

Background: Nucleosome assembly mediated by histone chaperones and DNA methylation play important roles in gene regulation.
Results: Dynamics of nucleosome assembly with and without CpG methylation was investigated.
Conclusion: CpG methylation increases the efficiency of nucleosome assembly.
Significance: CpG methylation accelerates nucleosome formation that may facilitate gene packaging in chromatin.
Keywords: DNA Methylation, Fluorescence Resonance Energy Transfer (FRET), Histone Chaperone, Nucleosome, Single-molecule Biophysics
Abstract
The nucleosome is the fundamental packing unit of the eukaryotic genome, and CpG methylation is an epigenetic modification associated with gene repression and silencing. We investigated nucleosome assembly mediated by histone chaperone Nap1 and the effects of CpG methylation based on three-color single molecule FRET measurements, which enabled direct monitoring of histone binding in the context of DNA wrapping. According to our observation, (H3-H4)2 tetramer incorporation must precede H2A-H2B dimer binding, which is independent of DNA termini wrapping. Upon CpG methylation, (H3-H4)2 tetramer incorporation and DNA termini wrapping are facilitated, whereas proper incorporation of H2A-H2B dimers is inhibited. We suggest that these changes are due to rigidified DNA and increased random binding of histones to DNA. According to the results, CpG methylation expedites nucleosome assembly in the presence of abundant DNA and histones, which may help facilitate gene packaging in chromatin. The results also indicate that the slowest steps in nucleosome assembly are DNA termini wrapping and tetramer positioning, both of which are affected heavily by changes in the physical properties of DNA.
Introduction
Nucleosomes are the basic packing units of genes in eukaryotes. Nucleosomes linked in an array format compact into chromatin and eventually into chromosomes. This structural organization is implicated in various mechanisms of gene regulation and maintenance (1). A nucleosome core particle comprises ∼147 bp DNA and an octameric histone core containing two copies of H2A, H2B, H3, and H4 (2). Nucleosomes are assembled by histone chaperones and chromatin assembly factors in vivo (3, 4). For in vitro assembly, a salt gradient method (5) or a histone chaperone can be employed.
Nucleosome assembly protein 1 (Nap1)2 is a histone chaperone routinely used for in vitro nucleosome assembly under physiological ionic conditions (6, 7). Nap1 has been utilized to investigate the thermodynamics of nucleosome assembly (8). The binding rates of histones to DNA in the presence of Nap1 have also been reported (9). Nap1 mediates various important biological activities including H2A-H2B dimer transfer from cytoplasm to nucleus (10, 11) and nucleosome rearrangement during transcription (12). Nap1 binds strongly to histones in vivo (10, 13) to inhibit nonproductive random interactions between histones and DNA, which eventually promotes canonical nucleosome assembly (14). Yeast Nap1 (yNap1) binds strongly to both H2A-H2B dimer and (H3-H4)2 tetramer (the values of Kd are on the order of nm) (15). However, yNap1 competes with DNA for the dimer with a 10-fold higher affinity than for the tetramer (14). Based on these findings, it was suggested that a nucleosome core particle is assembled via tetramer deposition on DNA followed by dimer incorporation. Although these studies provided valuable insights into the thermodynamics of nucleosome assembly (8), the kinetics has yet to be unraveled.
Methylation at the C5 position of the cytosine in a CpG dinucleotide is an epigenetic modification associated with repressive chromatin structures (16, 17). CpG islands, regions characterized by a high content of CpG dinucleotides, are mostly hypomethylated in normal somatic cells, whereas they are often hypermethylated in the promoters of tumor suppressors in cancer cells (18–20). These findings support that erroneous CpG methylation is closely tied to carcinogenesis (21, 22).
Sequence-dependent physical properties of DNA play a key role in the positioning and structural dynamics of nucleosomes (23–25). Therefore, modifications on DNA that alter its physical properties will likely affect the dynamics of nucleosome assembly. CpG methylation is implicated in repressive chromatin structures mainly in two ways. Some transcriptional repressors or related factors bind to methylated CpG and subsequently trigger the formation of a repressive chromatin structure (26, 27). CpG methylation also rigidifies DNA, thereby directly contributing to the formation of a repressive chromatin structure (28). We reported that CpG methylation induces nucleosome compaction and DNA topology change likely due to rigidified DNA upon methylation (29, 30). A third possibility is that CpG methylation might affect histone-DNA interactions. Changes in the rotational positioning of nucleosomes upon CpG methylation in Arabidopsis have been reported (31). Similar effects have also been reported with human and mouse nucleosomes (32). These results suggest that CpG methylation may induce changes in the interactions between histones and DNA, which would affect the dynamics of nucleosome assembly.
We investigated Nap1-mediated nucleosome assembly and the effects of CpG methylation based on single nucleosome measurements. To observe stepwise nucleosome assembly in real time in a time-resolved manner, we employed a three-color single molecule FRET (smFRET) approach that enabled monitoring of histone binding in the context of DNA wrapping. Studying a multi-step process in an ensemble-averaging format would necessitate separate measurements of multiple sub-steps, which requires synchronization of the system to various sub-steps and a lot of materials. These requirements impose a high technical barrier especially to measurements with CpG-methylated nucleosomes because currently the only practical method to obtain controlled DNA methylation is to ligate chemically synthesized oligonucleotides. Single molecule methods are useful in this regard as they require only a small amount of reagents. More importantly, a three-color single molecule FRET method provided us with an efficient means to trace the dynamics of DNA wrapping and histone binding simultaneously. According to our observations, DNA can be completely wrapped up to the nucleosomal state before H2A-H2B dimers are properly incorporated. We revealed that DNA wrapping and (H3-H4)2 tetramer binding are facilitated and that canonical dimer binding is inhibited upon CpG methylation, and these results are in line with rigidified DNA and increased histone-DNA affinity. Based on these results, we suggest that CpG methylation expedites nucleosome assembly, which may help facilitate gene packaging in chromatin. Our results confirm that the physical properties of DNA are critical factors controlling the efficiency of nucleosome assembly.
EXPERIMENTAL PROCEDURES
DNA Preparation
DNA constructs labeled with Cy3 and Cy5.5 derived from the SELEX 601 sequence were prepared by ligating oligonucleotides as reported previously (33, 34). Briefly, six 40–80-base-long oligonucleotides were annealed at 95 °C and gradually cooled down to 5 °C during a 45-min period. Two of the oligonucleotides were labeled with Cy3 or Cy5.5 (Integrated DNA Technologies, Coralville, IA), and one was labeled with a biotin for surface immobilization. The DNA sequence and fluorophore positions are shown in Fig. 1. In the case of 601 + 16–63 DNA, Cy5.5 dye (KERAFAST, Boston, MA) was internally labeled via Uni-Link that contains a C6-linker (Integrated DNA Technologies). The annealed DNA was ligated with T4 DNA ligase at 16 °C for 16 h and then purified with a PCR purification kit (Qiagen). DNA constructs were further purified with a 2.25% agarose gel. The identical sequences methylated at all CpG dinucleotides were purchased from the same source (Integrated DNA Technologies) and were ligated and purified in the same way.
FIGURE 1.

