

Background: Insulin stimulates glucose uptake by triggering TUG proteolysis, which liberates intracellular storage vesicles containing GLUT4.
Results: TUG acetylation modulates its interaction with Golgi matrix proteins and enhances its function to trap GLUT4 storage vesicles within unstimulated cells. SIRT2 modulates TUG acetylation and controls insulin sensitivity in vivo.
Conclusion: TUG acetylation promotes GLUT4 accumulation in insulin-responsive vesicles.
Significance: Nutritional status modulates insulin-stimulated glucose uptake.
Keywords: Adipocyte, Glucose Transport, Glucose Transporter Type 4 (GLUT4), Insulin, Membrane Trafficking, Nutrition, Protein Translocation, Sirtuin, Skeletal Muscle Metabolism
Abstract
Insulin causes the exocytic translocation of GLUT4 glucose transporters to stimulate glucose uptake in fat and muscle. Previous results support a model in which TUG traps GLUT4 in intracellular, insulin-responsive vesicles termed GLUT4 storage vesicles (GSVs). Insulin triggers TUG cleavage to release the GSVs; GLUT4 then recycles through endosomes during ongoing insulin exposure. The TUG C terminus binds a GSV anchoring site comprising Golgin-160 and possibly other proteins. Here, we report that the TUG C terminus is acetylated. The TUG C-terminal peptide bound the Golgin-160-associated protein, ACBD3 (acyl-CoA-binding domain-containing 3), and acetylation reduced binding of TUG to ACBD3 but not to Golgin-160. Mutation of the acetylated residues impaired insulin-responsive GLUT4 trafficking in 3T3-L1 adipocytes. ACBD3 overexpression enhanced the translocation of GSV cargos, GLUT4 and insulin-regulated aminopeptidase (IRAP), and ACBD3 was required for intracellular retention of these cargos in unstimulated cells. Sirtuin 2 (SIRT2), a NAD+-dependent deacetylase, bound TUG and deacetylated the TUG peptide. SIRT2 overexpression reduced TUG acetylation and redistributed GLUT4 and IRAP to the plasma membrane in 3T3-L1 adipocytes. Mutation of the acetylated residues in TUG abrogated these effects. In mice, SIRT2 deletion increased TUG acetylation and proteolytic processing. During glucose tolerance tests, glucose disposal was enhanced in SIRT2 knock-out mice, compared with wild type controls, without any effect on insulin concentrations. Together, these data support a model in which TUG acetylation modulates its interaction with Golgi matrix proteins and is regulated by SIRT2. Moreover, acetylation of TUG enhances its function to trap GSVs within unstimulated cells and enhances insulin-stimulated glucose uptake.
Introduction
Insulin mobilizes GLUT4 (glucose transporter 4) glucose transporters from a sequestered intracellular compartment into a cell surface-recycling pathway in fat and muscle. This action results in the translocation of GLUT4 to the cell surface and accelerates glucose uptake (1). The increase in glucose uptake can be as much as 20- or 30-fold in primary adipocytes. The remarkable magnitude of this effect is made possible because GLUT4 is so efficiently sequestered away from the plasma membrane in unstimulated cells. Only a few percent of total cellular GLUT4 is present in plasma membranes of unstimulated adipocytes; therefore, insulin can increase this amount severalfold. Modulation of the mechanism that controls GLUT4 intracellular retention, in the basal or unstimulated state, may have profound effects on the magnitude of the subsequent insulin response.
Basal GLUT4 sequestration is accomplished by the targeting of GLUT4 to specialized intracellular vesicles, termed “GLUT4 storage vesicles” (GSVs).5 Also called “insulin-responsive vesicles” or the “GLUT4 storage compartment,” GSVs are formed in a cell type-specific manner and are mobilized to the cell surface within minutes of insulin stimulation (2–5). These nonendosomal vesicles are thought to exist as a preformed pool within unstimulated cells (2, 6). They are small (∼60-nm diameter) and have a characteristic set of cargo proteins, including the insulin-regulated aminopeptidase, IRAP. Some data suggest that GLUT4 and IRAP do not accumulate in a pool of insulin-responsive GSVs in unstimulated muscle and adipose of individuals with insulin resistance (5, 7, 8). Nevertheless, despite the medical importance of understanding GSVs, the regulated trafficking of these vesicles has remained enigmatic. One impediment to progress has been the cell type specificity of basal GLUT4 sequestration, which is incompletely replicated in cultured cell lines and can be reduced further by experimental manipulations (9–11). A second issue is that GLUT4 is distributed among multiple intracellular compartments and to two different exocytic pathways arising from GSVs and endosomes (12, 13). The small size of GSVs makes these vesicles difficult to detect using total internal reflection fluorescence microscopy, so that endosome-derived GLUT4 may be taken for that present in GSVs (14). Altogether, the nature and regulation of GSVs continues to be debated.
The TUG (tether containing a UBX domain for GLUT4) protein, encoded by the Aspscr1 gene, was proposed as the first molecular marker for GSVs and was proposed to regulate the basal intracellular retention and insulin-stimulated release of these vesicles (12, 15–18). TUG binds GLUT4 through its N-terminal region and can simultaneously bind the Golgi proteins, PIST and Golgin-160, through its C-terminal region (17). It is thus thought to trap GSVs intracellularly by linking them to the cis-Golgi matrix in unstimulated cells (17, 19). Insulin then liberates GSVs by triggering site-specific endoproteolytic cleavage of TUG, which separates regions that bind GLUT4 from regions that bind Golgi proteins (17, 18). Data imply that this action accounts for a substantial portion of the acute effect of insulin in fat and muscle (14). During ongoing insulin exposure, GLUT4 and other GSV cargos recycle to the cell surface from endosomes and bypass a TUG-regulated compartment (12). This explains how insulin causes a dose-dependent discharge of GSVs from a sequestered pool into a cell surface-recycling pathway (20). Insulin signals acutely through the TC10α GTPase and its effector, PIST, to stimulate TUG cleavage (17, 18). This signal may be transient, and it is coordinated with signals through Akt2 to AS160/Tbc1D4 and Tbc1D1 (1, 14). These proteins modulate Rab GTPases to direct GSV trafficking and to return endocytosed GLUT4 to the cell surface directly from endosomes during ongoing stimulation (12, 13). This model shows how sustained insulin action can occur without ongoing TUG protein cleavage and new synthesis. Moreover, understanding how TUG controls GSVs will be critical to learn how insulin sensitivity may be modulated.
The present work clarifies how TUG sequesters GSVs in an insulin-responsive configuration. We report that the TUG C-terminal peptide is acetylated, which modulates the interaction of TUG with Golgi proteins, including ACBD3 (acyl-CoA binding domain-containing 3). Acetylation enhances insulin-responsive trafficking of GSV cargos. TUG acetylation is regulated by SIRT2, a NAD+-dependent deacetylase, which controls TUG proteolytic processing and glucose homeostasis in mice. The data support the notion that GSVs are controlled by acetylated TUG and that SIRT2 modulates insulin sensitivity by regulating this pool of GSVs.
