

Background: PSG1 is secreted by placental cells and regulates TGF-β1.
Results: The B2 domain of PSG1 activates latent TGF-β1, whereas macrophage induction of latent TGF-β1 relies on the LYHY amino acid sequence within the N-terminal domain.
Conclusion: PSG1 exerts bifunctional activity through two distinct domains, resulting in an increase in active TGF-β1.
Significance: These data describe a mechanism by which PSG1 modulates TGF-β1 during pregnancy.
Keywords: Heparan Sulfate, Macrophage, Placenta, Pregnancy, Surface Plasmon Resonance (SPR), Latency-associated Peptide, Latent TGF-β1
Abstract
Pregnancy-specific glycoproteins (PSGs) are a family of Ig-like proteins secreted by specialized placental cells. The PSG1 structure is composed of a single Ig variable region-like N-terminal domain and three Ig constant region-like domains termed A1, A2, and B2. Members of the human and murine PSG family have been shown to induce anti-inflammatory cytokines from monocytes and macrophages and to stimulate angiogenesis. We recently showed that recombinant forms of PSG1 (PSG1-Fc and PSG1-His) and PSG1 purified from the serum of pregnant women are associated with the immunoregulatory cytokine TGF-β1 and activated latent TGF-β1. Here, we sought to examine the requirement of specific PSG1 domains in the activation of latent TGF-β1. Plasmon surface resonance studies showed that PSG1 directly bound to the small latent complex and to the latency-associated peptide of TGF-β1 and that this binding was mediated through the B2 domain. Furthermore, the B2 domain alone was sufficient for activating the small latent complex. In separate experiments, we found that the PSG1-mediated induction of TGF-β1 secretion in macrophages was dependent on the N-terminal domain. Mutagenesis analysis revealed that four amino acids (LYHY) of the CC′ loop of the N-terminal domain were required for induction of latent TGF-β1 secretion. Together, our results show that two distinct domains of PSG1 are involved in the regulation of TGF-β1 and provide a mechanistic framework for how PSGs modulate the immunoregulatory environment at the maternal-fetal interface for successful pregnancy outcome.
Introduction
Pregnancy-specific glycoproteins (PSGs)3 are a large family of soluble proteins secreted by the syncytiotrophoblasts of the placenta during pregnancy. There are 10 protein-coding human PSG genes, PSG1–PSG9 and PSG11 (PSG10 is a noncoding pseudogene) arrayed within a genomic region of 0.55 Mb in chromosome 19 (Ensembl) (1). PSGs share membership in the Ig superfamily with the structurally related carcinoembryonic antigen-related cell adhesion molecules (CEACAMs). The human PSG structure consists of an Ig variable region-like N-terminal domain, followed by two or three Ig constant region type 2-like domains termed A1, A2, and B2. The human PSG structure differs from murine PSGs in that most of the murine PSGs contain three Ig variable region-like N-terminal domains and a single Ig constant region type 2-like A domain (2). Because the PSG1 mRNA is relatively highly expressed compared with other PSGs in the first trimester of pregnancy and in the term placenta, we concentrated our studies on this member of the family (3). Several studies support the hypothesis that PSGs contribute to the immunoregulatory environment required for fetal tolerance. PSG1, PSG6, and PSG11 induce the secretion of cytokines IL-10, IL-6, and TGF-β1 in human monocytes, and the PSG6 N-terminal domain alone is sufficient for the induction of TGF-β1 secretion (4). PSGs also regulate dendritic and macrophage cell function, thereby directing T cell activation (5, 6). In clinical studies, low serum PSG concentrations during the first trimester of pregnancy correlated with adverse pregnancy outcomes, including preterm delivery and small birth weight (7). Investigation of potential receptors for PSGs revealed that the tetraspanin CD9 binds murine PSG17, but not PSG23; however, both PSG17 and PSG23 were found to bind to cellular proteoglycans via the N-terminal domain (8, 9). Human PSG1 also binds cell-surface proteoglycans (10) and the platelet integrin αIIbβ3 (3).
TGF-β1 is a pleiotropic cytokine produced by a variety of hematopoietic and non-hematopoietic cells and is critical for angiogenesis, embryogenesis, cell matrix synthesis, and immune system regulation, including lymphocyte differentiation and inhibition of macrophage and dendritic cell activation (11). TGF-β1 plays a role in many biological processes, and its activity is highly regulated. TGF-β1 is produced in a biologically inactive form, covalently associated with the latency-associated peptide (LAP), and cannot signal unless this TGF-β1·LAP complex, also known as the small latent complex (SLC), is activated. Activation of latent TGF-β1 results in the proteolysis, dissociation, or altered binding of LAP to TGF-β1, allowing free TGF-β1 to bind its receptors (12). Factors that activate latent TGF-β1 include plasmin, metalloproteases, selected integrins, neuropilin-1, and thrombospondin-1 (TSP-1), as well as heat, low pH, and shear stress (12–14). We recently showed that both full-length PSG1 and PSG1-Fc, which contains three domains (N-terminal, A2, and B2), activate SLC of TGF-β1 (15). This finding led to the hypothesis that PSG1 could have therapeutic potential as a novel anti-inflammatory therapy. In vivo studies showed that PSG1-Fc treatment reduced inflammation in a TGF-β1-dependent manner in a model of dextran sodium sulfate-induced colitis (15).
To study the activation of SLC by PSG1, we performed surface plasmon resonance (SPR) experiments to characterize the interaction between these proteins. These experiments showed that PSG1 directly bound LAP in the presence or absence of the TGF-β1 polypeptide. To examine the contribution of specific domains of PSG1-Fc to activation of SLC, we expressed PSG1 domains independently fused to the Fc portion of human IgG1 (PSG1-N-Fc, PSG1-A2-Fc and PSG1-B2-Fc). We used the PSG1 single-domain constructs in SPR experiments to characterize their binding to LAP and SLC and determined that LAP binding was mediated via the Ig constant region type 2-like B2 domain. In addition, the B2 domain was found to be sufficient for latent TGF-β1 activation.
To date, few factors have been described that induce the secretion of TGF-β1 in distinct cell populations. In addition to PSGs, the induction of TGF-β1 has been described in macrophages treated with apoptotic lymphocytes (16, 17), Mycobacterium tuberculosis lipoarabinomannan (18), and lipopolysaccharide (19). We re-examined the induction of TGF-β1 by PSG1 and found that the N-terminal domain alone is sufficient for macrophage TGF-β1 production. In addition, we identified the region within the N-terminal domain required for TGF-β1 induction: a four-amino acid sequence (LYHY) located in the CC′ loop. With these studies, we characterized the binding and activation of TGF-β1 via PSG1 and defined two domains of PSG1 capable of regulating TGF-β1 biological activity.