Experimental setup to observe Nap1-mediated nucleosome assembly with three-color single molecule FRET. A, 601 + 1–73 DNA construct labeled with Cy3 and Cy5.5 mixed with the (H3-H4)2 tetramer labeled with Atto647N. See the box in B for the identities of the colored diamond fluorophore labels. B, 601 + 16–63 DNA construct labeled with Cy3 and Cy5.5 mixed with a histone octamer. The histone H2A-H2B dimer is labeled with Atto647N. C, Left panel, positions of the dyad, +1st, and −73rd nucleotides. Right panel, positions of the dyad, +1st, and −63rd nucleotides (Protein Data Bank (PDB) ID: 3MVD).
Protein Preparation
His6-Yeast NAP1 histone chaperone (yNAP1) was expressed and purified using nickel-nitrilotriacetic acid resin as described elsewhere (35). Histones were purified and labeled according to the published protocols (15, 36).
Single Molecule Three-color FRET Setup
We employed a one-donor/two-acceptor scheme (37, 38) with Cy3 (donor), Atto647N (acceptor 1), and Cy5.5 (acceptor 2). Fluorescence intensities from the three fluorophores (Cy3, Atto647N, and Cy5.5) were spectrally separated into three regions by dichroic mirrors (Chroma Technology, Bellows Falls, VT) (see Fig. 2). The cutoff wavelengths of the dichroic mirrors are: (i) 620 nm (see D1 in Fig. 2A) to separate Cy3 emission from the others, and (ii) 690 nm (see D2 and D3 in Fig. 2A) to separate Atto647N emission from Cy5.5 emission. To block the scattered light from the laser and reduce the leaks between channels, additional emission filters were installed, which are HQ655LP (see F1 in Fig. 2A) for the Atto647N/Cy5.5 channels and HQ 590/70m (see F2 in Fig. 2A) for the Cy3 channel.
FIGURE 2.

Three-color smFRET measurement scheme. A, instrumental setup. Prism-coupled total internal reflection (TIR) was employed on a commercial microscope (TE2000, Nikon, Tokyo Japan) with customization. Excitation of the FRET donor (Cy3) was achieved with a laser beam at 532 nm (Laser Quantum, Stockport, Cheshire, UK). Dichroic mirrors and filters (Chroma Technology) were used to split fluorescence signals according to the spectra, which were imaged on an electron multiplying CCD camera (iXon+897, Andor Technology, Belfast, Ireland, UK). Symbols: D1, long pass dichroic mirror 620DC; D2 and D3, long pass dichroic mirrors 690DCXR; M, mirror; F1, long pass filter HQ655LP; F2, band pass filter HQ590/70m; and EMCCD, electron multiplying CCD camera. B, normalized absorption spectra (dashed lines), emission spectra (solid lines), and detection ranges of Cy3 (green), Atto647N (red), and Cy5.5 (magenta). C, fluorescence signals detected in the three channels contributed by the three fluorophores. Values of the parameters for inter-channel fluorescence leak correction are listed. I3′, I647N′, and I5.5′ are the three fluorescence channel intensities measured in the three ranges shown in B. I3, I647N, and I5.5 are the leak-corrected intensities of Cy3, Atto647N, and Cy5.5, respectively. α is the leak ratio of I3 to the Atto647N channel, β is the leak ratio of I647N to the Cy5.5 channel, γ is the leak ratio of I5.5 to the Atto647N channel, and δ is the leak ratio of I3 to the Cy5.5 channel.
We corrected the fluorescence signals for inter-channel leaks. The leaks and corresponding correction parameters are summarized in Fig. 2. We determined the leak parameters (α, β, γ, and δ) by measuring the fluorescence intensities in the three channels with only a single fluorophore present. With the predetermined leak parameters, the corrected intensities of the three fluorophores (I3, I647N, and I5.5) are calculated by solving the following three linear equations.
where I3, I5.5, and I647N are the leak-corrected fluorescence intensities of Cy3, Atto647N, and Cy5.5, respectively. These corrected intensities still contain systematic errors due to the wavelength-dependent quantum efficiency of the detector and the constant attenuation by the emission filters. Therefore, they cannot be directly used for distance estimation.
Single Molecule FRET Measurements for Nucleosome Assembly
Surfaces of quartz microscope slides were silanized and subsequently coated with polyethylene glycol (PEG, 5000 MW, Laysan Bio, Arab, AL), which contains one biotinylated PEG (5000 MW, Laysan Bio) out of a hundred. We passivated a surface with BSA before immobilizing DNA via biotin-streptavidin conjugation. For nucleosome assembly measurements, a FRET pair (Cy3-Cy5.5)-labeled unmethylated or methylated DNA was immobilized on a slide. We typically saw 100–200 DNA constructs in an area of 35 × 100 μm on the slide surface. The histone-Nap1 complex was subsequently injected into the slide. Histone octamer (0.9 μm) and Nap1 (10.8 μm) were incubated at 30 °C for 1 h in a buffer containing 10 mm Tris-HCl (pH 7.5), 135 mm NaCl, 0.5 mm MgCl2, 1 mm DTT, and 3% glycerol and diluted to 0.06 μm histone octamer and 0.72 μm Nap1 in a buffer containing 10 mm Tris-HCl (pH 7.5), 100 mm NaCl, 0.5 mm MgCl2, 1 mm DTT, 0.1 mg/ml BSA, and 3% glycerol before injection. A mixture of protocatechuate dioxygenase (0.016 unit, Sigma-Aldrich) and protocatechuic acid (1 mm, Sigma-Aldrich) in addition to methyl viologen (1 mm) was added in the buffer to elongate the fluorophore photobleaching lifetime and stabilize the emission. We started with 0.9 μm histone tetramer and 5.4 μm Nap1 for the tetrasome assembly reactions. They were diluted to 0.06 and 0.36 μm, respectively, for tetramer and Nap1 before injection. The buffer conditions and the concentrations of all the other components were maintained constant.
Fluorescence signals from individual fluorophores were imaged on an electron multiplying CCD camera (see Fig. 2). A series of fluorescence images at a frame rate of 1/50 ms was recorded until most of the fluorophores in the field of view were photobleached, which took 20 min. The intensities of Cy3, Atto647N, and Cy5.5 fluorescence at a given time point are the brightness of the corresponding groups of pixels on the movie frame. The time series of fluorescence intensities of Cy3, Atto647N, and Cy5.5 was obtained from the series of fluorescence images contained in a movie, and inter-channel leak-corrected fluorescence intensities were plotted against the time elapsed from the injection of histone-Nap1 complex (see upper charts in Fig. 3, A–C). The FRET efficiencies at each time point were approximated with the formula FRET647N = I647N/(I3 + I647N + I5.5) and FRET5.5 = I5.5 /(I3 + I647N + I5.5), where I3, I647N, and I5.5 are the leak-corrected fluorescence intensities of Cy3, Atto647N, and Cy5.5, respectively (see lower charts in Fig. 3, A–C).
FIGURE 3.

