

Background: Hyperinsulinemia and increased blood levels of free fatty acids (FFA) trigger non-alcoholic steatohepatitis (NASH).
Results: Insulin induces EGFR-activation and proliferation. In presence of FFA, however, insulin triggers apoptosis as EGFR triggers CD95-activation JNK-dependently.
Conclusion: Insulin-induced EGFR-activation triggers proliferation, but shifts to CD95-dependent apoptosis when a JNK-signal is provided by FFA.
Significance: The study provides new insights into the pathogenesis of NASH.
Keywords: apoptosis, c-Jun N-terminal kinase (JNK), epidermal growth factor receptor (EGFR), fatty acid, insulin, CD95
Abstract
Insulin is known to induce hepatocyte swelling, which triggers via integrins and c-Src kinase an activation of the epidermal growth factor receptor (EGFR) and subsequent cell proliferation (1). Free fatty acids (FFAs) are known to induce lipoapoptosis in liver cells in a c-Jun-NH2-terminal kinase (JNK)-dependent, but death receptor-independent way (2). As non-alcoholic steatohepatitis (NASH) is associated with hyperinsulinemia and increased FFA-blood levels, the interplay between insulin and FFA was studied with regard to hepatocyte proliferation and apoptosis in isolated rat and mouse hepatocytes. Saturated long chain FFAs induced apoptosis and JNK activation in primary rat hepatocytes, but did not activate the CD95 (Fas, APO-1) system, whereas insulin triggered EGFR activation and hepatocyte proliferation. Coadministration of insulin and FFAs, however, abolished hepatocyte proliferation and triggered CD95-dependent apoptosis due to a JNK-dependent association of the activated EGFR with CD95, subsequent CD95 tyrosine phosphorylation and formation of the death-inducing signaling complex (DISC). JNK inhibition restored the proliferative insulin effect in presence of FFAs and prevented EGFR/CD95 association, CD95 tyrosine phosphorylation and DISC formation. Likewise, in presence of FFAs insulin increased apoptosis in hepatocytes from wild type but not from Alb-Cre-FASfl/fl mice, which lack functional CD95. It is concluded that FFAs can shift insulin-induced hepatocyte proliferation toward hepatocyte apoptosis by triggering a JNK signal, which allows activated EGFR to associate with CD95 and to trigger CD95-dependent apoptosis. Such phenomena may contribute to the pathogenesis of NASH.
Introduction
Apart from its metabolic effects, insulin stimulates cell proliferation in the liver and in other organs (for a review, see Refs. 3–7). As shown recently in rat hepatocytes, insulin leads to an activating epidermal growth factor receptor (EGFR)2 tyrosine phosphorylation at positions Tyr845 and Tyr1173, which is triggered by insulin-induced hepatocyte swelling and activation of a recently described swelling-induced osmosignaling pathway, which involves osmosensing by β1 integrins and c-Src activation (1, 8–10).
Free fatty acids (FFAs) are known to induce lipoapoptosis in hepatocytes (2). The molecular mechanisms leading to hepatocyte lipoapoptosis are not fully defined; however, recent studies suggest that FFAs induce lipoapoptosis in hepatocytes in a c-Jun NH2-terminal kinase (JNK)-dependent, but death receptor-independent way (2). JNKs have also been implicated in the pathogenesis of non-alcoholic steatohepatitis (NASH) in both, murine models (11, 12) and humans (13, 14). In NASH patients however, up-regulation of death receptors, such as TNF-related apoptosis-inducing ligand (TRAIL) receptors 1 and 2 (15, 16) and CD95 (Fas, APO-1), was also discussed to sensitize hepatocytes toward the extrinsic pathway of apoptosis (17).
JNKs also play a crucial role in CD95-dependent hepatocyte apoptosis triggered by CD95 ligand (CD95L), hydrophobic bile acids or hyperosmolarity (for a review, see Ref. 18). Here, proapoptotic stimuli induce the generation of reactive oxygen species (ROS) leading to both, a Yes kinase-mediated EGFR transactivation and JNK activation. The JNK signal then allows the activated EGFR to associate with CD95 and to tyrosine-phosphorylate CD95, which triggers CD95 oligomerization, translocation of the EGFR/CD95 complex to plasma membrane and formation of the death-inducing signaling complex (DISC) (for a review, see Ref. 18).
The aim of the present study was to elucidate, whether FFAs can shift insulin-induced hepatocyte proliferation to hepatocyte apoptosis in a CD95-dependent manner. This may be relevant in the pathogenesis of non-alcoholic fatty liver disease (NAFLD), such as NASH, which is frequently accompanied by peripheral insulin resistance and elevated serum concentrations of FFA (19–23), and hyperinsulinemia (24–26). The present study shows that the FFA-induced JNK-activation directs insulin-induced EGFR activation from proliferation to EGFR/CD95-association, EGFR-mediated CD95-tyrosine phosphorylation and subsequent activation of the CD95-mediated apoptotic machinery. Thus, FFA-induced JNK-activation provides a switch from insulin-induced hepatocyte proliferation toward CD95-mediated apoptosis.
EXPERIMENTAL PROCEDURES
Materials
The materials used were purchased as follows: William's Medium E from Biochrom (Berlin, Germany), penicillin/streptomycin, and FBS (fetal bovine serum) from Invitrogen GmbH (Darmstadt, Germany); soluble CD95L was obtained from Enzo Life Sciences (Lörrach, Germany) and was always employed with a 10-fold amount of enhancer protein as provided by the supplier. SP600125 was purchased from Tocris/Biozol (Eching, Germany). Collagenase, insulin, FFA-free low endotoxin BSA (bovine serum albumin), caprylate, palmitate, stearate, oleate, and linoleate were from Sigma Aldrich (Munich, Germany). 5-(and 6)-Chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA), ProLong® Gold Antifade reagent with DAPI (4′, 6-diamidino-2-phenylindole, dihydrochloride) was from Invitrogen GmbH (Darmstadt, Germany), horseradish peroxidase-conjugated anti-mouse IgG and anti-rabbit IgG from Bio-Rad Laboratories (Munich, Germany) and Dako (Hamburg, Germany). The antibodies used were purchased as follows: antibodies recognizing p38 mitogen-activated protein kinase (p38MAPK), caspase 8, Fas-associated death domain (FADD), CD95, and EGFR (immunoprecipitation) were from Santa Cruz Biotechnology (Heidelberg, Germany), extracellular-regulated kinase (Erk)-1/-2, EGFR, and phospho-tyrosine from Merck-Millipore (Darmstadt, Germany), phospho-JNK-1/-2 from Invitrogen GmbH (Darmstadt, Germany), JNK-1/-2 from BD Bioscience (Heidelberg, Germany), phospho-Erk-1/-2, phospho-p38MAPK, phospho-c-Jun (Ser-63), and c-Jun from Cell Signaling Technology, Inc. (Danvers). All other chemicals were from Merck-Millipore (Darmstadt, Germany), at the highest quality available.