EXPERIMENTAL PROCEDURES
Reagents, Cell Culture, and Molecular Biology
Antisera directed to the C terminus of TUG were described previously (15). Antisera to the N-terminal region of TUG were raised in rabbits using a peptide comprising residues 64–92 of murine TUG protein. Antisera to IRAP were similarly generated using a peptide containing residues 17–33 of murine IRAP. Other antibodies used include those directed to HA and Myc epitope tags (Covance or Thermo Scientific), GFP (Covance, MMS-118P), FLAG M2 (Agilent or Sigma), Acetyl-lysine (rabbit polyclonal antibody from Cell Signaling Technology), ACBD3 (mouse monoclonal from Santa Cruz Biotechnology, Inc.), GAPDH (Millipore), PIST (Abcam, ab109119), Golgin-160 (Santa Cruz Biotechnology and also a kind gift from Dr. C. Machamer), transferrin receptor (Sigma), Hsp90 (BD Transduction Laboratories), GST (Millipore), and SIRT2 (rabbit polyclonal, catalog no. 09-843, Millipore).
Cells were cultured in high glucose DMEM GlutaMAX medium (Invitrogen) containing 10% bovine growth serum (HyClone). Transient transfection of HEK293 cells was done using Lipofectamine 2000 (Invitrogen). Cells were typically lysed 36 h after transfection. Stable 3T3-L1 cell lines were generated by retroviral infection of preadipocytes. A shRNA was used to partially deplete endogenous TUG, and shRNA-resistant wild type or mutated TUG proteins were expressed at similar abundances (16, 17). A GLUT4 reporter was expressed, as described (9). To deplete ACBD3, the pSIREN-RetroQ vector (Clontech) was used for retroviral infection of 3T3-L1 cells. Virus production was done by transient transfection of 293 cells, together with pCL-Eco helper plasmid (16, 21). The shRNAs targeted the sequences CGGCATATGTTGCGTCCCA (shRNA 10) and GGTTAAAAGCAGTTCGGAA (shRNA 12). A nontargeting control shRNA was also used, and virus production, infection, and puromycin selection were done in parallel. 3T3-L1 adipocytes were differentiated in 10% fetal bovine serum containing supplements as described previously (16), except that 2 μm rosiglitazone (Cayman) was added to the differentiation medium.
A plasmid encoding pEGFPN1-ACBD3, used for transient overexpression in 293 cells, was a kind gift from Dr. Weimin Zhong (22). A plasmid encoding human FLAG-SIRT2 was obtained from Addgene (plasmid 13813) (23). To stably overexpress ACBD3, HA-ACBD3, or untagged SIRT2 in 3T3-L1 cells, cDNAs were cloned in the pBICD4 retrovirus vector, and infection and selection using a FACS were done as described previously (15, 17, 24). Similar effects on GLUT4 trafficking were observed using ACBD3 with and without the HA tag.
Immunoprecipitation, Immunoblotting, and Subcellular Fractionation
TUG antibody was cross-linked to protein A-Sepharose beads (GE Healthcare) using disuccinimidyl suberate (Pierce) essentially as described (Crosslink IP kit, Pierce catalog no. 26147). Affinity matrices to precipitate GFP- or FLAG-tagged proteins were purchased from Chromotek and Sigma, respectively. For coprecipitation, TNET lysis buffer (20 mm Tris, pH 7.4, 150 mm NaCl, 2 mm EDTA, 1% Triton X-100) was used with Complete protease inhibitors (Roche Applied Science). For some experiments, 3.5 mg/ml iodoacetamide and PMSF (Sigma) were added to the lysis buffer. To coimmunoprecipitate endogenous TUG and ACBD3, TNET lysis buffer was diluted 2-fold. To detect TUG acetylation, cells were lysed in radioimmune precipitation buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Nonidet P-40) with Complete protease inhibitors. Where indicated, cells were pretreated overnight using 3 mg/ml nicotinamide (Sigma). For all immunoprecipitations, cells were lysed on ice for 20 min with intermittent vortexing, and insoluble material was pelleted at 16,000 × g for 10 min. at 4 °C. Affinity matrices were added for a few h to overnight and then pelleted at 1000 × g and washed 3–6 times with lysis buffer. Proteins were eluted in 1× LDS NuPAGE sample buffer (Invitrogen) for 20 min at 37 °C and then reduced with 5% 2-mercaptoethanol (Sigma) and heated to 65 °C for 15 min. prior to SDS-PAGE, except when immunoblotting GLUT4.
For immunoblots, proteins were separated on 4–12% bis-tris polyacrylamide gels (Invitrogen) in MOPS buffer and then transferred to nitrocellulose using a semidry transfer apparatus (Bio-Rad) with NuPAGE transfer buffer (Invitrogen). Proteins were detected on film using chemiluminescence or on a LI-COR Odyssey imaging system using infrared fluorescence. Subcellular fractionation to isolate light and heavy microsomes and plasma membrane fractions was performed as described previously (9, 16).
Confocal Microscopy
3T3-L1 adipocytes were serum-starved and then treated with or without 160 nm insulin for 8 min. Cells were fixed for 25 min using 4% paraformaldehyde (Electron Microscopy Sciences) and then permeabilized for 5 min. using 0.1% Triton X-100. Nonspecific staining was blocked using 4% normal goat serum for 30 min, and then ACBD3 was stained using a mouse monoclonal antibody (Santa Cruz Biotechnology) at 1:200 for 1 h. After a brief wash using phosphate-buffered saline (PBS), 0.1% Tween 20, cells were blocked again using normal goat serum and then incubated for 45 min with a 1:200 dilution of Dylight549-conjugated goat anti-mouse IgG secondary antibody (Jackson Immunoresearch). Cells were washed again and then mounted using Prolong Gold (Invitrogen). Images were obtained using a Zeiss 510 Meta confocal microscope using a ×63/1.20 water immersion objective with the pinhole set for 1.0 Airy unit. For microscopy of unpermeabilized cells, to detect Myc-tagged GLUT4 at the cell surface, fixation was limited to 5 min, and the permeabilization step was omitted. After blocking with 4% normal goat serum, cell surface Myc was detected using 9E10 antibody (Covance) at 1:200 for 1 h. Cells were washed using PBS, blocked again, and then incubated with Dylight549-conjugated goat anti-mouse IgG secondary antibody for 40 min. After a final wash in PBS, cells were mounted and imaged as above.
Flow Cytometry
Measurement of GLUT4 translocation using a GLUT4–7myc-GFP reporter was done essentially as described (9). 3T3-L1 cells were differentiated in 10-cm dishes, plated to 96-well tissue culture dishes (Corning, Inc.) on day 6 of differentiation, and starved overnight in serum-free medium. Cells were stimulated as indicated using 160 nm insulin for 7 min and then washed with ice-cold PBS and fixed for 5 min using 4% paraformaldehyde. Cells were stained for cell surface Myc using 9E10 antibody (Covance or Thermo) at 1:200 in PBS containing 4% normal goat serum for 1 h. Cells were washed three times in PBS, blocked for 10 min in 4% normal goat serum, and stained using phycoerythrin-conjugated anti-mouse IgG secondary antibody (Jackson Immunoresearch) at a 1:200 dilution. After washing, cells were treated with 1 mg/ml collagenase (Sigma) in PBS for 7–10 min at 37 °C. The collagenase was diluted in 10 volumes of PBS, and the cell suspension was analyzed using BD FACSCalibur flow cytometers. Adipocytes were gated using FloJo essentially as described (11), and the FL4 signal was used to exclude autofluorescent debris. For each data point, the fluorescent signals from 2–4 replicate wells were averaged and counted as a single experiment. Typically, 3–5 of these experiments were used to calculate the overall mean, S.E., and statistical tests (based on the number of experiments).