Representative fluorescence intensity time traces and smFRET time traces reporting the process of tetrasome and nucleosome assembly. A sub-step of the nucleosome assembly shown in Fig. 1, A and B, is shown for each process time marked in the smFRET traces. A, tetrasome assembly with 601 + 1–73 DNA and Atto647N-labeled (H3-H4)2 tetramer. During the tetramer positioning time, a tetramer is positioned at the dyad of DNA, resulting in a rise in FRET647N (red trace). These times were collected from a set of ∼100 DNA molecules to construct a population growth histogram of an intermediate as shown at the bottom (in this case, a histogram for the number of DNA with a tetramer properly positioned during time 0 to t). Each tetramer positioning time observed (τ) increases a single count in the corresponding n(τ) function, and integrating the n(τ) function over 0 to t results in the histogram column height at t. During the termini wrapping time, DNA wraps around the tetramer to form a tetrasome, resulting in a rise in FRET5.5 (blue trace). B and C, nucleosome assembly with 601 + 16–63 DNA and histone octamer containing Atto647N-labeled H2A-H2B dimer. In B, the nucleosome is assembled via tetrasome formation followed by dimer incorporation. During the tetramer positioning and termini wrapping time, a tetramer is positioned at the dyad followed by termini wrapping, resulting in a rise in FRET5.5 (blue trace). During the dimer incorporation time, dimers are incorporated to the tetrasomal state, resulting in a rise in FRET647N (red trace). In C, the nucleosome is assembled via histone core formation followed by DNA termini wrapping. During the tetramer positioning and dimer incorporation time, an octameric histone core is formed, resulting in a rise in FRET647N (red trace). FRET5.5 also rises to a low level, indicating that the distances among fluorophores are within their Förster radii even without the termini wrapped up. The faint FRET arrows in this panel and Fig. 1B illustrate this FRET. During the termini wrapping time, DNA wraps around the histone octamer to form a nucleosome, resulting in the full rise in FRET5.5 (blue trace).
We repeated the single molecule measurements until we collected data from 1500 DNA constructs per case. The cases are tetrasome assembly with (i) unmethylated and (ii) methylated 601 + 1–73 DNA and nucleosome assembly with (iii) unmethylated and (iv) methylated 601 + 16–63 DNA. Nucleosome assembly takes place via two different paths; consequently, we had to collect data twice as often (∼3000 DNA constructs) in the 601 + 16–63 cases (iii and iv). Therefore, the total number of DNA constructs observed was ∼9000 (= 2 × 1500 for unmethylated and methylated tetrasome assembly with 601 + 1–73 DNA + 2 × 3000 for unmethylated and methylated nucleosome assembly with 601 + 16–63 DNA). This amount of data collection requires ∼60 stopped-flow single molecule measurements. The filtering criteria were (i) no photobleaching of fluorophores before they report nucleosome assembly and (ii) signal-to-noise ratio greater than 4.0 at 50 ms signal integration. This filtering yields only 4–8 useful traces out of 100 collected. Dye photobleaching lifetime and signal-to-noise ratio are independent of nucleosome assembly, thereby assuring random sampling of data.
Reconstitution of Nucleosomes and Histone/DNA Binding
Xenopus laevis histone octamer was incubated with yNap1 at 30 °C for 1 h in buffer A (10 mm Tris-HCl (pH 7.5), 135 mm NaCl, 0.5 mm MgCl2, 1 mm DTT, and 3% glycerol). The solution was subsequently mixed with DNA and incubated for an additional 4 h at 30 °C in buffer B (10 mm Tris-HCl (pH 7.5), 100 mm NaCl, 0.5 mm MgCl2, 1 mm DTT and 3% glycerol). The product was analyzed with 5% native PAGE in a 0.5× Tris borate-EDTA buffer. To examine H2A-H2B dimer binding on free DNA, H2A-H2B dimer in the presence of Nap1 was incubated in buffer A at 30 °C for 1 h and then mixed with either unmethylated or methylated DNA for an additional 4 h in buffer B.
RESULTS
Experimental System Construction for Three-color Single Molecule FRET
We designed two nucleosomal DNA constructs derived from the 147-bp SELEX 601 sequence containing 15 CpG dinucleotides. The 601 + 1–73 DNA is labeled with Cy3 at the +1st nucleotide from the dyad and with Cy5.5 at the −73rd nucleotide (Fig. 1A). An Atto647N labeled at histone H4 E63C forms a FRET pair with a Cy3 upon proper binding and positioning of the (H3-H4)2 tetramer on DNA (the first step in Fig. 1A). The FRET efficiency between Cy3 and Atto647N is referred to as FRET647N. Cy3 and Cy5.5 form a FRET pair upon DNA termini wrapping, and this FRET efficiency is referred to as FRET5.5. This setup enabled simultaneous monitoring of histone tetramer binding (FRET647N) and DNA termini wrapping (FRET5.5).
To monitor H2A-H2B dimer binding to DNA during nucleosome assembly, Cy3 was labeled at the +16th nucleotide from the dyad and Cy5.5 was labeled at the −63rd nucleotide (601 + 16–63 DNA shown in Fig. 1B). The histone dimer was labeled with Atto647N at H2B T112C. The value of FRET647N increases upon proper binding of a dimer to DNA (Fig. 1B), and the value of FRET5.5 increases upon DNA termini wrapping. By following the time traces of these two FRET efficiencies, we were able to monitor histone binding in the context of DNA wrapping.
Single Molecule Measurements Enabled Detection of Histone Binding in the Context of DNA Wrapping
Approximately 100–200 DNA constructs were monitored simultaneously with wide-field illumination (Fig. 2). Upon injection of labeled histones and Nap1, FRET signals in both Atto647N and Cy5.5 channels arise to report histone binding and DNA wrapping. The FRET efficiencies plotted in Fig. 3 are calculated from fluorescence intensities of the three fluorophores (FRET647N ≅ I647N/(I3 + I647N + I5.5), FRET5.5 ≅ I5.5/(I3 + I647N + I5.5), where Idye is the fluorescence intensity of the designated dye corrected for inter-channel leaks; see “Experimental Procedures” for further details).
As shown in Fig. 3, FRET time traces from individual DNA constructs reveal histone binding in the context of DNA wrapping. We collected data only from DNA showing stable signals in both Atto647N and Cy5.5 channels to ensure complete canonical nucleosome assembly within our observation time (20 min maximum). The observation time was limited by fluorophore photobleaching. We collected data from ∼100 nucleosomes per case.
Histone (H3-H4)2 Tetramer Is Required for Stable H2A-H2B Dimer Incorporation
No FRET signal was observed from Atto647N or Cy5.5 during the measurements with 601 + 16–63 DNA and Atto647N-labeled H2A-H2B dimers in the absence of (H3-H4)2 tetramer, indicating that dimers cannot bind to DNA stably (with >500-ms lifetime; typical short-term blinking is in the range of 100–200 ms) in the absence of the tetramer. No sign of DNA wrapping further supports this conclusion. We would have observed FRET signals with a wide range of efficiencies if dimers randomly but stably bound to DNA, which would result in DNA random coiling. This result strongly suggests that a tetramer must bind DNA before dimers can be stably incorporated, which is in good agreement with previous studies (14, 39).
A Tetrasome Is Assembled via Random Binding of a Tetramer to DNA Followed by Proper Positioning at the Dyad
Fig. 3A shows a typical real-time tetrasome assembly process. The smFRET time traces calculated from the corresponding donor and acceptor fluorescence intensities reveal stepwise FRET signal changes. The stepwise increase in FRET647N represents tetramer positioning at the dyad of DNA followed by DNA termini wrapping, which is indicated by a stepwise increase in FRET5.5. As tetramer binding affinity for DNA is 10-fold higher than that to Nap1 (14), a tetramer-Nap1 complex interacting with DNA results in tetramer retention on DNA (40). A tetramer may bind to DNA initially at a random position and move to the dyad after some time.
