA produced in a mixture containing 50 mm HEPES-NaOH, pH 7.4, 5 mm MgCl2, 100 μm substrate, 0.1 mg/ml bovine serum albumin. The lipids were extracted using the Bligh and Dyer method (20) under acidic conditions (pH 2–3) three times for the efficient recovery of acyl-LPA and alkyl-LPA. The pooled organic layer was evaporated and dissolved in chloroform/methanol (2:1). The lipids were separated using TLC chromatography (silica gel 60 plate, Merck). The solvent system consisted of chloroform, methanol, 20% ammonia (60:35:8). The phospholipids were visualized by iodine vapor or modified Dittmer-Lester reagent. The acyl-LPA and alkyl-LPA produced by the enzyme reaction were quantified using ImageJ software. For the determination of kinetic parameters, the released choline was obtained from the aqueous phase of Bligh and Dyer extraction and was quantified using the choline quantification kit. The kinetic data were analyzed using GraphPad Prism (GraphPad Software, San Diego, CA).
Immunofluorescence Microscopy
COS-7 cells were transfected with pEGFP-GDE4 or GDE7 constructs. Twenty-four h after transfection, the cells were fixed, and anti-calnexin antibody was visualized using Cy3-labeled goat anti-mouse IgG. COS7 cells were transfected with GDE4 cDNA in the pcDNA3.1/Myc expression vector and the pEGFP-phospholipase A2 IV α (PLA2IVα) construct. After 24 h, the cells were fixed with 4% paraformaldehyde and 4% sucrose in 0.2 m sodium phosphate buffer (pH 7.2) for 30 min and blocked with 1.5% normal goat serum in PBS for 1 h at room temperature, followed by incubation with an anti-calnexin antibody for 18 h at 4 °C. The primary antibody was visualized with Cy3-labeled goat anti-mouse IgG (GE Healthcare). The epididymal adipose tissue from db/db mice was fixed with neutral buffered formalin and embedded in paraffin. An immunohistochemical study was carried out using 4-μm-thick paraffin-embedded sections. For the secondary antibody, Alexa Fluor 488-labeled anti-rat IgG (Cell Signaling Technology) or Cy3-labeled anti-rabbit IgG (GE Healthcare) was used. Epifluorescent images were captured with a CCD camera (Hamamatsu) mounted on a Nikon Eclipse E600 microscope. Small interfering RNA (siRNA) duplex oligoribonucleotides against mouse GDE4 were synthesized by Sigma. The sequences were as follows: sense, 5′-GAAGGUAACUGGCAACUGATT-3′; antisense, 5′-UCAGUUGCCAGUUACCUUCTT-3′. Universal negative control siRNA (Sigma) was used as a control siRNA. 3T3-L1 cells were transfected with these siRNAs to a final concentration of 20 nm using Lipofectamine RNAimax (Invitrogen).
Quantitative Analyses of LPA, Lyso-PC, and Choline Metabolites
The extraction method of Bligh and Dyer (20) was modified according to the procedure by Aaltonen et al. (21). The UPLC-MS analysis was carried out using an Acquity UPLC system (Waters, Milford, MA) coupled to an Acquity TQD tandem quadrupole mass spectrometer (Waters) with electrospray ionization in the negative modes. Sample solution was injected onto a BEH C18 column (Waters; 2.1 × 50 mm, 1.7 μm) at a flow rate of 0.4 ml/min using gradient elution according to the procedure (21). Analyte detection was performed using multiple-reaction monitoring with the following transition: m/z 409 → 153 for 16:0 LPA. Cone voltage was 30 V, and collision energy was 2 V. Dwell time was 50 ms. An internal standard (17:0 LPA) was used for quantification.
Lipid metabolites from 3T3-L1 adipocytes were extracted using the Bligh and Dyer method. Their organic phases were evaporated and resuspended in chloroform/methanol (2:1). The lipids were subjected to the UPLC-MS with positive ion mode electrospray ionization. Samples were injected onto a BEH C18 column (Waters; 2.1 × 50 mm, 1.7 μm) at a flow rate of 0.4 ml/min, using gradient elution with 0.1% formic acid (A) and 0.1% formic acid in acetonitrile (B) as follows: 0–1.0 min 25% B, 1.0–6.0 min 25% B → 95% B, 6.0–7.0 min 95% B. Analyte detection was performed using multiple-reaction monitoring with the following transition: m/z 496.4 → 184 for 16:0 lyso-PC. Cone voltage was 30 V, and collision energy was 25 V. Dwell time was 50 ms. The upper aqueous phase was subjected to UPLC-MS with positive ion mode electrospray ionization. Samples were injected onto a BEH HILIC column (Waters; 2.1 × 50 mm, 1.7 μm) at a flow rate of 0.4 ml/min using gradient elution with 3 mm ammonium acetate (pH 8.0) in 80% acetonitrile (A) and 2.3 mm ammonium acetate (pH 8.0) in 5% acetonitrile (B) as follows: 0–0.1 min 100% A, 0.1–2.75 min 0% B → 60% B, 2.75–5.0 min 100% B. Analyte detection was performed using single ion recording: m/z 104 for choline and m/z 184.5 for phosphocholine. Cone voltage was 25 V. Dwell time was 50 ms. Peak area ratios of the analyte were calculated as a function of the concentration ratios of the analyte (QuanLynx, Waters).
Preparation of Anti-GDE4 and Anti-GDE7 Antibodies and Western Blotting
A cDNA encoding part of mouse GDE7 (amino acids 210–332) was subcloned into pMAL-c (New England Biolabs, Beverly, MA), to generate pMAL-GDE7. Polyclonal antibodies were obtained by injecting rabbits with the maltose-binding protein-GDE7 fusion protein. These antibodies were affinity-purified using an antigen-coupled Sepharose column (GE Healthcare). An anti-GDE4 rabbit polyclonal antibody was obtained from Hokudo (Hokkaido, Japan) using the purified mouse GDE4 protein as antigen. Mouse tissues were homogenized as described previously (17). The homogenates were centrifuged at 10,000 × g for 15 min. Ten μg of the supernatants were subjected to Western blotting as described previously (17).
Animals
Male ICR (CD-1; 7 weeks old), db/db (BKS.Cg-m+/+ Lepr db/J; 7 weeks old), and db/+ (7 weeks old) mice were maintained in accordance with the Hiroshima University Guidelines for the Care and Use of Laboratory Animals. The animal study was approved by the Hiroshima University Animal Committee (permit number C13-3).
RT-PCR Analyses
Stromal vascular fraction cells and adipocytes were isolated as described previously (22). Total RNA from mouse tissues and cells was prepared using RNeasy kits (Qiagen). The reverse transcriptase reaction and semiquantitative PCR analysis were carried out as described previously (8). The primers were as follows: GDE1, forward (5′-CAGGCAGCTAAGAATGGAGCAACAGGTGTG-3′) and reverse (5′-CCTTCTGCATGAGGAAGGCTGAAATCCCAC-3′); GDE2, forward (5′-TCATGCATGACACTACCCTGAGGCG-3′) and reverse (5′-ATGCATAGTCCCTGAGCTCCTGGTG-3′); GDE3, forward (5′-CCTTCTACCGCATCCACCCAAGAGG-3′) and reverse (5′-GGGTATTCTCAGGGGCCAGCATGGG-3′); GDE4, forward (5′-CAGCGATTCCTCAGTAGGCACATCTCTCAC-3′) and reverse (5′-GGTTCTTTTAGCTTCAGTATGATGGAGGGC-3′); GDE5, forward (5′-TTTGATGTCCACCTTTCAAAGGAC-3′) and reverse (5′-ACAGTTCTTAAGCTCTGGCAATTC-3′); GDE6, forward (5′-ACAAATGCTGCATCACCTTCCTGAC-3′) and reverse (5′-GTGCACGAAGGGAGCAAAGATCTGC-3′); GDE7, forward (5′-CCTGTCCCGCCAGTCAGGCCTAAATAAGG-3′) and reverse (5′-GCCCCAGGTAGTAGAGCAGCAGTATCCAG-3′); Emr1, forward (5′-ATTGTGGAAGCATCCGAGAC-3′) and reverse (5′-GTAGGAATCCCGCAATGATG-3′); Msr1, forward (5′-TCAAACTCAAAAGCCGACCT-3′) and reverse (5′-ACGTGCGCTTGTTCTTCTTT-3′); β-actin, forward (5′-TTGGGTATGGAATCCTGTGGCATC-3′) and reverse (5′-CGGACTCATCGTACTCCTGCTTGC-3′); adiponectin, forward (5′-ACAGGAGATGTTGGAATGACAG-3′) and reverse (5′-CTGCATAGAGTCCATTGTGGTC-3′).