EXPERIMENTAL PROCEDURES
Protein Production and Purification
PSG1-Fc and CEACAM9-Fc were generated as described previously (15). The PSG1 single-domain mutants were generated as follows. The cDNA consisting of the PSG1 leader peptide followed by the indicated domain was synthesized by GenScript USA Inc. (Piscataway, NJ) and subcloned into the EcoRI-BglII sites of the pFUSE-IgG1-e3-Fc1 vector (InvivoGen, San Diego, CA), resulting in the in-frame addition of the hinge region and CH2 and CH3 domains (Fc tag) of the IgG1 heavy chain. PSG1-His has a His6 tag at the N- and C-terminal ends of the protein and was purified from the supernatant of stably transfected CHO-K1 cells grown on a C2003 hollow fiber cell culture cartridge (FiberCell Systems, Frederick, MD) on an anti-PSG1 monoclonal antibody 4 column. Monoclonal antibody 4 was obtained from Dr. S. Jonjic (University of Rijeka, Rijeka, Croatia) and was previously used to purify PSG1 from the serum of pregnant women (15). Recombinant proteins generated by transient transfection were purified from cell culture medium (Opti-MEM I) of CHO-K1 cells transfected with Lipofectamine 2000 (Invitrogen) 72 h post-transfection. The protein consisting of the Fc tag (used as control for the treatment of macrophages) was generated by transient transfection of CHO-K1 cells with the pFUSE-hIgG1-Fc2 vector (InvivoGen). The four mutant N-terminal domain proteins were obtained by replacing the wild-type sequence in the cDNA encoding the leader peptide and the N-terminal domain of PSG1 cloned into the pFUSE-IgG1-e3-Fc1 vector with a synthesized cDNA fragment encoding the indicated mutations at the unique EcoRI-XhoI sites. For the generation of the recombinant B2 protein in bacteria, the B2 domain of PSG1 was PCR-amplified with primers 5′-GACGACGACAAGATGGGTCCAGACCTCCCCAG and 5′-GAGGAGAAGCCCGGTTAAGAGACTTCGACTGTCATG using the B2 domain cloned into pFUSE-IgG1-e3-Fc1 as the template. The PCR product was cloned into the pET-41 Ek/LIC vector (Novagen, Billerica, MA). The proper insert orientation and sequence were confirmed by sequencing with the S-tag primer as recommended by the manufacturer. Expression of the B2 cDNA cloned into the pET-41 Ek/LIC vector in Rosetta-gami 2(DE3) bacteria renders a polypeptide containing GST, His, and S-tag at the N terminus, and its expression was induced by addition of 1 mm isopropyl β-d-thiogalactopyranoside for 2 h. Following induction, the soluble and insoluble fractions were analyzed for the presence of the protein (designated as PSG1-GST-B2) by separation on a 4–12% NuPAGE Bis-Tris gel (Invitrogen), followed by staining of the gel with GelCode Blue (Thermo Scientific, Waltham, MA). The PSG1-GST-B2 protein, which had a predicted molecular mass of 40 kDa, was present in the insoluble fraction, and its identity was confirmed by immunoblot analysis with anti-PSG1 B2 peptide polyclonal antibody NBP1-58028 (Novus Biologicals, Littleton, CO), followed by HRP-conjugated goat anti-rabbit antibody. To generate purified PSG1-GST-B2 protein, Rosetta-gami 2(DE3) cells expressing PSG1-GST-B2 were disrupted using lysozyme (1 kilo-unit/ml) and Benzonase (25 units/ml) in BugBuster protein extraction reagent (Novagen) with cOmplete EDTA-free protease inhibitor mixture (Roche Applied Science), and the insoluble fraction was recovered by centrifugation, followed by several washes with BugBuster as suggested by the manufacturer. The bacterial pellet (containing the inclusion bodies) was solubilized in binding buffer (6 m guanidine HCl, 20 mm Tris-HCl (pH 8.0), 500 mm NaCl, and 5 mm imidazole), and the protein was purified following the HisTrap on-column refolding protocol (GE Healthcare). Recombinant GST (GenScript USA Inc.) was used as the control protein in experiments testing PSG1-GST-B2 activation of SLC.
All recombinant proteins generated for the studies described in this work were purified with the indicated column: protein A for Fc-tagged proteins and HisTrap for the bacterially produced B2 domain in an ÄKTAprime system (GE Healthcare) unless specified otherwise. Elution fractions containing the protein were pooled, concentrated, and buffer-exchanged with PBS in an Amicon Ultra-10K centrifugal filter unit (Millipore). For quantitation purposes, the purified proteins were separated on 4–20% NuPAGE Bis-Tris gels at different dilutions alongside known concentrations of BSA (Thermo Scientific), used as a standard. After staining with GelCode Blue, the proteins were quantitated by densitometry. Antibodies to the specific domains, including monoclonal antibody 4 (which reacts with the N-terminal domain), monoclonal antibody BAP1 (Aldevron Freiburg GmbH, Freiburg, Germany; which reacts with the A2 domain), and anti-B2 peptide polyclonal antibody, were used to confirm the identity of the single-domain mutants by Western blotting (data not shown).
SPR
Protein-protein interactions were analyzed by the SPR technique using a Biacore 3000 instrument (GE Healthcare). Recombinant human LAP and human latent TGF-β1 (catalog numbers 246-LP (carrier-free) and 299-LT, respectively; R&D Systems, Minneapolis, MN), were coupled via a standard amine-coupling procedure to the flow cells of a CM5 sensor chip until a level of 1000 resonance units was reached. A control cell was prepared in the same way but without protein. Tested proteins were injected separately into the flow cell at a flow rate of 10 μl/min at 25 °C. Each interaction was analyzed at least three times with different protein preparations.
Several analyte concentrations (8, 6, 5, 4, 2, 1, and 0.5 μm) were injected during the association phase for 3 min with HBS-EP (0.01 m HEPES (pH 7.4), 0.15 m NaCl, 3 mm EDTA, and 0.005% (v/v) Surfactant P20) as the running buffer. The dissociation phase, initiated by passage of HBS-EP buffer alone, was carried out over a period of 2 min. The chip surfaces were regenerated by a 60-s injection of 10 mm glycine HCl (pH 2.0). Kinetic data were analyzed using BIAevaluation version 4.1.1. The association (Ka) and dissociation (Kd) constants were calculated assuming a 1:1 Langmuir binding model. All binding curves were corrected for background and bulk refractive index contribution by subtraction of the reference flow cells.
For LSKL peptide inhibition studies, 2 μm PSG1-Fc was incubated with the LSKL peptide (GenScript USA Inc.) or a control peptide (SLLK) at 20 and 200 μm final concentrations for 1 h at 37 °C before injection at 10 μl/min over a biosensor chip with immobilized LAP. LAP was coupled via a standard amine-coupling procedure to the flow cell of a CM5 sensor chip until a level of 2000 resonance units was reached. A reference cell was prepared in the same way but without protein. The association and dissociation phases were perform as described above. The percentages of PSG1-Fc-LAP binding in the presence of the LSKL or SLLK peptide were calculated in comparison with PSG1-LAP binding in the absence of peptide (100%). Each interaction was analyzed at least three times, and all binding curves were corrected for background and bulk refractive index contribution by subtraction of the reference flow cell.
