

Background: Stra6 is a trans-membrane retinol transporter involved in retinoic acid (RA) signaling.
Results: The epigenetic signature of the Stra6 gene reveals an RA-responsive element.
Conclusion: An intragenic RARE drives RA-responsive expression of two different Stra6 isoforms.
Significance: The novel, shorter Stra6 transcript may encode a functionally different retinol transporter.
Keywords: Cell Differentiation, CRISPR/Cas, Embryonic Stem Cell, Epigenetics, Retinoic Acid, Retinoid-binding Protein, Vitamin A
Abstract
Cellular uptake of vitamin A (retinol) is essential for many biological functions. The Stra6 protein binds the serum retinol-binding protein, RBP4, and acts in conjunction with the enzyme lecithin:retinol acyltransferase to facilitate retinol uptake in some cell types. We show that in embryonic stem (ES) cells and in some tissues, the Stra6 gene encodes two distinct mRNAs transcribed from two different promoters. Whereas both are all-trans-retinoic acid (RA)-responsive in ES cells, the downstream promoter contains a half-site RA response element (RARE) and drives an ∼13-fold, RA-associated increase in luciferase reporter activity. We employed CRISPR-Cas9 genome editing to show that the endogenous RARE is required for RA-induced transcription of both Stra6 isoforms. We further demonstrate that in ES cells, 1) both RARγ and RXRα are present at the Stra6 RARE; 2) RA increases co-activator p300 (KAT3B) binding and histone H3 Lys-27 acetylation at both promoters; 3) RA decreases Suz12 levels and histone H3 Lys-27 trimethylation epigenetic marks at both promoters; and 4) these epigenetic changes are diminished in the absence of RARγ. In the brains of WT mice, both the longer and the shorter Stra6 transcript (Stra6L and Stra6S, respectively) are highly expressed, whereas these transcripts are found only at low levels in RARγ−/− mice. In the brains of vitamin A-deficient mice, both Stra6L and Stra6S levels are decreased. In contrast, in the vitamin A-deficient kidneys, the Stra6L levels are greatly increased, whereas Stra6S levels are decreased. Our data show that kidneys respond to retinol deficiency by differential Stra6 promoter usage, which may play a role in the retention of retinol when vitamin A is low.
Introduction
The plasma retinol-binding protein (RBP4)2 binds to a thyroxine transthyretin (TTR)-binding protein in a 1:1 stoichiometry to deliver and distribute vitamin A (all-trans-retinol) to many cell types in the body (for a review, see Ref. 1). An RBP4 receptor protein, stimulated by retinoic acid 6 (Stra6), binds RBP4 and allows uptake of the liganded, holo-RBP complex into some cell types (2, 3). A second RBP4 receptor (RBPR2) with structural similarities to Stra6 was recently identified in liver (4). In addition to Stra6, the enzyme lecithin:retinol acyltransferase (LRAT) was reported to play a key role in the uptake of retinol into certain cell types (2, 5–11). Uptake of retinol from RBP4 requires a functional interaction between LRAT and Stra6 (10).
Stra6 transcripts and protein are expressed in many tissues involved in retinol actions, such as the placenta, testis, skin, kidney, eye, brain, and choroid plexus (12, 13). Stra6 is involved in cell proliferation control, and knockdown of Stra6 in skin epithelia leads to aberrant hyperproliferation (14). The Stra6 protein can function as a cytokine receptor that activates Jak/Stat signaling in response to retinol-RBP4 complexes. Thus, retinol and RBP4 can influence transcription via Stra6 and subsequent activation of the transcription factor Stat5 (15, 16). Additionally, Stra6, when bound to the retinol-RBP4 complex, allows recruitment of an intracellular binding protein for retinol, the cellular retinol binding protein 1 (CRBP-1) (17). Stra6 also functions to regulate other biological activities, such as adipogenesis (19), lipid metabolism (15), and p53-induced apoptosis after DNA damage (18). Because TTR prevents Stra6 from associating with the RBP4-retinol complex, Stra6 is activated by retinol only when the plasma RBP4 level is higher than the level of TTR (17). This finding implicates TTR in the regulation of Stra6 signaling. In adipocyte precursor cells, Stra6 mediates bidirectional retinol transport, depending on whether RBP4 is retinol-bound (19).
Mutations in STRA6 can result in the Matthew-Wood syndrome, which consists of severe microphthalmia, pulmonary agenesis, bilateral diaphragmatic eventration, duodenal stenosis, pancreatic malformations, and growth retardation (13, 20, 21). Some mutations in STRA6 in humans involve either a homozygous insertion/deletion in exon 2 or a homozygous insertion in exon 7, predicting a premature stop codon (20). Several STRA6 mutations associated with human disease have been shown to limit or abolish retinol uptake into cells (23, 24). Three groups have independently reported marked ocular defects in Stra6 null mice (25–27). Genetic ablation of Stra6 results in a reduced retinoid content in the retinal pigment epithelium and neurosensory retina (greater than a 95% reduction in retinyl esters) with consequently fewer cone photoreceptor cells and diminished cone b-wave amplitude (25). Under the evaluated conditions, knock-out of Stra6 did not impair the physiological functions of retinoids in tissues other than the eye (26). Under vitamin A-deficient conditions, Stra6 was reported to facilitate redistribution from storage tissue (e.g. liver and lungs) to the eye (27), possibly in conjunction with the recently identified RBPR2, another RBP4 receptor (4). Ablation of Stra6 can also protect animals from the insulin-resistant state induced by feeding a high fat, high sucrose diet (28), which may be a result of impaired Jak/Stat signaling (29). Why humans with certain mutations in STRA6 exhibit a more severe morphological phenotype (25–27) than the Stra6 knock-out mice is currently not understood.
Vitamin A acts through its biologically active metabolite, all-trans-retinoic acid (RA), and the retinoic acid receptors (RARs) to regulate large numbers of genes at the transcriptional level in various cell types (30–32). On a molecular level, RA mediates major changes in epigenetic marks on specific target genes (33–37). We recently showed that Stra6 transcripts are increased by RA treatment of embryonic stem (ES) cells and that this induction depends on RARγ (38).
There is a great interest in the cellular uptake of vitamin A, yet the mechanisms by which the Stra6 gene is regulated at the transcriptional level are not clearly defined. In this report, we demonstrate that the Stra6 gene possesses two different promoters, both of which are induced by RA through a single intragenic RARE. We characterize the Stra6 retinoic acid-responsive DNA element (RARE) and demonstrate that RXRα/RARγ heterodimers directly associate with this cis-acting element of Stra6 to activate RA-dependent transcription. Finally, we identify the differential regulation of the two Stra6 promoters in the kidneys of vitamin A-deficient animals, which points to physiologically distinct functions of the two Stra6 isoforms.
EXPERIMENTAL PROCEDURES
Cell Culture and Retinoic Acid Treatment of ES Cells
The WT (CCE) and RARγ−/− ES cell lines were derived and cultured as described (33). RA (Sigma) was added to the cells 24 h after plating (1 μm final concentration), and ethanol (EtOH, 0.1%) served as a vehicle control. For time course evaluations, ES cells were plated in gelatin-coated tissue culture dishes and treated with RA (1 μm) at various time points up to 72 h prior to harvesting.
Vitamin A-deficient Mice
WT C57Bl/6 and LRAT null C57Bl/6 mice were given vitamin A-sufficient (VAS) or vitamin A-deficient (VAD) chow for 10 weeks, as described (6). The treatment of the mice was approved by the Institutional Animal Care and Use Committee at Weill Cornell. Tissues were harvested, RNA was isolated, and semiquantitative RT-PCR was performed as described (6). The brains were dissected without the eyes. Serum and liver retinoid levels were measured by HPLC as described (6), demonstrating that the mice on the VAD chow were vitamin A-deficient.
RNA Isolation and Reverse Transcription
Total RNA was isolated from F9 and ES cells using TRIzol reagent (Invitrogen), and RNA was quantitated by optical density at 260 nm. The RNA (1 μg) was reverse transcribed (Quanta Biosciences, Gaithersburg, MD) and then diluted 1:10 with H2O. The cDNA obtained was diluted 10-fold, and 3 μl of this cDNA was utilized for PCRs.