We selected FRET traces that show only a stable (>500 ms lifetime before photobleaching) and high FRET647N signal (>0.5, an approximate FRET efficiency for singly labeled tetramer binding) followed by a complete DNA wrapping signature (FRET5.5). We measured the time interval between the injection and the starting point of a stable high FRET647N signal (tetramer positioning time) and the time interval between the starting point of a stable FRET647N signal and the complete DNA wrapping signal (termini wrapping time) (Fig. 3A). The tetramer positioning time should contain information on both random initial binding time (denoted by t1′) and stable positioning time (denoted by t1). The termini wrapping time is the time during which DNA termini wrap completely to form a tetrasome (denoted by t2). A set of tetramer positioning times was collected from ∼100 traces to construct a histogram (Fig. 4A). This histogram represents the population growth trajectory of the intermediate with a tetramer properly positioned on DNA at the dyad. A pictorial description and explanations for how this histogram is constructed are given in Fig. 3A. The histogram was fit with a biexponential function to obtain the values of t1′ and t1. We did not observe unbinding or any change in the tetramer positioning once a stable high FRET647N signal was observed. The termini wrapping time histogram was fit with a single exponential function to obtain the value of t2 (Fig. 4B).
FIGURE 4.

Measurements of the process times in Nap1-mediated nucleosome assembly. The histograms of the population growth of the intermediates constructed by analyzing the FRET traces (Fig. 3A) were fit with relevant population growth equations and the time constants. The fitting equations and the time constants are shown within the histograms. UDNA and MDNA denote unmethylated and methylated DNA, respectively. A, the population growth histogram for an intermediate with tetramer properly positioned on DNA at the dyad. The histogram was constructed with the Tetramer positioning time depicted in Fig. 3A. This population growth time convolves the random binding time t1′ and the tetramer positioning time t1. B, the population growth histogram starting from the tetramer-bound state to reach the tetrasome state with termini wrapped up in the absence of the dimers. The histogram was constructed with the Termini wrapping time depicted in Fig. 3A. The growth time constant is denoted by t2. C, the population growth histogram starting from the tetrasomal state to reach the final nucleosomal state by adding dimers. The histogram was constructed with the Dimer incorporation time depicted in Fig. 3B. This growth time constant is denoted by t3. D, the population growth histogram for an intermediate with tetramer/dimer incorporated without the termini wrapped up. This histogram was constructed by the Tetramer & dimer incorporation time depicted in Fig. 3C. The curve was fit with two growth components with the constants t1′ and t2′. E, the population growth histogram starting from the tetramer/dimer-bound state to reach the termini wrapped state. This histogram was constructed with the Termini wrapping time depicted in Fig. 3C. This time constant is denoted by t3′.
Tetrasome Formation Is Facilitated upon CpG Methylation
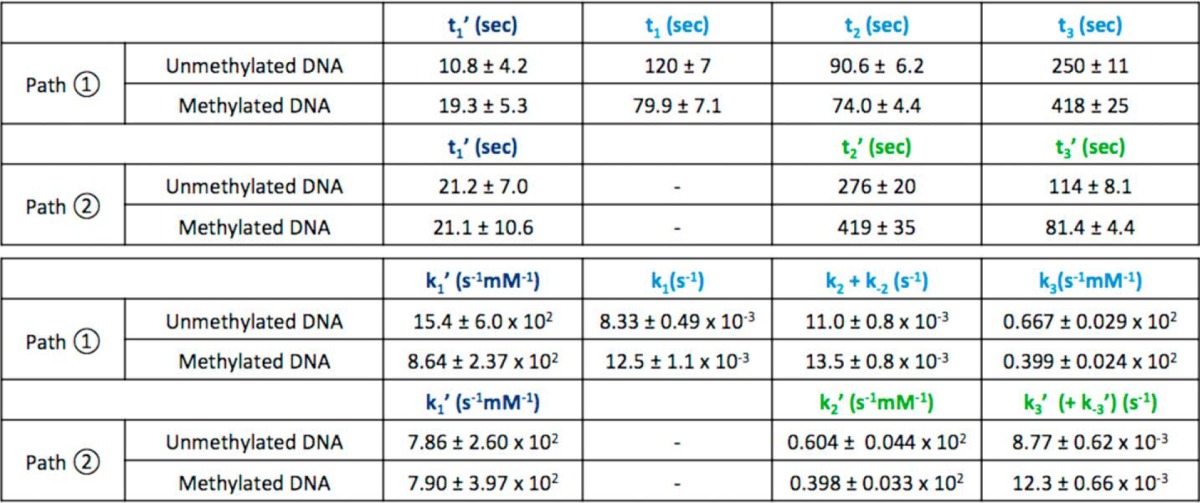
We repeated the measurements with CpG-methylated DNA and obtained the time constants t1′, t1, and t2. According to the results (Fig. 4, A and B), proper binding and positioning of a tetramer on DNA are facilitated by 1.5-fold upon methylation (t1, 120 ± 7 s versus 79.9 ± 7.1 s), whereas the efficiency of random binding (t1′) stays the same within error. The DNA termini wrapping time is also facilitated upon methylation (t2, 90.6 ± 6.2 s versus 74.0 ± 4.4 s). This result agrees well with Fig. 4E, which also shows efficient termini wrapping upon methylation. The plots in Fig. 4 are naturally normalized to the total amount of DNA that we monitored, which is 1500 constructs. Therefore, the two different levels of assembled nucleosomes at any time point in Fig. 4E directly reflect the different efficiencies of termini wrapping between the methylated and unmethylated cases. These results indicate that tetrasome formation becomes facilitated upon CpG methylation.
H2A-H2B Dimers Incorporate Before or After Complete Termini Wrapping
Next, we monitored the assembly with 601 + 16–63 DNA, (H3-H4)2/Nap1, and Atto647N-labeled (H2A-H2B)/Nap1. Fig. 3, B and C, show a set of representative smFRET time traces, revealing that the dimers can be incorporated into two pre-nucleosomal intermediates. The high FRET647N states (histone-bound states) are short-lived in many smFRET traces. According to our analysis, the total emission from a single ATTO647N fluorophore follows an exponential decay with an equivalent lifetime of 8.2 ± 0.6 s (R2 of fitting = 0.99). This short photobleaching lifetime is typical when a fluorophore is labeled at protein and excited at a high power for single molecule measurements. In some rare traces, we see very long-lived ATTO647N because the lifetime distribution follows an exponential decay. If the extinction of the FRET647N signal is a sign of histone unbinding, we should see repetitive rebinding and unbinding, which was not the case in our observation timescale. These analyses verify that the extinction of the FRET647N signal is largely due to photobleaching.
In one set of nucleosomes, dimers are incorporated after complete tetrasome formation, as is shown in Fig. 3B. Due to the low labeling efficiency of the dimers (23%), the vast majority of FRET traces show only a single dimer-binding event. We plotted the time intervals between DNA wrapping and dimer incorporation (Fig. 4, C and D). This time interval should reflect two stepwise incorporations of two dimers, if the events are far separated in time. However, the quality of the fitting with a single exponential function is reasonably good, suggesting that two dimers incorporate either simultaneously or very cooperatively. Another possibility is that the vast majority of formed nucleosomes are hexasomes. According to the dimer labeling efficiency (23%), we expect that about 5% of the total nucleosomes should show two-step photobleaching if each contains two dimers. We observed at least 4% of total nucleosomes showing two-step photobleaching, ruling out the possibility of hexasomes being the majority. It should be noted that the limited time resolution of our measurements might have made it impossible to observe sequential two dimer-binding events if they are separated by only a few seconds. The dimer incorporation time elapsed from tetrasome formation is denoted by t3 (Fig. 4C).