ELISAs
Active TGF-β1 was detected as described previously (15). Briefly, 50 ng/ml SLC (R&D Systems) was incubated with PSG1-A2-Fc, PSG1-N-Fc, PSG1-B2-Fc, PSG1-GST-B2, CEACAM9-Fc, or GST at the concentrations indicated in the figure legends (0.25–10.0 μg/ml) at 37 °C for 1 h in a final volume of 0.1 ml of PBS in siliconized tubes. The samples were transferred to a 96-well Nunc MaxiSorp plate that had been blocked with 0.5% BSA following an overnight coating step with either recombinant human TGF-β receptor II-Fc or anti-TGF-β1 antibody (R&D Systems). After 2 h, the plates were washed, and the presence of active TGF-β1 was detected with a biotinylated antibody specific for mature TGF-β1 from a DuoSet ELISA kit (R&D Systems).
Luciferase Assay
The presence of bioactive TGF-β was determined using the TGF-β-responsive plasminogen activator inhibitor-1 luciferase reporter mink lung epithelial cell (MLEC) line provided by Dr. D. Rifkin (New York University, New York, NY) as described previously (20). In brief, 1.6 × 104 MLECs were seeded onto a 96-well plate in serum-free DMEM supplemented with 0.1% insulin/transferrin/sodium selenite medium (Sigma-Aldrich). After 3 h, the MLECs were treated either with DMEM and 0.1% insulin/transferrin/sodium selenite medium or with 2.5 μg/ml PSG1-A2-Fc, PSG1-N-Fc, PSG1-GST-B2, GST, or CEACAM9-Fc that had been incubated at 37 °C for 1 h with or without 50 ng/ml SLC. After 16 h, the cells were rinsed with PBS and lysed with Passive Lysis Buffer (Promega, Madison, WI), and the lysate was analyzed on a GloMax luminometer (Promega) following addition of the luciferase substrate and reported as relative light units. Similarly, for LSKL peptide inhibition studies, 50 ng/ml SLC was incubated at 37 °C for 1 h with PSG1-GST-B2 in the presence of a 10-fold molar excess of LSKL or SKKL peptide (4.4 nm) and added to MLECs in triplicate as described above. The increase in luciferase was compared with PSG1-GST-B2- and GST control-treated samples as a measure of active TGF-β1.
Induction of Macrophage TGF-β1
RAW264.7 macrophages were plated at a density of 1.8 × 105 cells/well of a 96-well plate in triplicate in DMEM and 5% HyClone FetalClone III (GE Healthcare). After overnight incubation at 37 °C, the medium was replaced with medium containing the proteins at the indicated concentrations, and the supernatants were collected after 16–22 h. Supernatant (0.1 ml) was activated with 1 n HCl, followed by neutralization to measure total TGF-β1 by ELISA following the manufacturer's recommendations (R&D Systems). All treatments were performed in triplicate in at least three independent experiments.
Quantitative PCR
Total RNA was isolated from RAW264.7 cells with an RNeasy mini kit (Qiagen). RNA (1 μg) was used to generate cDNA with a high capacity cDNA reverse transcription kit (Applied Biosystems). The TGF-β1 mRNA levels were determined with the following primer-probe sets (Invitrogen): for TGF-β1, assay ID MM01178820_M1, and for the hypoxanthine phosphoribosyltransferase (HPRT) housekeeping gene, assay ID MM00446968_M1 using TaqMan Gene Expression Master Mix in a 20-μl final reaction volume as recommended by the manufacturer. The Ct values of TGF-β1 were normalized to those of hypoxanthine phosphoribosyltransferase, and the experiment was repeated three independent times using three wells per treatment.
Cell Culture
All cells were cultured in 5% CO2 and 95% air in 37 °C humidified incubators in the presence of 100 units/ml penicillin/streptomycin (Invitrogen) and 1× Normocin (InvivoGen). RAW264.7 cells were obtained from the American Type Culture Collection (Manassas, VA) and cultured in the medium and conditions recommended. Regular FBS was replaced with HyClone FetalClone III, which has very low levels of TGF-β1, in experiments designed to test the ability of different proteins to induce TGF-β1 secretion from RAW264.7 cells.
Generation of a Structural Model of the PSG1 N-terminal Domain
A structural model of the N-terminal domain of human PSG1 was generated using SWISS-MODEL with the crystal structure of the N-terminal domain of CEACAM5 as a reference (Protein Data Bank ID 2QSQ) (21,22). The model was refined using 3Drefine and evaluated with PROCHECK (23). The final model had 98.9% of the residues in the most favorable or additionally allowed regions (24). The root mean square deviation between the model and the CEACAM5 crystal structure was 0.2 Å for 107 residues.
Statistical Analysis
Statistical significance of raw data between two groups was evaluated using Student's unpaired t test. Results are expressed as the mean ± S.D. Statistical analysis was performed using GraphPad Prism software.
RESULTS
The PSG1 B2 Domain Directly Binds SLC of TGF-β1 and LAP
We showed previously that full-length PSG1 purified from the serum of pregnant women and a natural splice variant composed of three domains (N-terminal, A2, and B2) with an Fc tag at the C terminus, designated PSG1-Fc, activate latent TGF-β1 (15). The best characterized activators of TGF-β1, including selected integrins and TSP-1, bind to LAP of TGF-β1 in the absence of the mature TGF-β1 polypeptide (25, 26). Here, we analyzed whether PSG1 can bind to LAP in the absence of the mature TGF-β1 polypeptide and defined the kinetics of the interactions between PSG1 and LAP and between PSG1 and SLC of TGF-β1 using SPR analysis. For these experiments, we used PSG1-Fc, which is the protein we previously used for our in vivo studies, and PSG1-His, which is composed of the four PSG1 domains (N-terminal, A1, A2, and B2) (Fig. 1B). The SPR experiments revealed a direct interaction between PSG1 and LAP in the absence and presence of the mature TGF-β1 polypeptide (Fig. 2). The affinity of the PSG1-LAP interaction is in the micromolar range (Kd = 1.09–3.56 μm). Kinetic parameters of these interactions were calculated and are shown in Table 1. To determine which domain(s) of PSG1-Fc are involved in TGF-β1 binding, we generated PSG1 single-domain constructs (Fig. 1). Binding of the PSG1 single-domain proteins to SLC and LAP was also analyzed by SPR (Fig. 3). Proteins consisting of only the N-terminal or A2 domain did not bind to SLC or LAP more than the control protein CEACAM9-Fc. Interestingly, the B2 domain alone showed binding to SLC and LAP, revealing a direct interaction between the PSG1 B2 domain and LAP.
FIGURE 1.
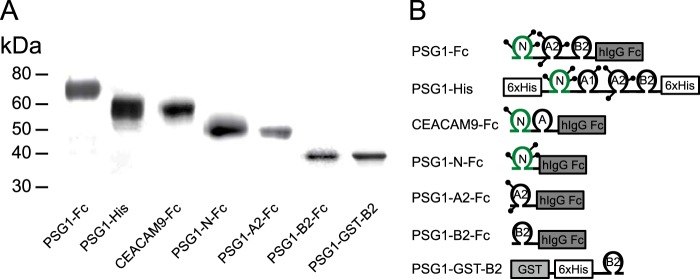
Schematic of recombinant PSG1 and PSG1 domain constructs. A, PSG1-Fc, PSG1-His, CEACAM9-Fc, PSG1-N-Fc, PSG1-A2-Fc, and PSG1-B2-Fc produced in CHO-K1 cells and PSG1-GST-B2 expressed in bacteria were purified and loaded onto a 4–20% NuPAGE gel. The gel was stained with GelCode Blue to visualize the proteins. B, schematic representation of the proteins listed in A. The position of protein tags (Fc or His) is indicated; lollipops represent potential N-linked glycosylation sites.