Quantitative PCR Analysis
Epididymal white adipose tissue of db/db, db/+, and ICR mice fed on a high fat diet (HFD; 60% calories as fat) for 8 weeks was isolated. Quantitative PCR analysis was carried out as described previously (9).
In Situ Hybridization
In situ hybridization using digoxigenin-labeled probes was performed as described previously (18).
Statistical Analyses
Statistical significance was determined by one-way analysis of variance and Duncan's multiple-range test. Differences were considered significant for p < 0.05 (*) and p < 0.01 (**).
RESULTS
Isolation of Two Novel Members of the Glycerophosphodiester Phosphodiesterase Family, GDE4 and GDE7
Using a bioinformatic approach, the nucleotide sequence corresponding to the GDE domain of mouse GDE3 was used to search the GenBankTM database with BLAST. Two homologous sequences containing the GP-PDE motif (pfam03009) were found in the mouse cDNA database (NM_025638 and NM_024228). Two novel mouse cDNAs, termed GDE4/gdpd1 and GDE7/gdpd3, were shown to encode proteins of 314 and 330 amino acids, respectively. On the basis of the amino acid sequence, mouse GDE4/gdpd1 showed 92% homology to human GDE4 (23), a so far poorly understood member of the GP-PDE protein family. The amino acid sequence of the GDE domain is conserved among GDE1, GDE4, and GDE7 and showed a cluster of strictly conserved residues (His-45, Arg-46, Glu-54, Asn-55, Glu-72, His-87, and Asp-88) in human GDE4 protein (Fig. 1A). As shown in Fig. 1B, hydropathy analysis also indicated that GDE4 and GDE7 have two putative transmembrane regions, as for GDE1. The amino acid identities of mouse GDE1 and GDE4 toward mouse GDE7 are 34.5 and 53.4%, respectively (Fig. 1B). The amino acid sequences of both the prokaryote and eukaryote GP-PDE protein families were aligned using ClustalW. The resulting phylogenetic tree showed the closest evolutionary relationship between GDE4 and GDE7 proteins (Fig. 1C). This study was designed to investigate the detailed expression profiles and enzymatic properties of mouse GDE4 and GDE7, uncharacterized members of the mammalian GP-PDE family.
FIGURE 1.
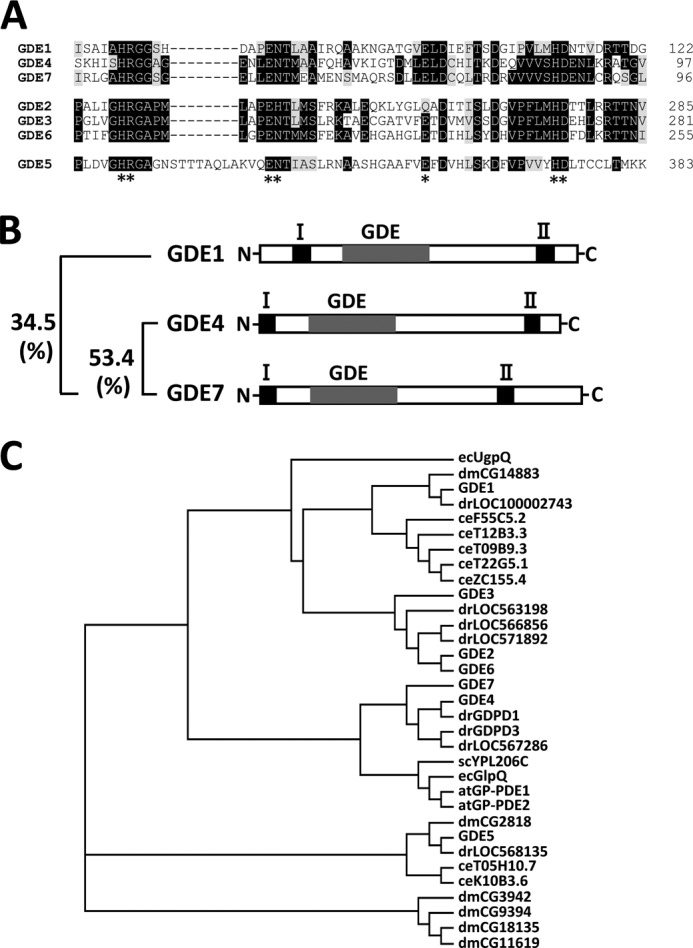
GDE4 and GDE7 are novel members of the mammalian GP-PDE family. A, deduced amino acid sequences of the conserved catalytic regions of human GP-PDEs. Black boxes, identical amino acids; gray boxes, similar amino acids. Conserved amino acids are indicated by asterisks. B, schematic diagrams illustrating the primary structures of these three mouse GP-PDEs. Putative transmembrane regions are numbered from the N terminus. GDE represents the putative GDE domain. The sequence identity of the amino acids among the putative catalytic regions of GDE1, GDE4, and GDE7 is shown. C, phylogenetic tree of GP-PDE family members. The GDE domains of putative GP-PDEs from both prokaryotes and eukaryotes with mouse GP-PDEs (GDE1–7) are aligned. The phylogenetic tree indicates the closest evolutionary relationship between GDE4 and GDE7 proteins. ec, Escherichia coli; dm, Drosophila melanogaster; dr, Danio rerio; ce, Caenorhabditis elegans; sc, Saccharomyces cerevisiae; at, Arabidopsis thaliana.
Tissue and Cellular Localization of GDE4 and GDE7
RT-PCR analyses were performed using total RNA derived from various mouse tissues (Fig. 2A). As reported previously (24), GDE1 mRNA was widely expressed in mouse tissues, including lung, heart, brain, kidney, and testis. In contrast, GDE7 mRNA was highly expressed in mouse stomach and kidney. GDE4 mRNA was detected in the gastrointestinal tract, including the colon and intestine, and testis and was widely seen in brain regions. These results suggest that mammalian GP-PDEs have different patterns of tissue distribution, and as such, each might have distinct physiological roles. We focused on the tissue expression pattern of GDE4 and GDE7 in the gastrointestinal tract. Western blotting showed that the GDE7 protein is predominantly expressed in the mouse stomach (Fig. 2B). To identify the cell types in which the GDE4 and GDE7 mRNAs are expressed, sections from a variety of adult mouse gastrointestinal tract and testis were hybridized in situ with an antisense RNA probe complementary to GDE4 or GDE7. GDE4 mRNA is expressed in intestinal epithelial cells and spermatids (Fig. 2C), whereas intense hybridization signals for GDE7 mRNA were detected in the glandular epithelium (Fig. 2D). Sense RNA was used as a control probe, and no positive staining was observed.
FIGURE 2.

mRNA and protein expression of the mammalian GDE family genes in mouse tissues. A, total RNAs from mouse tissues were prepared and subjected to semiquantitative PCR analyses. GDE4 mRNA was mainly detected in the gastrointestinal tract, testis, and widely in brain regions. GDE7 mRNA was highly expressed in mouse kidney and stomach tissues. B, Western blot analysis of GDE4 and GDE7 in the gastrointestinal tract. GDE4 was distributed all over the gastrointestinal tract, whereas GDE7 was mainly detected in the mouse stomach among these tissues. C, in situ hybridization showing GDE4 mRNA in intestinal epithelial cells and in spermatids in testis sections. D, in situ hybridization showing GDE7 mRNA highly expressed in gastroesophageal epithelial cells.