Cell Surface Biotinylation
Fully differentiated 3T3-L1 adipocytes were serum-starved overnight. Cells were washed in PBS and stimulated with or without insulin in KRP buffer (12.5 mm HEPES, 120 mm NaCl, 6 mm KCl, 1.2 mm MgSO4, 1.0 mm CaCl2, 0.6 mm Na2HPO4, 0.4 mm NaH2PO4, 2.5 mm d-glucose, pH 7.4) for 3 min at 37 °C, and then NHS-SS-Biotin (Thermo) was added to a final concentration of 0.5 mg/ml for an additional 12 min at 37 °C (similar to Ref. 25). Biotinylation was quenched by the addition of 1 m Tris stock to a final concentration of 50 mm. Plates were cooled to 4 °C, washed with quenching buffer (50 mm Tris, 10 mm EDTA, 150 mm NaCl), and then lysed using TNET buffer containing Complete protease inhibitors and PMSF (as above). Insoluble debris was pelleted by centrifugation at 16,000 × g for 10 min at 4 °C, and then biotinylated proteins were purified on Neutravidin beads (Thermo). Proteins were eluted in 1× NuPAGE sample buffer (Invitrogen) containing 50 mm DTT, with rotation at room temperature for 1 h. Samples were analyzed by immunoblotting as described above.
Recombinant Protein Expression and Pull-downs
GST-tagged ACBD3 constructs were a kind gift from Dr. Weimin Zhong (22) and were expressed using the pGEX-2T vector in BL21(DE3) GOLD E. coli (Agilent Technologies). Bacterial cultures at 37 °C were induced with 0.5 mm isopropyl-β-d-thiogalactopyranoside when A600 = 0.6. After 5 h, bacteria were lysed in NETN buffer (20 mm Tris-HCl, pH 8.0, 140 mm NaCl, 1 mm EDTA, 0.5% Nonidet P-40) containing Complete protease inhibitors (Roche Applied Science) and PMSF. Bacterial lysates were kept on ice and were sonicated for a total of 2.5 min in 10-s on/off cycles, and insoluble debris was pelleted at 16,000 × g for 30–60 min. Proteins in the supernatant were purified using glutathione-Sepharose 4B beads (GE Health Sciences) and washed in NETN buffer with 500 mm NaCl added. GST-tagged proteins were eluted in NETN containing 20 mm reduced glutathione.
For peptide pull-down experiments using recombinant proteins, purified GST-tagged proteins were mixed with biotinylated TUG C-terminal peptides that had been previously bound to streptavidin-agarose beads (Sigma). The peptides used were biotin-Ahx-GGTAQPVKRSLGKVPKWLKLPASKR-COOH, biotin-Ahx-GGTAQPVKRSLGKVPKWLKLPASK*R-COOH, and biotin-Ahx-GGTAQPVK*RSLGK*VPK*WLK*LPASK*R-COOH, where Ahx indicates an aminohexanoic acid linker and K* indicates acetyl-lysine. Peptide synthesis and purification were done both at the Yale Keck Biotechnology Resource facility and at Lifetein, Inc. (South Plainfield, NJ). After incubation overnight at 4 °C, streptavidin beads were washed, and bound proteins were eluted in 1× LDS sample buffer (Invitrogen) and analyzed by immunoblotting. Peptide pull-down experiments using cell lysates were done with the same peptides. These were incubated with 3T3-L1 adipocyte lysates prepared using 0.5% Nonidet P-40, 40 mm Tris, pH 7.4, 100 mm NaCl, 5 mm EDTA, 3 mg/ml nicotinamide, and protease inhibitors as above. Binding was permitted to occur overnight at 4 °C, and then beads were pelleted and washed five times using spin columns, and proteins were eluted in sample buffer and analyzed by SDS-PAGE and immunoblotting.
In Vitro Deacetylation
The acetylated, biotin-labeled biotinylated TUG C-terminal peptide above was used at 1.8 mg/ml final concentration. As a control, acetylated, biotin-labeled human p53 peptide (residues 368–393, containing AcK382; Anaspec) was used at similar concentration. Peptides were incubated with 6 μg of recombinant SIRT2 or 6 units of recombinant SIRT1 (Cayman Chemical, catalog nos. 10011191 and 10011190, respectively) in final reaction volumes of 50 μl. The reactions were done in 25 mm Tris, pH 7.4, 1 mm MgCl2, 75 mm NaCl, and 10 mm NAD+ at 37 °C for the indicated times. Aliquots (5 μl) were spotted onto nitrocellulose membranes, which were then blocked for 30 min using 5% nonfat milk in PBS, 0.1% Tween 20. Membranes were probed to detect acetyl-lysine as described above for immunoblotting, with detection on film. Control membranes were blocked using 5% BSA, and peptides were detected on film using HRP-conjugated streptavidin (Pierce).
Animal Studies
All studies were approved by the Yale University Institutional Animal Care and Use Committee. Age-matched SIRT2 knock-out (KO) and control mice were obtained from Jackson Laboratories (stock numbers 012772 and 000664, respectively). All mice were male and were on a C57BL/6J background. Mice were maintained on regular chow (Harlan-Teklad 2018) in the Yale Animal Resources Center and were kept on a standard 12-h light/dark cycle. Intraperitoneal glucose tolerance tests were performed on 13-week old mice at the Yale Mouse Metabolic Phenotyping Center essentially as described (26). Mice were fasted for 6 h, and then 1 mg/kg body weight glucose (as 10% dextrose) was administered intraperitoneally. Blood samples were taken by tail bleeding at 0, 15, 30, 45, 60, 90, and 120 min after the injection and were used for measurement of glucose (using a YSI 2700D glucose analyzer, YSI Inc.) and insulin (using an ELISA, Millipore Inc.). Body composition analyses were performed using a Bruker Minispec analyzer, as described previously (18).
For measurement of TUG acetylation, 14-week-old mice were fasted 6 h and then sacrificed. Lysates were prepared from liver using radioimmune precipitation buffer and a Qiagen TissueLyser II. TUG was immunoprecipitated, and eluted protein was immunoblotted using acetyl-lysine and TUG antibodies. Densitometry was done on film using transillumination (Epson Perfection V700 scanner) or using a LI-COR Odyssey imaging system. ImageJ software was used for quantification.
For measurement of TUG processing and GLUT4 abundance, 12-week-old SIRT2 KO and WT control mice were fasted for 4–6 h and then treated by intraperitoneal injection of insulin (8 units/kg) and glucose (1 g/kg) or an equivalent volume (0.3 ml) of PBS. After 20 min, mice were anesthetized and sacrificed by cervical dislocation. Quadriceps muscles were excised, snap-frozen in liquid N2, and stored at −80 °C. Tissues were homogenized in ice-cold radioimmune precipitation buffer using a Qiagen TissueLyser II, as described (18). Immunoblotting and densitometry was performed as above.