Generation of cDNA, Semiquantitative, and Real-time PCR
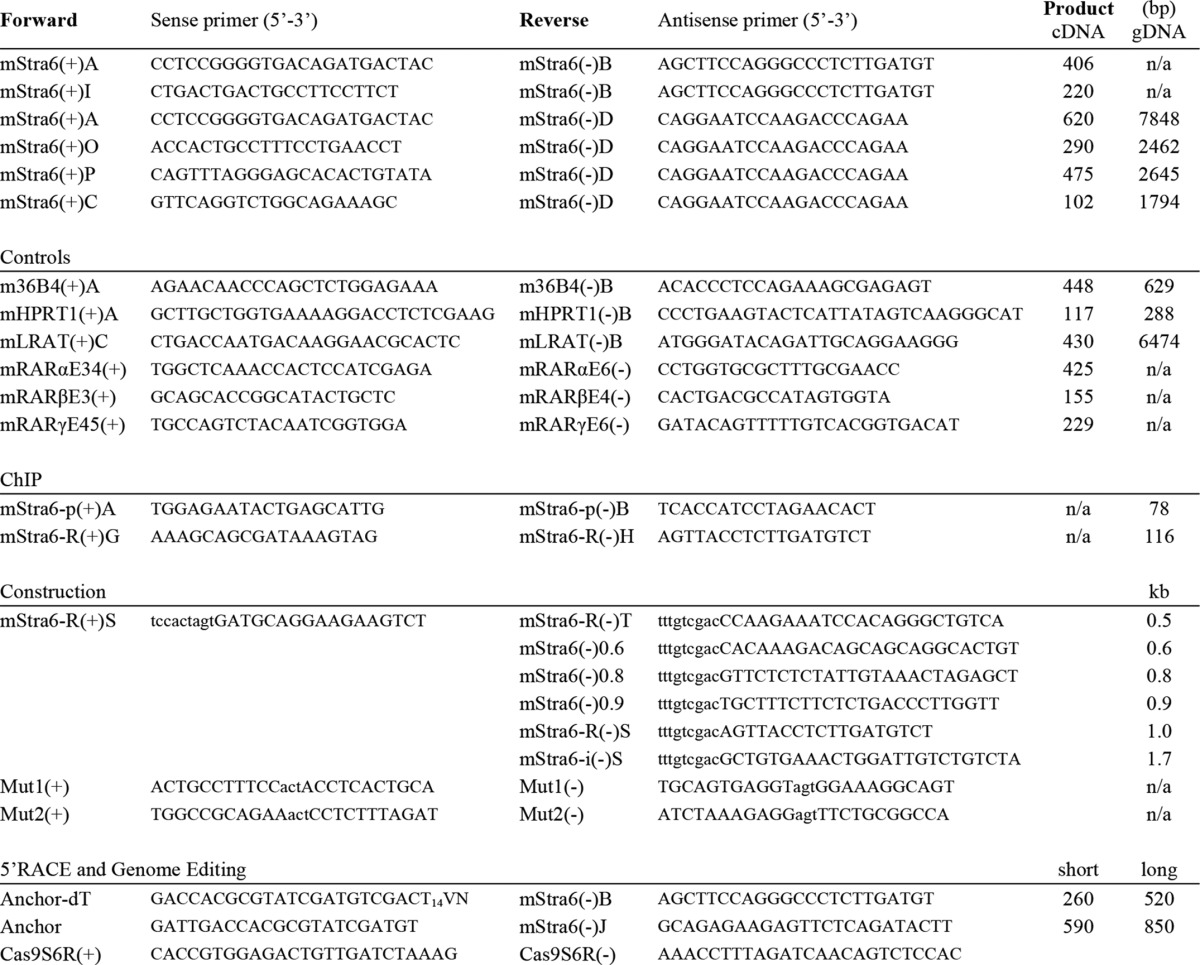
Real-time PCR was performed using SYBR Green Supermix (Quanta Biosciences) in a 15-μl reaction containing reaction mix (1×), a 0.2 μm concentration of each primer, and 3 μl of cDNA template. The reactions were run on a Bio-Rad MyiQTM single color real-time PCR detection system (Bio-Rad). Amplification in the linear range was demonstrated by a serial dilution of cDNA from RA-treated wild type (WT) cells included in each reaction (1:1, 1:5, 1:10, 1:50, 1:100, and 1:500). Reactions with H2O and template without reverse transcriptase, respectively, served as negative controls for primer-dimer and for amplification of residual genomic DNA. All real-time PCR primers were designed to span intronic regions. Primer sequences are listed (Table 1). Each expression analysis was performed at least three times (e.g. n≥3, independently propagated cells, experiment repeated three times). Within each PCR analysis, samples were run in triplicate. PCR products were verified by DNA sequencing.
TABLE 1.
Primer sequences
Mapping of Transcriptional Start Sites
5′-Rapid amplification of cDNA ends (RACE) was performed with the Roche Applied Science 5′/3′ RACE kit using mStra6(−)J for cDNA synthesis and mStra6(−)B for nested PCR amplification. The PCR products were purified using a PCR purification kit (Qiagen), and the eluted DNA was recovered by ligation into the pGEM-T easy vector (Promega, WI). The ligated products were recovered by bacterial transformation. Single colonies were picked, and plasmid DNA was isolated. Insert-containing plasmids were identified by restriction digestion and sequenced using T7(+) and SP6(+) primers.
Constructing and Assaying Stra6 Reporter Plasmids
Firefly luciferase reporter constructs were designed by cloning PCR fragments of various Stra6 genomic regions into the pGL3 reporter plasmid. In brief, PCR fragments were SpeI/SalI-digested and cloned into NheI/XhoI sites of the pGL3 basic plasmid (Promega, WI). The Stra6 reporter constructs and their genomic coordinates are shown in Table 2. Point mutations were introduced into the pGL3 mStra6–0.8 construct using Stratagene QuikChange (see Table 1 for primer sequences). The ES cells were transiently transfected with different luciferase reporter constructs using Lipofectamine LTX (Invitrogen). The pRL-TK reporter plasmid, Renilla luciferase-thymidine kinase (Promega, WI), served as a control for transfection efficiency. WT ES cells were transfected and then cultured in DME containing 1.0 μm RA for 8 or 24 h, as indicated. Firefly and Renilla luciferase activities were sequentially measured with a luminometer using the Dual-Luciferase reporter assay system (Promega).
TABLE 2.
Stra6 reporter constructs and luciferase data
Increase in luciferase activity upon 24-h RA treatment (relative to vehicle-treated cells). Shown are genomic coordinates for Stra6 reporter elements and the RARE (mm9). Note that pGL3 Stra6-PL/PS is a composite reporter construct that contains two Stra6 genomic regions (one upstream and one downstream of the luciferase coding region). These constructs are further described in the legend to Fig. 2A.
| pGL3 construct | RA-responsive (Luc RA/control) | Genomic coordinates |
|||
|---|---|---|---|---|---|
| Chromosome | Strand | Start | End | ||
| Fold increase | |||||
| Stra6-PL | 2.3 | 9 | + | 57976612 | 57977225 |
| Stra6-PL/PS | 1.4 | 9 | + | 57976612 | 57977225 |
| 57988013 | 57989474 | ||||
| Stra6-1.7 | 3.9 | 9 | + | 57987743 | 57989474 |
| Stra6-1.0 | 5.8 | 9 | + | 57987743 | 57988689 |
| Stra6-0.9 | 14.9 | 9 | + | 57987743 | 57988585 |
| Stra6-0.8 | 13.4 | 9 | + | 57987743 | 57988564 |
| Stra6-0.6 | 2.1 | 9 | + | 57987743 | 57988278 |
| Stra6-0.5 | 2.2 | 9 | + | 57987743 | 57988237 |
| RARE | Inverted repeat | 9 | + | 57988490 | 57988511 |
Genome Editing and Screening
Double-stranded DNA fragments targeting the Stra6-RARE (Table 1) were cloned into the pX330 vector (39). Integrity of the insert was confirmed by sequencing, and the construct was transfected into WT ES cells. Upon overnight recovery, the transfected cells were reseeded at low density (2 × 103 cells/15-cm dish) to obtain independent clonal lines. The colonies were screened by PCR (mStra6(+)P and mStra6-R(−)H primers) followed by MboI restriction digest. The PCR bands with modified MboI sites were sequenced to map the exact mutations introduced at the Stra6-RARE. Cell lines with single and double mutations of the Stra6-RARE were assayed in three independent experiments for transcriptional induction of Stra6 by RA.