Preparation and Culture of Primary Rat and Murine Hepatocytes
Hepatocytes were isolated from livers of male Wistar rats (160–180 g), Alb-Cre control mice (wild type (wt), 8–10 weeks; on C57BL/6 background), or Alb-Cre-FASfl/fl mice ((8–10 weeks, mice with death domain-encoding exon 9 of Fas, flanked by loxP sites, were generated to allow conditional inactivation of Fas through cell type-specific expression of Cre recombinase) C57BL/6-Fastm1Cgn/J, The Jackson Laboratory) by a collagenase perfusion technique in an adapted version as described previously (27). Animals were fed ad libitum with a standard diet. Aliquots of murine hepatocytes were plated on collagen-coated culture plates (BD Falcon, Heidelberg, Germany) and maintained in William‘s Medium E (Biochrom, Berlin, Germany), supplemented with 5 mmol/liter glutamine, 100 units/ml penicillin, 0.1 mg/ml streptomycin, 100 nmol/liter dexamethasone, and 10% FBS. Cells were incubated in a humidified atmosphere of 5% CO2 and 95% air at 37 °C. Aliquots of rat hepatocytes were plated on collagen-coated culture plates and maintained in bicarbonate-buffered Krebs-Henseleit medium (115 mmol/liter NaCl, 25 mmol/liter NaHCO3, 5.9 mmol/liter KCl, 1.18 mmol/liter MgCl2, 1.23 mmol/liter NaH2PO4, 1.2 mmol/liter Na2SO4, 1.25 mmol/liter CaCl2), supplemented with 6 mmol/liter glucose in a humidified atmosphere of 5% CO2 and 95% air at 37 °C. After 2 h, the medium was removed, and the cells were washed twice. Subsequently the culture was continued for 24 h in William‘s Medium E, supplemented with 2 mmol/liter glutamine, 100 nmol/liter insulin, 100 units/ml penicillin, 0.1 mg/ml streptomycin, 100 nmol/liter dexamethasone, and 5% FBS. After 24 h experimental treatments were performed using William‘s Medium E that contained 2 mmol/liter glutamine, 100 nmol/liter dexamethasone, and 1% FFA-free BSA. The viability of hepatocytes was more than 95% as assessed by trypan blue exclusion. The experiments were approved by the responsible local authorities.
Treatment of Hepatocytes with FFAs
Caprylate (C8), palmitate (C16), stearate (C18:0), oleate (C18:1), and linoleate (C18:2) were dissolved each in EtOH at 50 mmol/liter and 200 mmol/liter. The final working solutions were prepared by diluting stock solutions (1:1000) in culture medium supplemented with FFA-free BSA. Each experimental condition and each control contained 1% FFA-free BSA and 0.1% EtOH. Experimental treatment of the cells did not change pH of cell culture medium.
Immunoblot Analysis
At the end of the incubation period, the medium was removed and the cells were immediately lysed at 4 °C by using a lysis buffer containing 20 mmol/liter Tris-HCl (pH 7.4), 140 mmol/liter NaCl, 10 mmol/liter NaF, 10 mmol/liter sodium pyrophosphate, 1% (v/v) Triton X-100, 1 mmol/liter EDTA, 1 mmol/liter EGTA, 1 mmol/liter sodium vanadate, 20 mmol/liter β-glycerophosphate, and protease inhibitor mixture. The lysates were kept on ice for 10 min and then centrifuged at 8000 rpm for 8 min at 4 °C, and aliquots of the supernatant were taken for protein determination using the Bio-Rad protein assay (Bio-Rad Laboratories, Munich, Germany). Equal amounts of protein were subjected to sodium dodecyl sulfate (SDS)/polyacrylamide gel electrophoresis, and transferred onto nitrocellulose membranes using a semidry transfer apparatus (GE Healthcare, Freiburg, Germany). Membranes were blocked for 30 min in 5% (w/v) BSA containing 20 mmol/liter Tris (pH 7.5), 150 mmol/liter NaCl, and 0.1% Tween 20 (TBS-T) and exposed to primary antibodies overnight at 4 °C. After washing with TBS-T and incubation at room temperature for 2 h with horseradish peroxidase-coupled anti-mouse or anti-rabbit IgG antibody, respectively (all diluted 1:10 000), the immunoblots were washed extensively, and bands were visualized using the FluorChem E detection instrument from ProteinSimple (Santa Clara, CA). Semi-quantitative evaluation was carried out by densitometry using the Alpha View image acquisition and analysis software from ProteinSimple. Protein phosphorylation is given as the ratio of detected phospho-protein/total protein.
Immunoprecipitation
Liver samples were harvested in lysis buffer containing 136 mmol/liter NaCl, 20 mmol/liter Tris-HCl, 10% (v/v) glycerol, 2 mmol/liter EDTA, 50 mmol/liter β-glycerophosphate, 20 mmol/liter sodium pyrophosphate, 0.2 mmol/liter Pefablock, 5 mg/liter aprotinin, 5 mg/liter leupeptin, 4 mmol/liter benzamidine, 1 mmol/liter sodium vanadate, supplemented with 1% (v/v) Triton X-100. The protein amount was determined as described above. Samples containing equal protein amounts were incubated for 2 h at 4 °C with a polyclonal rabbit anti-CD95 or rabbit anti-EGFR antibody to immunoprecipitate CD95 or EGFR, respectively. Then protein A-/G-agarose (Santa Cruz Biotechnology, Heidelberg, Germany) was added and incubated at 4 °C overnight. Immunoprecipitates were washed three times with lysis buffer supplemented with 0.1% (v/v) Triton X-100 and then transferred to Western blot analysis as described above. The anti-phospho-tyrosine antibody was used to detect activating phosphorylation of CD95 or EGFR in the respective immunoprecipitates. Caspase 8 and FADD antibodies were used to detect association to CD95.
Immunofluorescence Staining
To detect EGFR and CD95 translocation, isolated hepatocytes were cultured for 24 h on collagen-coated glass coverslips (Ø 12 mm) in 24-well culture plates. After treatment, cells were fixed using paraformaldehyde (4% (w/v), 20 min, 4 °C), permeabilized using Triton X-100 (0.1% (v/v), 2 min, 4 °C) and blocked with FBS (5% (w/v), 30 min, room temperature). Then cells were exposed to a mouse anti-EGFR antibody and a rabbit anti-CD95 antibody (1:100 in PBS, overnight, 4 °C), washed off, and stained with an anti-mouse-FITC and an anti-rabbit Cy3-conjugated antibody (1:500 in PBS, 2 h, room temperature). Following immunofluorescence staining, samples were covered with ProLong® Gold Antifade reagent with DAPI and EGFR, respectively CD95 localization was visualized by confocal laser scanning microscopy using LSM510 META (Zeiss, Oberkochen, Germany).
Detection of ROS
Hepatocytes were seeded on collagen-coated 6-well culture plates (BD Falcon, Heidelberg, Germany) and cultured for 24 h. Cells were incubated with PBS containing 5 μmol/liter CM-H2DCFDA for 30 min at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. To detect ROS generation, CM-H2DCFDA-loaded cells were supplemented again with culture medium and then exposed to FFAs for the indicated time period. Thereafter cells were washed briefly using ice-cold PBS, and cells were lysed in 0.1% Triton X-100 (v/v) dissolved in aqua bidest. Lysates were centrifuged immediately (10 000 × g, 4 °C, 1 min), and fluorescence of the supernatant was measured at 515–565 nm using a luminescence spectrometer LS-5B (PerkinElmer Life Sciences, Rodgau, Germany) at a 488 nm excitation wavelength. Fluorescence intensity of controls was set to 1 and fluorescence found under the respective treatment is given relative to it.
Assessment of Hepatocyte Proliferation
Hepatocyte proliferation was measured using a colorimetric BrdU cell proliferation assay (5-bromo-2′-deoxyuridine-ELISA; Roche Applied Science, Mannheim, Germany). Therefore, primary rat hepatocytes were cultured on collagen-coated flat-bottomed 96-well microtiter plates for up to 24 h. The culture medium was removed and replaced by culture medium containing 1 μmol/liter BrdU for additional 48 h. For the respective samples, BrdU incorporation was determined according to the manufacturer's recommendations.
Assessment of Apoptosis
Apoptosis was detected using a terminal deoxynucleotidyl-transferase-mediated X-dUTP nick-end labeling (TUNEL) assay (In Situ Cell Death Detection Kit; Roche Applied Science, Mannheim, Germany). Therefore, primary rat hepatocytes were cultured on collagen-coated glass coverslips (Ø 12 mm) in 24-well culture plates for up to 18 h. The number of apoptotic cells was determined by counting the percentage of fluorescein-positive cells. At least 300 cells from three different cell preparations each were counted for each condition. Cells were visualized on an LSM510 META laser scanning microscope (Zeiss, Oberkochen, Germany).