Statistical Analysis
Values are expressed as mean ± S.E. Significance was assessed using an unpaired, two-tailed Student's t test for two groups or by one-way analysis of variance with Bonferroni correction for multiple comparisons (Prism, GraphPad Software). Differences were considered significant at p < 0.05.
RESULTS
Previous data imply that to mediate GLUT4 intracellular retention, the TUG C terminus must bind an “anchoring site” within cells (15). TUG C-terminal regions interact with two Golgi proteins, Golgin-160 and PIST, which bind each other and form part of this site (17, 18, 27). Data sets in the Phosphosite mass spectrometry database identified the penultimate residue in TUG, Lys-549 (murine numbering), as an acetylated residue. Acetylation often functions in metabolic sensing and regulation (28, 29). We considered that acetylation may modulate how TUG links GLUT4 to an anchoring site within unstimulated cells. However, preliminary data suggested that a C-terminally deleted form of TUG still coimmunoprecipitates Golgin-160 and PIST in transfected cells. Therefore, we hypothesized that the TUG C-terminal peptide binds an additional protein and that a complex of anchoring proteins may act together to control the accumulation of GLUT4 in an insulin-responsive compartment.
To test whether TUG is acetylated, we performed acetyl-lysine (AcK) immunoblots on TUG proteins that had been immunoprecipitated from cultured cells. As shown in Fig. 1A, endogenous TUG proteins purified from 293 cells and 3T3-L1 adipocytes contained detectable AcK. Treatment of the cells with nicotinamide, an inhibitor of sirtuin-type deacetylases (30), increased the AcK signal in 3T3-L1 adipocytes without affecting total TUG abundance. We did not observe any change in TUG acetylation when cells were treated with trichostatin A, another deacetylase inhibitor that inhibits non-sirtuin deacetylases (not shown). These data support the hypothesis that endogenous TUG proteins are acetylated in cultured cells.
FIGURE 1.
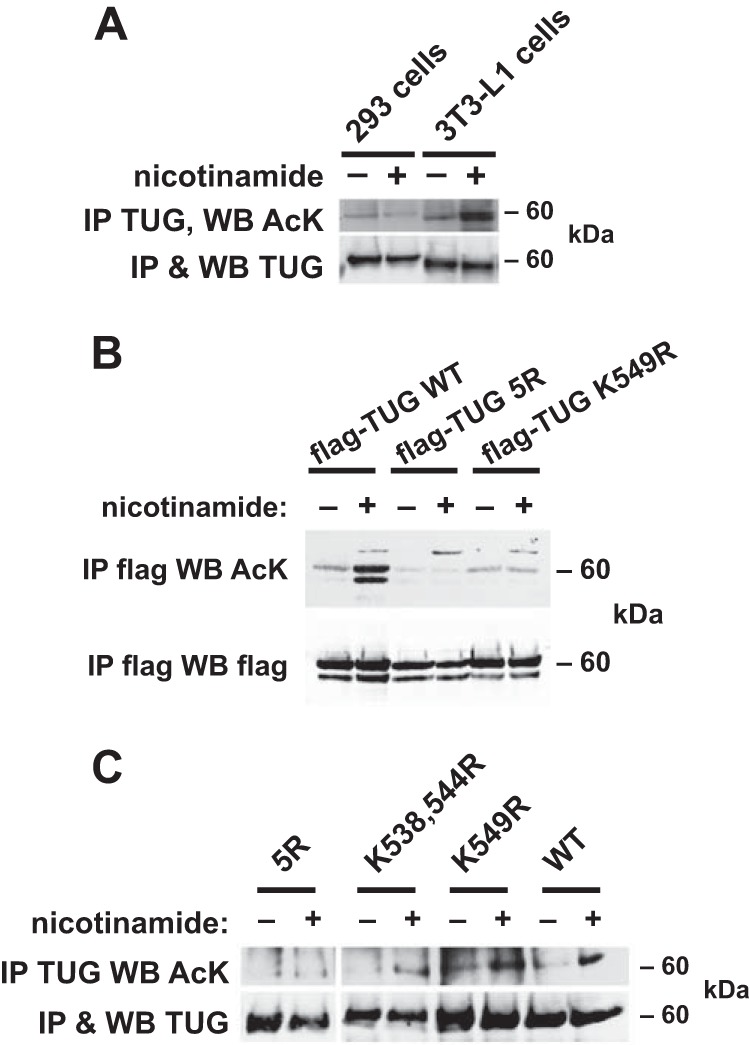
TUG C-terminal residues are acetylated. A, 293 cells or 3T3-L1 adipocytes were treated overnight with nicotinamide, as indicated, and then endogenous TUG protein was immunoprecipitated (IP). Immunoblots (WB) were done to detect AcK and TUG, as indicated. B, FLAG-tagged WT or mutated TUG proteins were transfected in 293 cells, immunoprecipitated after overnight treatment of the cells with nicotinamide, and immunoblotted to detect AcK, as indicated. TUG 5R contains arginine-to-lysine mutations at five positions in the C-terminal peptide; TUG K549R is mutated at one of these sites. The experiment was repeated three times with consistent results. C, immunoprecipitations were done using 3T3-L1 adipocytes in which the specified forms of TUG were expressed in cells partially depleted of endogenous TUG using an shRNA. Cells were treated overnight with nicotinamide, and eluates were immunoblotted to detect TUG and AcK as indicated. The experiment was repeated twice with consistent results.
To further define the acetylated residues, we transfected wild type and mutated TUG proteins in 293 cells. We initially mutated the lysine at position 549 to arginine, making TUG K549R. This mutation eliminates the potential acetylation site, without markedly altering the charge present when the residue is not acetylated. TUG contains five lysine residues within its C-terminal 18 amino acids, and bioinformatics suggested that these may be present on one face of an α-helix. Because acetylation can involve patches of lysine residues (31), we also made a mutated form of TUG in which all five of these lysines (Lys-533, -538, -541, -544, and -549) are mutated to arginine, termed “TUG 5R.” We expressed WT TUG, TUG 5R, and TUG K549R proteins, containing N-terminal FLAG tags and assessed acetylation by immunoprecipitation followed by immunoblotting. As shown in Fig. 1B, AcK was detected on wild type TUG and was markedly increased by nicotinamide treatment of the cells. By contrast, the AcK signal was undetectable on precipitated TUG 5R, even in cells treated with nicotinamide. Some AcK was detectable on TUG K549R, and the nicotinamide-induced increase was not observed. These data support the idea that TUG is acetylated in transfected 293 cells and that acetylation occurs on Lys-549 and to a lesser extent on other lysines present within the TUG C-terminal peptide. Overexpression may account for the effect of nicotinamide on wild type TUG in transfected 293 cells (Fig. 1B), but not on endogenous TUG in 293 cells (Fig. 1A), if it alters the balance between acetylation and deacetylation. For example, the acetylation machinery may be overwhelmed by the increased TUG abundance in transfected cells, thereby increasing the proportion of deacetylated TUG and leading to a greater effect when deacetylases are inhibited. Excess TUG may also engage sirtuin deacetylases in a promiscuous manner, which could also increase the effect of nicotinamide treatment.