To address the intracellular distribution of the GDE4 and GDE7 proteins, COS-7 cells were transiently transfected with pEGFP-GDE4 or pEGFP-GDE7 constructs. The predominant signals for both the green fluorescent protein (GFP)-tagged GDE4 and GDE7 were localized to the perinuclear region (Fig. 3A). The intracellular localizations of GDE4 and GDE7 were further examined by comparing their staining patterns in COS-7 cells with that of the endoplasmic reticulum marker calnexin. Here, recombinant GDE4 and GDE7 partially co-localized with calnexin (Fig. 3A). The intracellular localization of GDE4 was further examined by comparing the staining patterns in COS-7 cells with PLA2IVα, which under these conditions showed a diffuse localization (cytosolic) as well as a perinuclear staining compatible with a Golgi localization (Fig. 3B), in agreement with the reported localization of this enzyme (25). The results showed that heterologous GDE4 partially co-localized with overexpressed PLA2IVα, suggesting its prevalent Golgi localization (Fig. 3B). Further examinations are needed to clarify the intracellular localizations of GDE4 and GDE7 using specific markers of the Golgi complex compartments.
FIGURE 3.
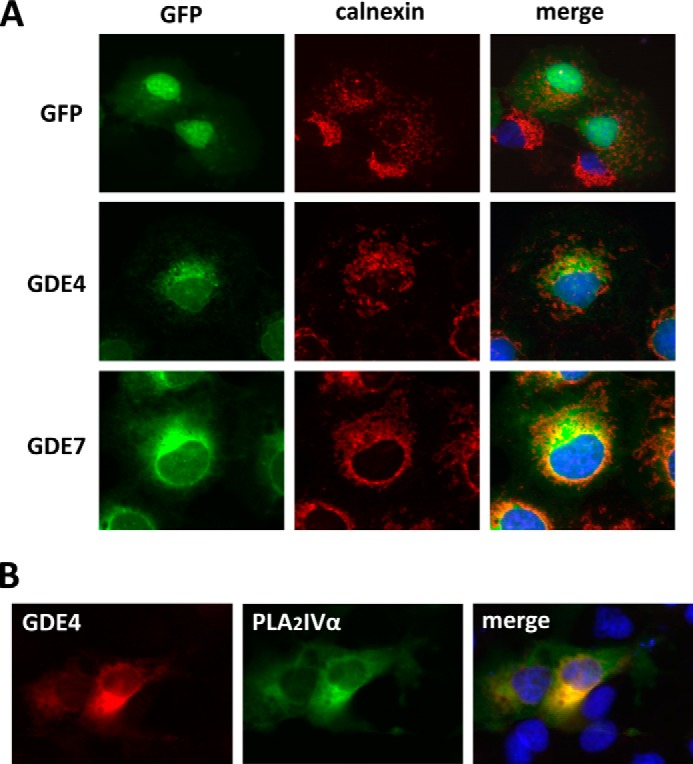
Intracellular localization of mouse GDE4 and GDE7. A, COS-7 cells were transiently transfected with constructs for GFP tag alone or GFP-tagged GDE4 and GDE7. Twenty-four h after transfection, the cells were fixed, and anti-calnexin antibody was visualized by Cy3-labeled goat anti-mouse IgGs. B, COS-7 cells were transiently transfected with Myc-tagged GDE4 with GFP-tagged PLA2IVα. Twenty-four h after transfection, the cells were fixed, and the primary antibody (anti-Myc monoclonal antibody 9E10) was visualized using Cy3-labeled goat anti-mouse IgGs.
GDE4 and GDE7 Enzymatic Activities
Enzymatic assays using protein samples from HEK293T cells transfected with pEGFP-GDE4 or pEGFP-GDE7 constructs indicated that neither GroPIns nor GroPCho was hydrolyzed by GDE4 and GDE7 (Fig. 4). Interestingly, GDE4 and GDE7 contain a C-terminal region that is conserved with secreted sphingomyelinase D (SMaseD) (amino acids 320–385) from the arachnid Loxosceles laeta (Fig. 5A). A previous study by van Meeteren et al. (26) examined the substrate specificity of L. laeta SmaseD and showed that it has lysophospholipase D activity toward lyso-PC. Thus, we analyzed the lysophospholipase activity of GDE4 and GDE7 toward lyso-PC. We found that the released choline was elevated during the enzyme assay using postnuclear preparations of the HEK293T cells expressing GDE4 or GDE7 (Fig. 5B). However, no significant changes in choline release were observed for GDE1 and GDE5.
FIGURE 4.
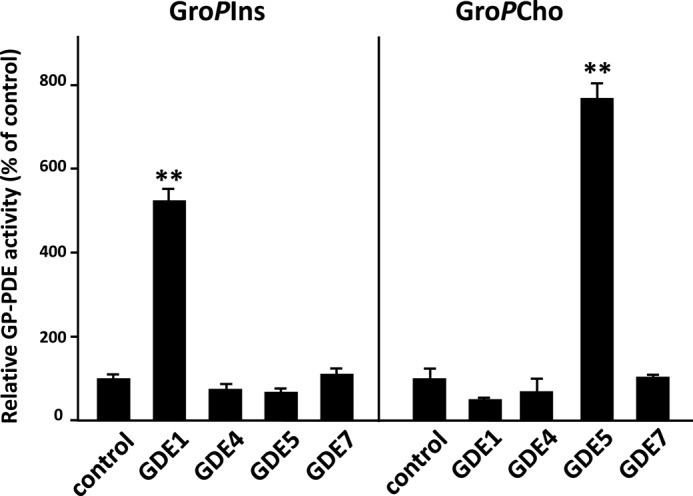
GDE4 and GDE7 cannot hydrolyze either glycerophosphoinositol or glycerophosphocholine. HEK293T cells were transiently transfected with constructs for the mammalian GDEs. control, empty vector-transfected sample. Total protein extracts from transfected HEK293 cells were subjected to enzymatic assays for GroPCho and GroPIns PDE activity as described under “Experimental Procedures.” The data are from a single experiment carried out in triplicate (mean ± S.D. (error bars)) and are representative of two independent experiments.
FIGURE 5.

GDE4 and GDE7 can hydrolyze lyso-PC. A, the GDE4 and GDE7 proteins contain a C-terminal domain that shares a common motif with secreted sphingomyelinases, L. laeta clone H17 sphingomyelinase (AY093599) and Loxosceles boneti (Lb) sphingomyelinases (AY559844 and AY559845). B, HEK293 cells were transiently transfected with constructs for the mammalian GDEs. Total protein extracts from transfected HEK293 cells were subjected to an enzymatic assay for lyso-PC PDE activity by measuring choline release. Control represents the empty vector-transfected sample. The data are from a single experiment carried out in triplicate (mean ± S.D. (error bars)) and are representative of two independent experiments.
To characterize the substrate specificity for GDE4, the double His- and V5-tagged mouse GDE4 protein was overexpressed in insect Sf9 cells and purified by Ni2+-chelate affinity chromatography. The purity of this mouse recombinant GDE4 was determined by SDS-PAGE. A single protein band was observed at 37 kDa, which corresponds to the calculated molecular mass of the His-tagged GDE4 (Fig. 6A). Using the purified enzyme, we did not detect GDE4-mediated hydrolysis of GroPCho, GroPIns, GroPSer, or GroPEth (data not shown). Next, lyso-PC, PC (egg yolk), and sphingomyelin were tested as substrates of this purified GDE4 by measuring the released choline. GDE4 preferentially hydrolyzed lyso-PC (Fig. 6B).
FIGURE 6.