In the other set of nucleosomes, dimers bind stably to tetrasomal intermediates before the DNA termini are fully wrapped (Fig. 3C). The tetramer/dimer-bound intermediate without termini wrapping has a low non-negligible FRET5.5 signal (0.2–0.3). This is because the distances among the fluorophores in this state are within their Förster radii even without the termini wrapped up. In Figs. 1B and 3C, the small faint arrows indicate this low level of FRET5.5. Only when the termini wrap fully around the histone core does FRET5.5 reach the maximum value. We constructed a histogram of the time intervals between the injection and stable dimer binding (dimer binding time, Fig. 4D) and a histogram of the time intervals between dimer binding and DNA wrapping (denoted by t3′) (Fig. 4E). These plots were fit respectively with a biexponential function and a single exponential function. The dimer binding time should contain information on both tetramer binding and dimer binding. We ignored the reverse processes (i.e. histone dissociation) because we did not detect any histone dissociation event. From the fitting, we obtained 21.2 ± 7.0 s for tetramer binding and 276 ± 20 s for dimer binding (t2′). This tetramer binding time is the same within error as the time for random tetramer binding (t1′), suggesting that dimers are incorporated mostly to the first intermediate where tetramers are randomly bound to DNA. We did not observe any sign of separate binding events of two dimers per nucleosome, likely because of the same reason as explained above.
Based on the sequence of the assembly steps observed, we constructed a scheme of the kinetics (Fig. 5). In the scheme, nucleosome assembly takes place mainly via two paths. In one path (path 1 in Fig. 5), a tetramer binds to and positions on DNA at the dyad, followed by DNA termini wrapping and subsequent dimer incorporation. In the other path (path 2 in Fig. 5), all the core histones are incorporated to a DNA template before the termini wrap up.
FIGURE 5.
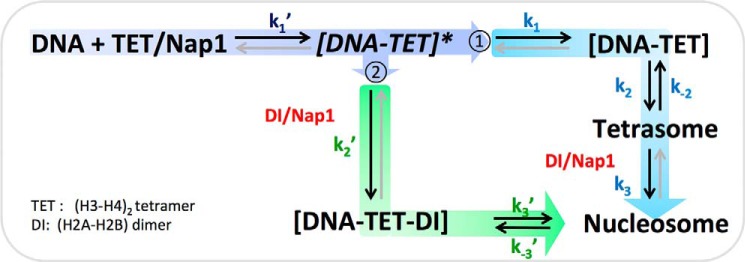
Kinetic scheme for Nap1-mediated nucleosome assembly according to our observation. TET and DI denote (H3-H4)2 tetramer and H2A-H2B dimer, respectively. The process of nucleosome assembly is divided into two paths according to the time point of H2A-H2B dimer incorporation relative to the tetrasome formation (i.e. complete DNA termini wrapping). Along the first path (path 1), Nap1 transfers a (H3-H4)2 tetramer to DNA, resulting in the formation of a stably positioned [DNA-TET] complex through an unstable [DNA-TET]* intermediate, followed by DNA termini wrapping to form a tetrasome. Incorporation of H2A-H2B dimers into the tetrasome completes nucleosome assembly. Along the second path (path 2), H2A-H2B dimers are incorporated to the [DNA-TET]* intermediate, resulting in a ternary [DNA-TET-DI] complex, followed by DNA termini wrapping to complete nucleosome assembly. Gray arrows are the processes that we could not observe with our setup due to the time limit of observation (20 min). The process times t1′, t1, t2, t3, t2′, and t3′ measured in Fig. 4 correspond to 1/k1′, 1/k1, 1/(k2 + k−2), 1/k3, and 1/(k3′ + k−3′), respectively.
H2A-H2B Dimer Incorporation Becomes Inefficient upon CpG Methylation
We repeated the measurements with CpG-methylated 601 + 16–63 DNA. In the first set of data where dimers are incorporated after complete tetrasome formation, t3 becomes longer (418 ± 25 s versus 250 ± 11 s) upon methylation, indicating that dimer incorporation becomes substantially inefficient. Once we see stable binding, the reverse process was not detected within our time limit. Based on the measured kinetic rates and the published equilibrium constants (14), stably bound dimers dissociate in >1000 s on average (>1300 s for a tetramer), which is a timescale rarely accessible in our system due to premature photobleaching of fluorophores labeled at histones.
In the other set of data where dimers bind before the termini wrap up, dimer incorporation time (t2′) is also elongated to 419 ± 35 s from 276 ± 20 s. The similarity between the values of t2′ and t3 indicates that stable dimer incorporation time is not greatly affected by the termini structure of a tetrasome intermediate to which dimers bind. The DNA termini wrapping time elapsed from dimer incorporation (t3′) becomes shorter upon CpG methylation (114 ± 8 s versus 81.4 ± 4.4 s). This is in good agreement with the faster termini wrapping during tetrasome formation (shorter t2 upon methylation). DNA unwrapping is negligibly slow; therefore, t3′ is ∼1/k3′, the process time for DNA wrapping elapsed from dimer binding.
Inefficient Dimer Incorporation upon CpG Methylation Is due to Increased DNA Affinity for Histones
Inefficient dimer incorporation upon CpG methylation is in apparent contradiction to facilitated tetrasome formation. Because the primary difference between dimer and tetramer interaction with DNA is in their binding strengths, we examined how the strength of dimer binding to DNA changes upon CpG methylation. Dimers were added to DNA with and without CpG methylation in the presence of Nap1. The reaction products were analyzed with native PAGE (Fig. 6A). Dimers mixed with methylated DNA yielded notable aggregation, revealing a weaker DNA band and stronger aggregate bands. The total intensity of the bands on the methylated DNA lane is apparently weaker than that on the unmethylated DNA lane, suggesting more precipitation with methylated DNA. After brief centrifugation (5 min at 14,000 × g), the A260 values of the supernatants (0.322 and 0.213, respectively, for unmethylated and methylated DNA) in combination with the A260/A280 ratios (1.49 and 1.16, respectively, for unmethylated and methylated DNA) confirm significantly more precipitation with methylated DNA. These results suggest that the slow dimer incorporation upon CpG methylation is likely due to increased nonproductive dimer binding to DNA resulting in less efficient Nap1 function as a histone chaperone.
FIGURE 6.