To define what residues are required for TUG acetylation in 3T3-L1 adipocytes, we expressed wild type and mutated TUG proteins using retroviruses. We used 3T3-L1 cells containing an shRNA to partially deplete endogenous TUG proteins, so that the exogenous, shRNA-resistant wild type or mutated TUG proteins were expressed at similar abundances and without epitope tags (16, 17). We termed these cells “TUG WT” cells, “TUG 5R” cells, etc. We treated these cells in the absence or presence of nicotinamide and then immunoprecipitated TUG and immunoblotted AcK on the purified proteins. As shown in Fig. 1C, acetylation was present on TUG and was increased by nicotinamide in TUG WT cells. Similar results were obtained in cells containing TUG K538R,K544R. In cells containing K549R, the nicotinamide-induced increase in acetylation was reduced but not completely eliminated. In TUG 5R 3T3-L1 adipocytes, no acetylation was detectable, even after nicotinamide treatment. We conclude that although Lys-549 may be the main acetylation site, alternative sites can be used when this site is not available. Mutation of all five lysines in the TUG C-terminal peptide was required to eliminate TUG acetylation in 3T3-L1 adipocytes.
We hypothesized that the TUG C terminus binds an intracellular anchoring protein in addition to Golgin-160 and PIST, as noted above. A candidate was ACBD3, a ubiquitously expressed protein that interacts with Golgin-160 and that is also known as GCP60, PAP7, GOCAP1, and GOLPH1 (32–35). We noted that the C terminus of TUG resembles a peptide in the Numb protein that binds directly to ACBD3 (22). To test whether endogenous TUG and ACBD3 proteins interact in 3T3-L1 adipocytes, we used an antibody directed to the TUG N terminus to immunoprecipitate TUG and associated proteins from 3T3-L1 adipocyte lysates. Fig. 2A shows that ACBD3 was copurified with TUG when immune sera were used and not when control, preimmune sera were used. To further test whether ACBD3 colocalizes with GLUT4, we used confocal microscopy to image endogenous ACBD3 and a GFP-tagged GLUT4 reporter protein. As shown in Fig. 2B, punctate colocalization of ACBD3 and GLUT4 was observed in 3T3-L1 adipocytes in the basal state. In insulin-stimulated cells, there seemed to be fewer colocalized punctae. Together, these data support the idea that ACBD3 acts together with Golgin-160 to retain TUG-bound, GLUT4-containing vesicles within 3T3-L1 adipocytes.
FIGURE 2.

TUG binds ACBD3 in 3T3-L1 adipocytes, and its C terminus can mediate this interaction. A, 3T3-L1 adipocyte lysates were subjected to immunoprecipitation (IP) using an immobilized antibody directed to the TUG N terminus or preimmune control. Eluates were immunoblotted (WB) to detect TUG and endogenous ACBD3. B, confocal immunofluorescence microscopy was performed on basal and insulin-stimulated 3T3-L1 adipocytes to detect endogenous ACBD3 and GFP-tagged GLUT4. Colocalized punctae are indicated by arrows. Scale bar, 10 μm. C, recombinant GST-tagged ACBD3 proteins were incubated with streptavidin beads alone or with beads loaded with biotinylated, synthetic peptides corresponding to the TUG C terminus (residues 528–550). Peptides were unacetylated, acetylated at lysine 549, or acetylated at all five lysines present in the peptide (5Ac). Eluted proteins were immunoblotted for GST to detect binding. The experiment was repeated three times with similar results. D, the peptides used in C were immobilized on streptavidin beads and incubated with 3T3-L1 cell lysates. Beads were pelleted and washed, and eluted proteins were analyzed by SDS-PAGE and immunoblotting as indicated. The experiment was repeated twice with similar results. E, wild type TUG or TUG K549R was coexpressed with GFP-tagged Golgin-160 by transient transfection of 293 cells, as indicated. Golgin-160 was immunoprecipitated, and eluted proteins were immunoblotted to detect TUG. The experiment was repeated twice with similar results.
We next tested whether ACBD3 binds directly to the TUG C-terminal peptide. We produced recombinant GST fusion proteins containing ACBD3 residues 1–60 or 1–277 and incubated these with streptavidin beads alone or with streptavidin beads loaded with biotinylated TUG peptides. The peptides corresponded to the C terminus of TUG (residues 528–550) in either unmodified form or with acetyl-lysine at residue 549 (AcK549) or at all five lysines present in the peptide (5Ac). As shown in Fig. 2C, the unmodified TUG peptide bound both recombinant ACBD3 (residues 1–277) and ACBD3 (residues 1–60) proteins. Binding was reduced when the TUG AcK549 peptide was used and was further reduced in pull-downs using the TUG 5Ac peptide. To further test what proteins interact with the acetylated peptide, we used the peptides to bind proteins in 3T3-L1 adipocyte lysates. ACBD3 was detected in pull-down experiments using the unmodified peptide (Fig. 2D). This interaction was reduced when the TUG AcK549 peptide was used and was essentially absent when the TUG 5Ac peptide was used, consistent with data in Fig. 2C. Remarkably, Golgin-160 and PIST were also purified using the unmodified and AcK549 peptides. These proteins were not present in pull-downs using the 5Ac peptide or in the negative control in which beads without peptide were used. Acetylation of Lys-549 did not reduce the amounts of Golgin-160 or PIST that were purified but slightly increased the amount of Golgin-160 that was purified.
Previous data imply that Golgin-160 binds TUG residues 377–550 (17). To further test whether acetylation of TUG Lys-549 might modulate this interaction, we used GFP-tagged Golgin-160 to coimmunoprecipitate wild type TUG or TUG K549R from transfected 293 cells. Fig. 2E shows that the K549R mutation reduced, but did not eliminate, the interaction of TUG and Golgin-160. In addition to precipitating intact TUG, Golgin-160 purified 42- and 54-kDa proteins detected using an antibody to the TUG C terminus. These forms correspond to previously described C-terminal products observed in 3T3-L1 adipocytes (17), which may be produced when TUG is expressed at high levels in transfected 293 cells. The Lys-549 mutation disrupted these bands, suggesting that acetylation at this residue may be required for TUG proteolytic processing. This hypothesis is further supported by data shown below. The main point is that ACBD3 binds to the unmodified TUG C terminus and that acetylation at Lys-549 may shift the binding of TUG away from ACBD3 and toward Golgin-160.
We next studied how TUG acetylation and ACBD3 may regulate GLUT4 trafficking in 3T3-L1 adipocytes. Initially, we performed subcellular fractionation experiments to isolate plasma membrane (PM) and light microsome (LM) fractions. As shown in Fig. 3A, insulin stimulated the translocation of GLUT4 out of the LM fraction and to the PM fraction in cells expressing wild type TUG. In cells containing TUG 5R, the degree to which insulin stimulated GLUT4 translocation was markedly reduced. This was mostly due to reduced abundance of GLUT4 in PM fractions in the insulin-stimulated state. However, we also noted reduced amounts of GLUT4 in the basal LM fraction, which contains GSVs. GLUT4 abundance was also reduced in heavy microsomes (not shown), consistent with previous data showing that GLUT4 stability is reduced in cells with disrupted TUG function (16). The data support the idea that GLUT4 retention in GSVs is impaired in unstimulated TUG 5R cells, compared with TUG WT controls, which accounts for the reduced effect of insulin to stimulate an increase in GLUT4 at the plasma membrane.