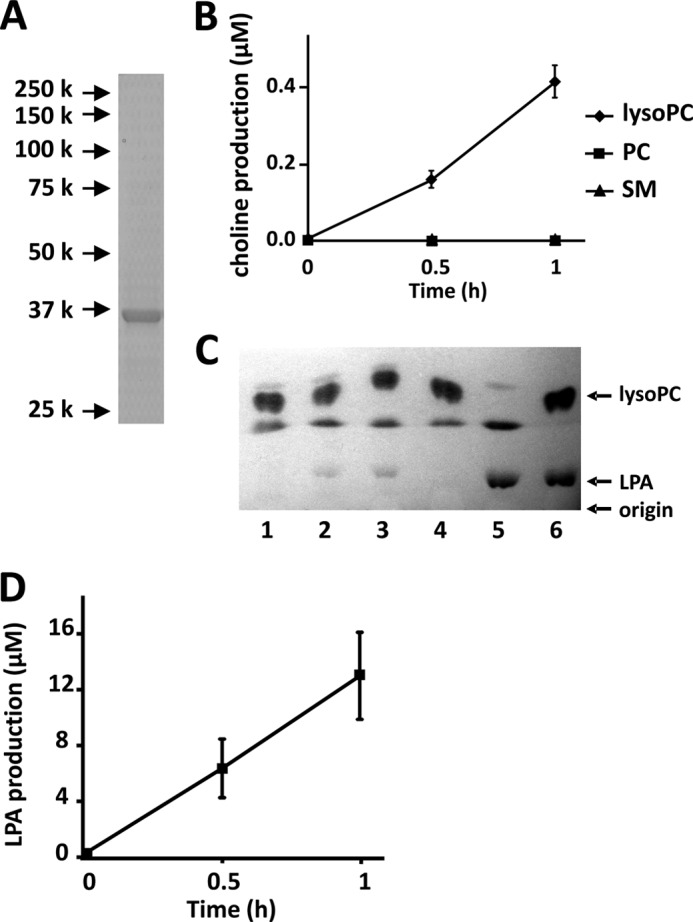
GDE4 hydrolyzes lyso-PC into LPA and choline. A, His-tagged mouse GDE4 protein was overexpressed in Sf9 cells. Purified recombinant mouse GDE4 was visualized by Coomassie Brilliant Blue (CBB) staining. A single protein band was observed at 37 kDa. B, substrate specificity of mouse GDE4. The substrates (10 μg/ml) used for the enzyme reaction were lyso-PC, PC, and sphingomyelin (SM). Lysophospholipase D activity was examined in the presence of GDE4 (4 μg/ml) by measuring the released choline. The data are from a single experiment carried out in triplicate (mean ± S.D. (error bars)) and are representative of three independent experiments. C, enzyme activity of GDE4 (5 μg) toward lyso-PC (18:1) was measured using thin layer chromatography (TLC). The enzyme reaction was stopped at 0 min (lane 1), 30 min (lane 2), and 60 min (lane 3). The reaction mixture was incubated for 60 min without mouse GDE4 (lane 4). 20 nmol of LPA (18:1) was incubated for 60 min instead of lyso-PC in the presence of mouse GDE4 to verify the recovery of LPA by the acidic Bligh and Dyer method (lane 5). Twenty nmol of LPA and 50 nmol of lyso-PC were spotted on TLC as standard. Phospholipids were visualized by iodine vapor. Data are representative of three independent experiments. D, the amount of LPA produced by the enzyme reaction shown in C was quantified using ImageJ software. Data are means of triplicate experiments (mean ± S.E.).
We also characterized the enzymatic activity of the purified GDE4 protein by measuring LPA production. GDE4 produced LPA from lyso-PC in a time-dependent manner (Fig. 6, C and D). Because the possibility of contamination by insect proteins could not be excluded, we further purified the GDE4, obtained from the Ni2+-chelate affinity chromatography, immunoprecipitating it with an antibody against the V5 tag, and we confirmed its lyso-PC hydrolytic activity (data not shown). The enzyme activities of GDE4 toward lyso-PC, lyso-PAF, lyso-PE, lyso-PI, and lyso-PS were analyzed by measuring LPA production. Among these lysophospholipids, lyso-PAF was the preferred substrate (Fig. 7, A and B). The enzyme activity toward lyso-PC was about half that toward lyso-PAF (Fig. 7, A and B). Lyso-PI, lyso-PE, and lyso-PS were hydrolyzed by GDE4 to a lesser extent (Fig. 7B). GDE4 did not hydrolyze 1,2-dibutyryl-sn-glycero-3-phosphatidylcholine (dibutyryl-PC) (Fig. 7C). Even short hydrocarbon chains in PC are not permissive for this enzyme reaction, suggesting that GDE4 activity is specific for lysophospholipids and that it is not dependent on the water solubility of the substrates. The enzyme activity of GDE4 toward lyso-PC was approximately one-fourth of that of autaxin, a well known lysophospholipase D (27), in the same assay conditions (Fig. 7D). The Km, kcat, and kcat/Km were calculated from kinetic experiments and shown in Table 1. Km and kcat/Km values for lyso-PC are 1.0 mm and 0.31 × 102 m−1 s−1, whereas those for lyso-PAF are 0.32 mm and 2.4 × 102 m−1 s−1, respectively. These observations indicate that GDE4 has a lysophospholipase D activity toward lysophospholipids, especially toward lyso-PAF. As shown in Fig. 3A, GDE4 protein is localized to the perinuclear region. HEK293T cells transfected with pEGFP-GDE4 showed an about 1.5-fold increase in intracellular LPA (16:0) level as compared with those transfected with pEGFP (Fig. 7E). These observations raised the possibility that GDE4 is a candidate for directly regulating the intracellular LPA level. A further proof that GDE4 expression increases intracellular LPA levels comes from experiments demonstrating that GDE4 acts as a peroxisome proliferator-activated receptor γ (PPARγ) activator. Co-expression of an established chimera system, in which the ligand-binding domain of PPARγ was fused to the DNA-binding domain of the yeast transcription factor GAL4, together with GDE4 overexpression resulted in a 7-fold increase in the reporter activity (Fig. 7F), suggesting that LPA produced by GDE4 acts as a potent activator of PPARγ. To confirm the involvement of GDE4 in intracellular lyso-PC metabolism, we performed siRNA-mediated GDE4 knockdown in 3T3-L1 cells. The reduction in GDE4 mRNA and protein levels was assessed by quantitative RT-PCR (Fig. 8A) and Western blotting, respectively (Fig. 8B). Although intracellular LPA levels in adipocytes were under the detection limits, GDE4 silencing resulted in an increase in intracellular lyso-PC (16:0), which is a good substrate for GDE4 in vitro. Intracellular choline levels remained unchanged in GDE4-interfered cells; however, another component of the aqueous choline pool, the phosphocholine, decreased in levels (Fig. 8C). These data strongly suggest that GDE4 actually regulates intracellular lyso-PC/choline metabolism.
FIGURE 7.

GDE4 hydrolyzes lyso-PAF into alkyl-LPA. A, enzyme activity of purified mouse GDE4 (6 μg/ml) was measured using TLC. The substrates used were lyso-PC (16:0), lyso-PAF, and PAF. The enzyme reaction was stopped at 0, 60, and 120 min. Phospholipids were visualized using the modified Dittmer-Lester reagent. Data are representative of three independent experiments. B, the enzyme activity of mouse GDE4 (6 μg/ml) toward lysophospholipids was measured using TLC. The substrates used were lyso-PAF, lyso-PC, lyso-PI, lyso-PE, and lyso-PS. Data are means of triplicate experiments (mean ± S.E. (error bars)). C, enzyme activities of GDE4 (20 μg/ml) toward 0.1 mm lyso-PC and 0.1 mm 1,2-dibutyryl-sn-glycero-3-phosphatidylcholine (dibutyryl-PC) were measured. The enzyme activities were examined by measuring the released choline. Data are means of triplicate experiments (mean ± S.E.). D, the enzyme activity of GDE4 is compared with that of autotaxin. The enzyme activity was measured in the presence of 1 μg/ml autotaxin or 20 μg/ml GDE4. Lyso-PC (0.1 mm) was used as a substrate. The lysophospholipase D activities were examined by measuring of the released choline. Data are means of triplicate experiments (mean ± S.E.). E, HEK293T cells were transiently transfected with constructs for EGFP or EGFP-tagged GDE4. After 2 days of the transfection, intracellular LPA levels were measured as described under “Experimental Procedures.” Data are means of triplicate experiments (mean ± S.E.). F, HEK293T were transiently transfected with constructs for EGFP or EGFP-tagged GDE4 and pGAL4-PPARγ chimera receptor and a luciferase reporter (6xUAS-luc) plasmids. After 24 h, the cells were scraped and subjected to luciferase as described under “Experimental Procedures.” *, p < 0.05; **, p < 0.01.
TABLE 1.
Kinetic parameters of GDE4
Enzyme activity was determined by measuring the released choline.
| Substrate | Km | kcat | kcat/Km |
|---|---|---|---|
| mm | s−1 | 102m−1 s−1 | |
| Lyso-PC | 1.0 | 0.032 | 0.31 |
| Lyso-PAF | 0.32 | 0.077 | 2.4 |
FIGURE 8.