Chromatin Immunoprecipitation (ChIP) Assays
A one-step ChIP protocol that utilizes formaldehyde cross-linking was employed for histone ChIP assays. For active RNA polymerase II (PolII) and transcription factor ChIP assays (PolII-CTD, Suz12, and Ring1B), we used a two-step ChIP protocol where the formaldehyde cross-linking is preceded by a disuccinimidyl glutarate cross-linking step (40–42). ChIP assays were performed on 5.0 × 105 sonicated ES cells using 2 μg of antibody/immunoprecipitation. The assays were performed on at least three samples from independent experiments (e.g. independently propagated cells). Antibodies used were as follows: H3K27me3 (07-449, Millipore); H3K27ac (07-360, Millipore); PolII-CTD (MMS-134R, Covance, Princeton, NJ); RARγ (ab12012, Abcam, MA); RXRα (D-20, sc-553, Santa Cruz Biotechnology, Inc.); Suz12 (3737S, Millipore, MA); p300 (N-15, sc-584, Santa Cruz Biotechnology); rabbit IgG (sc-2027, Santa Cruz Biotechnology). The ChIP-on-chip assays were performed as described (33).
Data Analysis and Statistics
Data from at least three independent experiments were analyzed using one-way analysis of variance in the expression and ChIP analyses. The S.E. was determined for each of the data sets (at least three biological, independent repeats, each in triplicate, plotted as error bars in the graphs), and analysis of variance values of p < 0.05 among compared samples were assigned statistical significance.
RESULTS
The Increase in Stra6 Transcripts in RA-treated ES Cells Is Associated with Extensive Epigenetic Changes
We employed WT and RARγ−/− ES cells (38) to delineate the dynamics of the increase in Stra6 transcripts associated with RA treatment of murine ES cells in culture. We demonstrated that there is a 24-fold increase in Stra6 transcripts 48 h after RA addition in WT ES cells and that this increase does not occur in the RARγ−/− ES cells (Fig. 1A). We next determined the epigenetic changes that occur along the entire Stra6 genomic region in response to RA treatment of ES cells. Transcriptional activation is strongly associated with Lys-9/14 acetylation (H3K9/14ac) and Lys-4 trimethylation (H3K4me3) of histone 3 (43). We therefore assessed the dynamics of these epigenetic marks in RA-treated WT and RARγ−/− ES cells using ChIP-chip analysis (Fig. 1B). At the RefSeq promoter, which we named PromoterLong (PL), the increase in H3K9/14ac was much greater in WT than in RARγ−/− cells (Fig. 1B). At a region ∼12 kb downstream of the PL (RefSeq) promoter, we again observed an RA-dependent increase in the H3K9/14ac epigenetic marks in WT but not in RARγ−/− ES cells (Fig. 1B). Because this may reflect a downstream promoter driving expression of a shorter transcript, we named this region PromoterShort (PS). The H3K4me3 mark was present in both WT and RARγ−/− cells at PL but not at PS (Fig. 1B). We detected increased Stra6 transcript levels after RA addition only in the WT cells. Thus, the presence of the H3K9/14ac mark correlated with transcriptional activation, whereas the increase in the H3K4me3 mark specifically at PL was insufficient to induce transcription because the increase in the H3K4me3 mark occurred even in the RARγ−/− cells (Fig. 1B). In contrast, H3K27me3, a repressive histone modification introduced by the Polycomb repressive complex 2 (PRC2), was decreased at both Stra6 promoters PL and PS in response to RA treatment in the WT but not in the RARγ−/− ES cells (Fig. 1B). The dynamic changes in these three key histone epigenetic marks revealed two distinct, genomic regions of the Stra6 gene, which in WT ES cells are remodeled in response to RA.
FIGURE 1.

RA-responsiveness of Stra6 in WT and RARγ−/− ES cells. A, in WT but not in RARγ−/− ES cells, Stra6 transcript levels (total) increase with the duration of RA exposure (1 μm). B, heat map of Stra6 epigenetic signatures (H3K9/14ac, H3K27me3, and H3K4me3) in WT and RARγ−/− ES cells upon RA exposure for 1, 8, and 24 h (relative to vehicle-treated control). The intensity of the heat map specifies the levels of epigenetic marks (represented by the scale bar at the top right, red/white, highest). The promoter regions PL and PS correspond to the PRefSeq and to the novel promoter proximal to the Stra6 RARE, respectively. Note that the heat map is aligned with the schematics in D below. C, detection of Stra6 transcripts from PL and PS promoters as well as total Stra6 transcript levels in untreated and RA-treated WT and RARγ−/− ES cells by RT-PCR (top). The primers utilized are specified by letters. RARγ transcript levels and 36B4 (loading control) are also shown. Bottom, mapping the 5′-end of RA-induced Stra6 transcripts by 5′-RACE. D, Stra6 genomic structure (top), reporter constructs (middle), and promoter diagram (bottom). Alternative transcripts and Stra6 genomic regions are depicted at the top. The RefSeq promoter (PL) and the novel RA-responsive promoter (PS) identified are shown here. Squares, exons (open, untranslated). Stra6 reporter constructs were evaluated for RA induction of PL and PS (-fold induction by RA is noted to the right). The alternative Stra6 transcripts correspond to NM_001162475.1 (6.1), NM_009291.2 (6.2), NM_001162476.1 (6.3), NM_001162479.1 (6.4), and a novel short isoform (6.5; GenBankTM number AK092227.1). Stra6 promoter regions (PL and PS) and exon usage of the 5′-mRNAs are shown. The locations of primer binding sites are specified by single letters below or above the arrowheads (forward and reverse primers, respectively). Error bars, S.E.
The Stra6 Gene Encodes Two Transcripts Expressed from Two Distinct Promoters
The data from our ChIP-chip experiments (Fig. 1B) pointed to a second regulatory region in the Stra6 intragenic region, termed PS. A short form of Stra6, which encodes an amino-terminally truncated Stra6 protein, has been identified in human samples (GenBankTM AK092227.1). We hypothesized that the PS epigenetic “hotspot” located 12 kb downstream of the PL (RefSeq) reflects transcriptional initiation of a short Stra6 isoform (Stra6S) expressed in mouse ES cells in response to RA.
We designed primers specific for the long and the putative short isoform, respectively, and evaluated the transcript levels of these two Stra6 isoforms in WT and RARγ−/− ES cells in response to RA treatment. The transcript levels of both isoforms were dramatically increased by RA treatment of WT cells, whereas no transcripts were detected in the RARγ−/− ES cells (Fig. 1C). As a positive control for the induction of Stra6 by RA, we employed pan-primers to evaluate the total Stra6 transcript levels (all isoforms) (Fig. 1C). To confirm the genotype of the RARγ knock-out ES cells, we evaluated the RARγ transcript levels in the WT and the RARγ−/− ES cells (Fig. 1C). The 36B4 transcript levels were used as a reference gene (Fig. 1C).
We next determined that the Stra6 short isoform was generated by a downstream promoter. We used 5′-RACE to identify transcriptional start sites (TSSs) of Stra6 transcription in WT ES cells before and after treatment with RA. We identified two TSSs ∼560 and ∼300 bp upstream of the Stra6(−)B primer binding site (Fig. 1C, right). The 5′-RACE PCR products are slightly larger than the expected sizes of the long and the short isoform of Stra6 (521 and 261 bp) due to the addition of a 5′-linker (41 bp). Thus, our data show that the two TSSs give rise to the long and a short isoforms of Stra6, both of which increased in WT but not in RARγ−/− ES cells after RA addition (Fig. 1C). We conclude that in WT ES cells, a long and a short Stra6 transcript are generated from two different promoters, the PL and the PS promoter, respectively (shown in Fig. 1B, red bars at the bottom). The levels of both transcripts increase upon RA treatment of WT ES cells (Fig. 1C). The Stra6 transcripts originating from the PL and the PS promoters are designated Stra6L (Stra6.1) and Stra6S (Stra6.5), respectively (Fig. 1D). Transcript levels of Stra6.2, Stra6.3, and Stra6.4 were below detection in ES cells (data not shown).