FIGURE 3.

TUG C-terminal lysines and ACBD3 act together to enhance GLUT4 translocation. A, PM and LM fractions were isolated from basal and insulin-stimulated 3T3-L1 adipocytes expressing the indicated forms of TUG. GLUT4 was immunoblotted to detect translocation. B, HA-tagged ACBD3 was expressed using a retrovirus in 3T3-L1 adipocytes containing WT or 5R TUG. Whole cell lysates were immunoblotted (WB) using anti-ACBD3 and anti-HA antibodies. C, flow cytometry of a GLUT4 reporter was used to measure ratios of surface/total GLUT4 in basal and insulin-stimulated 3T3-L1 adipocytes containing WT or 5R TUG, with or without ACBD3 overexpression, as indicated (mean ± S.E.; n = 5 in each group). D, cell surface proteins were biotinylated in basal and insulin-stimulated 3T3-L1 adipocytes, as indicated, and then purified on streptavidin beads. Immunoblots were done to detect cell surface and total IRAP and, as a control, cell surface TfR. The insulin-stimulated increase in surface IRAP, normalized to TfR, is indicated. The experiment was repeated twice, with similar results. E, cell surface and total GLUT4 reporter were imaged using confocal microscopy to detect externalized Myc tag and GFP, respectively, in the indicated 3T3-L1 adipocytes. Scale bar, 10 μm. F and G, flow cytometry was used to measure GLUT4 translocation in undifferentiated 3T3-L1 preadipocytes. Cells containing WT or 5R TUG and TUG WT control- and ACBD3-overexpressing cells were analyzed, as indicated (mean ± S.E. (error bars), n = 3–4 in each group).
To better understand how TUG and ACBD3 may cooperate to control GLUT4 trafficking, we used a retrovirus to overexpress ACBD3 in TUG WT or TUG 5R 3T3-L1 adipocytes. Fig. 3B shows that, compared with cells containing only endogenous ACBD3, the ACBD3-overexpressing cells had about a 2-fold increase in ACBD3 abundance. Flow cytometry of a marker encoded by the retrovirus vector showed that nearly all of the cells were infected. Therefore, the increase in ACBD3 abundance is similar in each of the infected cells and does not result from marked overexpression of ACBD3 in a minority of cells within the population.
We performed flow cytometry to measure GLUT4 translocation, using a previously described dual-tagged reporter protein (9). As shown in Fig. 3C, expression of TUG 5R reduced the ability of insulin to stimulate translocation. The decreased translocation was primarily due to reduced cell surface GLUT4 in the insulin-stimulated state, although we occasionally noted increased cell surface GLUT4 in the basal state. Translocation was consistently impaired, similar to data obtained using subcellular fractionation, above, and supporting the idea that TUG 5R impairs GLUT4 accumulation in an insulin-responsive pool of GSVs in unstimulated cells. In TUG WT cells overexpressing ACBD3, we observed an increased effect of insulin to stimulate GLUT4 translocation. This was due to both enhanced intracellular retention of GLUT4 in unstimulated cells and increased translocation to the cell surface after insulin stimulation. These data are consistent with the idea that the pool of insulin-responsive GSVs is enlarged. In TUG 5R cells overexpressing ACBD3, enhanced GLUT4 translocation was not observed. Rather, the effect of insulin to stimulate GLUT4 translocation was reduced, similar to TUG 5R cells not overexpressing ACBD3. Together, the data support the idea that ACBD3 overexpression enhances insulin-responsive GLUT4 translocation in 3T3-L1 adipocytes and that highly insulin-responsive translocation is disrupted by the mutation of acetylation sites at the TUG C terminus.
To test whether ACBD3 overexpression increases the translocation of another GSV cargo, we assessed IRAP translocation using a cell surface biotinylation assay. As shown in Fig. 3D, IRAP translocation was enhanced in 3T3-L1 adipocytes overexpressing ACBD3, compared with control cells, as long as wild type TUG was present. In cells containing TUG 5R, IRAP translocation was reduced, both in the absence and presence of ACBD3 overexpression. The effect of TUG 5R on IRAP translocation is further quantified in data shown below. To further study effects of ACBD3 overexpression on GLUT4 translocation, we used confocal microscopy to image cell surface and total GLUT4 reporter proteins. Fig. 3E shows that GLUT4 translocation was enhanced in 3T3-L1 adipocytes overexpressing ACBD3 and containing wild type TUG. In cells containing TUG 5R, insulin-stimulated translocation was diminished, regardless of whether ACBD3 was overexpressed. These data are consistent with data in Fig. 3, C and D. Together, the data imply that ACBD3 overexpression increases the size of the insulin-responsive GSV pool and that this effect occurs only if acetylatable residues are present in the TUG C-terminal peptide. Because ACBD3 binds preferentially to the unacetylated TUG C terminus, the effect of ACBD3 overexpression to enhance translocation implies that ACBD3 acts together with other proteins, including Golgin-160 and PIST.
Insulin-responsive GSVs and TUG-regulated vesicles are both observed in a differentiation-dependent manner in 3T3-L1 adipocytes and not in undifferentiated preadipocytes, consistent with the idea that TUG regulates GSVs (3, 10, 12). We anticipated that GLUT4 trafficking would be unaffected in TUG 5R preadipocytes, compared with TUG WT preadipocytes. Data in Fig. 3F support this prediction, using the flow cytometry assay to measure GLUT4 trafficking. Similarly, we hypothesized that ACBD3 overexpression would not enhance GLUT4 translocation in undifferentiated 3T3-L1 preadipocytes, in contrast to its effect in differentiated 3T3-L1 adipocytes. Supporting this prediction, GLUT4 targeting was similar in TUG WT preadipocytes with and without ACBD3 overexpression, as shown in Fig. 3G. In both of these experiments, insulin stimulated a 2–2.5-fold increase in cell surface GLUT4 in undifferentiated 3T3-L1 cells, similar to previous data using this assay (9). The data are consistent with the idea that TUG and ACBD3 regulate the intracellular accumulation of a differentiation-dependent pool of insulin-responsive GSVs in 3T3-L1 cells.