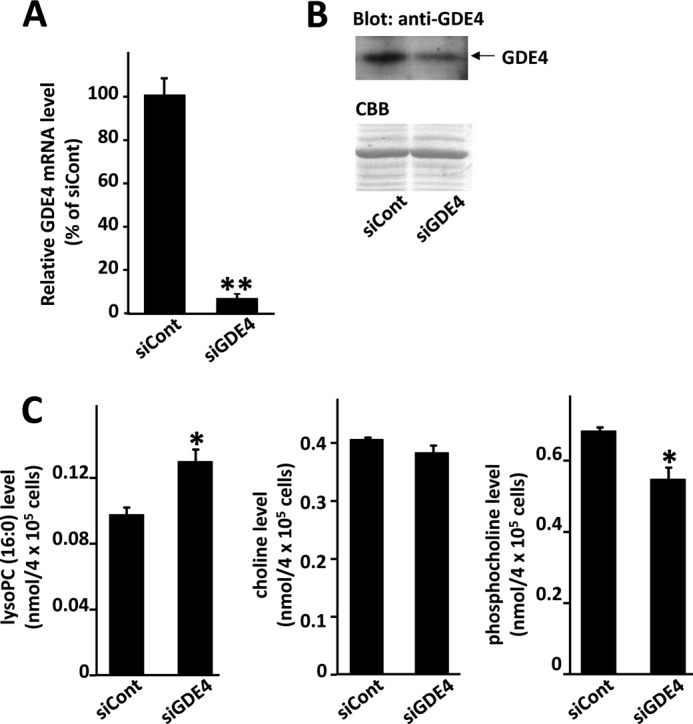
Effects of GDE4 interference on intracellular lyso-PC metabolism. 3T3-L1 adipocytes were transfected with control siRNA (siCont) or GDE4 siRNA (siGDE4). A, 24 h after transfection, total RNAs were extracted and subjected to quantitative PCR analyses to examine expression levels of GDE4 mRNA. The level of β-actin transcript was used as a control. B, 48 h after transfection, whole cell lysates were obtained, and total protein extracts (10 μg/lane) were subjected to SDS-PAGE followed by Western blotting using an anti-GDE4 antibody. The filter was stained with Coomassie Brilliant Blue (CBB) as a control of protein loading. C, 48 h after transfection, intracellular lyso-PC (16:0), phosphocholine, and choline levels were measured as described under “Experimental Procedures.” Data are means of triplicate experiments (mean ± S.E.). Statistical significance was determined by unpaired Student's t test. *, p < 0.05; **, p < 0.01.
To examine the substrate specificity for GDE7, we also obtained the His-tagged mouse GDE7 protein expressed in Sf9 cells. Mouse recombinant GDE7 was purified to near homogeneity as determined by SDS-PAGE (Fig. 9A). We also tested lyso-PC, PC, and sphingomyelin as substrates using the purified GDE7 protein and showed that GDE7 is specific for lyso-PC, similarly to GDE4 (Fig. 9B). The enzyme activities of GDE7 toward various lysophospholipids were also analyzed by measuring LPA production (Fig. 9C). The Km, kcat, and kcat/Km of GDE7 protein are shown in Table 2. The enzyme activity of GDE7 toward lyso-PC was the highest among these lysophospholipids and comparable with that toward lyso-PAF (Table 2 and Fig. 9C), different from that of GDE4.
FIGURE 9.
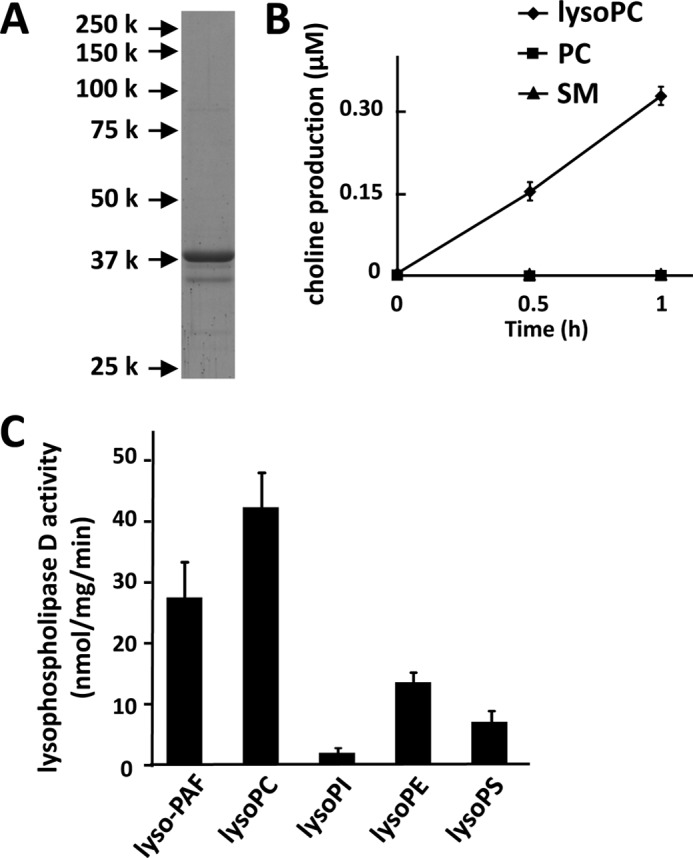
GDE7 is a phosphodiesterase of both lyso-PAF and lyso-PC. A, His-tagged mouse GDE7 protein was overexpressed in Sf9 cells. Purified mouse GDE7 was visualized by Coomassie Brilliant Blue staining. A major protein band was detected at 39 kDa. B, substrate specificity of mouse GDE7. The substrates used were lyso-PC, PC, and sphingomyelin (SM). The enzyme activity was examined in the presence of GDE7 (0.05 μg/ml) by measuring the released choline. The data are from a single experiment carried out in triplicate (mean ± S.D. (error bars)) and are representative of two independent experiments. C, the enzyme activity of mouse GDE7 (6 μg/ml) toward lysophospholipids was measured using TLC as described above. The substrates used were lyso-PAF, lyso-PC, lyso-PI, lyso-PE, and lyso-PS. Data are means of triplicate experiments (mean ± S.E. (error bars)).
TABLE 2.
Kinetic parameters of GDE7
Enzyme activity was determined by measuring the released choline.
| Substrate | Km | kcat | kcat/Km |
|---|---|---|---|
| mm | s−1 | 103m−1 s−1 | |
| Lyso-PC | 0.61 | 0.81 | 1.3 |
| Lyso-PAF | 0.26 | 0.77 | 2.9 |
GDE4 mRNA Expression in Macrophages and Obese Adipose Tissue
Finally, we asked whether GDE4 or GDE7 mRNA expression is differentially regulated in several disease models. In the present study, we have focused on GDE4 mRNA and observed that its expression in mouse macrophage RAW264.7 cells is finely modulated because it is up-regulated by lipopolysaccharide (LPS) treatment (Fig. 10A). We examined GDE4 mRNA expression in white adipose tissue of obese mice because recent reports have emphasized macrophage accumulation in obese adipose tissue with chronic low grade inflammation (28, 29). We analyzed mRNA expression in adipose tissues of the leptin receptor-deficient obese mouse model (db/db) and HFD obese mice. As expected, mRNA expression of macrophage marker genes, such as Emr1 and Msr1, in the db/db white adipose tissue is up-regulated (Fig. 10B). We found that GDE4 mRNA expression is markedly increased in the white adipose tissue of both db/db (Fig. 10B) and HFD (Fig. 10C) obese mice. We further confirmed that GDE4 mRNA is expressed in the stromal-vascular fraction from epididymal white adipose tissue in HFD mice (Fig. 10D). Finally, immunohistological analysis demonstrated that the GDE4 protein is strongly expressed in F4/80-positive cells (macrophages) in the db/db white adipose tissue (Fig. 10E). Taken together, these observations suggest that GDE4 mRNA up-regulation is closely associated with macrophage infiltration into adipose tissue.
FIGURE 10.