Transcriptional Activation Assays Reveal an RARE Proximal to the Promoter of the Stra6S Isoform
We next cloned putative promoter regions of Stra6 upstream of the luciferase reporter gene in order to identify the genomic sequences involved in the induction of Stra6 by RA (Fig. 1D). We performed transient transfection assays in WT ES cells and evaluated conserved regions proximal to PL (0.6 kb) and PS (1.7 and 1.0 kb) for RA-responsive transcriptional activity (1 μm RA, 24 h). In order to evaluate potential enhancer activity of PS, we included a construct in which PS is located downstream of the PL TSS, thereby modeling transcription of the long Stra6 isoform. We found that PS was induced to a greater extent than PL by RA (∼5-fold versus 2-fold, respectively), whereas PS failed to enhance the promoter activity of PL (1.3-fold) (Fig. 1D). These transient transfection assays confirmed the presence of an unannotated RA-inducible promoter (PS), which is located ∼12 kb downstream of the RefSeq promoter (PL) identified in the NCBI database (Fig. 1, B and D).
We next cloned various regions proximal to the Stra6 PS upstream of the reporter luciferase gene and performed transient transfection assays in WT and RARγ−/− ES cells. The cells were cultured in either the absence or presence of RA for 8 or 24 h (Fig. 2). We utilized a variety of deletion constructs in the 1.7-kb region proximal to PS to demonstrate that the greatest increase in transcriptional activation after RA treatment of WT ES cells occurred when a small, 286-bp region was included (e.g. Stra6-0.9, Stra6-0.8, bracketed in Fig. 2C). The majority of the RA inducibility was lost when we deleted this region (Stra6-0.6, Stra6-0.5) (Fig. 2B). In the RARγ−/− ES cells, the Stra6-0.8 construct did not result in RA induction of the luciferase reporter (Fig. 2B), indicating that this RA-responsive region requires RARγ for transcriptional activity. We analyzed the DNA sequence of the RA-responsive region (Fig. 2C, bracketed) and identified a conserved RARE half-site, ATGACC (boldface type indicating conserved nucleotides), located in this intronic region (Fig. 2C, double-boxed sequence). When the RARE half-site was mutated, the RA-responsiveness of the Stra6-0.8 reporter construct was abrogated (Fig. 2B). The Stra6-m2 contains a triple mutation in which the ATGAC (double-boxed in Fig. 2C) is mutated to AACTC. We also tested another RARE half-site (CTGAA; boxed in Fig. 2C). Again, we introduced a triple mutation (CTGAA to CACTA), but this mutation did not affect the RA-responsiveness of the Stra6–0.8 reporter construct (Fig. 2B). Thus, the ATGAC half-site is responsible for the RA-responsiveness of the PS Stra6 promoter (Fig. 2A).
FIGURE 2.

Mapping the RARE of Stra6 using reporter assays. A, schematic representation of the Stra6 genomic region and reporter constructs. Promoters for the long and the novel short forms of Stra6 transcripts are designated as PL and PS, respectively. The arrow indicates the epigenetic “hotspot” observed in the heat map (Fig. 1). R, conserved RARE half-site. A gray bar below the schematic reporter construct marks the RA-responsive region shown in C. B, Stra6 reporter assays in WT and RARγ−/− ES cells upon RA treatment. Deletion analysis of the RA-responsive Stra6-1.7 construct is shown at the top. Point-mutated Stra6-0.8 constructs (m1 and m2), the PL construct, and Stra6-0.8 construct in RARγ−/− ES cells (boxed) are shown at the bottom. Stra6-m1 and Stra6-m2 indicate mutations in RARE half-sites (see details below). All luciferase assays were performed at least three times, starting with fresh cells. *, p < 0.05 relative to vehicle control. C, key elements of the Stra6 short isoform proximal to the essential region identified through the reporter assays (bracketed): the transcriptional start site (i.e. the 5′-end of the transcript) (>), the conserved RARE half-site with the inverted repeat shown in boldface type (double boxed, m2, TGA → ACT), the non-essential RARE half-site (boxed, m1, TGA → ACT), and the putative start codon (boldface type). Exon sequences are depicted in capital letters with gray shades indicating exonic regions present in both Stra6S and Stra6L. The ▵, splice acceptor site for splicing of the longer isoforms (in which case the upstream region of this exon is spliced out). ▴, splice acceptor site common for the short (6.5) and the longer (6.1) Stra6 isoforms. D, diagram of the screening strategy and sequences of CRISPR modified ES clones. The CRISPR targeting sequence is underlined, and the site of double-stranded breakage is indicated by a gray triangle. The genomic sequences of three clones are depicted (one biallelic and two monoallelic mutations). Note that only the mutated allele is shown. E, PCR screening and expression of Stra6 isoforms in ES cell lines with mutated RAREs. F, transcriptional induction of Stra6 in ES cell lines with mutated RAREs. The induction of Stra6 by RA was impaired in cell lines with monoallelic mutations and strongly impaired in the cell line with biallelic mutations in the Stra6 RARE (*, p < 0.05 relative to RA-treated WT cells). Stra6 transcript levels were normalized to the 36B4 reference gene. Error bars, S.E.
In summary, we found that a 0.8-kb region proximal to PS was sufficient to drive a large increase (∼13-fold) in luciferase reporter gene expression in response to RA (Fig. 2) and that this increase required RARγ. We further identified an RARE half-site, which, when mutated, abolished the RA-responsiveness of the reporter construct.
The Stra6 RARE Is Required for Induction of Endogenous Stra6 by RA in ES Cells
We next wanted to address the role of the endogenous RARE in RA-induced transcription of Stra6. We employed the CRISPR-Cas9 technology to introduce a targeted deletion of the Stra6 RARE (Fig. 2D) in WT ES cells. We evaluated independent clones for RA-responsive transcription of Stra6 and found that ablating the Stra6 RARE resulted in impaired induction of both Stra6S and Stra6L (Fig. 2E). The cell lines harboring monoallelic or biallelic deletion of the Stra6 RARE exhibited ∼45 and 95% reduction, respectively, in the total Stra6 transcript levels (all isoforms) relative to RA-stimulated WT ES cells (Fig. 2F).
The Stra6 Promoters, PS and PL, Show Differential Epigenetic Responses to RA in WT Versus RARγ−/− ES Cells
The consensus RARE element is composed of two direct repeats (RGKTSAN2/5RGKTSA). In contrast, the RARE half-site identified in the reporter assays is part of an inverted repeat, (gaaTGACCTctttAGATCAaca, half-sites in capital letters; see the legend to Fig. 2 for details). We therefore evaluated the RARγ and RXRα association with the Stra6 PS and PL regions by ChIP. We show that RARγ and RXRα associate with the PS promoter region in the WT cells but not in the RARγ−/− ES cells (Fig. 3, A and B). H3K27ac, generally a mark of enhancers/transcriptional activation (44, 45), occurred at higher levels at both the PS and PL regions following RA addition to the WT cells but not the RARγ−/− cells (Fig. 3C). The acetylation of H3K27 correlated with coactivator protein p300 association (Fig. 3D), which indicates a role for p300 in introducing this histone modification (46).
FIGURE 3.
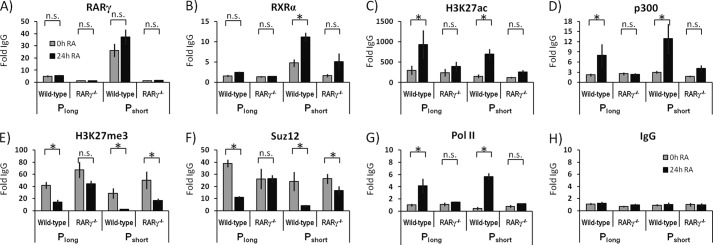
Epigenetic signatures of the Stra6 gene in WT and RARγ−/− ES cells upon RA treatment. Chromatin association with Stra6 PL and Stra6 PS regions in WT and RARγ−/− ES cells treated with vehicle control or RA. A, RARγ; B, RXRα; C, H3K27ac; D, p300; E, H3K27me3; F, Suz12; G, RNA polymerase II; H, IgG (negative control). All ChIP experiments were performed at least three times, starting with fresh cells (n ≥ 3). Gray bars, untreated cells; black bars, cells after 24 h of RA treatment. The IgG ChIP is a negative control. See Fig. 1 for the genomic locations of the ChIP regions. Note the variable x axes in different panels. *, p < 0.05; n.s., not statistically significant. Error bars, S.E.