To test whether ACBD3 is required for full intracellular retention of GSV cargos in unstimulated cells, we used retroviruses to express shRNAs in 3T3-L1 adipocytes. Two different shRNAs, designated shRNAs 10 and 12, effectively depleted ACBD3 (Fig. 4A). We assessed the targeting of GLUT4 and IRAP in these cells using cell surface biotinylation (25). This approach enabled us to study the endogenous IRAP protein as well as the GLUT4 reporter (which can be biotinylated on lysine residues within the exofacial Myc tag). As shown in Fig. 4B, ACBD3 depletion caused an increase in surface-exposed GLUT4, both in the basal and insulin-stimulated states. Independent replicates of this experiment were quantified and plotted in Fig. 4C. Data in Fig. 4D show that IRAP was also targeted to the surface in ACBD3-depleted cells. Independent replicates of this experiment were quantified and plotted, as shown in Fig. 4E. The increase in cell surface IRAP was less dramatic than that observed for the GLUT4 reporter, which may in part reflect the observation that total IRAP abundance was unaffected, whereas GLUT4 reporter abundance was increased. Regardless, the effects were similar to those observed in 3T3-L1 adipocytes in which TUG or Golgin-160 is depleted using RNAi (12, 16, 27). As in these instances, some additional effect of insulin was still present in the ACBD3-depleted cells, which may reflect insulin-stimulated vesicle fusion at the plasma membrane. The cell surface redistribution of GLUT4 and IRAP was observed using two different shRNAs, implying that it is not due to an off-target effect. Moreover, there was no effect on transferrin receptor (TfR) targeting to the cell surface. Thus, ACBD3 depletion specifically affected GSV and not endosomal cargos, similar to TUG disruption. Of note, ACBD3 knockdown may disrupt TUG function both directly and indirectly through Golgin-160 (33, 34) and possibly other proteins that, together, anchor TUG-bound vesicles within cells. We conclude that ACBD3 is required for the intracellular retention of GSV cargo proteins, GLUT4 and IRAP, within unstimulated 3T3-L1 adipocytes.
FIGURE 4.

ACBD3 is required for intracellular retention of GSV cargos in 3T3-L1 adipocytes. A, ACBD3 was immunoblotted in 3T3-L1 adipocytes containing two distinct shRNAs to deplete ACBD3 or a scrambled control shRNA, as indicated. Hsp90 was immunoblotted as a loading control. B, surface-exposed GLUT4 reporter was biotinylated in basal and insulin-stimulated 3T3-L1 adipocytes containing shRNAs as indicated. Proteins were purified on immobilized streptavidin, and eluted proteins were immunoblotted to detect GLUT4 and TfR. Control immunoblots of whole cell lysates were done to detect total GLUT4 and Hsp90. C, quantification of the experiment shown in B (mean ± S.E. (error bars), n = 3). D, 3T3-L1 adipocytes were treated as in B, and eluates were immunoblotted to detect endogenous IRAP and TfR at the cell surface. Whole cell lysates were immunoblotted to detect total cellular IRAP and Hsp90, as indicated. E, quantification of the experiment shown in D (mean ± S.E., n = 5).
We next considered whether a sirtuin-type deacetylase might regulate TUG acetylation to control the accumulation of GLUT4 in GSVs. We focused on SIRT2, the main cytosolic sirtuin isoform (36). We first tested whether SIRT2 could bind and deacetylate TUG. We expressed FLAG-tagged SIRT2 in 293 cells and precipitated TUG proteins using an immobilized TUG antibody. As shown in Fig. 5A, SIRT2 coimmunoprecipitated with TUG and was not present in control samples in which TUG was not immunoprecipitated. These data support the idea that SIRT2 can associate with TUG. To test whether SIRT2 is able to deacetylate TUG, we immunoblotted AcK in TUG immunoprecipitates prepared from SIRT2-transfected cells and untransfected control cells. As shown in Fig. 5B, overexpression of SIRT2 reduced TUG acetylation, supporting the idea that SIRT2 can act catalytically to deacetylate TUG proteins.
FIGURE 5.

SIRT2 binds and deacetylates TUG. A, lysates from 293 cells transfected with FLAG-tagged SIRT2 were subjected to immunoprecipitation (IP) using beads with or without a cross-linked anti-TUG antibody, as indicated. Eluted proteins were immunoblotted (WB) to detect SIRT2 and TUG, as indicated. The experiment was repeated at least twice with consistent results. B, endogenous TUG proteins were immunoprecipitated from untransfected or SIRT2-transfected 293 cells. Eluted proteins were immunoblotted to detect AcK. The experiment was repeated twice with consistent results. C, synthetic peptides corresponding to TUG(528–550), with AcK549, or p53(368–393), with AcK382, were incubated in the presence of recombinant SIRT2 or SIRT1. All peptides had an N-terminal biotin. After incubation for the indicated times, peptides were spotted on nitrocellulose membranes and immunoblotted to detect AcK or total biotinylated peptide. Experiments were repeated three times with similar results.
To further test the sirtuin isoform specificity of TUG deacetylation and to show that this action is direct, we used recombinant SIRT2 and SIRT1 enzymes. As shown in Fig. 5C, we incubated SIRT2 or SIRT1 with the Lys-549-acetylated TUG C-terminal peptide for various amounts of time and used dot blots to assay AcK (using an anti-AcK antibody) and total peptide (using a biotin moiety on the N terminus of the peptide). SIRT2 catalyzed the removal of AcK from the TUG peptide within 1–2 h. In the SIRT1 reactions, substantial AcK remained on the peptide at 4 h and even 24 h after the incubation was started. As a positive control for SIRT1 activity, we used an acetylated peptide derived from p53, a well established SIRT1 substrate (37). SIRT1, but not SIRT2, rapidly deacetylated this p53 peptide. Thus, SIRT2 and SIRT1 demonstrated specificity toward the TUG and p53 peptide, respectively, which is noteworthy, given previous data suggesting that SIRT1 lacks substrate specificity in vitro (38). The main point is that that SIRT2 binds TUG and deacetylates the TUG C-terminal peptide.
We predicted that SIRT2-mediated deacetylation of the TUG C terminus would disrupt insulin-responsive trafficking of GLUT4 and IRAP. To test this idea, we expressed untagged SIRT2 using a retrovirus in TUG WT and 5R 3T3-L1 cells. Immunoblots using a SIRT2 antibody revealed that the exogenous SIRT2 protein was robustly expressed compared with endogenous SIRT2 (which was difficult to detect) in 3T3-L1 adipocytes (shown below). We then used cell surface biotinylation to assess targeting of the endogenous IRAP protein. As shown in Fig. 6A, basal targeting of IRAP to the plasma membrane was increased in SIRT2-overexpressing 3T3-L1 adipocytes, and insulin further increased the abundance of IRAP present at the cell surface. These effects required the presence of acetylatable residues at the TUG C terminus, because they were not observed in cells containing TUG 5R. Possibly, the SIRT2 effect requires cycling of acetylation on and off the TUG C terminus, which is not replicated by the mutated TUG 5R protein. Data from four independent replicates of the experiment shown in Fig. 6A were quantified and plotted in Fig. 6B. Of note, the effect of TUG 5R to reduce IRAP translocation is similar to that observed for GLUT4 in Fig. 3C and greater than that observed for IRAP in Fig. 3D (reflecting the improved insulin response in the control cells in Fig. 6, A and B). The data show that the effect of SIRT2 to increase cell surface targeting of IRAP requires lysine residues present in the wild type TUG C terminus. The data support the idea that SIRT2-mediated deacetylation of TUG reduces its ability to sequester IRAP in an insulin-responsive pool of GSVs.
FIGURE 6.