Caspase 8 activity in primary hepatocytes was determined by an in vitro caspase assay (Invitrogen GmbH, Darmstadt, Germany) according to the manufacturer's recommendations.
Statistical Analysis
Results from at least three independent experiments are expressed as mean values ± S.E. n refers to the number of independent experiments. For each experimental treatment and time point analyzed, a separate control experiment was carried out. Differences between experimental groups were analyzed by Student's t test or one-way analysis of variance following Dunnett's multiple comparison post hoc test where appropriate (GraphPad Prism; La Jolla, CA; Microsoft Excel for Windows, Redmond, WA). p < 0.05 was considered statistically significant.
RESULTS
Effect of Insulin and FFAs on Proliferation and Apoptosis
To analyze the proliferative and apoptotic effects of insulin and FFAs on primary hepatocytes, BrdU-uptake measurements and TUNEL staining were performed. In line with previous data (1), insulin (100 nmol/liter) led within 48 h to a 2.4 ± 0.2-fold (n = 6) increase of BrdU-uptake suggestive for hepatocyte proliferation (Fig. 1A), but had no effect on hepatocyte apoptosis as determined by TUNEL assays (numbers of TUNEL-positive cells after 18 h were 2.3% ± 0.6% (n = 6) versus 2.5% ± 0.4% (n = 6) in insulin-treated and control cells, respectively) (Fig. 1B). In contrast to insulin, the saturated FFAs caprylate, palmitate, and stearate (50 μmol/liter each) had no effect on hepatocyte proliferation (caprylate 0.9 ± 0.1-fold of control (n = 6); palmitate 0.8 ± 0.1-fold of control (n = 6); stearate 0.7 ± 0.1-fold of control (n = 3)) (Fig. 1, A and C), but the number of apoptotic cells measured by TUNEL staining increased within 18 h (caprylate 7.7% ± 1.5% (n = 6); palmitate 23.3% ± 4.1% (n = 6); stearate 32.0% ± 10.5% (n = 3)) (Fig. 1, B and D). The unsaturated FFAs oleate and linoleate however had no significant effect on apoptosis induction (oleate 4.6% ± 3.8%; linoleate 2.0% ± 1.8% (n = 3)) (Fig. 1D). When hepatocytes were treated with a combination of insulin and saturated FFAs, the insulin-induced proliferation was completely abolished (Fig. 1A), whereas the number of apoptotic cells increased to 21.2% ± 3.1% by caprylate plus insulin (n = 6), 41.9% ± 5.1% by palmitate plus insulin (n = 6) and 47.1% ± 10.3% by stearate plus insulin (n = 3) (Fig. 1, B and D). In contrast, the combination of insulin and unsaturated FFA induced no enhancement of apoptosis (Fig. 1D); however proliferation increased compared with treatment with the unsaturated FFAs alone (oleate 1.0 ± 0.1-fold of control; oleate plus insulin 1.4 ± 0.1-fold of control; linoleate 1.1 ± 0.1-fold of control; linoleate plus insulin 1.5 ± 0.3-fold of control (each n = 3)) (Fig. 1C). Thus, the proliferative effect of insulin was preserved, but blunted in presence of unsaturated FFAs.
FIGURE 1.

Effects of insulin and FFAs on proliferation and apoptosis. A and C, after a culture period of 24 h, culture medium was removed from rat liver hepatocytes and replaced by medium containing BrdU. Then, hepatocytes were treated with insulin (100 nmol/liter), caprylate, palmitate, stearate, oleate, or linoleate (50 μmol/liter, each) or the combination of insulin and FFA for 48 h and analyzed for BrdU incorporation. BrdU uptake by hepatocytes kept in control medium was set to 1. Statistical analyses of at least three independent experiments for each condition are shown. *, p < 0.05 denotes statistical significance compared with the unstimulated control. B and D, in another set of experiments, hepatocytes were cultured for 24 h and thereafter stimulated with insulin (100 nmol/liter), caprylate, palmitate, stearate, oleate, or linoleate (each 50 μmol/liter) or a combination of insulin and FFA for 18 h and the number of apoptotic cells was determined using TUNEL technique. Statistical analyses of at least three independent experiments for each condition are shown. *, p < 0.05 versus insulin plus FFA treatment.
In rat hepatocytes, insulin triggered phosphorylation of the EGFR at positions Tyr845 and Tyr1173 (Fig. 2, A and B), which is known to induce an activation of the EGFR-tyrosine kinase activity (28, 29). No phosphorylation at Tyr1045 was observed (Fig. 2A), indicating ligand-independent activation of the EGFR by insulin (30). Insulin-induced EGFR tyrosine phosphorylation was blunted by about 50% when FFAs were coadministered with insulin, however, EGFR activation was still significantly increased compared with the control. (Fig. 2, A and B). No activating EGFR phosphorylation was observed in response to caprylate or palmitate in the absence of insulin (Fig. 2, A and B).
FIGURE 2.
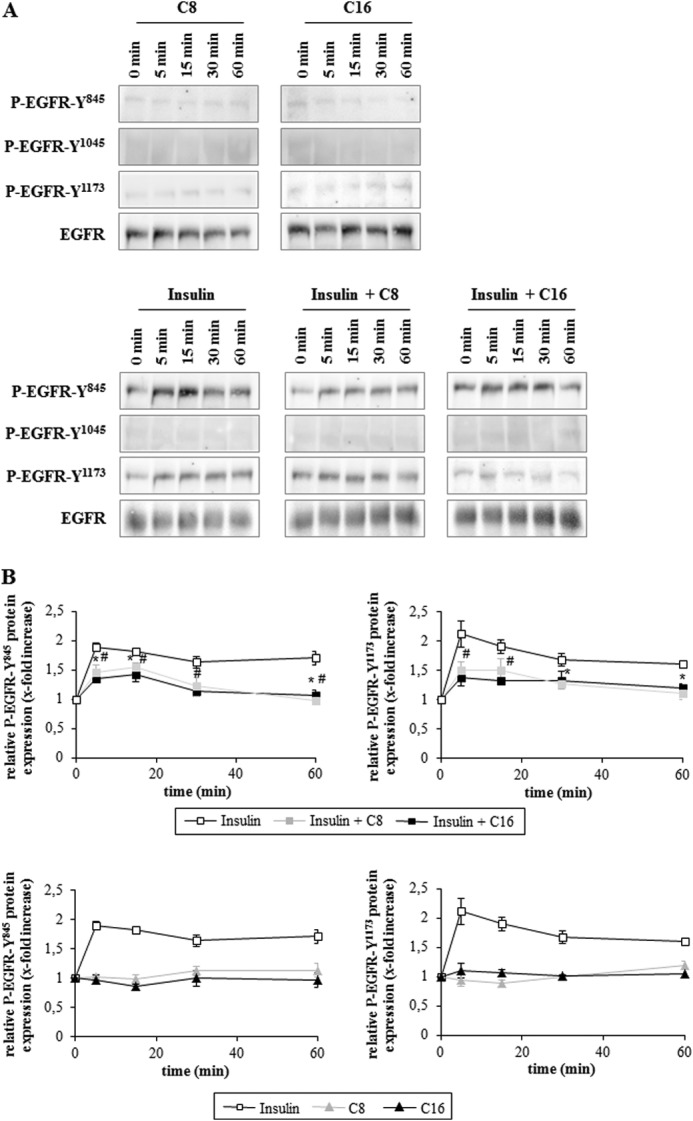
Inhibition of insulin-induced EGFR activation by FFAs. Rat hepatocytes were cultured for 24 h and thereafter stimulated with caprylate or palmitate (50 μmol/liter each), insulin (100 nmol/liter) or a combination of insulin and FFA for up to 60 min. Samples were taken at the indicated time points. Activating EGFR-tyrosine phosphorylation was analyzed by Western blotting using phospho-specific antibodies. Total EGFR served as respective loading control. A, representative immunoblots of three independent experiments. B, values from densitometric analyses of three independent experiments were normalized to the level of total EGFR and expressed as the mean-fold increase over control ± S.E. For the individual time points control was set to 1. Open squares, insulin; closed gray squares, insulin plus C8; closed black squares, insulin plus C16; closed gray triangle, C8; closed black triangle, C16. *, p < 0.05 statistical significance between insulin and insulin plus C8. #, p < 0.05 between insulin and insulin plus C16. EGFR activation at positions Tyr845 and Tyr1173 was significantly increased by insulin at each analyzed time point. Insulin plus FFAs significantly increased EGFR phosphorylation within the first 20 min at Tyr845 and after 5 min at Tyr1173.