We detected the H3K27me3 epigenetic mark in both WT and RARγ−/− ES cells; this mark showed a dramatic decrease upon RA addition at both the Stra6 PS and PL regions in WT, whereas higher levels were observed in the RARγ−/− cells relative to WT (Fig. 3E). The trimethylation of H3K27 correlated with the levels of the Polycomb group protein Suz12 (Fig. 3D), a key component of the Polycomb repressive complex 2 that trimethylates H3K27 (47).
Finally, we evaluated the levels of PolII at the Stra6 PS and PL regions. We demonstrate that at both the Stra6 PS and PL regions, the PolII association increased greatly in WT cells upon RA addition (Fig. 3G). This increase was not seen in the RARγ−/− ES cells (Fig. 3G). The IgG ChIP, a negative control, demonstrates that the background levels of nonspecific ChIP signals do not change in response to RA (Fig. 3H).
Thus, from our ChIP-chip and ChIP-qPCR assays, we conclude that among the evaluated histone marks, the greatest difference between Stra6 promoters PL and PS relates to the H3K4me3 mark. Specifically, the H3K4me3 levels increased at PL in an RA-dependent manner in both WT and RARγ−/− ES cells, whereas at PS, the H3K4me3 levels did not change in response to RA (Fig. 1B). Importantly, whereas we detected no RARγ and RXRα association with PL, both receptors showed strong association with the PS proximal promoter region (Fig. 3, A and B). The epigenetic signatures of Stra6 PS and PL are summarized in Table 3.
TABLE 3.
Transcription factors and epigenetic marks associated with Stra6 promoters in ES cells
The table summarizes the effects of RA on the epigenetic signatures of Stra6L and Stra6S promoters in WT and RARγ−/− ES cells (the data are presented in Figs. 1B and 4).
| Epigenetic mark | Stra6 PL | Stra6 PS | Approach |
|---|---|---|---|
| H3K4me3 | RA-dependent increase in WT and RARγ−/− | No detectable changes | ChIP-chip |
| H3K9/K14ac | RA-dependent increase in WT and to a lesser extent in RARγ−/− | RA-dependent increase in WT and to a lesser extent in RARγ−/− | ChIP-chip |
| p300 | RA dependent increase in WT but not in RARγ−/− | RA-dependent increase in WT but not in RARγ−/− | ChIP-qPCR |
| H3K27ac | RA-dependent increase in WT and to a lesser extent in RARγ−/− | RA-dependent increase in WT and to a lesser extent in RARγ−/− | ChIP-qPCR |
| H3K27me3 | RA-dependent depletion in WT but less in RARγ−/− | RA-dependent depletion in WT but not in RARγ−/− | ChIP-chip, ChIP-qPCR |
| Suz12 | RA-dependent depletion in WT but not in RARγ−/− | RA-dependent depletion in WT but less in RARγ−/− | ChIP-qPCR |
| RARγ | No detectable changes | RA-dependent increase in WT | ChIP-qPCR |
| RXRα | No detectable changes | RA-dependent increase in WT and to a lesser extent in RARγ−/− | ChIP-qPCR |
| PolII-CTD | RA-dependent increase in WT but not in RARγ−/− | RA-dependent increase in WT but not in RARγ−/− | ChIP-qPCR |
The Stra6 Short Isoform mRNA Is Highly Expressed in Brain and Is Reduced in Vitamin A Deficiency
We next asked if the short Stra6 isoform is expressed in animals and, if so, in what tissues. We determined the levels of Stra6S and Stra6L transcripts by RT-PCR in various tissues of WT mice (Fig. 4A). We detected Stra6L transcripts in the heart, intestine, and brain (Fig. 4A). Importantly, we also observed high levels of the Stra6S transcripts in the brain, uterus, kidney, and heart (Fig. 4A). We assessed the RARγ transcript levels in these tissues (Fig. 4A) and found that the Stra6S and Stra6L transcripts are present in a subset of the tissues that are positive for RARγ transcripts. This supports a role for RARγ in regulating the transcription of both Stra6 isoforms.
FIGURE 4.

Tissue-specific expression of Stra6 alternative splice isoforms. A, tissue-specific expression of Stra6L and Stra6S in WT mouse tissues. The brain exhibits the highest levels of both transcripts of all of the tissues we examined. RARγ transcripts are detected in several tissues, yet Stra6 is detected only in a subset of these. Transcript levels of the 36B4 reference gene are shown at the bottom. Representative data are presented. B, vitamin A deficiency is associated with a shift in Stra6 promoter usage. Transcript levels of the Stra6L isoform are increased in the kidneys of vitamin A-deficient mice. Transcript levels of the Stra6S isoform are decreased in the LRAT null animals on normal diet and below detection in vitamin A-deficient LRAT null animals. No change in RARγ transcript levels was observed in response to vitamin A deficiency. Transcript levels of LRAT (genotyping control on liver samples) and HPRT1 (loading control) are shown at the bottom. Representative data from eight animals are shown. The relative levels of the Stra6S and Stra6L isoforms are plotted to the right. C, transcript levels of both the Stra6L and Stra6S isoforms are decreased in the brains of vitamin A-deficient mice. The transcript levels of RARγ (and RARβ) did not change, whereas RARα transcript levels decreased slightly in response to vitamin A deficiency. Transcript levels of the 36B4 reference gene are included. Data from 12 animals are shown. D, Stra6L and Stra6S transcript levels are decreased in the brains of RARγ null mice. The knockout of RARγ was associated also with minor decreases in RARβ and RARα transcript levels in the brain. Transcript levels of the 36B4 reference gene are included. Data from six animals are shown. Error bars, S.E.
Vitamin A-deficient Mice Display Aberrant Stra6 Transcript Levels in Brain and Kidneys
We investigated whether the Stra6S and Stra6L transcript levels were differentially regulated in response to vitamin A deficiency. We tested the effects of vitamin A deficiency on Stra6 transcript levels by placing WT and LRAT−/− C57Bl/6 male and female mice on a vitamin A-deficient diet for 10 weeks and compared these mice with controls fed a normal (vitamin A-sufficient) chow diet. We previously showed that LRAT−/− mice become VAD more rapidly because they are unable to store retinol in the ester form in the liver and other tissues that act as storage depots for retinyl esters (6). We found greatly increased Stra6L transcript levels in kidneys of WT and LRAT−/− mice that were deprived of vitamin A (Fig. 4B) and decreased levels of Stra6S transcripts in the kidneys of LRAT−/− mice relative to the WT mice. This difference was enhanced by vitamin A deficiency, which almost abolished the Stra6S transcript levels in LRAT−/− mice (Fig. 4B). The RARγ transcript levels in the kidneys did not change in response to vitamin A deficiency (Fig. 4B), suggesting that the changes in Stra6 transcript levels are not caused by altered levels of RARγ. In vitamin A-sufficient conditions (WT-VAS) the Stra6S is the isoform predominantly expressed in the kidney, but with increasing severity of vitamin A depletion (either genetic, dietary, or combined), we observed a shift from Stra6S to Stra6L expression (Fig. 4B). We conclude that the Stra6 gene exhibits isoform-specific expression (e.g. differential promoter usage) in response to the levels of retinol and/or retinol metabolites in the kidney.
In the brains of vitamin A-deficient animals, the levels of both long and short Stra6 isoforms are decreased (Fig. 4C), whereas the RARβ and RARγ transcript levels did not change in response to vitamin A deficiency in WT and LRAT−/− mice (Fig. 4C). Also, we detected decreased levels of both the long and short Stra6 isoforms in the brains of RARγ null mice on a chow diet that contained vitamin A (Fig. 4D). Thus, RARγ is necessary for maintaining the normal levels of both Stra6S and Stra6L transcripts in the brain.