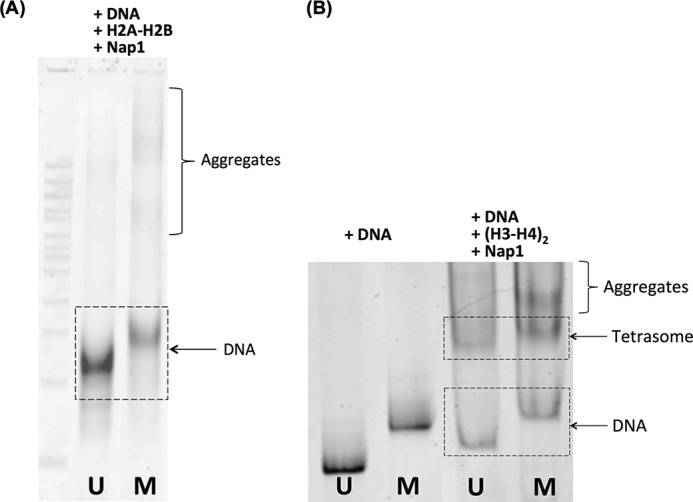
Native PAGE analyses for histone-DNA interactions in the presence of Nap1. U and M denote unmethylated and methylated DNA, respectively. A, a fixed amount (200 ng) of DNA was incubated with 6× molar excess of H2A-H2B dimer in the presence of Nap1 (3× excess to the dimer) for 4 h at 30 °C at 0.1 m NaCl in 25μl total volume. The reaction products were analyzed with native PAGE. The gel was stained with a high sensitivity silver staining kit (SYBR Gold nucleic acid gel stain, GE Healthcare) and scanned with a high sensitivity scanner (Typhoon 9410, GE Healthcare). Thinner DNA and stronger aggregate bands are evident in the methylated DNA case. The total amount of DNA revealed on the gel is apparently smaller with methylated DNA. To quantify any precipitate trapped in the well, the reaction products were spun down for 5 min at 14,000 × g followed by UV-visible absorbance measurements. The A260 values of the supernatants (0.322 and 0.213, respectively, for unmethylated and methylated DNA) in combination with the A260/A280 ratios (1.49 and 1.16, respectively, for unmethylated and methylated DNA) confirm significantly more precipitation with methylated DNA. B, a fixed amount (80 ng) of DNA was incubated with 3× molar excess of (H3-H4)2 tetramer in the presence of Nap1 (6× excess to the tetramer) for 4 h at 30 °C at 0.1 m NaCl. The tetrasome/DNA band intensity ratios are 1.2 and 1.6, respectively, for unmethylated and methylated DNA, indicating more efficient tetrasome assembly upon CpG methylation.
Tetramer Positioning and DNA Termini Wrapping Are the Slowest Steps of Nucleosome Assembly
We converted all the measured process times into rate constants (Table 1). As we have not observed any sign of two-step dimer binding, we assumed for the calculations of k2′ and k3 that two dimers are incorporated in a single step within our time resolution. According to the rate constants, tetramer positioning (k1) and DNA termini wrapping (k2 + k−2 and k3′) are the slowest steps when the histone concentration is high: >1000-fold slower than histone binding at 1 mm histone concentration. Therefore, despite inhibited dimer incorporation, CpG methylation should overall facilitate nucleosome assembly in the presence of abundant DNA and histones. We attempted to observe more efficient nucleosome assembly with methylated DNA in a bulk reaction at high DNA and histone concentrations. However, the assembly reaction becomes very efficient under these conditions, which also make the assembled nucleosomes very stable. Consequently, most DNA constructs were assembled to nucleosomes in 4 h regardless of DNA methylation status, making it very difficult to reveal any difference in the assembly efficiency. We reduced the reaction time to 30 min, but did not observe any significant difference. The tetrasome, on the other hand, is much less stable than the nucleosome. Given a long reaction time, the tetrasome would reach equilibrium with DNA in the presence of Nap1. By measuring the population density of the tetrasome relative to DNA, we would be able to probe the assembly efficiency. We assembled tetrasomes at ∼1000-fold increased DNA concentration (as compared with the single molecule measurements) with 3× molar excess of tetramer and Nap1 (6× molar excess to tetramer) for 4 h. As shown in Fig. 6B, the band intensity of the tetrasome relative to DNA is stronger with methylated DNA (tetrasome/DNA band intensity ratio = 1.2 and 1.6, respectively, for unmethylated and methylated DNA), suggesting that DNA methylation facilitates tetrasome assembly despite the aggregation due to random binding (an aggregate band in the methylated case is evident in Fig. 6B). This result supports the suggestion of more efficient nucleosome assembly upon DNA methylation.
TABLE 1.
Kinetic rates of Nap1-mediated nucleosome assembly
The kinetic scheme is shown in Fig. 5. Kinetic rates (k) are either the reciprocals of the process times (t1, t2 and t3′), or the reciprocals of the process times (t1′, t2′, and t3) divided by histone concentration.
DISCUSSION
Multiple studies have revealed the effects of CpG methylation on the physical properties of DNA (41–43). In particular, CpG methylation reduces the flexibility of DNA (42) and increases its stiffness (43). Our previous results (29, 33) also support that CpG methylation rigidifies DNA and induces tight wrapping around the nucleosome. Low flexibility of DNA has dual roles in nucleosome assembly: (i) raising the height of the free energy barrier to DNA bending/twisting and (ii) decreasing the entropy cost of DNA wrapping (24). Our study reveals that DNA termini wrapping is facilitated upon CpG methylation during the assembly of a tetrasome (t2) and a nucleosome (t3′), suggesting that a lower entropy cost dominates the process. More efficient wrapping of rigidified DNA upon CpG methylation would provide two advantages in forming a repressive chromatin structure. First, DNA termini unwrapping that allows factor binding would become less efficient upon CpG methylation. This structural solidification effect will also facilitate the formation of repressive chromatin structures triggered by factors such as methyl-CpG-binding proteins (44, 45). Second, facilitated DNA wrapping will enable nucleosomes to recover quickly from partially disassembled states during transcription (46).
Mixed effects of DNA methylation on histone-DNA interactions and nucleosome assembly have been reported with results often dependent upon the position and extent of methylation. Some of the studies demonstrated altered translational positioning of nucleosomes (47–49) and inhibited nucleosome assembly (50, 51) upon methylation. A recent study reported that DNA affinity for histone octamer is increased upon DNA methylation (47, 52). Another study showed that DNA methylation enhances nucleosome occupancy by increasing DNA affinity for histone octamer (32). Our present study also supports that DNA methylation increases DNA affinity for histones.
Our results suggest that the tetramer-positioning rate would not be greatly affected by random histone-DNA interactions because tetramer positioning is extremely slow as compared with histone binding. On the other hand, dimer incorporation is much faster than tetramer positioning, and consequently, the rate is determined mainly by how efficiently Nap1 inhibits nonproductive histone-DNA interactions (14). Therefore, random histone-DNA interactions promoted upon CpG methylation would inhibit proper dimer incorporation to their canonical positions, making nucleosome assembly less efficient. As we revealed that tetramer positioning and DNA wrapping are the slowest steps during nucleosome assembly, the overall effect of CpG methylation is still to make nucleosome assembly more efficient. According to our observations, dimers can be incorporated before or after the complete tetrasome formation. The frequencies of events showing dimer incorporation before and after termini wrapping are similar to each other within error (Fig. 7). This result indicates that dimers bind to a tetrasomal intermediate regardless of the DNA termini conformation, verifying that the competition between Nap1 and DNA for dimer is what limits the rate of dimer incorporation.
FIGURE 7.

The ratios of the number of nucleosomes assembled via dimer incorporation after tetrasome formation (path 1 in Fig. 5) to that before tetrasome formation (path 2 in Fig. 5).