GDE4 mRNA expression is up-regulated in obese white adipose tissue. A, mouse macrophage RAW264.7 cells were stimulated with 1 μg/ml LPS for 18 h, and then total RNAs were subjected to quantitative PCR. All values are normalized to β-actin levels. The data (mean ± S.E. (error bars)) are representative of two independent experiments. *, p < 0.05 compared with those of control RAW264.7 cells (control). B, total RNAs from individual mice (n = 3) were subjected to quantitative PCR. Values are normalized to β-actin levels. *, p < 0.05 compared with results for control mice (db/+). The data (mean ± S.E.) are representative of two independent experiments. C, mice were divided into two groups (n = 4) and fed a basal diet (control) or HFD for 8 weeks (n = 4). The relative mRNA expression level of each gene was determined by quantitative PCR and normalized to β-actin level. *, p < 0.05; **, p < 0.01 compared with results for mice with basal diet (control). The data (mean ± S.E.) are representative of two independent experiments. D, mature adipocytes and the stromal-vascular fraction were isolated from white adipose tissue of HFD mice. The mRNA expression level of each gene was analyzed by RT-PCR. E, an immunohistochemical study was carried out using 4-μm-thick paraffin-embedded sections of the epididymal adipose tissue from db/db mice for the macrophage marker F4/80 (Serotec, Oxford, UK), followed by Cy3-labeled goat anti-rabbit IgG. The GDE4 antibody was visualized with Cy3-labeled goat anti-rabbit IgG.
DISCUSSION
GP-PDEs are widely found in prokaryotes and eukaryotes, although their distinct physiological roles remain unclear, particularly in eukaryotes (5, 6). Mammalian GP-PDEs have more selective substrate specificities toward GPs, which prompts us to assume that their enzymatic activities are mainly devoted to modulate GroPIns and/or GroPCho concentrations. The present study was initially aimed at investigating the physiological roles of uncharacterized mammalian GP-PDEs and, especially, at elucidating how these enzymes might control the levels of the GPs as signaling molecules in mammalian cells. Among the GP-PDE family proteins, three of the mammalian GP-PDEs, GDE1, GDE4, and GDE7, have similar domain composition with two putative transmembrane regions. In addition, the subcellular localizations of GDE4 and GDE7 are similar to that of GDE1 (data not shown). However, unlike GDE1 (11), recombinant mouse GDE4 and GDE7 do not hydrolyze GroPIns. Moreover, GDE4 and GDE7 cannot hydrolyze GroPCho, which is otherwise the preferred substrate of GDE2 and GDE5 (9, 10). We found that the amino acid sequences of the C-terminal regions of GDE4 and GDE7 are highly conserved with that of L. laeta SMaseD, which is a spider venom. van Meeteren et al. (26) showed that L. laeta SMaseD not only has SMaseD activity but also has lysophospholipase D activity toward lyso-PC. Thus, we hypothesized that the substrates for GDE4 and GDE7 could be the lipids, in particular the lysophospholipids. In the present study, we purified the recombinant GDE4 and GDE7 proteins and demonstrate that both of these enzymes show lysophospholipase D activity toward lyso-PC. Furthermore, we have proven that GDE4 is actually involved in the intracellular lyso-PC metabolism in 3T3-L1 cells. GDE4 also shows a preferred substrate specificity toward lyso-PAF. Previous reports have shown that PLA2IVα translocates into the perinuclear region when cells are stimulated in a Ca2+-dependent manner (30, 31). Considering the localization of GDE4 and GDE7 in the perinuclear region, it is possible that they are involved in the metabolism of the lysophospholipids or of lyso-PAF, probably in association with PLA2IVα.
LPA has been characterized as a mediator of a variety of cellular events, including cell proliferation and platelet activation, through its actions on a family of G-protein-coupled receptors, the LPA receptors. Interestingly, PPARγ has been recently reported as an intracellular receptor for LPA (32), which suggests that LPA produced intracellularly can activate PPARγ and is involved in glucose metabolism in cells, including skeletal muscle cells and adipocytes. This study shows that GDE4 can actually regulate the intracellular LPA level and PPARγ activity, suggesting that intracellular LPA produced by GDE4 acts as a PPARγ ligand. Otherwise, among the extracellular LPA-producing enzymes, autotaxin is well studied to function as a lysophospholipase D that converts lyso-PC into LPA in the plasma (33–36). Although LPA has been shown to be produced extracellularly by autotaxin, we cannot rule out the possibility that the LPA produced intracellularly by GDE4 (or GDE7) can be transported across the plasma membrane and act on LPA receptors on the surface of cells. To date, it has been suggested that dietary LPA is a ligand for the LPA2 receptor in the mucosa in the stomach and can promote proliferation of epithelial cells (37, 38). Interestingly, in this study, GDE7 is shown to be expressed in gastric epithelial cells where the LPA2 receptor is also present, suggesting the possibility of an autocrine regulation of these cells.
LPA can have different linkages of the radyl chain to the glycerol moiety, which results in either acyl-LPA or alkyl-LPA. As mentioned above, acyl-LPA is the most abundant form in the plasma, and it is produced by the plasma lysophospholipase D, autotaxin. Alkyl-LPA preferentially binds to LPA5, one of the LPA receptors, with greater specificity than acyl-LPA (39). Alkyl-LPA has also been shown to stimulate migration and proliferation of ovarian cancer cells and platelet aggregation (40, 41). In addition, like acyl-LPA, alkyl-LPA is also a high affinity agonist of PPARγ (Kd(app) = 60 nm) (42). The mechanisms of alkyl-LPA biosynthesis are not well characterized; however, two possibilities have been proposed. One is that 1-O-alkyl-2-acetyl-sn-glycerol is deacetylated, and the resulting 1-O-alkyl-sn-glycerol is phosphorylated to yield alkyl-LPA (43). Indeed, a recent study showed a specific diacylglycerol kinase that phosphorylates 1-O-alkyl-sn-glycerol, producing alkyl-LPA (44). The other pathway appears to be mediated by autotaxin, which hydrolyzes lyso-PAF with a lower affinity than lyso-PC (27). In the present study, our findings that GDE4 can produce alkyl-LPA may be valuable for the definition of an alternative mechanism of alkyl-LPA biosynthesis and the physiological roles of alkyl-LPA.
PAF (1-O-alkyl-2-acetyl-sn-glycero-3-phosphocholine) is a potent pro-inflammatory phospholipid that is synthesized by macrophages (45, 46). PAF and LPS have related pathological activities in several human syndromes (46). This includes sepsis, which is a severe inflammatory disease, which indicates that PAF is one of the key downstream intermediates produced after exposure to LPS. On the other hand, LPS administered to experimental animals results in up-regulation of the plasma form of a PAF acetylhydrolase that is secreted from macrophages (47, 48). Newly synthesized PAF is rapidly degraded by this PAF acetylhydrolase, which yields lyso-PAF (49). Our observations suggest that in activated macrophages, GDE4 is involved in the conversion of lyso-PAF into alkyl-LPA.
Recent reports have focused on macrophage accumulation in adipose tissue during the development of obesity and have further shown that the influence of adipose tissue macrophages on energy homeostasis and inflammatory responses is related to obesity-induced insulin resistance. Previous studies have demonstrated that LPA has antiadipogenic activity via the LPA1 receptor and tonic inhibition of glucose tolerance, which results possibly from inhibition of insulin secretion (50). This suggests that GDE4 expressed in adipose macrophages is involved in systemic glucose metabolism via LPA and/or alkyl-LPA production.
In summary, the present study provides valuable insight into the roles of mammalian GP-PDEs that are independent from PDE activity toward GPs. Further studies should clarify whether GDE4 and GDE7 are involved in the novel biosynthetic pathway for LPA as a ligand for LPA receptors.
Acknowledgments
We thank Y. Sanady and T. Yamamoto-yama for technical support.
This work was supported in part by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (to N. Y.) and from the Italian Association for Cancer Research (to D. C.) and PRIN Project 2012CK5RPF_05 (to S. M.).
- GP
- glycerophosphodiester
- DDM
- dodecyl-β-d-maltoside
- GP-PDE
- glycerophosphodiester phosphodiesterase
- GroPCho
- glycerophosphocholine
- GroPEth
- glycerophosphoethanolamine
- GroPIns
- glycerophosphoinositol
- GroPSer
- glycerophosphoserine
- HFD
- high fat diet
- ICR
- imprinting control region
- PAF
- platelet-activating factor (1-O-alkyl-2-acetyl-sn-glycero-3-phosphocholine)
- PC
- phosphatidylcholine
- PS
- phosphatidylserine
- PE
- phosphatidylethanolamine
- PI
- phosphatidylinositol
- LPA
- lysophosphatidic acid
- PLA2
- phospholipase A2
- SMaseD
- sphingomyelinase D
- PPARγ
- peroxisome proliferator-activated receptor γ
- EGFP
- enhanced GFP.