DISCUSSION
Stra6 is involved in numerous cell signaling pathways and mediates the uptake of retinol from holo-RBP (2). Thus, there is increasing interest in the regulation of Stra6 expression, the focus of this research. Murine Stra6 was originally cloned by Bouillet et al. (12), who determined that a genomic region 4.5 kb upstream of the RefSeq promoter does not recapitulate the RA inducible transcription of Stra6. We have mapped a Stra6 RARE to a region 12 kb downstream of the RefSeq promoter, and we demonstrate here that two different transcripts are generated from the Stra6 gene in cultured ES cells and in various murine tissues. In WT ES cells, RA induces transcription of both Stra6S and Stra6L through a single RARE (Figs. 1 and 2). Similarly, in the brain, Stra6L and Stra6S levels both decrease in response to vitamin A deprivation, but intriguingly, in the kidney, the Stra6S and Stra6L transcripts are regulated in an opposing fashion (Fig. 4).
Consistent with the increase in Stra6L and Stra6S transcript levels in RA-treated WT ES cells (Fig. 1), we observed RA-dependent increases in PolII levels at both Stra6L and Stra6S promoters (Fig. 3). In contrast, RARγ and RXRα showed strong association only with the Stra6S promoter (Fig. 3B). The absence of RAREs proximal to Stra6-PL is supported by our analysis of published ChIP-seq data (48) and is consistent with previous findings (12). We employed CRISPR-Cas9 technology to demonstrate that in ES cells, the Stra6 RARE, which is located proximal to the Stra6S promoter, is required for RA-responsive transcription of both Stra6S and Stra6L (Fig. 2E).
We focused on the epigenetic changes associated with the transcriptional activation of the Stra6 gene by RA. We found that both Stra6L and Stra6S promoters are transcriptionally activated by RA, as evident by an RA-dependent increase in histone acetylation (H3K9/14ac, H3K27ac, and p300) and Polycomb depletion (H3K27me3 and Suz12). Importantly, these marks exhibited no changes when RARγ−/− ES cells were treated with RA (Fig. 3), demonstrating the requirement for RARγ in RA-dependent Polycomb removal and histone acetylation of the Stra6 genomic region and in the transcriptional activation of Stra6 in ES cells.
In contrast to histone acetylation, which increased at both Stra6 PL and PS, we observed an RA-associated increase in H3K4me3 levels at the Stra6L promoter only; notably, the H3K4me3 levels increased even in RARγ−/− ES cells (Fig. 1B). The RA-associated increase in H3K4me3 at the Stra6L promoter is consequently independent of RARγ, and this modification alone is insufficient to induce transcriptional activation of Stra6. LRAT and Meis1 display similar transcriptional regulation; RARγ is required for the RA-dependent increase in H3K9/14ac marks, whereas the RA-induced increase in H3K4me3 takes place even in RARγ−/− ES cells (38). In contrast, the Cyp26a1 promoter exhibited lower levels of both H3K4me3 and H3K9/14ac marks in RA-treated RARγ−/− relative to WT ES cells (38). The H3K4me3 modification is deposited by mammalian homologs of the Drosophila Trithorax, termed myeloid/lymphoid or mixed-lineage leukemia (MLL) proteins, and in particular, the MLL2 complex plays an important role in differentiation of ES cells (49).
Our data from WT mice on VAS versus VAD diets show differences in the regulation of Stra6L and Stra6S transcripts in the kidneys (Fig. 4B). When retinol is supplied daily in the diet (Fig. 4B, VAS), only the Stra6S transcript is expressed. However, when retinol is not present in the diet for 10 weeks, the LRAT−/− mice become vitamin A-deficient (6), and only the Stra6L transcript is expressed in the kidneys (Fig. 4B, VAD, LRAT−/−). In WT mice that are on a VAD diet for 10 weeks and are only partially vitamin A-deficient (6), both the Stra6L and Stra6S transcripts are expressed (Fig. 4B, Wildtype, VAD). These data show that Stra6S mRNA is expressed in kidneys when retinol is present in the body but that as retinol stores are depleted in the livers of WT mice during the 10 weeks on the vitamin A-deficient diet, expression of the Stra6L transcript is activated in the kidneys although retinol is still present in the WT mice. Our data strongly suggest that when circulating retinol is abundant (from dietary uptake), Stra6S is the predominant isoform expressed in the murine kidneys. In contrast, as retinol stores are depleted as a result of dietary vitamin A deficiency, expression of the Stra6L isoform increases in the kidneys. The differential regulation of the two Stra6 transcript isoforms in the kidney in response to vitamin A deprivation points to physiologically distinct functions of the different isoforms in the regulation of retinoid homeostasis.
In the brain, unlike the kidney, both the Stra6L and Stra6S transcripts are reduced in both WT and LRAT−/− mice on a vitamin A-deficient diet (Fig. 4C). Thus, in the brain, we do not observe the major switch from the Stra6S to Stra6L transcripts during vitamin A deficiency that we detected in the kidneys (Fig. 4, B versus C). Furthermore, we also show that both the Stra6L and Stra6S transcript levels are reduced in the brains of RARγ−/− mice compared with the WT mice (Fig. 4D). This is in marked contrast to the elevated levels of Stra6 observed in RA-treated F9 RARα null cells (35) and in the testicular tubules of RARα null mice (12). Consequently, whereas RARγ plays a direct role in activating Stra6 expression, RARα may function to repress transcription of Stra6. Whether loss of RARα results in elevated levels of one or multiple Stra6 isoforms is not known. The presence of RARγ transcripts in each of the tissues where either Stra6S or Stra6L transcripts are present (uterus, kidney, heart, brain, and intestine) further supports the dependence of Stra6 expression on RARγ.
Stra6 expression is directly regulated by RA (Fig. 3A) and responds to vitamin A availability in vivo (Fig. 4). However, because we observed elevated levels of Stra6L in the kidneys of vitamin A-deficient animals, we conclude that Stra6L can be expressed in a manner independent of RA. This may reflect an additional layer of regulation whereby the kidneys are able to increase Stra6L levels when faced with vitamin A scarcity.
Our studies identify a shorter Stra6.5 isoform (Fig. 1), which encodes a putative amino-terminally truncated Stra6 protein (532 amino acids). In contrast, Stra6.1, Stra6.2, Stra6.3, and Stra6.4 isoforms all encode identical, larger Stra6 proteins (670 amino acids, NP_001155947.1). The putative Stra6S protein may share functional properties with the B-chain of human RBPR2, whose protein coding sequence is split onto two separate genes, one encoding the amino-terminal A-chain and one the carboxyl-terminal B-chain, respectively (4). It is interesting to note that RBP4 interactions and retinol uptake are impaired by insertions at residues 16, 84, and 133 in the amino-terminal region of the larger Stra6 protein (50), a region that is absent from Stra6S (and from the B-chain of RBP2). In contrast, mutations at residues 323, 357, and 548, shared by both Stra6 isoforms, completely abolished the function of the larger Stra6 protein (50). It is tempting to speculate that the carboxyl-terminal region of Stra6 has a structural role in RBP4 interaction, whereas the amino-terminal region of Stra6 may serve a regulatory function, possibly by distinguishing between apo- and holo-RBP4 (unliganded and liganded, respectively). Importantly, that distinction could be involved in regulating the direction of retinol transport (22). This model suggests that decreasing Stra6S levels in the kidneys may be a physiological response to vitamin A deprivation to reduce the loss of vitamin A to the urine. Further studies will be needed to characterize the functional differences between the Stra6 proteins encoded by the Stra6L and Stra6S transcripts, but our data indicate that the kidneys respond to vitamin A deficiency by differential Stra6 promoter usage, which may play a major role in the retention of retinol when vitamin A is scarce.
Acknowledgments
We thank Carlos Rodriguez for kidney cDNA, Steven Trasino for assistance in harvesting of mouse tissues and critically reading the manuscript, Tamara Weissman and Dan Stummer for editorial assistance, Dr. Pierre Chambon for the RARγ knock-out mice, and the Gudas laboratory for useful discussions.
This work was supported, in whole or in part, by National Institutes of Health, NCI, Grant R01 CA043796 (to L. J. G.). This work was also supported by Weill Cornell funds.