Activation of Mitogen-activated Protein Kinases (MAPK) by FFAs
At concentrations of 200 μmol/liter or above, FFAs can induce so-called lipoapoptosis and JNKs were identified as important mediators of FFA-induced lipoapoptosis (2). While FFA concentrations of 200 μmol/liter significantly increased JNK-1/-2 phosphorylation after 60 min, FFAs at a concentration of 50 μmol/liter induced JNK-1/-2 activation already within the first 20 min of caprylate or palmitate addition (Fig. 3). Caprylate and palmitate (50 μmol/liter each) increased within 15 min carboxy-H2-DCFDA fluorescence by 1.6 ± 0.2-fold and 1.6 ± 0.1-fold (n = 3), respectively (data not shown), raising the possibility that FFA-induced oxidative stress may trigger activation of JNKs.
FIGURE 3.

Concentration-dependent JNK-1/-2 activation by FFAs. Rat hepatocytes were cultured for 24 h and thereafter treated with the indicated concentration of caprylate or palmitate for up to 60 min. Samples were taken at the indicated time points. Phosphorylation of JNK-1/-2 was analyzed by Western blot using specific antibodies and subsequent densitometric analysis. Total JNK-1/-2 served as respective loading control. Representative blots are shown. Closed black squares, 50 μmol/liter; closed gray squares, 200 μmol/liter. Phosphorylation at t = 0 was set to 1. Data represent the mean ± S.E. of >three independent experiments, *, p < 0.05 statistical significance compared with the unstimulated control.
Coadministration of insulin had no significant effect on FFA-induced JNK-1/-2 phosphorylation (Fig. 4, A and B) and no activation of JNK-1/-2 were observed in response to insulin alone (Fig. 4A). Compared with saturated FFAs, if at all only a weak JNK-1/-2 activation was induced by unsaturated (oleate and linoleate) FFAs (Fig. 4B).
FIGURE 4.

A and B, effects of insulin and FFA on JNK-1/-2 activation. Rat hepatocytes were cultured for 24 h and thereafter stimulated with insulin (100 nmol/liter), caprylate, palmitate, stearate, oleate, or linoleate (50 μmol/liter each) or a combination of insulin and FFA for up to 360 min. Samples were taken at the indicated time points. Phosphorylation of JNK-1/-2 was analyzed by Western blot using specific antibodies and subsequent densitometric analysis. Total JNK-1/-2 served as respective loading control. Closed black squares, FFA; closed gray squares, insulin plus FFA; open squares, insulin. Phosphorylation at t = 0 was set to 1. Data represent the mean ± S.E. of ≥three independent experiments, *, p < 0.05 statistical significance compared with the unstimulated control. No statistical significance between FFA alone and insulin plus FFA with regard to JNK activation.
Both, insulin and FFAs induced a transient activation of Erk-1/-2, which was not significantly affected when insulin and FFAs were added together (Fig. 5). There was also no statistically significant difference between insulin and insulin plus FFA with regard to p38MAPK phosphorylation.
FIGURE 5.

A and B, effects of insulin and FFA on Erk-1/-2 and p38MAPK activation. Rat hepatocytes were cultured for 24 h and thereafter stimulated with insulin (100 nmol/liter), caprylate, palmitate (50 μmol/liter each) or a combination of insulin and FFA for up to 60 min. Samples were taken at the indicated time points. Phosphorylation of Erk-1/-2 and p38MAPK was analyzed by Western blot using specific antibodies and subsequent densitometric analysis. Total Erk-1/-2 and p38MAPK served as respective loading control. Closed black squares, FFA; closed gray squares, insulin plus FFA; open squares, insulin. Phosphorylation at t = 0 was set to 1. Data represent the mean ± S.E. of ≥5 independent experiments, *, p < 0.05 statistical significance compared with the unstimulated control. No statistical significance between insulin, FFA, and insulin plus FFA with regard to Erk-1/-2 and p38MAPK activation.
A role of JNKs in mediating proapoptotic and antiproliferative signaling following coadministration of insulin and FFAs is also suggested by the finding that two different JNK inhibitors, i.e. SP600125 and L-JNKI-1 could largely restore the proliferative effect of insulin in presence of caprylate (insulin plus caprylate 0.9 ± 0.05-fold of control, in presence of SP600125 2.0 ± 0.4-fold of control; in presence of L-JNKI-1 1.8 ± 0.3-fold of control (n = 3 for each condition)) and palmitate (insulin plus palmitate 1.0 ± 0.1-fold of control, in presence of SP600125 1.7 ± 0.1-fold of control; in presence of l-JNKI-1 1.6 ± 0.1-fold of control (n = 3 for each condition)) (Fig. 6A). Both, SP600125 and L-JNKI-1 also counteracted the insulin-induced apoptosis in presence of FFAs (insulin plus caprylate 16.2% ± 1.9%, in presence of SP600125 10.8% ± 1.0%, or L-JNKI-1 9.3% ± 2.0% and insulin plus palmitate 35.4% ± 2.5%, in presence of SP600125 24.8% ± 1.7%, or L-JNKI-1 25.3% ± 2.3% (n = 3 for each condition)) (Fig. 6B).
FIGURE 6.

Effects of JNK-inhibition on proliferation and apoptosis in isolated rat hepatocytes. A, after a culture period of 24 h, culture medium was replaced by medium containing BrdU. Then, hepatocytes were treated with insulin (100 nmol/liter), FFAs (each 50 μmol/liter) or a combination of both for another 48 h and analyzed for BrdU incorporation. When indicated, cells were pretreated (30 min) with SP600125 (100 μmol/liter) or L-JNKI-1 (5 μmol/liter). BrdU uptake by hepatocytes kept in control medium was set to 1. Statistical analyses of at least three independent experiments for each condition are shown. *, p < 0.05 denotes statistical significance between insulin plus FFA and after administration of JNK inhibitors. B, in another set of experiments, hepatocytes were stimulated with insulin (100 nmol/liter), FFAs (50 μmol/liter), or a combination of both for 18 h and the number of apoptotic cells was determined using TUNEL technique. Where indicated, cells were pretreated (30 min) with SP600125 (100 μmol/liter) or L-JNKI-1 (5 μmol/liter). Statistical analyses of at least three independent experiments for each condition are shown. *, p < 0.05 versus insulin plus FFA treatment.
Apoptosis Induction Involves Activation of the CD95 System
To determine whether the proapoptotic effect of combined treatment with FFAs and insulin involves an activation of the CD95 system, experiments were performed in primary hepatocytes isolated from wild type and Alb-Cre-FASfl/fl mice, which lack functional CD95 (31). Similar to rat hepatocytes, insulin had no effect on hepatocyte apoptosis in wild type as well as Alb-Cre-FASfl/fl-mice and the number of TUNEL-positive cells in presence of insulin (wt 4.6% ± 0.6% (n = 3), Alb-Cre-FASfl/fl 3.6% ± 1.1% (n = 3)) did not significantly differ from those found in controls (wt 6.8% ± 1.5% versus Alb-Cre-FASfl/fl 3.1% ± 1.5% (each n = 3)) (Fig. 7). Likewise, caprylate (wt 20.1% ± 2.9% versus Alb-Cre-FASfl/fl 18.6% ± 2.1% (each n = 3)) and palmitate (wt 34.6% ± 2.9% versus Alb-Cre-FASfl/fl 31.1% ± 1.5% (each n = 3)) induced apoptotic cell death within 18 h to a similar extent in both mice strains. However, whereas coadministration of insulin and FFA roughly doubled the amount of apoptotic cells in wild type mice (caprylate plus insulin 43.3% ± 2.1%, palmitate plus insulin 62.3% ± 3.3% (each n = 3)), the proapoptotic effect of insulin in presence of FFA was no longer observed in mice lacking functional CD95 (Fig. 7), indicating an involvement of CD95 during apoptosis induction by coadministered insulin/FFAs. Therefore, the signaling events leading to CD95 activation and DISC formation were studied.