REFERENCES
- 1. Baburina I., Jackowski S. (1999) Cellular responses to excess phospholipid. J. Biol. Chem. 274, 9400–9408 [DOI] [PubMed] [Google Scholar]
- 2. Gallazzini M., Burg M. B. (2009) What's new about osmotic regulation of glycerophosphocholine. Physiology 24, 245–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Corda D., Zizza P., Varone A., Filippi B. M., Mariggiò S. (2009) The glycerophosphoinositols: cellular metabolism and biological functions. Cell Mol. Life Sci. 66, 3449–3467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mariggiò S., Sebastià J., Filippi B. M., Iurisci C., Volonté C., Amadio S., De Falco V., Santoro M., Corda D. (2006) A novel pathway of cell growth regulation mediated by a PLA2α-derived phosphoinositide metabolite. FASEB J. 20, 2567–2569 [DOI] [PubMed] [Google Scholar]
- 5. Yanaka N. (2007) Mammalian glycerophosphodiester phosphodiesterases. Biosci. Biotechnol. Biochem. 71, 1811–1818 [DOI] [PubMed] [Google Scholar]
- 6. Corda D., Mosca M. G., Ohshima N., Grauso L., Yanaka N., Mariggiò S. (2014) The emerging physiological roles of the glycerophosphodiesterase family. FEBS J. 281, 998–1016 [DOI] [PubMed] [Google Scholar]
- 7. Burg M. B. (1995) Molecular basis of osmotic regulation. Am. J. Physiol. 268, F983–F996 [DOI] [PubMed] [Google Scholar]
- 8. Gallazzini M., Ferraris J. D., Burg M. B. (2008) GDPD5 is a glycerophosphocholine phosphodiesterase that osmotically regulates the osmoprotective organic osmolyte GPC. Proc. Natl. Acad. Sci. U.S.A. 105, 11026–11031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Topanurak S., Ferraris J. D., Li J., Izumi Y., Williams C. K., Gucek M., Wang G., Zhou X., Burg M. B. (2013) High NaCl- and urea-induced posttranslational modifications that increase glycerophosphocholine by inhibiting GDPD5 phosphodiesterase. Proc. Natl. Acad. Sci. U.S.A. 110, 7482–7487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Okazaki Y., Ohshima N., Yoshizawa I., Kamei Y., Mariggiò S., Okamoto K., Maeda M., Nogusa Y., Fujioka Y., Izumi T., Ogawa Y., Shiro Y., Wada M., Kato N., Corda D., Yanaka N. (2010) A novel glycerophosphodiester phosphodiesterase, GDE5, controls skeletal muscle development via a non-enzymatic mechanism. J. Biol. Chem. 285, 27652–27663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zheng B., Berrie C. P., Corda D., Farquhar M. G. (2003) GDE1/MIR16 is a glycerophosphoinositol phosphodiesterase regulated by stimulation of G protein-coupled receptors. Proc. Natl. Acad. Sci. U.S.A. 100, 1745–1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Corda D., Kudo T., Zizza P., Iurisci C., Kawai E., Kato N., Yanaka N., Mariggiò S. (2009) The developmentally regulated osteoblast phosphodiesterase GDE3 is glycerophosphoinositol-specific and modulates cell growth. J. Biol. Chem. 284, 24848–24856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Simon G. M., Cravatt B. F. (2008) Anandamide biosynthesis catalyzed by the phosphodiesterase GDE1 and detection of glycerophospho-N-acyl ethanolamine precursors in mouse brain. J. Biol. Chem. 283, 9341–9349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Simon G. M., Cravatt B. F. (2010) Characterization of mice lacking candidate N-acyl ethanolamine biosynthetic enzymes provides evidence for multiple pathways that contribute to endocannabinoid production in vivo. Mol. Biosyst. 6, 1411–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Park S., Lee C., Sabharwal P., Zhang M., Meyers C. L., Sockanathan S. (2013) GDE2 promotes neurogenesis by glycosylphosphatidylinositol-anchor cleavage of RECK. Science 339, 324–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ohshima N., Yamashita S., Takahashi N., Kuroishi C., Shiro Y., Takio K. (2008) Escherichia coli cytosolic glycerophosphodiester phosphodiesterase (UgpQ) requires Mg2+, Co2+, or Mn2+ for its enzyme activity. J. Bacteriol. 190, 1219–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yanaka N., Imai Y., Kawai E., Akatsuka H., Wakimoto K., Nogusa Y., Kato N., Chiba H., Kotani E., Omori K., Sakurai N. (2003) Novel membrane protein containing glycerophosphodiester phosphodiesterase motif is transiently expressed during osteoblast differentiation. J. Biol. Chem. 278, 43595–43602 [DOI] [PubMed] [Google Scholar]
- 18. Nogusa Y., Fujioka Y., Komatsu R., Kato N., Yanaka N. (2004) Isolation and characterization of two serpentine membrane proteins containing glycerophosphodiester phosphodiesterase, GDE2 and GDE6. Gene 337, 173–179 [DOI] [PubMed] [Google Scholar]
- 19. Okazaki Y., Furuno M., Kasukawa T., Adachi J., Bono H., Kondo S., Nikaido I., Osato N., Saito R., Suzuki H., Yamanaka I., Kiyosawa H., Yagi K., Tomaru Y., Hasegawa Y., Nogami A., Schönbach C., Gojobori T., Baldarelli R., Hill D. P., Bult C., Hume D. A., Quackenbush J., Schriml L. M., Kanapin A., Matsuda H., Batalov S., Beisel K. W., Blake J. A., Bradt D., Brusic V., Chothia C., Corbani L. E., Cousins S., Dalla E., Dragani T. A., Fletcher C. F., Forrest A., Frazer K. S., Gaasterland T., Gariboldi M., Gissi C., Godzik A., Gough J., Grimmond S., Gustincich S., Hirokawa N., Jackson I. J., Jarvis E. D., Kanai A., Kawaji H., Kawasawa Y., Kedzierski R. M., King B. L., Konagaya A., Kurochkin I. V., Lee Y., Lenhard B., Lyons P. A., Maglott D. R., Maltais L., Marchionni L., McKenzie L., Miki H., Nagashima T., Numata K., Okido T., Pavan W. J., Pertea G., Pesole G., Petrovsky N., Pillai R., Pontius J. U., Qi D., Ramachandran S., Ravasi T., Reed J. C., Reed D. J., Reid J., Ring B. Z., Ringwald M., Sandelin A., Schneider C., Semple C. A., Setou M., Shimada K., Sultana R., Takenaka Y., Taylor M. S., Teasdale R. D., Tomita M., Verardo R., Wagner L., Wahlestedt C., Wang Y., Watanabe Y., Wells C., Wilming L. G., Wynshaw-Boris A., Yanagisawa M., Yang I., Yang L., Yuan Z., Zavolan M., Zhu Y., Zimmer A., Carninci P., Hayatsu N., Hirozane-Kishikawa T., Konno H., Nakamura M., Sakazume N., Sato K., Shiraki T., Waki K., Kawai J., Aizawa K., Arakawa T., Fukuda S., Hara A., Hashizume W., Imotani K., Ishii Y., Itoh M., Kagawa I., Miyazaki A., Sakai K., Sasaki D., Shibata K., Shinagawa A., Yasunishi A., Yoshino M., Waterston R., Lander E. S., Rogers J., Birney E., Hayashizaki Y., FANTOM Consortium, and RIKEN Genome Exploration Research Group Phase I & II Team (2002) Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420, 563–573 [DOI] [PubMed] [Google Scholar]
- 20. Bligh E. G., Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 21. Aaltonen N., Laitinen J. T., Lehtonen M. (2010) Quantification of lysophosphatidic acids in rat brain tissue by liquid chromatography-electrospray tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 878, 1145–1152 [DOI] [PubMed] [Google Scholar]
- 22. Sanada Y., Kumoto T., Suehiro H., Nishimura F., Kato N., Hata Y., Sorisky A., Yanaka N. (2013) RASSF6 expression in adipocytes is down-regulated by interaction with macrophages. PLoS One 8, e61931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chang P. A., Shao H. B., Long D. X., Sun Q., Wu Y. J. (2008) Isolation, characterization and molecular 3D model of human GDE4, a novel membrane protein containing glycerophosphodiester phosphodiesterase domain. Mol. Membr. Biol. 25, 557–566 [DOI] [PubMed] [Google Scholar]