FIGURE 2.
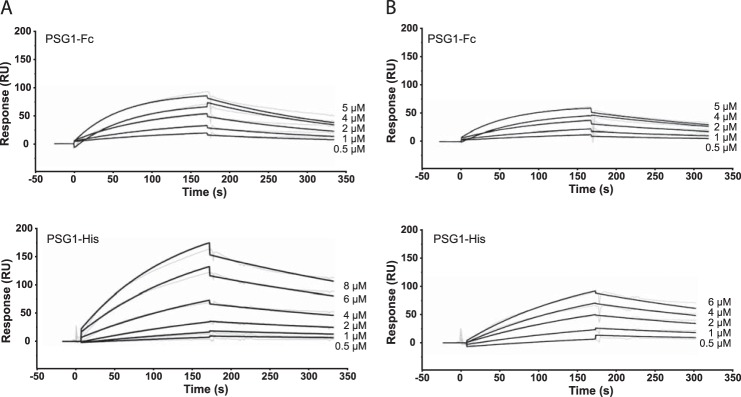
PSG1 interacts with SLC and LAP of TGF-β1. Shown are the results from SPR analysis of the interaction of PSG1-Fc and PSG1-His with SLC (A) or LAP (B) of TGF-β1. A protein concentration that ranged from 8 to 0.5 μm was analyzed across blank, SLC, and LAP biosensor surfaces. The data were analyzed using a simultaneous fit algorithm to calculate the kinetic parameters presented in Table 1. Representative SPR sensorgrams for each response are shown as gray lines, and fit analyses are shown as black lines. RU, response units.
TABLE 1.
Kinetic analysis of PSG1 interaction with the SLC and LAP using a 1:1 Langmuir binding model
The kinetic data shown in Fig. 2 for the interaction of PSG1-Fc and PSG1-His with the SLC and LAP of TGF-β1 were fit using a 1:1 Langmuir binding model for estimation of the association (ka) and dissociation (kd) rates and the dissociation constant (Kd = kd/ka).
| Kd | ka (mean ± S.D.) | kd (mean ± S.D.) | |
|---|---|---|---|
| μm | m−1 s−1 | s−1 | |
| SLC | |||
| PSG1-Fc | 2.56 | (3.10 ± 0.82) × 103 | (7.39 ± 0.83) × 10−3 |
| PSG1-His | 3.56 | (2.13 ± 0.86) × 103 | (7.56 ± 0.27) × 10−3 |
| LAP | |||
| PSG1-Fc | 2.05 | (2.32 ± 0.86) × 103 | (4.76 ± 0.42) × 10−3 |
| PSG1-His | 1.09 | (8.07 ± 0.31) × 103 | (8.81 ± 0.42) × 10−3 |
FIGURE 3.
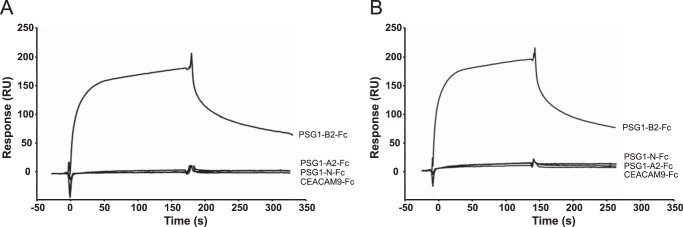
The B2 domain of PSG1 interacts with SLC and LAP of TGF-β1. Show are Biacore sensorgrams for the interaction of PSG1-Fc domains and CEACAM9-Fc at 2 μm with SLC (A) and LAP (B) of TGF-β1. Purified CEACAM9-Fc and PSG1-Fc proteins containing the N-terminal, A2, and B2 domains of PSG1 were injected over a CM5 sensor chip with immobilized SLC or LAP of TGF-β1. A flow cell to which no protein was coupled served as a surface control. Representative sensorgrams are shown. RU, response units.
The PSG1 B2 Domain Is Sufficient to Activate SLC of TGF-β1
PSG1 B2 domain binding to SLC and LAP suggested that the B2 domain alone may activate SLC. Therefore, we tested the single-domain proteins (PSG1-N-Fc, PSG1-A2-Fc, and PSG1-GST-B2) in a cell-free assay for their ability to activate SLC of TGF-β1 (15). We used two ELISA protocols to detect active TGF-β1: one in which plates were coated with an antibody that recognizes the active form of TGF-β1 (Fig. 4A) and one in which samples were added to plates coated with TGF-β receptor II (Fig. 4, B and C). Generation of the B2 domain construct (PSG1-B2-Fc) was made difficult by the very low yield of protein following transfection of CHO-K1 or HEK293T cells. The B2 domain is the only domain of PSG1 that does not have potential N-linked glycosylation sites (27). Therefore, we employed a bacterial expression system that allowed us to generate sufficient quantities of the B2 domain (PSG1-GST-B2) to further test the function of this domain in activation assays. As shown in Fig. 4A, PSG1-GST-B2 activated SLC, generating significant levels of active TGF-β1 compared with the GST control protein. The PSG1 A2 and N-terminal single-domain proteins used at the same concentration (2.5 μg/ml) also had the capacity to activate SLC compared with the CEACAM9-Fc control protein but always generated significantly less active TGF-β1 than SLC treated with PSG1-GST-B2. To further examine the ability of the single-domain proteins to activate SLC, we performed experiments with higher protein concentrations. As shown in Fig. 4B, the A2 and N-terminal single-domain proteins activated only a small percentage (∼10%) of SLC even when tested at 10 μg/ml. In contrast, the PSG1 B2 domain activated a higher percentage of SLC at 1 μg/ml, with a percent activation consistently over 50% when used at 5 μg/ml (Fig. 4C).
FIGURE 4.

The B2 domain of PSG1 is sufficient for activation of SLC of TGF-β1. A, 2.5 μg/ml CEACAM9-Fc, PSG1-A2-Fc, PSG1-N-Fc, PSG1-GST-B2, or GST was incubated with 50 ng/ml SLC for 1 h at 37 °C and then analyzed for active TGF-β1 by ELISA as described under “Experimental Procedures.” B, increasing concentrations of PSG1-N-Fc, PSG1-A2-Fc, and CEACAM9-Fc were incubated with 50 ng/ml SLC for 1 h at 37 °C, and the percentage of SLC activated was measured with a TGF-β receptor II-Fc capture ELISA. 100% SLC activation was defined as the amount of active TGF-β1 measured following acid activation of 50 ng/ml SLC. C, increasing concentrations of PSG1-GST-B2 or GST were incubated with 50 ng/ml SLC for 1 h at 37 °C, and the percent SLC activated was measured as described for B. D, 2.5 μg/ml CEACAM9-Fc, PSG1-A2-Fc, PSG1-N-Fc, PSG1-GST-B2, GST, or medium alone (med) was incubated with 50 ng/ml SLC in DMEM and 0.1% insulin/transferrin/sodium selenite medium for 1 h at 37 °C and then added in triplicate to luciferase reporter cells (MLECs). The treated MLECs were incubated for 16 h at 37 °C and lysed, and the luciferase activity was analyzed on a GloMax luminometer. The relative light units (RLU) reflect the dose-dependent increase in luciferase activity following active TGF-β signaling. For A–D, all treatments were performed in triplicate for a minimum of three independent experiments. *, p ≤ 0.02 by Student's t test for data in A–D.