- RBP4
- serum retinol-binding protein (NCBI gene ID: 19662)
- 36B4
- ribosomal protein large P0 (NCBI gene ID: 11837)
- H3K27
- histone H3 Lys-27
- H3K27ac
- H3K27 acetylation
- H3K27me3
- H3K27 trimethylation
- H3K4me3
- histone H3 Lys-4 trimethylation
- H3K9/14ac
- histone H3Lys-9/14 acetylation
- CRBP-1
- cellular retinol-binding protein 1 (NCBI gene ID: 19659)
- ES
- embryonic stem
- HPRT1
- hypoxanthine guanine phosphoribosyl transferase 1 (NCBI gene ID: 15452)
- LRAT
- lecithin:retinol acyltransferase (NCBI gene ID: 79235)
- p300
- KAT3B (NCBI gene ID: 328572)
- PL
- PromoterLong
- PS
- PromoterShort
- RA
- retinoic acid
- RAR
- retinoic acid receptor
- RARE
- retinoic acid response element
- RARα
- retinoid acid receptor α (NCBI gene ID: 19401)
- RARβ
- retinoid acid receptor β (NCBI gene ID: 218772)
- RARγ
- retinoid acid receptor γ (NCBI gene ID: 5916)
- RBPR2
- serum retinol-binding protein receptor 2 (NCBI gene ID: 74152)
- RXRα
- retinoid X receptor α (NCBI gene ID: 20181)
- RefSeq
- NCBI reference sequence
- Ring1B
- ring finger protein 1B (NCBI gene ID: 19821)
- Stra6
- stimulated by retinoic acid 6 (NCBI gene ID: 20897)
- Suz12
- suppressor of zeste 12 homolog (NCBI gene ID: 52615)
- TSS
- transcriptional start site
- TTR
- thyroxine transthyretin (NCBI Gene ID: 22139)
- VAD
- vitamin A (retinol)-deficient diet
- VAS
- vitamin A (retinol)-sufficient diet
- MLL
- mixed-lineage leukemia
- PolII
- RNA polymerase II.
REFERENCES
- 1. O'Byrne S. M., Blaner W. S. (2013) Retinol and retinyl esters: biochemistry and physiology: Thematic Review Series: fat-soluble vitamins: vitamin A. J. Lipid Res. 54, 1731–1743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kawaguchi R., Yu J., Honda J., Hu J., Whitelegge J., Ping P., Wiita P., Bok D., Sun H. (2007) A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 315, 820–825 [DOI] [PubMed] [Google Scholar]
- 3. Zhong M., Kawaguchi R., Ter-Stepanian M., Kassai M., Sun H. (2013) Vitamin a transport and the transmembrane pore in the cell-surface receptor for plasma retinol binding protein. PLoS One 8, e73838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alapatt P., Guo F., Komanetsky S. M., Wang S., Cai J., Sargsyan A., Rodríguez Díaz E., Bacon B. T., Aryal P., Graham T. E. (2013) Liver retinol transporter and receptor for serum retinol-binding protein (RBP4). J. Biol. Chem. 288, 1250–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim Y. K., Wassef L., Hamberger L., Piantedosi R., Palczewski K., Blaner W. S., Quadro L. (2008) Retinyl ester formation by lecithin:retinol acyltransferase is a key regulator of retinoid homeostasis in mouse embryogenesis. J. Biol. Chem. 283, 5611–5621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu L., Gudas L. J. (2005) Disruption of the lecithin:retinol acyltransferase gene makes mice more susceptible to vitamin A deficiency. J. Biol. Chem. 280, 40226–40234 [DOI] [PubMed] [Google Scholar]
- 7. Ghyselinck N. B., Båvik C., Sapin V., Mark M., Bonnier D., Hindelang C., Dierich A., Nilsson C. B., Håkansson H., Sauvant P., Azaïs-Braesco V., Frasson M., Picaud S., Chambon P. (1999) Cellular retinol-binding protein I is essential for vitamin A homeostasis. EMBO J. 18, 4903–4914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Napoli J. L., Boerman M. H., Chai X., Zhai Y., Fiorella P. D. (1995) Enzymes and binding proteins affecting retinoic acid concentrations. J. Steroid Biochem. Mol. Biol. 53, 497–502 [DOI] [PubMed] [Google Scholar]
- 9. O'Byrne S. M., Wongsiriroj N., Libien J., Vogel S., Goldberg I. J., Baehr W., Palczewski K., Blaner W. S. (2005) Retinoid absorption and storage is impaired in mice lacking lecithin:retinol acyltransferase (LRAT). J. Biol. Chem. 280, 35647–35657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Amengual J., Golczak M., Palczewski K., von Lintig J. (2012) Lecithin:retinol acyl transferase is critical for cellular uptake of vitamin A from serum retinol binding protein. J. Biol. Chem. 287, 24216–24227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu L., Ross A. C. (2010) Acidic retinoids synergize with vitamin A to enhance retinol uptake and STRA6, LRAT, and CYP26B1 expression in neonatal lung. J. Lipid Res. 51, 378–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bouillet P., Sapin V., Chazaud C., Messaddeq N., Décimo D., Dollé P., Chambon P. (1997) Developmental expression pattern of Stra6, a retinoic acid-responsive gene encoding a new type of membrane protein. Mech. Dev. 63, 173–186 [DOI] [PubMed] [Google Scholar]
- 13. Pasutto F., Sticht H., Hammersen G., Gillessen-Kaesbach G., Fitzpatrick D. R., Nürnberg G., Brasch F., Schirmer-Zimmermann H., Tolmie J. L., Chitayat D., Houge G., Fernández-Martínez L., Keating S., Mortier G., Hennekam R. C., von der Wense A., Slavotinek A., Meinecke P., Bitoun P., Becker C., Nürnberg P., Reis A., Rauch A. (2007) Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am. J. Hum. Genet. 80, 550–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Skazik C., Amann P. M., Heise R., Marquardt Y., Czaja K., Kim A., Rühl R., Kurschat P., Merk H. F., Bickers D. R., Baron J. M. (2014) Downregulation of STRA6 expression in epidermal keratinocytes leads to hyperproliferation-associated differentiation in both in vitro and in vivo skin models. J. Invest. Dermatol. 134, 1579–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Berry D. C., Jin H., Majumdar A., Noy N. (2011) Signaling by vitamin A and retinol-binding protein regulates gene expression to inhibit insulin responses. Proc. Natl. Acad. Sci. U.S.A. 108, 4340–4345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen C. H., Hsieh T. J., Lin K. D., Lin H. Y., Lee M. Y., Hung W. W., Hsiao P. J., Shin S. J. (2012) Increased unbound retinol-binding protein 4 concentration induces apoptosis through receptor-mediated signaling. J. Biol. Chem. 287, 9694–9707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Berry D. C., O'Byrne S. M., Vreeland A. C., Blaner W. S., Noy N. (2012) Cross-talk between signalling and vitamin A transport by the retinol-binding protein receptor STRA6. Mol. Cell. Biol. 32, 3164–3175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carrera S., Cuadrado-Castano S., Samuel J., Jones G. D., Villar E., Lee S. W., Macip S. (2013) Stra6, a retinoic acid-responsive gene, participates in p53-induced apoptosis after DNA damage. Cell Death Differ. 20, 910–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Muenzner M., Tuvia N., Deutschmann C., Witte N., Tolkachov A., Valai A., Henze A., Sander L. E., Raila J., Schupp M. (2013) Retinol-binding protein 4 and its membrane receptor STRA6 control adipogenesis by regulating cellular retinoid homeostasis and retinoic acid receptor α activity. Mol. Cell. Biol. 33, 4068–4082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Golzio C., Martinovic-Bouriel J., Thomas S., Mougou-Zrelli S., Grattagliano-Bessieres B., Bonniere M., Delahaye S., Munnich A., Encha-Razavi F., Lyonnet S., Vekemans M., Attie-Bitach T., Etchevers H. C. (2007) Matthew-Wood syndrome is caused by truncating mutations in the retinol-binding protein receptor gene STRA6. Am. J. Hum. Genet. 80, 1179–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chassaing N., Golzio C., Odent S., Lequeux L., Vigouroux A., Martinovic-Bouriel J., Tiziano F. D., Masini L., Piro F., Maragliano G., Delezoide A. L., Attié-Bitach T., Manouvrier-Hanu S., Etchevers H. C., Calvas P. (2009) Phenotypic spectrum of STRA6 mutations: from Matthew-Wood syndrome to non-lethal anophthalmia. Hum. Mutat. 30, E673–E681 [DOI] [PubMed] [Google Scholar]