FIGURE 7.
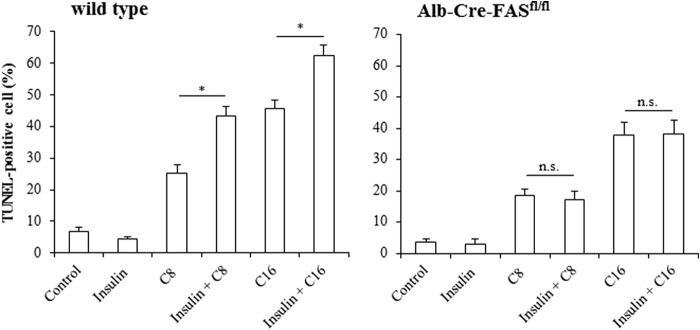
FFA-induced apoptosis in wild type and Alb-Cre-FASfl/fl-mice. Primary hepatocytes from wild type (Alb-Cre control mice on C57BL/6 background) and Alb-Cre-FASfl/fl-mice were cultured for 24 h and thereafter treated with insulin (100 nmol/liter), FFAs (each 50 μmol/liter) or a combination of both for 18 h and the number of apoptotic cells was determined using TUNEL technique. Insulin stimulates apoptosis in presence of FFA in wild type, but not in hepatocytes devoid of CD95. Statistical analyses of at least three independent experiments for each condition are shown. *, p < 0.05 versus FFA treatment alone. Not significant (n.s.), p > 0.05 versus FFA treatment alone.
CD95 Activation and Formation of DISC in Presence of FFAs and Insulin
Immunoprecipitation studies revealed that in response to insulin or FFA treatment alone, no activation of the CD95 system was detectable with respect to CD95/EGFR association, CD95 tyrosine phosphorylation and DISC formation, i.e. association of FADD and caspase 8 to the death receptor (Fig. 8). Only upon coadministration of FFAs with insulin CD95/EGFR association, CD95 tyrosine phosphorylation and DISC formation occurred as it was also observed in response to CD95L (Fig. 8). In line with this, caspase 8 activity increased in presence of insulin and FFA (Fig. 9) indicating an activation of the CD95 signaling pathway.
FIGURE 8.

FFA-induced JNK activation and insulin-induced EGFR activation can trigger CD95 activation and formation of the death-inducing signaling complex in rat hepatocytes. Rat hepatocytes were cultured for 24 h and subsequently stimulated with caprylate or palmitate (50 μmol/liter each), insulin (100 nmol/liter), or CD95L (100 ng/ml) for up to 60 min. Where indicated, cells were pretreated for 30 min with SP600125 (100 μmol/liter) or L-JNKI-1 (5 μmol/liter). Samples were taken at the time points indicated. EGFR and CD95 were immunoprecipitated and analyzed by Western blotting. Activating EGFR-tyrosine phosphorylation (P-EGFR-Y) and JNK-1/-2 activation were detected 15 min after stimulation, CD95-tyrosine phosphorylation (P-CD95-Y) and c-Jun phosphorylation after 30 min, caspase 8/CD95 and FADD/CD95 association (i.e. DISC formation) 60 min after addition of FFAs, insulin, or CD95L. Total EGFR, JNK-1/-2, c-Jun, and CD95 served as respective loading controls. Representative immunoblots of three independent experiments are depicted.
FIGURE 9.
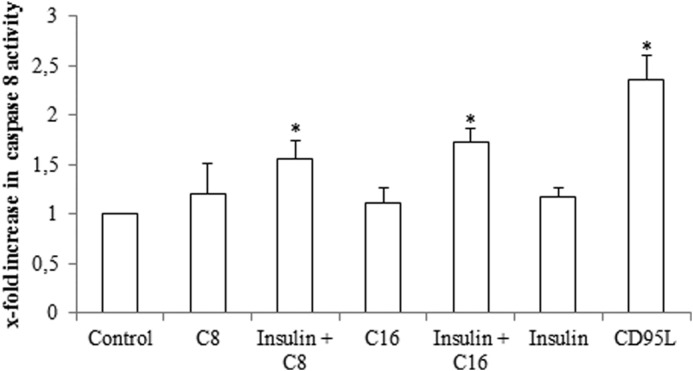
Effect of FFA on caspase 8 activation. Rat hepatocytes were cultured for 24 h and subsequently stimulated with caprylate or palmitate (50 μmol/liter, each), insulin (100 nmol/liter), or CD95L (100 ng/ml) for 180 min. Caspase 8 activity was determined as described in “Experimental Procedures” and are expressed relative to the activity found in unstimulated controls. Statistical analyses of three independent experiments for each condition are shown. *, p < 0.05 denotes statistical significance compared with the unstimulated control. Caspase 8 activation is only found when FFA and insulin were added together, but not when added separately.
To assess the role of JNK-1/-2 in triggering CD95/EGFR association, inhibitor studies were performed. Both, SP600125 and L-JNKI-1, inhibited not only c-Jun phosphorylation, but also largely blocked the association of EGFR with CD95, CD95-tyrosine phosphorylation as well as DISC formation in response to combined treatment with insulin and FFAs (Fig. 8).
The subcellular localization of EGFR and CD95 was studied by immunofluorescence analysis in primary rat hepatocytes. EGFR and CD95 colocalizing pixels were quantified by calculation of the weighted colocalization coefficient according to a preset threshold. As shown in Fig. 10, insulin and EGF, but not FFAs induced within 90 min an internalization of the EGFR. Coadministration of insulin and FFAs for 90 min prevented the insulin-induced EGFR internalization and even triggered an enhancement of EGFR and CD95 immunoreactivity at the plasma membrane (Figs. 10 and 11). Under these conditions the number of colocalizing EGFR- and CD95-positive pixels in the plasma membrane was significantly increased (insulin plus C8 59.2% ± 7.7%; insulin plus C16 63.2% ± 9.6% (each n = 3)) (Fig. 11) in line with the reported translocation of the EGFR/CD95 complex to the plasma membrane following CD95 activation (32). SP600125 and l-JNKI-1 largely inhibited the colocalization of EGFR and CD95 in presence of insulin and FFA (SP600125 23.0% ± 3.6% and L-JNKI-1 31.3% ± 2.0% in the presence of insulin plus C8; SP600125 27.7% ± 5.7% and L-JNKI-1 26.4% ± 4.9% in the presence of insulin plus C16 (n = 3 for each condition)). No significant CD95 translocation to the plasma membrane was observed after treatment with insulin or FFA alone (insulin 8.3% ± 0.9%; C8 13.3% ± 5.2%; C16 9.7% ± 3.0% compared with control 7.7% ± 1.8% (each n = 3)) (Fig. 11).
FIGURE 10.