PSG1-B2-Fc Activation of Latent TGF-β1 Generates Biologically Active TGF-β1
To detect the presence of biologically active TGF-β following treatment with the specific PSG1 domains, we used a well characterized luciferase reporter cell line (MLEC) that allows quantification of mature TGF-β (20). For this two-step assay, SLC was first incubated with individual PSG1 domains at 37 °C for 1 h (as in Fig. 4, A–C) and then added to wells containing MLECs. After 16 h at 37 °C, the MLEC monolayers were lysed, and the luciferase signal was measured on a luminometer. MLECs responded to the presence of active TGF-β following incubation of SLC with PSG1-A2-Fc and PSG1-N-Fc compared with active TGF-β detected following incubation with the protein control. However, significantly higher TGF-β bioactivity was generated when SLC of TGF-β was incubated with the B2 domain before treatment of the cells (Fig. 4D). These results are in agreement with the increased capacity of the B2 domain to activate latent TGF-β1 in the cell-free activation assays. MLECs treated with PSG1 domains in the absence of SLC did not show enhanced luciferase activity compared with control proteins (mean relative light units ± S.D. for PSG1-A2-Fc, 0.7 ± 0.04; PSG1-N-Fc, 0.41 ± 0.04; PSG1-GST-B2, 0.39 ± 0.02; and control proteins alone, 0.79 ± 0.11).
PSG1 Does Not Interact with Latent TGF-β1 through the LSKL Sequence in LAP
The matrix glycoprotein TSP-1 binds and activates latent TGF-β1 through a mechanism dependent upon the LSKL sequence in LAP and the KRFK sequence in TSP-1 (26). Several studies have shown that the LSKL peptide competes with TSP-1 and inhibits the ability of this protein to trigger activation of latent TGF-β1 (26, 28). Therefore, we tested the ability of LSKL to inhibit the interaction of PSG1 with LAP using SPR. LSKL and a scrambled peptide control at 10- and 100-fold molar excess did not reduce binding of PSG1 to LAP compared with binding in the absence of peptides (Fig. 5A). These results were confirmed in the cell-dependent assay, in which the induction of luciferase activity as a measure of TGF-β levels was unchanged in the reporter MLECs following incubation of PSG1-GST-B2 and SLC in the presence of LSKL (Fig. 5B).
FIGURE 5.
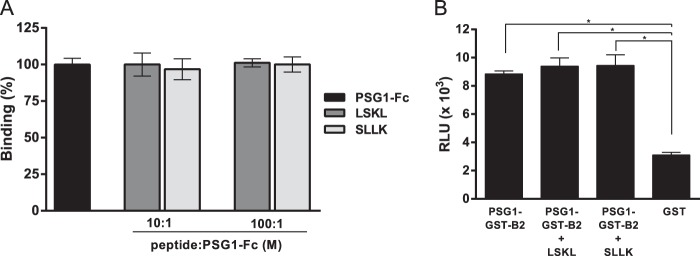
The LSKL peptide does not inhibit PSG1-Fc binding to LAP or the activation of SLC via PSG1-GST-B2. A, PSG1-Fc protein (2 μm) was incubated with the LSKL peptide or a control peptide (SLLK) at a 10:1 or 100:1 peptide/protein molar ratio at 37 °C for 1 h and injected at 10 μl/min over a LAP biosensor surface. The percentage of binding of PSG1-Fc in the presence of peptide is shown relative to PSG1-Fc binding to LAP in the absence of peptide (100%). The mean ± S.D. for three independent experiments is shown. B, SLC (50 ng/ml) was incubated with PSG1-GST-B2 alone or with PSG1-GST-B2 in the presence of a 10-fold molar excess of the LSKL or SLLK peptide at 37 °C for 1 h and added in triplicate to luciferase reporter cells (MLECs). After 16 h at 37 °C, the cells were lysed, and the luciferase activity was analyzed on a luminometer. The relative light units (RLU) reflect the increase in luciferase activity following active TGF-β signaling. *, p < 0.002 with Student's t test.
The PSG1 N-terminal Domain Induces TGF-β1 in RAW264.7 Macrophages
TGF-β1 is a ubiquitous cytokine produced by many cells types, but the identification of factors that induce its production has been limited. Previously, we reported the induction of TGF-β1 from monocytes following treatment with PSG1, PSG6, and PSG11 (4). The N-terminal domains of the 10 human PSG proteins consist of 109 amino acids, and they share ∼58.7% sequence identity. Treatment of monocytes with the N-terminal domain of PSG6 was sufficient for TGF-β1 induction, but the ability of the N-terminal domain of PSG1 or PSG11 to induce TGF-β1 has not been tested. Therefore, we examined the capacity of the N-terminal domain of PSG1 to induce TGF-β1 and also the possible role of the A2 and B2 domains of this protein in TGF-β1 induction. Due to the low yield of the B2 single-domain protein following expression from mammalian cells and the presence of endotoxin, a known inducer of TGF-β1 by macrophages in the B2 domain protein generated in bacteria, we generated a PSG1-A2-B2-Fc construct to evaluate the possible role of the B2 domain in TGF-β1 induction (Fig. 6A). We tested the PSG1-A2-B2-Fc protein alongside the N-terminal and A2 single-domain proteins for their ability to induce TGF-β1 in RAW264.7 macrophages. As shown in Fig. 6B, TGF-β1 was significantly induced following treatment with the PSG1-N-Fc protein, but not with the PSG1-A2-Fc or PSG1-A2-B2-Fc protein after overnight culture (18–24 h). Time course studies with PSG1-N-Fc and the control protein at 3 μg/ml indicated that the earliest time point post-treatment at which we detected a significant difference in total TGF-β1 in the conditioned medium was 8 h (1.5 ± 0.17-fold increase). The difference in total TGF-β1 between treatments was greater when the conditioned medium was collected 18–24 h post-treatment, in which case, we consistently observed a >2-fold difference. Significant induction of TGF-β1 mRNA by quantitative real-time PCR (normalized to hypoxanthine phosphoribosyltransferase (HPRT)) was detected at 4 h post-treatment (2.18 ± 0.17-fold induction over control protein-treated cells). In addition, bone marrow-derived macrophages treated with the N-terminal domain of PSG1 secreted TGF-β1 compared with the control protein-treated cells, whereas the PSG1-A2-Fc and PSG1-A2-B2-Fc proteins did not (data not shown). To examine the amino acid sequence in the PSG1 N-terminal domain required for TGF-β1 production by macrophages, we mutated the amino acids in the CC′ and C′C″ loops to the corresponding amino acids in CEACAM5 (formerly known as CEA) (Fig. 6C) (29). PSG1-N-Fc with the amino acids mutated in the C′C″ loop remained capable of inducing significant TGF-β1 from macrophages; however, the protein with the amino acids mutated in the CC′ loop failed to induce TGF-β1 even when the macrophages were treated with 12 μg/ml protein (Fig. 6D, black and gray bars, respectively). To further characterize the amino acids in the CC′ loop required for TGF-β1 induction, we generated two distinct mutants by replacing either three amino acids (Y42N/H43R/Y44Q) or four amino acids (L41G/Y42N/H43R/Y44Q) with the corresponding amino acids present at the same positions in human CEACAM5 (Fig. 6, A and C). As shown in Fig. 6E, the PSG1 N-terminal Y42N/H43R/Y44Q mutant retained the capacity to induce TGF-β1 secretion, albeit its activity was significantly lower compared with the wild-type PSG1 N-terminal protein, whereas the PSG1 N-terminal L41G/Y42N/H43R/Y44Q mutant failed to induce TGF-β1. This result indicates that the CC′ loop of the N-terminal domain of PSG1 is critical for PSG1 induction of TGF-β1 from macrophages and that amino acids LYHY are required for this function. We generated a structural model for the N-terminal domain of PSG1 based on the CEACAM5 crystal structure (Protein Data Bank ID 2QSQ) to examine whether the CC′ and C′C″ loop regions are located in solvent-exposed extracellular loops (21, 22). As expected, the CC′ and C′C″loops are highly exposed in the structural model for the N-terminal domain of PSG1, and Leu-41 is accessible for ligand binding (Fig. 6F).