The reverse process of nucleosome assembly has been suggested as a model for nucleosome disassembly. In the model, the dimers are released from DNA following tetramer/dimer interface opening. Subsequent dissociation of the tetramer from DNA completes the disassembly (53). This model implicates an open conformational intermediate, where dimers are partially dissociated from the tetramer. We observed some FRET traces showing changes in dimer-DNA FRET in a stably bound state (Fig. 3C), suggesting that a dimer may sample multiple positions before it finds the most stable one. The intermediate state denoted in our model as [DNA-TET-DI] (where TET and DI denote (H3-H4)2 tetramer and H2A-H2B dimer) would, therefore, convolve multiple states, potentially including ones with partially open dimer-tetramer interface.
We did not observe any clear sign of two separate binding events of dimers per nucleosome. This result suggests that two dimers are incorporated either simultaneously or very cooperatively within a few seconds. Because of the second scenario, we cannot rule out hexasome existence in our system. The stoichiometry between H2A-H2B dimer and Nap1 was reported to be 1:1 as one yNap1 dimer binds two histone dimers (15). A recent study confirmed that Nap1 dimer is a physiological conformation (54). On the contrary, hexasome existence has been confirmed based on a mass spectrometric analysis (55). The hexasome has been suggested as a physiological intermediate during nucleosome disassembly. Our results suggest that at least during nucleosome assembly dimer binding is very cooperative and that hexasome states are not stable enough to be detected under our experimental conditions.
The present single molecule study on Nap1-mediated nucleosome assembly dissected the steps of histone bindings in the context of DNA wrapping and revealed or confirmed diverse intermediates and aspects of nucleosome assembly. Our observations unraveled three effects of CpG methylation on the efficiency of nucleosome assembly: (i) facilitated (H3-H4)2 tetramer positioning, (ii) promoted DNA termini wrapping, and (iii) decelerated dimer incorporation. We suggest that the first two changes are caused by rigidified DNA, and that the third change is due to increased histone-DNA affinity. These changes would make the overall nucleosome assembly more efficient, which may help facilitate gene packaging in chromatin. Our results indicate that the physical properties of DNA are critical in controlling the overall efficiency of nucleosome assembly.
Acknowledgments
We thank Drs. Pamela Dyer and Karolin Luger for providing a part of the labeled histones used in the measurements.
This work was supported, in whole or in part, by National Institutes of Health Grant GM097286 (to T. H. L.).
- Nap1
- nucleosome assembly protein 1
- yNap1
- yeast Nap1
- smFRET
- single molecule FRET
- EMCCD
- electron multiplying CCD camera.
REFERENCES
- 1. Kornberg R. D., Klug A. (1981) The nucleosome. Sci. Am. 244, 52–64 [DOI] [PubMed] [Google Scholar]
- 2. Luger K., Mäder A. W., Richmond R. K., Sargent D. F., Richmond T. J. (1997) Crystal structure of the nucleosome core particle at 2.8 Ä resolution. Nature 389, 251–260 [DOI] [PubMed] [Google Scholar]
- 3. Akey C. W., Luger K. (2003) Histone chaperones and nucleosome assembly. Curr. Opin. Struct. Biol. 13, 6–14 [DOI] [PubMed] [Google Scholar]
- 4. Rocha W., Verreault A. (2008) Clothing up DNA for all seasons: histone chaperones and nucleosome assembly pathways. FEBS Lett. 582, 1938–1949 [DOI] [PubMed] [Google Scholar]
- 5. Wilhelm F. X., Wilhelm M. L., Erard M., Duane M. P. (1978) Reconstitution of chromatin: assembly of the nucleosome. Nucleic Acids Res. 5, 505–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fujii-Nakata T., Ishimi Y., Okuda A., Kikuchi A. (1992) Functional analysis of nucleosome assembly protein, NAP-1: the negatively charged COOH-terminal region is not necessary for the intrinsic assembly activity. J. Biol. Chem. 267, 20980–20986 [PubMed] [Google Scholar]
- 7. Fyodorov D. V., Kadonaga J. T. (2003) Chromatin assembly in vitro with purified recombinant ACF and NAP-1. Methods Enzymol. 371, 499–515 [DOI] [PubMed] [Google Scholar]
- 8. Andrews A. J., Luger K. (2011) A coupled equilibrium approach to study nucleosome thermodynamics. Methods Enzymol. 488, 265–285 [DOI] [PubMed] [Google Scholar]
- 9. Mazurkiewicz J., Kepert J. F., Rippe K. (2006) On the mechanism of nucleosome assembly by histone chaperone NAP1. J. Biol. Chem. 281, 16462–16472 [DOI] [PubMed] [Google Scholar]
- 10. Ito T., Bulger M., Kobayashi R., Kadonaga J. T. (1996) Drosophila NAP-1 is a core histone chaperone that functions in ATP-facilitated assembly of regularly spaced nucleosomal arrays. Mol. Cell. Biol. 16, 3112–3124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mosammaparast N., Ewart C. S., Pemberton L. F. (2002) A role for nucleosome assembly protein 1 in the nuclear transport of histones H2A and H2B. EMBO J. 21, 6527–6538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McQuibban G. A., Commisso-Cappelli C. N., Lewis P. N. (1998) Assembly, remodeling, and histone binding capabilities of yeast nucleosome assembly protein 1. J. Biol. Chem. 273, 6582–6590 [DOI] [PubMed] [Google Scholar]
- 13. Mosammaparast N., Jackson K. R., Guo Y., Brame C. J., Shabanowitz J., Hunt D. F., Pemberton L. F. (2001) Nuclear import of histone H2A and H2B is mediated by a network of karyopherins. J. Cell Biol. 153, 251–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Andrews A. J., Chen X., Zevin A., Stargell L. A., Luger K. (2010) The histone chaperone Nap1 promotes nucleosome assembly by eliminating nonnucleosomal histone DNA interactions. Mol. Cell 37, 834–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Andrews A. J., Downing G., Brown K., Park Y. J., Luger K. (2008) A thermodynamic model for Nap1-histone interactions. J. Biol. Chem. 283, 32412–32418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Riggs A. D., Pfeifer G. P. (1992) X-chromosome inactivation and cell memory. Trends Genet. 8, 169–174 [DOI] [PubMed] [Google Scholar]
- 17. Takai D., Jones P. A. (2002) Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl. Acad. Sci. U.S.A. 99, 3740–3745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bird A. P. (1986) CpG-rich islands and the function of DNA methylation. Nature 321, 209–213 [DOI] [PubMed] [Google Scholar]
- 19. Esteller M. (2007) Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 8, 286–298 [DOI] [PubMed] [Google Scholar]
- 20. Robertson K. D. (2002) DNA methylation and chromatin: unraveling the tangled web. Oncogene 21, 5361–5379 [DOI] [PubMed] [Google Scholar]
- 21. Klein C. B., Costa M. (1997) DNA methylation, heterochromatin and epigenetic carcinogens. Mutat. Res. 386, 163–180 [DOI] [PubMed] [Google Scholar]
- 22. Bird A. (2002) DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21 [DOI] [PubMed] [Google Scholar]