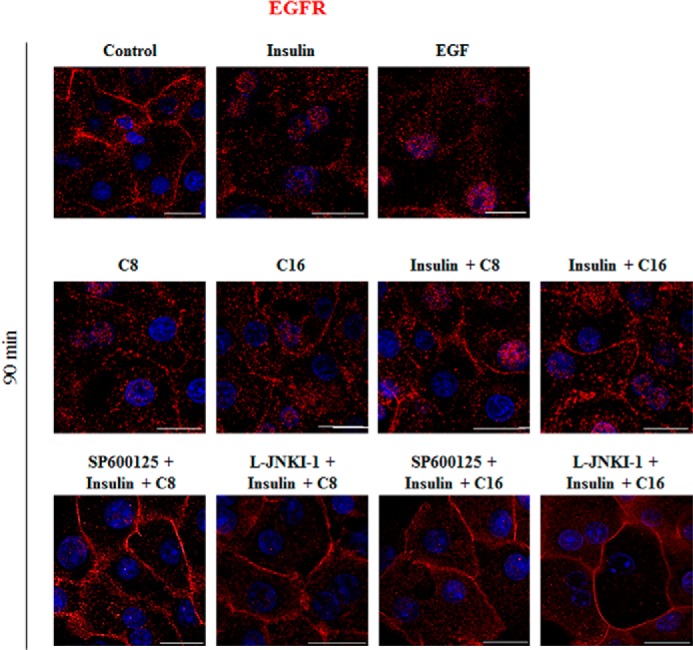
EGFR expression in primary parenchymal cells. Primary hepatocytes were cultured on glass coverslips for 24 h, stimulated for 90 min with the indicated agents and immunostained for EGFR expression. EGFR localization was visualized by confocal laser scanning microscopy. Insulin (100 nmol/liter) and EGF (10 ng/ml) induced internalization of the EGFR, whereas caprylate or palmitate (50 μmol/liter each) induced no internalization. The scale bar corresponds to 20 μm. Representative pictures of three independent experiments are depicted.
FIGURE 11.

Immunofluorescence staining of CD95 and EGFR in insulin- and FFA-treated primary hepatocytes. A, primary hepatocytes were cultured on glass coverslips for 24 h, stimulated with insulin (100 nmol/liter), caprylate, or palmitate (each 50 μmol/liter) or the combination of insulin and FFA for 90 min and immunostained for CD95 and EGFR expression. Localization of both receptors was visualized by confocal laser scanning microscopy. Greyscale images show colocalizing pixels of the red and green channel. The scale bar corresponds to 20 μm. Representative pictures of three independent experiments are depicted. B, colocalizing pixels were quantified by calculation of the weighted colocalization coefficient using LSM510 Meta 4.2 Software (Zeiss, Jena, Germany). *, p < 0.05 denotes statistical significance compared with the unstimulated control; #, p < 0.05 versus insulin plus FFA treatment.
DISCUSSION
The major finding of the current study is that JNK activation by saturated FFAs not only ameliorates insulin-induced EGFR activation, but also shifts insulin-induced EGFR activation and proliferation toward CD95 activation and apoptosis, as schematically depicted in Fig. 12. Unsaturated fatty acids, however, do not activate JNKs and do not induce apoptosis (Figs. 1D and 4B) (33). Saturated FFAs rapidly trigger generation of ROS, which may explain JNK activation. Mitochondria (34, 35) and NADPH oxidase were discussed as predominant sources of palmitate-induced ROS generation in the liver (36, 37). The FFA-induced JNK signal directs activated EGFR from proliferative signaling toward association with CD95 and proapoptotic signaling.
FIGURE 12.
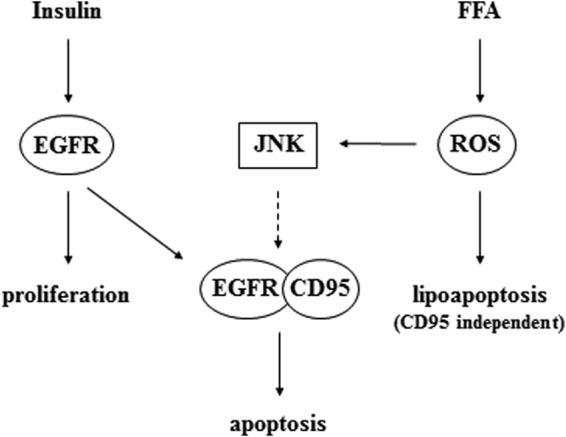
Schematic presentation of insulin and FFA effects on hepatocyte proliferation and apoptosis in hepatocytes. Insulin induces activation of the EGFR and proliferation, while FFAs induce ROS generation, which triggers death receptor-independent lipoapoptosis and JNK activation. When EGFR is simultaneously activated by insulin, this JNK signal allows for EGFR coupling to CD95 and initiation of CD95-dependent apoptotic cell death.
Insulin induces proliferation of hepatocytes by NKCC1- and Na+/H+ antiporter-driven cell swelling (38), which induces EGFR activation through an integrin- and c-Src kinase-dependent osmosensing/signaling pathway (1). In line with the literature, insulin caused an activating phosphorylation of the EGFR at positions Tyr845 and Tyr1173 (Fig. 2), thereby inducing hepatocyte proliferation (Fig. 1A). Activation of the insulin receptor leads to tyrosine phosphorylation of the insulin receptor substrate (IRS)-1, thereby triggering downstream signaling pathways such as the phosphatidylinositide 3-kinase/Akt pathway, which mediate metabolic actions of insulin (39) and insulin-induced hepatocyte swelling (40). JNK can induce insulin resistance through serine/threonine phosphorylation of the IRS-1 and −2 and thereby attenuate downstream insulin signaling (41–44). This is also reflected by the attenuation of insulin-induced EGFR activation in presence of FFA.
Saturated FFAs such as palmitate can induce lipoapoptosis in hepatocytes (2) and other cell types, such as pancreatic β cells (45) or cardiac myocytes (46). Induction of lipoapoptosis involves an endoplasmic reticulum (ER) stress response with activation of inositol-requiring protein-1 and protein kinase-like ER kinase. The resulting activation of JNK and induction of the transcription factor C/EBP-homologous protein, leads to the up-regulation of pro-apoptotic proteins such as Bim and PUMA. The following activation of Bax results in activation of effector caspases and death receptor-independent apoptosis (14, 47–49). Whereas recent studies suggest that FFAs induce lipoapoptosis in hepatocytes in a JNK-dependent but death receptor-independent way (2), up-regulation of TRAIL receptors 1 and 2 (15, 16) and CD95 were discussed to sensitize hepatocytes to the extrinsic pathway of apoptosis (17).
JNKs also play a crucial role in CD95-mediated hepatocyte apoptosis. Here, pro-apoptotic stimuli, trigger ROS generation via NADPH oxidase(s), which leads to a Yes kinase-mediated EGFR transactivation and JNK activation, mediating the association of the EGFR with CD95, which initiates CD95-dependent apoptotic cell death (for a review, see Ref. 50). A special situation is found in hepatic stellate cells (HSC), which represent a hepatic stem/progenitor cell compartment (51, 52). Here, CD95L triggers in quiescent HSC the activation of the EGFR, stimulates proliferation and induces CD95 tyrosine nitration (53), which leads to apoptosis resistance (54). However, in activated HSC no inhibitory CD95 tyrosine nitration occurs, whereas EGFR activation is preserved. If JNK activation is induced in these cells, EGFR couples to CD95 and the mitogenic signal is shifted to an apoptotic one. Thus JNK activation provides a switch between EGFR-mediated proliferation and CD95-mediated apoptosis (53, 55). A similar phenomenon is shown in the present study in rat hepatocytes: insulin-induced proliferation is shifted toward apoptosis when a JNK signal is provided by FFAs. The mechanism how JNKs promote the association between CD95 and EGFR is still unclear. JNK itself is not found in the protein complex (data not shown) therefore it is likely that JNK phosphorylates a yet unknown protein, such as the receptor interacting protein (RIP), a serine/threonine kinase, that may function as an adaptor protein to CD95, which is required for CD95/EGFR protein assembly (56). The identity of such a hypothetical adapter protein is not yet known and requires further investigation.
This insulin-dependent proapoptotic action of FFAs is almost completely mediated by CD95, as evidenced by our studies with Alb-Cre-FASfl/fl mice (Fig. 7), which lack functional CD95 (31). Due to this result it is unlikely that other FFA-triggered apoptosis pathways are influenced by insulin. In line with this, the palmitate-induced up-regulation and cell surface expression of TRAIL-death receptor R2 (DR5) was not affected by insulin (data not shown).