FIGURE 6.

Amino acids in the CC′ loop region of the N-terminal domain of PSG1 are essential for induction of TGF-β1 from RAW264.7 macrophages. A, recombinant proteins used for these studies were separated on a 4–20% NuPAGE gel and stained with GelCode Blue. Lane 1, PSG1-A2-B2-Fc; lane 2, PSG1-N-Fc; lane 3, PSG1-N-CC′-Fc; lane 4, PSG1-N-C′C″-Fc; lane 5, PSG1-N-YHY-Fc; lane 6, PSG1-N-LYHY-Fc. B, TGF-β1 secretion following incubation of RAW264.7 murine macrophages with 3 μg/ml control Fc protein (cntrl-Fc), PSG1-A2-Fc, and PSG1-N-Fc or 6 μg/ml PSG1-A2-B2-Fc to achieve concentrations equimolar to the single-domain proteins. C, sequence alignment of the CC′ and C′C″ loop regions of CEACAM5, PSG1, and PSG1 N-terminal domain mutants. Residues conserved in CEACAM5 and PSG1 are highlighted in gray. The C, C′, and C″ β-strands are indicated with green arrows above the corresponding sequence. Residues in the PSG1 N-terminal domain mutated to the residues corresponding to the same position in CEACAM5 are colored red. D, TGF-β1 secretion of RAW264.7 macrophages following incubation with increasing concentrations of PSG1-N-CC′-Fc, PSG1-N-C′C″-Fc, or control Fc protein. E, RAW264.7 macrophages were incubated with 12 μg/ml wild-type PSG1 N-terminal domain (WT), PSG1-N-Y42N/H43R/Y44Q mutant (YHY→NRQ), PSG1-N-L41G/Y42N/H43R/Y44Q mutant (LYHY→GNRQ), or control Fc protein. For B–E, TGF-β1 following acid activation was measured in the supernatants by ELISA as described under “Experimental Procedures.” *, p ≤ 0.05 by Student's t test; ns, not significant. F, ribbon representation of the structural model of the PSG1 N-terminal domain. β-Sheets are shown in green and labeled with uppercase white letters. The α-helix between strands E and F is shown in orange, and loops are shown in gray. Residues 41LYHY44 of the CC′ loop are labeled and highlighted in red, and their side chains are shown in stick representation. The CC′ and C′C″ loops and the N and C termini of the protein domain are indicated.
DISCUSSION
PSGs were first described as abundant serum markers of early pregnancy that increased in concentration as pregnancy progressed (30). PSGs are secreted by placental syncytiotrophoblasts, which also secrete human chorionic gonadotropin, progesterone, and estrogen hormones (31). Several studies suggest that PSGs influence fetal tolerance; however, syncytiotrophoblasts may also play a role in pathogen recognition and inflammation, as trophoblast cells were found to express Toll-like receptors (32). Recent descriptions of the bacterial microbiome found in the placenta suggest the presence of immunoregulatory systems in place at the onset of placentation (33, 34). Our results obtained upon examination of the role of PSG1 in the induction and activation of a major immunoregulatory cytokine, TGF-β1, help advance our understanding of how fetal-derived cells of the placenta and their proteins influence maternal and fetal immune networks during pregnancy.
SPR technology has been used successfully to study the interactions of TGF-β with its cognate receptors and the mature TGF-β1 polypeptide with LAP (35, 36). In this study, we used SPR to examine the interaction of PSG1 with SLC of TGF-β1, which is composed of LAP and the mature TGF-β1 polypeptide, as well as the interaction of PSG1 with recombinant LAP. These experiments revealed a direct interaction of PSG1 with SLC and with LAP. Two different PSG1 constructs were tested in the SPR experiments: PSG1-His and PSG1-Fc. PSG1-His is composed of the four domains of PSG1 (N-terminal, A1, A2, and B2), whereas PSG1-Fc lacks the A1 domain. Both PSG1 proteins showed the same binding kinetics for SLC and LAP, indicating that PSG1-LAP binding is not affected by the absence of the A1 domain or the presence of TGF-β1 in SLC. PSG1-His is mainly a monomeric protein based on analysis by size exclusion chromatography on a Superose 12 column (GE Healthcare). In contrast, the majority of the PSG1-Fc protein is dimeric due to the presence of the Fc tag (data not shown). Thus, the presence of the Fc tag and the oligomerization state of PSG1 did not alter the affinity of the PSG1-LAP interaction.
We consistently observed by ELISA that higher levels of TGF-β1 co-purified with the PSG1-B2-Fc protein expressed in CHO-K1 cells compared with the concentration of TGF-β1 associated with PSG1-N-Fc and PSG1-A2-Fc generated under the same conditions. Similarly, the concentration of human LAP measured by ELISA when the proteins were transiently expressed in HEK293T cells was always highest in the PSG1-B2-Fc preparations compared with the amount of LAP associated with equimolar concentrations of PSG1-N-Fc and PSG1-A2-Fc (data not shown). Together with the results of the SPR analysis presented here, these observations support the conclusion that the B2 domain, which is present in all PSG1 splice variants, plays a central role in the ability of PSG1 to activate latent TGF-β1. Although we could not detect binding of the N-terminal or A2 domain of PSG1 to SLC or LAP using SPR, our functional studies indicate that a potential contribution of the N-terminal and A2 domains to the ability of PSG1-Fc to activate latent TGF-β1 cannot be completely excluded. Also, preliminary results indicate that the A1 domain of PSG1 can activate SLC in the cell-free activation assay and generate bioactive TGF-β1 in the luciferase-based MLEC assays to a similar extent as the N-terminal domain.