- 24. Zheng B., Chen D., Farquhar M. G. (2000) MIR16, a putative membrane glycerophosphodiester phosphodiesterase, interacts with RGS16. Proc. Natl. Acad. Sci. U.S.A. 97, 3999–4004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. San Pietro E., Capestrano M., Polishchuk E. V., DiPentima A., Trucco A., Zizza P., Mariggiò S., Pulvirenti T., Sallese M., Tete S., Mironov A. A., Leslie C. C., Corda D., Luini A., Polishchuk R. S. (2009) Group IV phospholipase A2α controls the formation of inter-cisternal continuities involved in intra-Golgi transport. PLoS Biol. 7, e1000194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van Meeteren L. A., Frederiks F., Giepmans B. N., Pedrosa M. F., Billington S. J., Jost B. H., Tambourgi D. V., Moolenaar W. H. (2004) Spider and bacterial sphingomyelinases D target cellular lysophosphatidic acid receptors by hydrolyzing lysophosphatidylcholine. J. Biol. Chem. 279, 10833–10836 [DOI] [PubMed] [Google Scholar]
- 27. Tokumura A., Majima E., Kariya Y., Tominaga K., Kogure K., Yasuda K., Fukuzawa K. (2002) Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem. 277, 39436–39442 [DOI] [PubMed] [Google Scholar]
- 28. Weisberg S. P., McCann D., Desai M., Rosenbaum M., Leibel R. L., Ferrante A. W., Jr. (2003) Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 112, 1796–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dalmas E., Clément K., Guerre-Millo M. (2011) Defining macrophage phenotype and function in adipose tissue. Trends Immunol. 32, 307–314 [DOI] [PubMed] [Google Scholar]
- 30. Schievella A. R., Regier M. K., Smith W. L., Lin L. L. (1995) Calcium-mediated translocation of cytosolic phospholipase A2 to the nuclear envelope and endoplasmic reticulum. J. Biol. Chem. 270, 30749–30754 [DOI] [PubMed] [Google Scholar]
- 31. Hirabayashi T., Kume K., Hirose K., Yokomizo T., Iino M., Itoh H., Shimizu T. (1999) Critical duration of intracellular Ca2+ response required for continuous translocation and activation of cytosolic phospholipase A2. J. Biol. Chem. 274, 5163–5169 [DOI] [PubMed] [Google Scholar]
- 32. McIntyre T. M., Pontsler A. V., Silva A. R., St Hilaire A., Xu Y., Hinshaw J. C., Zimmerman G. A., Hama K., Aoki J., Arai H., Prestwich G. D. (2003) Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARγ agonist. Proc. Natl. Acad. Sci. U.S.A. 100, 131–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van Meeteren L. A., Moolenaar W. H. (2007) Regulation and biological activities of the autotaxin-LPA axis. Prog. Lipid Res. 46, 145–160 [DOI] [PubMed] [Google Scholar]
- 34. Jonkers J., Moolenaar W. H. (2009) Mammary tumorigenesis through LPA receptor signaling. Cancer Cell 15, 457–459 [DOI] [PubMed] [Google Scholar]
- 35. Nakanaga K., Hama K., Aoki J. (2010) Autotaxin: an LPA producing enzyme with diverse functions. J. Biochem. 148, 13–24 [DOI] [PubMed] [Google Scholar]
- 36. Okudaira S., Yukiura H., Aoki J. (2010) Biological roles of lysophosphatidic acid signaling through its production by autotaxin. Biochimie 92, 698–706 [DOI] [PubMed] [Google Scholar]
- 37. Tanaka T., Morito K., Kinoshita M., Ohmoto M., Urikura M., Satouchi K., Tokumura A. (2013) Orally administered phosphatidic acids and lysophosphatidic acids ameliorate aspirin-induced stomach mucosal injury in mice. Dig. Dis. Sci. 58, 950–958 [DOI] [PubMed] [Google Scholar]
- 38. Tanaka T., Horiuchi G., Matsuoka M., Hirano K., Tokumura A., Koike T., Satouchi K. (2009) Formation of lysophosphatidic acid, a wound-healing lipid, during digestion of cabbage leaves. Biosci. Biotechnol. Biochem. 73, 1293–1300 [DOI] [PubMed] [Google Scholar]
- 39. Williams J. R., Khandoga A. L., Goyal P., Fells J. I., Perygin D. H., Siess W., Parrill A. L., Tigyi G., Fujiwara Y. (2009) Unique ligand selectivity of the GPR92/LPA5 lysophosphatidate receptor indicates role in human platelet activation. J. Biol. Chem. 284, 17304–17319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Simon M. F., Chap H., Douste-Blazy L. (1982) Human platelet aggregation induced by 1-alkyl-lysophosphatidic acid and its analogs: a new group of phospholipid mediators? Biochem. Biophys. Res. Commun. 108, 1743–1750 [DOI] [PubMed] [Google Scholar]
- 41. Tokumura A., Sinomiya J., Kishimoto S., Tanaka T., Kogure K., Sugiura T., Satouchi K., Waku K., Fukuzawa K. (2002) Human platelets respond differentially to lysophosphatidic acids having a highly unsaturated fatty acyl group and alkyl ether-linked lysophosphatidic acids. Biochem. J. 365, 617–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tsukahara T., Tsukahara R., Yasuda S., Makarova N., Valentine W. J., Allison P., Yuan H., Baker D. L., Li Z., Bittman R., Parrill A., Tigyi G. (2006) Different residues mediate recognition of 1-O-oleyllysophosphatidic acid and rosiglitazone in the ligand binding domain of peroxisome proliferator-activated receptor γ. J. Biol. Chem. 281, 3398–3407 [DOI] [PubMed] [Google Scholar]
- 43. Chiang K. P., Niessen S., Saghatelian A., Cravatt B. F. (2006) An enzyme that regulates ether lipid signaling pathways in cancer annotated by multidimensional profiling. Chem. Biol. 13, 1041–1050 [DOI] [PubMed] [Google Scholar]
- 44. Gellett A. M., Kharel Y., Sunkara M., Morris A. J., Lynch K. R. (2012) Biosynthesis of alkyl lysophosphatidic acid by diacylglycerol kinases. Biochem. Biophys. Res. Commun. 422, 758–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Prescott S. M., Zimmerman G. A., Stafforini D. M., McIntyre T. M. (2000) Platelet-activating factor and related lipid mediators. Annu. Rev. Biochem. 69, 419–445 [DOI] [PubMed] [Google Scholar]
- 46. Ishii S., Shimizu T. (2000) Platelet-activating factor (PAF) receptor and genetically engineered PAF receptor mutant mice. Prog. Lipid Res. 39, 41–82 [DOI] [PubMed] [Google Scholar]
- 47. Howard K. M., Olson M. S. (2000) The expression and localization of plasma platelet-activating factor acetylhydrolase in endotoxemic rats. J. Biol. Chem. 275, 19891–19896 [DOI] [PubMed] [Google Scholar]
- 48. Wu X., Zimmerman G. A., Prescott S. M., Stafforini D. M. (2004) The p38 MAPK pathway mediates transcriptional activation of the plasma platelet-activating factor acetylhydrolase gene in macrophages stimulated with lipopolysaccharide. J. Biol. Chem. 279, 36158–36165 [DOI] [PubMed] [Google Scholar]
- 49. Ohshima N., Ishii S., Izumi T., Shimizu T. (2002) Receptor-dependent metabolism of platelet-activating factor in murine macrophages. J. Biol. Chem. 277, 9722–9727 [DOI] [PubMed] [Google Scholar]
- 50. Simon M. F., Daviaud D., Pradère J. P., Grès S., Guigné C., Wabitsch M., Chun J., Valet P., Saulnier-Blache J. S. (2005) Lysophosphatidic acid inhibits adipocyte differentiation via lysophosphatidic acid 1 receptor-dependent down-regulation of peroxisome proliferator-activated receptor γ2. J. Biol. Chem. 280, 14656–14662 [DOI] [PubMed] [Google Scholar]