Taken together, the present study provides new insights into the pathogenesis of hepatocyte apoptosis in response to FFAs and insulin, which may be relevant for the pathogenesis of NASH. Here, insulin resistance, hyperinsulinemia and increased levels of circulating FFAs play an important role in the development and progression of this disease (for a review, see Ref. 57).
Acknowledgments
We thank Elisabeth Winands, Lisa Knopp, and Janina Thies for expert technical assistance.
This work was supported by Deutsche Forschungsgemeinschaft through Collaborative Research Center 974 (Düsseldorf) “Communication and System Relevance in Liver Injury and Regeneration.”
- EGFR
- epidermal growth factor receptor
- CD95
- CD95 receptor (Fas, APO-1)
- CD95L
- CD95 ligand, CM-H2DCFDA, 5-(and 6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate
- DISC
- death inducing signaling complex
- ER
- endoplasmic reticulum
- FADD
- Fas-associated death domain protein
- FFA
- free fatty acid
- HSC
- hepatic stellate cell
- IP
- immunoprecipitation
- IRS
- insulin receptor substrate
- NAFLD
- non-alcoholic fatty liver disease
- NASH
- non-alcoholic steatohepatitis
- ROS
- reactive oxygen species
- TRAIL
- TNF-related apoptosis-inducing ligand.
REFERENCES
- 1. Reinehr R., Sommerfeld A., Häussinger D. (2010) Insulin induces swelling-dependent activation of the epidermal growth factor receptor in rat liver. J. Biol. Chem. 285, 25904–25912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Malhi H., Bronk S. F., Werneburg N. W., Gores G. J. (2006) Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J. Biol. Chem. 281, 12093–12101 [DOI] [PubMed] [Google Scholar]
- 3. Diehl A. M., Rai R. M. (1996) Liver regeneration: Regulation of signal transduction during liver regeneration. FASEB J. 10, 215–227 [DOI] [PubMed] [Google Scholar]
- 4. Hunter T. (1997) Oncoprotein networks. Cell 88, 333–346 [DOI] [PubMed] [Google Scholar]
- 5. Avruch J. (1998) Insulin signal transduction through protein kinase cascades. Mol. Cell Biochem. 182, 31–48 [PubMed] [Google Scholar]
- 6. Nystrom F. H., Quon M. J. (1999) Insulin signaling: metabolic pathways and mechanisms for specificity. Cell Signal 11, 563–574 [DOI] [PubMed] [Google Scholar]
- 7. Taha C., Klip A. (1999) The insulin signaling pathway. J. Membr. Biol. 169, 1–12 [DOI] [PubMed] [Google Scholar]
- 8. Häussinger D., Kurz A. K., Wettstein M., Graf D., vom Dahl S., Schliess F. (2003) Involvement of integrins and Src in tauroursodeoxycholate-induced and swelling-induced choleresis. Gastroenterology 124, 1476–1487 [DOI] [PubMed] [Google Scholar]
- 9. Vom Dahl S., Schliess F., Reissmann R., Görg B., Weiergräber O., Kocalkova M., Dombrowski F., Häussinger D. (2003) Involvement of integrins in osmosensing and signaling toward autophagic proteolysis in rat liver. J. Biol. Chem. 278, 27088–27095 [DOI] [PubMed] [Google Scholar]
- 10. Schliess F., Reissmann R., Reinehr R., vom Dahl S., Häussinger D. (2004) Involvement of integrins and Src in insulin signaling toward autophagic proteolysis in rat liver. J. Biol. Chem. 279, 21294–21301 [DOI] [PubMed] [Google Scholar]
- 11. Schattenberg J. M., Singh R., Wang Y., Lefkowitch J. H., Rigoli R. M., Scherer P. E., Czaja M. J. (2006) JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology 43, 163–172 [DOI] [PubMed] [Google Scholar]
- 12. Wang Y., Ausman L. M., Russell R. M., Greenberg A. S., Wang X. D. (2008) Increased apoptosis in high-fat diet-induced nonalcoholic steatohepatitis in rats is associated with c-Jun NH2-terminal kinase activation and elevated proapoptotic Bax. J. Nutr. 138, 1866–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Puri P., Mirshahi F., Cheung O., Natarajan R., Maher J. W., Kellum J. M., Sanyal A. J. (2008) Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 134, 568–576 [DOI] [PubMed] [Google Scholar]
- 14. Cazanave S. C., Mott J. L., Elmi N. A., Bronk S. F., Werneburg N. W., Akazawa Y., Kahraman A., Garrison S. P., Zambetti G. P., Charlton M. R., Gores G. J. (2009) JNK1-dependent PUMA expression contributes to hepatocyte lipoapoptosis. J. Biol. Chem. 284, 26591–26602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Malhi H., Barreyro F. J., Isomoto H., Bronk S. F., Gores G. J. (2007) Free fatty acids sensitise hepatocytes to TRAIL mediated cytotoxicity. Gut. 56, 1124–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Volkmann X., Fischer U., Bahr M. J., Ott M., Lehner F., Macfarlane M., Cohen G. M., Manns M. P., Schulze-Osthoff K., Bantel H. (2007) Increased hepatotoxicity of tumor necrosis factor-related apoptosis-inducing ligand in diseased human liver. Hepatology 46, 1498–1508 [DOI] [PubMed] [Google Scholar]
- 17. Feldstein A. E., Canbay A., Angulo P., Taniai M., Burgart L. J., Lindor K. D., Gores G. J. (2003) Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 125, 437–443 [DOI] [PubMed] [Google Scholar]
- 18. Reinehr R., Häussinger D. (2012) CD95 death receptor and epidermal growth factor receptor (EGFR) in liver cell apoptosis and regeneration. Arch. Biochem. Biophys. 518, 2–7 [DOI] [PubMed] [Google Scholar]
- 19. Sanyal A. J., Campbell-Sargent C., Mirshahi F., Rizzo W. B., Contos M. J., Sterling R. K., Luketic V. A., Shiffman M. L., Clore J. N. (2001) Non-alcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 120, 1183–1192 [DOI] [PubMed] [Google Scholar]
- 20. Angulo P., Lindor K. D. (2002) Non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 17, S186–S190 [DOI] [PubMed] [Google Scholar]
- 21. Akbar D. H., Kawther A. H. (2006) Non-alcoholic fatty liver disease and metabolic syndrome: what we know and what we don't know. Med. Sci. Monit. 12, RA23–R26 [PubMed] [Google Scholar]
- 22. Susca M., Grassi A., Zauli D., Volta U., Lenzi M., Marchesini G., Bianchi F. B., Ballardini G. (2001) Liver inflammatory cells, apoptosis, regeneration and stellate cell activation in non-alcoholic steatohepatitis. Dig. Liver Dis. 33, 768–777 [DOI] [PubMed] [Google Scholar]
- 23. Ribeiro P. S., Cortez-Pinto H., Solá S., Castro R. E., Ramalho R. M., Baptista A., Moura M. C., Camilo M. E., Rodrigues C. M. (2004) Hepatocyte apoptosis, expression of death receptors, and activation of NF-κB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am. J. Gastroenterol. 99, 1708–1717 [DOI] [PubMed] [Google Scholar]
- 24. Matteoni C. A., Younossi Z. M., Gramlich T., Boparai N., Liu Y. C., McCullough A. J. (1999) Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology 116, 1413–1419 [DOI] [PubMed] [Google Scholar]
- 25. Clark J. M., Diehl A. (2003) Nonalcoholic fatty liver disease: an underrecognized cause of cryptogenic cirrhosis. JAMA 289, 3000–3004 [DOI] [PubMed] [Google Scholar]
- 26. Zafrani E. S. (2004) Non-alcoholic fatty liver disease: an emerging pathological spectrum. Virchows Arch. 444, 3–12 [DOI] [PubMed] [Google Scholar]
- 27. Meijer A. J., Gimpel J. A., Deleeuw G. A., Tager J. M., Williamson J. R. (1975) Role of anion translocation across the mitochondrial membrane in the regulation of urea synthesis from ammonia by isolated rat hepatocytes. J. Biol. Chem. 250, 7728–7738 [PubMed] [Google Scholar]