- 23. Anselmi C., Bocchinfuso G., De Santis P., Savino M., Scipioni A. (2000) A theoretical model for the prediction of sequence-dependent nucleosome thermodynamic stability. Biophys. J. 79, 601–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Widom J. (2001) Role of DNA sequence in nucleosome stability and dynamics. Q. Rev. Biophys. 34, 269–324 [DOI] [PubMed] [Google Scholar]
- 25. Segal E., Fondufe-Mittendorf Y., Chen L., Thåström A. C., Field Y., Moore I. K., Wang J. P. Z., Widom J. (2006) A genomic code for nucleosome positioning. Nature 442, 772–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jones P. L., Veenstra G. J. C., Wade P. A., Vermaak D., Kass S. U., Landsberger N., Strouboulis J., Wolffe A. P. (1998) Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 19, 187–191 [DOI] [PubMed] [Google Scholar]
- 27. Nan X., Ng H. H., Johnson C. A., Laherty C. D., Turner B. M., Eisenman R. N., Bird A. (1998) Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393, 386–389 [DOI] [PubMed] [Google Scholar]
- 28. Tate P. H., Bird A. P. (1993) Effects of DNA methylation on DNA-binding proteins and gene expression. Curr. Opin. Genet. Dev. 3, 226–231 [DOI] [PubMed] [Google Scholar]
- 29. Choy J. S., Wei S., Lee J. Y., Tan S., Chu S., Lee T. H. (2010) DNA methylation increases nucleosome compaction and rigidity. J. Am. Chem. Soc. 132, 1782–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee J. Y., Lee T. H. (2012) Effects of histone acetylation and CpG methylation on the structure of nucleosomes. Biochim. Biophys. Acta 1824, 974–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chodavarapu R. K., Feng S., Bernatavichute Y. V., Chen P. Y., Stroud H., Yu Y., Hetzel J. A., Kuo F., Kim J., Cokus S. J., Casero D., Bernal M., Huijser P., Clark A. T., Krämer U., Merchant S. S., Zhang X., Jacobsen S. E., Pellegrini M. (2010) Relationship between nucleosome positioning and DNA methylation. Nature 466, 388–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Collings C. K., Waddell P. J., Anderson J. N. (2013) Effects of DNA methylation on nucleosome stability. Nucleic Acids Res. 41, 2918–2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee J. Y., Lee T. H. (2012) Effects of DNA methylation on the structure of nucleosomes. J. Am. Chem. Soc. 134, 173–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee J. Y., Wei S., Lee T. H. (2011) Effects of histone acetylation by Piccolo NuA4 on the structure of a nucleosome and the interactions between two nucleosomes. J. Biol. Chem. 286, 11099–11109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tóth K. F., Mazurkiewicz J., Rippe K. (2005) Association states of nucleosome assembly protein 1 and its complexes with histones. J. Biol. Chem. 280, 15690–15699 [DOI] [PubMed] [Google Scholar]
- 36. Dyer P. N., Edayathumangalam R. S., White C. L., Bao Y., Chakravarthy S., Muthurajan U. M., Luger K. (2004) Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 375, 23–44 [DOI] [PubMed] [Google Scholar]
- 37. Horsey I., Furey W. S., Harrison J. G., Osborne M. A., Balasubramanian S. (2000) Double fluorescence resonance energy transfer to explore multicomponent binding interactions: a case study of DNA mismatches. Chem. Commun. 1043–1044 [Google Scholar]
- 38. Watrob H. M., Pan C.-P., Barkley M. D. (2003) Two-step FRET as a structural tool. J. Am. Chem. Soc. 125, 7336–7343 [DOI] [PubMed] [Google Scholar]
- 39. Nakagawa T., Bulger M., Muramatsu M., Ito T. (2001) Multistep chromatin assembly on supercoiled plasmid DNA by nucleosome assembly protein-1 and ATP-utilizing chromatin assembly and remodeling factor. J. Biol. Chem. 276, 27384–27391 [DOI] [PubMed] [Google Scholar]
- 40. Sheinin M. Y., Li M., Soltani M., Luger K., Wang M. D. (2013) Torque modulates nucleosome stability and facilitates H2A/H2B dimer loss. Nat. Commun. 4, 2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nathan D., Crothers D. M. (2002) Bending and flexibility of methylated and unmethylated EcoRI DNA. J. Mol. Biol. 316, 7–17 [DOI] [PubMed] [Google Scholar]
- 42. Geahigan K. B., Meints G. A., Hatcher M. E., Orban J., Drobny G. P. (2000) The dynamic impact of CpG methylation in DNA. Biochemistry 39, 4939–4946 [DOI] [PubMed] [Google Scholar]
- 43. Derreumaux S., Chaoui M., Tevanian G., Fermandjian S. (2001) Impact of CpG methylation on structure, dynamics and solvation of cAMP DNA responsive element. Nucleic Acids Res. 29, 2314–2326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jones P. A., Liang G. (2009) Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 10, 805–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jeong S., Liang G., Sharma S., Lin J. C., Choi S. H., Han H., Yoo C. B., Egger G., Yang A. S., Jones P. A. (2009) Selective anchoring of DNA methyltransferases 3A and 3B to nucleosomes containing methylated DNA. Mol. Cell Biol. 29, 5366–5376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kulaeva O. I., Hsieh F. K., Studitsky V. M. (2010) RNA polymerase complexes cooperate to relieve the nucleosomal barrier and evict histones. Proc. Natl. Acad. Sci. U.S.A. 107, 11325–11330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hogan M. E., Rooney T. F., Austin R. H. (1987) Evidence for kinks in DNA folding in the nucleosome. Nature 328, 554–557 [DOI] [PubMed] [Google Scholar]
- 48. Davey C., Pennings S., Allan J. (1997) CpG methylation remodels chromatin structure in vitro. J. Mol. Biol. 267, 276–288 [DOI] [PubMed] [Google Scholar]
- 49. Davey C. S., Pennings S., Reilly C., Meehan R. R., Allan J. (2004) A determining influence for CpG dinucleotides on nucleosome positioning in vitro. Nucleic Acids Res. 32, 4322–4331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang Y. H., Griffith J. (1996) Methylation of expanded CCG triplet repeat DNA from fragile X syndrome patients enhances nucleosome exclusion. J. Biol. Chem. 271, 22937–22940 [PubMed] [Google Scholar]
- 51. Godde J. S., Kass S. U., Hirst M. C., Wolffe A. P. (1996) Nucleosome assembly on methylated CGG triplet repeats in the fragile X mental retardation gene 1 promoter. J. Biol. Chem. 271, 24325–24328 [DOI] [PubMed] [Google Scholar]
- 52. Pennings S., Allan J., Davey C. S. (2005) DNA methylation, nucleosome formation and positioning. Brief. Funct. Genomic. Proteomic. 3, 351–361 [DOI] [PubMed] [Google Scholar]
- 53. Böhm V., Hieb A. R., Andrews A. J., Gansen A., Rocker A., Tóth K., Luger K., Langowski J. (2011) Nucleosome accessibility governed by the dimer/tetramer interface. Nucleic Acids Res. 39, 3093–3102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. D'Arcy S., Martin K. W., Panchenko T., Chen X., Bergeron S., Stargell L. A., Black B. E., Luger K. (2013) Chaperone Nap1 shields histone surfaces used in a nucleosome and can put H2A-H2B in an unconventional tetrameric form. Mol. Cell 51, 662–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Azegami N., Saikusa K., Todokoro Y., Nagadoi A., Kurumizaka H., Nishimura Y., Akashi S. (2013) Conclusive evidence of the reconstituted hexasome proven by native mass spectrometry. Biochemistry 52, 5155–5157 [DOI] [PubMed] [Google Scholar]