The SPR results show that PSG1 and the PSG1 B2 domain alone bound directly to LAP in the absence of the mature TGF-β1 polypeptide. Because we have previously shown that PSG1 activates latent TGF-β1 in the presence of protease inhibitors, our results suggest that the mechanism by which PSG1 mediates latent TGF-β1 activation likely involves a PSG1-induced conformational change in LAP, as was shown previously for TSP-1 and integrin αvβ6 (15, 37). Our results suggest that amino acids other than the LSKL sequence in LAP are involved in the interaction between the B2 domain of PSG1 and LAP. The matrix glycoprotein tenascin X (TNX) has recently been described as an activator of TGF-β (28). The fibrinogen-like domain of TNX activates SLC of TGF-β in a cell adhesion-dependent manner, which requires expression of integrin α11β1 as a receptor for TNX. Although activation of SLC by PSG1 is cell-adhesion independent, there may be similarities in the mechanism by which PSG1 and TNX activate SLC; the B2 domain of PSG1 and the fibrinogen-like domain of TNX share conserved amino acids that may potentially be responsible for the interaction of these proteins with LAP.
PSGs are the only secreted proteins in the CEACAM family that consist of several membrane-bound proteins with diverse functions, including cell adhesion, immunomodulation, pathogen binding, and angiogenesis (38). Early studies by Zhou and Hammarström (29) examining the putative functional regions of human CEACAM and PSG molecules identified sequences within the N-terminal domain that are highly conserved, non-glycosylated, and possibly free to interact with potential PSG ligand(s). For some members of the CEACAM family, the CC′ loop in the N-terminal domain contains critical amino acids for interaction with putative molecules, as this is the region of binding for pathogens, including murine hepatitis virus (39), Neisseria spp., and Haemophilus influenza (38). We found that amino acids in the CC′ loop are essential for the ability of the N-terminal domain of PSG1 to induce the secretion of TGF-β1 in the RAW264.7 macrophage cell line. Mutation of amino acids LHYH in the CC′ loop to the corresponding amino acids in the CC′ loop of CEACAM5 resulted in a protein that did not induce secretion of TGF-β1. Interestingly, we showed previously that PSG6 and PSG11 can also induce the secretion of TGF-β1; together with six other human PSGs, they have the LYHY sequence in the N-terminal domain in a conserved position (4). Although PSG4 has the VYHY sequence in this position, substitution of the similarly charged valine for leucine at position 41 may still render a protein with the same function, suggesting that all 10 PSGs may share the ability to induce TGF-β1 secretion by macrophages. At this time, the macrophage receptor(s) for the N-terminal domain of PSG1 remain to be identified. Although we could not test whether the PSG1-B2-Fc single-domain protein induces TGF-β1 secretion due to the low protein yield and high content of associated TGF-β1, our results using a mutant consisting of the PSG1 A2 and B2 domains suggest that neither the A2 nor B2 domain is involved in this PSG1 function. Overall, the results presented here indicate that direct binding of the B2 domain of PSG1 to LAP of TGF-β1 results in the activation of SLC and that the interaction between PSG1 and SLC is not mediated through the LSKL sequence in LAP. In addition, we showed that amino acids LYHY in the CC′ loop of the N-terminal domain are involved in the ability of PSG1 to induce production of latent TGF-β1 by macrophages.
Acknowledgments
We thank Malgorzata G. Norton and Michael Kennedy Ph.D. (Center for Biologics Evaluation and Research, Food and Drug Administration) for the use of Biacore instruments and Michael Flora (Uniformed Services University of the Health Sciences) for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Award R21AI101979 from NIAID.
- PSG
- pregnancy-specific glycoprotein
- CEACAM
- carcinoembryonic antigen-related cell adhesion molecule
- LAP
- latency-associated peptide
- SLC
- small latent complex
- TSP-1
- thrombospondin-1
- SPR
- surface plasmon resonance
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- MLEC
- mink lung epithelial cell
- TNX
- tenascin X.
REFERENCES
- 1. Teglund S., Olsen A., Khan W. N., Frängsmyr L., Hammarström S. (1994) The pregnancy-specific glycoprotein (PSG) gene cluster on human chromosome 19: fine structure of the 11 PSG genes and identification of 6 new genes forming a third subgroup within the carcinoembryonic antigen (CEA) family. Genomics 23, 669–684 [DOI] [PubMed] [Google Scholar]
- 2. McLellan A. S., Fischer B., Dveksler G., Hori T., Wynne F., Ball M., Okumura K., Moore T., Zimmermann W. (2005) Structure and evolution of the mouse pregnancy-specific glycoprotein (Psg) gene locus. BMC Genomics 6, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shanley D. K., Kiely P. A., Golla K., Allen S., Martin K., O'Riordan R. T., Ball M., Aplin J. D., Singer B. B., Caplice N., Moran N., Moore T. (2013) Pregnancy-specific glycoproteins bind integrin αIIbβ3 and inhibit the platelet-fibrinogen interaction. PLoS ONE 8, e57491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Snyder S. K., Wessner D. H., Wessells J. L., Waterhouse R. M., Wahl L. M., Zimmermann W., Dveksler G. S. (2001) Pregnancy-specific glycoproteins function as immunomodulators by inducing secretion of IL-10, IL-6 and TGF-β1 by human monocytes. Am. J. Reprod. Immunol. 45, 205–216 [DOI] [PubMed] [Google Scholar]
- 5. Martínez F. F., Knubel C. P., Sánchez M. C., Cervi L., Motrán C. C. (2012) Pregnancy-specific glycoprotein 1a activates dendritic cells to provide signals for Th17-, Th2-, and Treg-cell polarization. Eur. J. Immunol. 42, 1573–1584 [DOI] [PubMed] [Google Scholar]
- 6. Motrán C. C., Díaz F. L., Gruppi A., Slavin D., Chatton B., Bocco J. L. (2002) Human pregnancy-specific glycoprotein 1a (PSG1a) induces alternative activation in human and mouse monocytes and suppresses the accessory cell-dependent T cell proliferation. J. Leukoc. Biol. 72, 512–521 [PubMed] [Google Scholar]
- 7. Pihl K., Larsen T., Laursen I., Krebs L., Christiansen M. (2009) First trimester maternal serum pregnancy-specific β-1-glycoprotein (SP1) as a marker of adverse pregnancy outcome. Prenat. Diagn. 29, 1256–1261 [DOI] [PubMed] [Google Scholar]
- 8. Waterhouse R., Ha C., Dveksler G. S. (2002) Murine CD9 is the receptor for pregnancy-specific glycoprotein 17. J. Exp. Med. 195, 277–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sulkowski G. N., Warren J., Ha C. T., Dveksler G. S. (2011) Characterization of receptors for murine pregnancy specific glycoproteins 17 and 23. Placenta 32, 603–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lisboa F. A., Warren J., Sulkowski G., Aparicio M., David G., Zudaire E., Dveksler G. S. (2011) Pregnancy-specific glycoprotein 1 induces endothelial tubulogenesis through interaction with cell surface proteoglycans. J. Biol. Chem. 286, 7577–7586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li M. O., Wan Y. Y., Sanjabi S., Robertson A. K., Flavell R. A. (2006) Transforming growth factor-β regulation of immune responses. Annu. Rev. Immunol. 24, 99–146 [DOI] [PubMed] [Google Scholar]