- 28. Boerner J. L., Biscardi J. S., Silva C. M., Parsons S. J. (2005) Transactivating agonists of the EGF receptor require Tyr 845 phosphorylation for induction of DNA synthesis. Mol. Carcinog. 44, 262–273 [DOI] [PubMed] [Google Scholar]
- 29. Biscardi J. S., Maa M. C., Tice D. A., Cox M. E., Leu T. H., Parsons S. J. (1999) c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J. Biol. Chem. 274, 8335–8343 [DOI] [PubMed] [Google Scholar]
- 30. Poppleton H. M., Wiepz G. J., Bertics P. J., Patel T. B. (1999) Modulation of the protein tyrosine kinase activity and autophosphorylation of the epidermal growth factor receptor by its juxtamembrane region. Arch. Biochem. Biophys. 363, 227–236 [DOI] [PubMed] [Google Scholar]
- 31. Hao Z., Hampel B., Yagita H., Rajewsky K. (2004) T cell-specific ablation of Fas leads to Fas ligand-mediated lymphocyte depletion and inflammatory pulmonary fibrosis. J. Exp. Med. 199, 1355–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Eberle A., Reinehr R., Becker S., Häussinger D. (2005) Fluorescence resonance energy transfer analysis of proapoptotic CD95/EGF receptor interactions in Huh7 cells. Hepatology 41, 715–726 [DOI] [PubMed] [Google Scholar]
- 33. Wei Y., Wang D., Topczewski F., Pagliassotti M. J. (2006) Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am. J. Physiol. Endocrinol. Metab. 291, 275–281 [DOI] [PubMed] [Google Scholar]
- 34. Nakamura S., Takamura T., Matsuzawa-Nagata N., Takayama H., Misu H., Noda H., Nabemoto S., Kurita S., Ota T., Ando H., Miyamoto K., Kaneko S. (2009) Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J. Biol. Chem. 284, 14809–14818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Egnatchik R. A., Leamy A. K., Noguchi Y., Shiota M., Young J. D. (2014) Palmitate-induced activation of mitochondrial metabolism promotes oxidative stress and apoptosis in H4IIEC3 rat hepatocytes. Metabolism 63, 283–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gao D., Nong S., Huang X., Lu Y., Zhao H., Lin Y., Man Y., Wang S., Yang J., Li J. (2010) The effects of palmitate on hepatic insulin resistance are mediated by NADPH Oxidase 3-derived reactive oxygen species through JNK and p38MAPK pathways. J. Biol. Chem. 285, 29965–29973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pereira S., Park E., Mori Y., Haber C. A., Han P., Uchida T., Stavar L., Oprescu A. I., Koulajian K., Ivovic A., Yu Z., Li D., Bowman T. A., Dewald J., El-Benna J., Brindley D. N., Gutierrez-Juarez R., Lam T. K., Najjar S. M., McKay R. A., Bhanot S., Fantus I. G., Giacca A. (2014) FFA-induced hepatic insulin resistance in vivo is mediated by PKCδ, NADPH oxidase, and oxidative stress. Am. J. Physiol. Endocrinol. Metab. 307, E34–E46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Häussinger D., Lang F. (1992) Cell volume and hormone action. Trends Pharmacol. Sci. 13, 371–373 [DOI] [PubMed] [Google Scholar]
- 39. Taniguchi C. M., Emanuelli B., Kahn C. R. (2006) Critical nodes in signaling pathways: insights into insulin action. Nat. Rev. Mol. Cell Biol. 7, 85–96 [DOI] [PubMed] [Google Scholar]
- 40. Schliess F., von Dahl S., Häussinger D. (2001) Insulin resistence induced by loop diuretics and hyperosmolarity in perfused rat liver. Biol. Chem. 382, 1063–1069 [DOI] [PubMed] [Google Scholar]
- 41. Aguirre V., Werner E. D., Giraud J., Lee Y. H., Shoelson S. E., White M. F. (2002) Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J. Biol. Chem. 277, 1531–1537 [DOI] [PubMed] [Google Scholar]
- 42. Solinas G., Naugler W., Galimi F., Lee M. S., Karin M. (2006) Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proc. Natl. Acad. Sci. U.S.A. 103, 16454–16459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ruddock M. W., Stein A., Landaker E., Park J., Cooksey R. C., McClain D., Patti M. E. (2008) Saturated fatty acids inhibit hepatic insulin action by modulating insulin receptor expression and post-receptor signaling. J. Biochem. 144, 599–607 [DOI] [PubMed] [Google Scholar]
- 44. Holzer R. G., Park E. J., Li N., Tran H., Chen M., Choi C., Solinas G., Karin M. (2011) Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell 141, 173–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shimabukuro M., Zhou Y. T., Levi M., Unger R. H. (1998) Fatty acid-induced beta cell apoptosis: a link between obesity and diabetes. Proc. Natl. Acad. Sci. U.S.A. 95, 2498–2502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kong J. Y., Rabkin S. W. (2000) Palmitate-induced apoptosis in cardiomyocytes is mediated through alterations in mitochondria: prevention by cyclosporin A. Biochim. Biophys. Acta 1485, 45–55 [DOI] [PubMed] [Google Scholar]
- 47. Feldstein A. E., Werneburg N. W., Canbay A., Guicciardi M. E., Bronk S. F., Rydzewski R., Burgart L. J., Gores G. J. (2004) Free fatty acids promote hepatic lipotoxicity by stimulating TNF-α expression via a lysosomal pathway. Hepatology 40, 185–194 [DOI] [PubMed] [Google Scholar]
- 48. Guicciardi M. E., Leist M., Gores G. J. (2004) Lysosomes in cell death. Oncogene 23, 2881–2890 [DOI] [PubMed] [Google Scholar]
- 49. Malhi H., Gores G. J. (2008) Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin. Liver Dis. 28, 360–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Häussinger D., Reinehr R. (2011) Osmotic regulation of bile acid transport, apoptosis and proliferation in rat liver. Cell. Physiol. Biochem. 28, 1089–1098 [DOI] [PubMed] [Google Scholar]
- 51. Kordes C., Sawitza I., Müller-Marbach A., Ale-Agha N., Keitel V., Klonowski-Stumpe H., Häussinger D. (2007) CD133+ hepatic stellate cells are progenitor cells. Biochem. Biophys. Res. Commun. 352, 410–417 [DOI] [PubMed] [Google Scholar]
- 52. Kordes C., Sawitza I., Götze S., Herebian D., Häussinger D. (2014) Hepatic stellate cells can contribute to liver regeneration via progenitor cell formation. J. Clin. Invest. 124, 5503–5515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Reinehr R., Sommerfeld A., Häussinger D. (2008) CD95 ligand is a proliferative and antiapoptotic signal in quiescent hepatic stellate cells. Gastroenterology 134, 1494–1506 [DOI] [PubMed] [Google Scholar]
- 54. Reinehr R., Görg B., Höngen A., Häussinger D. (2004) CD95-tyrosine nitration inhibits hyperosmotic and CD95 ligand-induced CD95 activation in rat hepatocytes. J. Biol. Chem. 279, 10364–10373 [DOI] [PubMed] [Google Scholar]
- 55. Sommerfeld A., Reinehr R., Häussinger D. (2009) Bile acid-induced epidermal growth factor receptor activation in quiescent rat hepatic stellate cells can trigger both proliferation and apoptosis. J. Biol. Chem. 284, 22173–22183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Duan H., Dixit V. M. (1997) RAIDD is a new 'death' adapter molecule. Nature 385, 86–89 [DOI] [PubMed] [Google Scholar]
- 57. Cohen J. C., Horton J. D., Hobbs H. H. (2011) Human fatty liver disease: old questions and new insights. Science 332, 1519–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]