- 22. Kawaguchi R., Zhong M., Kassai M., Ter-Stepanian M., Sun H. (2013) Differential and isomer-specific modulation of vitamin A transport and the catalytic activities of the RBP receptor by retinoids. J. Membr. Biol. 246, 647–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kawaguchi R., Yu J., Wiita P., Honda J., Sun H. (2008) An essential ligand-binding domain in the membrane receptor for retinol-binding protein revealed by large-scale mutagenesis and a human polymorphism. J. Biol. Chem. 283, 15160–15168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chassaing N., Ragge N., Kariminejad A., Buffet A., Ghaderi-Sohi S., Martinovic J., Calvas P. (2013) Mutation analysis of the STRA6 gene in isolated and non-isolated anophthalmia/microphthalmia. Clin. Genet. 83, 244–250 [DOI] [PubMed] [Google Scholar]
- 25. Ruiz A., Mark M., Jacobs H., Klopfenstein M., Hu J., Lloyd M., Habib S., Tosha C., Radu R. A., Ghyselinck N. B., Nusinowitz S., Bok D. (2012) Retinoid content, visual responses, and ocular morphology are compromised in the retinas of mice lacking the retinol-binding protein receptor, STRA6. Invest. Ophthalmol. Vis. Sci. 53, 3027–3039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Berry D. C., Jacobs H., Marwarha G., Gely-Pernot A., O'Byrne S. M., DeSantis D., Klopfenstein M., Feret B., Dennefeld C., Blaner W. S., Croniger C. M., Mark M., Noy N., Ghyselinck N. B. (2013) The STRA6 receptor is essential for retinol-binding protein-induced insulin resistance but not for maintaining vitamin A homeostasis in tissues other than the eye. J. Biol. Chem. 288, 24528–24539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Amengual J., Zhang N., Kemerer M., Maeda T., Palczewski K., Von Lintig J. (2014) STRA6 is critical for cellular vitamin A uptake and homeostasis. Hum. Mol. Genet. 23, 5402–5417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zemany L., Kraus B. J., Norseen J., Saito T., Peroni O. D., Johnson R. L., Kahn B. B. (2014) Downregulation of STRA6 in adipocytes and adipose stromovascular fraction in obesity and effects of adipocyte-specific STRA6 knockdown in vivo. Mol. Cell. Biol. 34, 1170–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Norseen J., Hosooka T., Hammarstedt A., Yore M. M., Kant S., Aryal P., Kiernan U. A., Phillips D. A., Maruyama H., Kraus B. J., Usheva A., Davis R. J., Smith U., Kahn B. B. (2012) Retinol-binding protein 4 inhibits insulin signaling in adipocytes by inducing proinflammatory cytokines in macrophages through a c-Jun N-terminal kinase- and Toll-like receptor 4-dependent and retinol-independent mechanism. Mol. Cell. Biol. 32, 2010–2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Urvalek A., Laursen K. B., Gudas L. J. (2014) The roles of retinoic acid and retinoic acid receptors in inducing epigenetic changes. Subcell. Biochem. 70, 129–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gudas L. J. (2012) Emerging roles for retinoids in regeneration and differentiation in normal and disease states. Biochim. Biophys. Acta 1821, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mongan N. P., Gudas L. J. (2007) Diverse actions of retinoid receptors in cancer prevention and treatment. Differentiation 75, 853–870 [DOI] [PubMed] [Google Scholar]
- 33. Kashyap V., Gudas L. J., Brenet F., Funk P., Viale A., Scandura J. M. (2011) Epigenomic reorganization of the clustered Hox genes in embryonic stem cells induced by retinoic acid. J. Biol. Chem. 286, 3250–3260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Laursen K. B., Mongan N. P., Zhuang Y., Ng M. M., Benoit Y. D., Gudas L. J. (2013) Polycomb recruitment attenuates retinoic acid-induced transcription of the bivalent NR2F1 gene. Nucleic Acids Res. 41, 6430–6443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Taneja R., Bouillet P., Boylan J. F., Gaub M. P., Roy B., Gudas L. J., Chambon P. (1995) Reexpression of retinoic acid receptor (RAR) γ or overexpression of RAR α or RAR β in RAR γ-null F9 cells reveals a partial functional redundancy between the three RAR types. Proc. Natl. Acad. Sci. U.S.A. 92, 7854–7858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boylan J. F., Lohnes D., Taneja R., Chambon P., Gudas L. J. (1993) Loss of retinoic acid receptor γ function in F9 cells by gene disruption results in aberrant Hoxa-1 expression and differentiation upon retinoic acid treatment. Proc. Natl. Acad. Sci. U.S.A. 90, 9601–9605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Faria T. N., Mendelsohn C., Chambon P., Gudas L. J. (1999) The targeted disruption of both alleles of RARβ(2) in F9 cells results in the loss of retinoic acid-associated growth arrest. J. Biol. Chem. 274, 26783–26788 [DOI] [PubMed] [Google Scholar]
- 38. Kashyap V., Laursen K. B., Brenet F., Viale A. J., Scandura J. M., Gudas L. J. (2013) RARγ is essential for retinoic acid induced chromatin remodeling and transcriptional activation in embryonic stem cells. J. Cell Sci. 126, 999–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., Zhang F. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gillespie R. F., Gudas L. J. (2007) Retinoid regulated association of transcriptional co-regulators and the Polycomb group protein SUZ12 with the retinoic acid response elements of Hoxa1, RARβ(2), and Cyp26A1 in F9 embryonal carcinoma cells. J. Mol. Biol. 372, 298–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kashyap V., Gudas L. J. (2010) Epigenetic regulatory mechanisms distinguish retinoic acid-mediated transcriptional responses in stem cells and fibroblasts. J. Biol. Chem. 285, 14534–14548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Laursen K. B., Wong P. M., Gudas L. J. (2012) Epigenetic regulation by RARα maintains ligand-independent transcriptional activity. Nucleic Acids Res. 40, 102–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Barski A., Cuddapah S., Cui K., Roh T. Y., Schones D. E., Wang Z., Wei G., Chepelev I., Zhao K. (2007) High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 [DOI] [PubMed] [Google Scholar]
- 44. Verdone L., Agricola E., Caserta M., Di Mauro E. (2006) Histone acetylation in gene regulation. Brief Funct. Genomic. Proteomic. 5, 209–221 [DOI] [PubMed] [Google Scholar]
- 45. Guillemette B., Drogaris P., Lin H. H., Armstrong H., Hiragami-Hamada K., Imhof A., Bonneil E., Thibault P., Verreault A., Festenstein R. J. (2011) H3 lysine 4 is acetylated at active gene promoters and is regulated by H3 lysine 4 methylation. PLoS Genet. 7, e1001354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Holmqvist P. H., Mannervik M. (2013) Genomic occupancy of the transcriptional co-activators p300 and CBP. Transcription 4, 18–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pasini D., Bracken A. P., Jensen M. R., Lazzerini Denchi E., Helin K. (2004) Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 23, 4061–4071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mendoza-Parra M. A., Walia M., Sankar M., Gronemeyer H. (2011) Dissecting the retinoid-induced differentiation of F9 embryonal stem cells by integrative genomics. Mol. Syst. Biol. 7, 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Denissov S., Hofemeister H., Marks H., Kranz A., Ciotta G., Singh S., Anastassiadis K., Stunnenberg H. G., Stewart A. F. (2014) Mll2 is required for H3K4 trimethylation on bivalent promoters in embryonic stem cells, whereas Mll1 is redundant. Development 141, 526–537 [DOI] [PubMed] [Google Scholar]
- 50. Kawaguchi R., Yu J., Wiita P., Ter-Stepanian M., Sun H. (2008) Mapping the membrane topology and extracellular ligand binding domains of the retinol binding protein receptor. Biochemistry 47, 5387–5395 [DOI] [PMC free article] [PubMed] [Google Scholar]