- 12. Annes J. P., Munger J. S., Rifkin D. B. (2003) Making sense of latent TGFβ activation. J. Cell Sci. 116, 217–224 [DOI] [PubMed] [Google Scholar]
- 13. Glinka Y., Prud'homme G. J. (2008) Neuropilin-1 is a receptor for transforming growth factor β-1, activates its latent form, and promotes regulatory T cell activity. J. Leukoc. Biol. 84, 302–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ohno M., Cooke J. P., Dzau V. J., Gibbons G. H. (1995) Fluid shear stress induces endothelial transforming growth factor β-1 transcription and production. Modulation by potassium channel blockade. J. Clin. Invest. 95, 1363–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Blois S. M., Sulkowski G., Tirado-González I., Warren J., Freitag N., Klapp B. F., Rifkin D., Fuss I., Strober W., Dveksler G. S. (2014) Pregnancy-specific glycoprotein 1 (PSG1) activates TGF-β and prevents dextran sodium sulfate (DSS)-induced colitis in mice. Mucosal Immunol. 7, 348–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fadok V. A., Bratton D. L., Konowal A., Freed P. W., Westcott J. Y., Henson P. M. (1998) Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-β, PGE2, and PAF. J. Clin. Invest. 101, 890–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xiong W., Frasch S. C., Thomas S. M., Bratton D. L., Henson P. M. (2013) Induction of TGF-β1 synthesis by macrophages in response to apoptotic cells requires activation of the scavenger receptor CD36. PLoS ONE 8, e72772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Toossi Z., Young T. G., Averill L. E., Hamilton B. D., Shiratsuchi H., Ellner J. J. (1995) Induction of transforming growth factor β1 by purified protein derivative of Mycobacterium tuberculosis. Infect. Immun. 63, 224–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Toossi Z., Hirsch C. S., Hamilton B. D., Knuth C. K., Friedlander M. A., Rich E. A. (1996) Decreased production of TGF-β1 by human alveolar macrophages compared with blood monocytes. J. Immunol. 156, 3461–3468 [PubMed] [Google Scholar]
- 20. Abe M., Harpel J. G., Metz C. N., Nunes I., Loskutoff D. J., Rifkin D. B. (1994) An assay for transforming growth factor-β using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal. Biochem. 216, 276–284 [DOI] [PubMed] [Google Scholar]
- 21. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 22. Guex N., Peitsch M. C., Schwede T. (2009) Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis 30, S162–S173 [DOI] [PubMed] [Google Scholar]
- 23. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 24. Bhattacharya D., Cheng J. (2013) 3Drefine: consistent protein structure refinement by optimizing hydrogen bonding network and atomic-level energy minimization. Proteins 81, 119–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Annes J. P., Chen Y., Munger J. S., Rifkin D. B. (2004) Integrin αVβ6-mediated activation of latent TGF-β requires the latent TGF-β binding protein-1. J. Cell Biol. 165, 723–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ribeiro S. M., Poczatek M., Schultz-Cherry S., Villain M., Murphy-Ullrich J. E. (1999) The activation sequence of thrombospondin-1 interacts with the latency-associated peptide to regulate activation of latent transforming growth factor-β. J. Biol. Chem. 274, 13586–13593 [DOI] [PubMed] [Google Scholar]
- 27. Osborne J. C., Jr., Rosen S. W., Nilsson B., Calvert I., Bohn H. (1982) Physicochemical studies of pregnancy-specific β1-glycoprotein: unusual ultracentrifugal and circular dichroic properties. Biochemistry 21, 5523–5528 [DOI] [PubMed] [Google Scholar]
- 28. Alcaraz L. B., Exposito J. Y., Chuvin N., Pommier R. M., Cluzel C., Martel S., Sentis S., Bartholin L., Lethias C., Valcourt U. (2014) Tenascin-X promotes epithelial-to-mesenchymal transition by activating latent TGF-β. J. Cell Biol. 205, 409–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou G. Q., Hammarström S. (2001) Pregnancy-specific glycoprotein (PSG) in baboon (Papio hamadryas): family size, domain structure, and prediction of a functional region in primate PSGs. Biol. Reprod. 64, 90–99 [DOI] [PubMed] [Google Scholar]
- 30. Lee J. N., Grudzinskas J. G., Chard T. (1979) Circulating levels of pregnancy proteins in early and late pregnancy in relation to placental tissue concentration. Br. J. Obstet. Gynaecol. 86, 888–890 [DOI] [PubMed] [Google Scholar]
- 31. Zhou G. Q., Baranov V., Zimmermann W., Grunert F., Erhard B., Mincheva-Nilsson L., Hammarström S., Thompson J. (1997) Highly specific monoclonal antibody demonstrates that pregnancy-specific glycoprotein (PSG) is limited to syncytiotrophoblast in human early and term placenta. Placenta 18, 491–501 [DOI] [PubMed] [Google Scholar]
- 32. Tangerås L. H., Stødle G. S., Olsen G. D., Leknes A. H., Gundersen A. S., Skei B., Vikdal A. J., Ryan L., Steinkjer B., Myklebost M. F., Langaas M., Austgulen R., Iversen A. C. (2014) Functional Toll-like receptors in primary first-trimester trophoblasts. J. Reprod. Immunol. 106, 88–99 [DOI] [PubMed] [Google Scholar]
- 33. Fardini Y., Chung P., Dumm R., Joshi N., Han Y. W. (2010) Transmission of diverse oral bacteria to murine placenta: evidence for the oral microbiome as a potential source of intrauterine infection. Infect. Immun. 78, 1789–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Aagaard K., Ma J., Antony K. M., Ganu R., Petrosino J., Versalovic J. (2014) The placenta harbors a unique microbiome. Sci. Transl. Med. 6, 237ra265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Radaev S., Zou Z., Huang T., Lafer E. M., Hinck A. P., Sun P. D. (2010) Ternary complex of transforming growth factor-β1 reveals isoform-specific ligand recognition and receptor recruitment in the superfamily. J. Biol. Chem. 285, 14806–14814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bailly S., Brand C., Chambaz E. M., Feige J. J. (1997) Analysis of small latent transforming growth factor-β complex formation and dissociation by surface plasmon resonance. Absence of direct interaction with thrombospondins. J. Biol. Chem. 272, 16329–16334 [DOI] [PubMed] [Google Scholar]
- 37. Shi M., Zhu J., Wang R., Chen X., Mi L., Walz T., Springer T. A. (2011) Latent TGF-β structure and activation. Nature 474, 343–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gray-Owen S. D., Blumberg R. S. (2006) CEACAM1: contact-dependent control of immunity. Nat. Rev. Immunol. 6, 433–446 [DOI] [PubMed] [Google Scholar]
- 39. Dveksler G. S., Dieffenbach C. W., Cardellichio C. B., McCuaig K., Pensiero M. N., Jiang G. S., Beauchemin N., Holmes K. V. (1993) Several members of the mouse carcinoembryonic antigen-related glycoprotein family are functional receptors for the coronavirus mouse hepatitis virus-A59. J. Virol. 67, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]