SIRT2 overexpression acts through the TUG C terminus to reduce intracellular retention of GSV cargos. A, cell surface-exposed IRAP was biotinylated in basal and insulin-stimulated 3T3-L1 adipocytes. Control and SIRT2-overexpressing cells containing WT or 5R TUG were used, as indicated. Biotinylated proteins were purified on streptavidin beads, and eluted proteins were immunoblotted to detect surface IRAP and TfR, as indicated. Control immunoblots of whole cell lysates are shown. B, the experiment in A was repeated and quantified (mean ± S.E. (error bars), n = 4). C, GLUT4 translocation was analyzed using flow cytometry of a reporter protein in 3T3-L1 adipocytes expressing WT or 5R TUG, with or without SIRT2 overexpression, as indicated. The relative ratios of surface/total GLUT4 are shown (mean ± S.E., n = 5). D, TUG acetylation was analyzed by immunoprecipitating (IP) TUG and immunoblotting (WB) AcK in 3T3-L1 adipocytes containing WT or 5R TUG, with or without SIRT2 overexpression. Precipitates and whole cell lysates were immunoblotted as indicated. The experiment was repeated at least twice with similar results.
To test the effect of SIRT2 overexpression on GLUT4 trafficking, we used flow cytometry to measure the ratio of surface to total GLUT4 reporter in 3T3-L1 adipocytes. As shown in Fig. 6C, insulin-stimulated GLUT4 translocation was reduced in TUG 5R cells, compared with TUG WT cells. This result is similar to data shown above in Figs. 3 (A and C) (for GLUT4) and 6B (for IRAP). SIRT2 overexpression caused an increase in both basal and insulin-stimulated GLUT4 targeting to the cell surface in TUG WT cells. These effects were not observed in SIRT2-overexpressing TUG 5R cells, in which GLUT4 targeting was similar to that in 5R cells not overexpressing SIRT2. These GLUT4 targeting data are similar to data obtained for IRAP in Fig. 6B and imply that the effect of SIRT2 requires acetylatable residues at the TUG C terminus. The magnitude of the increase in surface-exposed IRAP, detected using biotinylation (Fig. 6B), appears larger than that for surface GLUT4, detected using flow cytometry (Fig. 6C), particularly in the basal state. This may be because the biotinylation assay detects proteins that transit across the plasma membrane during the ∼12-min reaction rather than static amounts at the cell surface. The main conclusion, supported by both assays, is that SIRT2 overexpression increases the targeting of GSV cargos to the cell surface and that this effect requires TUG C-terminal residues that are deacetylated by SIRT2.
We further characterized the effects of SIRT2 overexpression in TUG WT and 5R 3T3-L1 adipocytes by immunoprecipitating TUG and immunoblotting AcK. As shown in Fig. 6D, AcK was detected in the eluates in TUG WT cells and was substantially reduced in TUG 5R cells and in both TUG WT and 5R cells overexpressing SIRT2. As noted above, TUG undergoes site-specific endoproteolytic cleavage, and the C-terminal cleavage product is observed both as a 42-kDa fragment and in modified form at 54 kDa (17). These products were observed in TUG WT cells; abundance of the 54-kDa form was similar to that of intact 60-kDa TUG protein, and the 42-kDa product was present in lower amounts. Intriguingly, the abundances of these products, but not of intact TUG, were reduced in TUG 5R cells and in TUG WT and 5R cells overexpressing SIRT2. The data support the idea that proteolytic processing is reduced in these cells, as may also be the case for TUG K549R in Fig. 2E. Together, the data suggest that TUG acetylation is required for proteolysis. This hypothesis is consistent with previous data showing that only membrane-associated, and not cytosolic, TUG proteins undergo proteolytic processing in 3T3-L1 adipocytes (17). Finally, immunoblots in Fig. 6D confirm that exogenous SIRT2 is well expressed and show that endogenous SIRT2 is not detected and is probably present at very low abundance in 3T3-L1 adipocytes. This is consistent with data showing that nicotinamide treatment only modestly increased the acetylation of endogenous TUG proteins in cultured cells. Together, the results support a model in which acetylated TUG anchors GSV cargos in an intracellular, insulin-responsive pool and in which the size of this pool is reduced by SIRT2-mediated deacetylation of TUG.
To understand the potential physiologic role of SIRT2 action for glucose homeostasis, we studied SIRT2 KO mice. We first tested whether TUG acetylation is increased in SIRT2 KO mice, compared with WT controls. We determined that TUG could be immunoprecipitated effectively from liver lysates, which were prepared from mice that had been fasted for 6 h. Precipitated TUG was immunoblotted for AcK and TUG, and densitometry was performed. As shown in Fig. 7, A and B, the ratio of AcK to TUG was increased ∼2-fold in SIRT2 KO mice compared with WT controls. These data support the notion that SIRT2 regulates TUG acetylation in vivo in mice.
FIGURE 7.

SIRT2 disruption in mice increases TUG acetylation and proteolytic processing and enhances insulin sensitivity. A, TUG was immunoprecipitated (IP) from liver lysates of WT and SIRT2 KO mice, and eluted proteins were immunoblotted (WB) for AcK and TUG. A representative immunoblot is shown. B, replicates of the experiment in A were quantified, and the ratio of AcK/TUG signal is plotted (mean ± S.E. (error bars), n = 3 in each group). C, WT and SIRT2 KO mice were treated by intraperitoneal injection of saline or insulin-glucose solution and sacrificed after 20 min. Quadriceps muscle lysates were immunoblotted to detect intact TUG (60 kDa) and C-terminal cleavage products (42 kDa). GAPDH indicates differences in loading. D, the ratio of TUG C-terminal product/intact TUG abundance is plotted for the experiment in C (mean ± S.E., n = 4 mice in each group). E, the abundance of intact TUG, relative to GAPDH, is plotted for the experiment in C (mean ± S.E., n = 4). F, quadriceps of mice used in the experiment in C were immunoblotted for GLUT4, and relative abundance is plotted for SIRT2 KO and WT control mice (mean ± S.E., n = 8). G–J, intraperitoneal glucose tolerance tests were performed on WT and SIRT2 KO mice. Glucose (G) and insulin (I) concentrations are plotted over time, and the areas under the curve (AUC) for glucose (H) and insulin (J) are shown. *, p < 0.05; **, p < 0.01; mean ± S.E., n = 8 mice in each group.
We next tested the prediction that proteolytic processing of TUG would be enhanced by SIRT2 deletion. Age-matched SIRT2 KO and WT mice were fasted 4–6 h and then given an intraperitoneal injection of insulin-glucose solution or saline control. Mice were sacrificed 20 min after the injection, and then quadriceps muscle lysates were prepared and immunoblotted. As shown in the example immunoblot in Fig. 7C, insulin stimulated the conversion of intact, 60-kDa TUG into its 42-kDa C-terminal product in the WT control mice. In SIRT2 KO mice, this conversion was already prominent in the saline-treated animals, and insulin stimulated a further conversion of intact TUG into its C-terminal product. Data from all mice are quantified in Fig. 7C, which plots the ratio of the C-terminal product to the intact TUG precursor. The effect of insulin to promote TUG proteolytic processing and the effect of SIRT2 deletion to enhance TUG processing are also evident from the depletion of intact TUG protein. Fig. 7E quantifies intact TUG relative to a GAPDH loading control. Insulin stimulated a reduction in intact TUG abundance in WT mice. In SIRT2 KO mice, intact TUG was markedly reduced in fasted animals and trended toward further depletion after insulin stimulation. These data are in accord with the significant effect of insulin to stimulate TUG processing in SIRT2 KO as well as WT mice (Fig. 7D). Together, the data support a model in which SIRT2 regulates TUG acetylation and in which acetylation is required for TUG to undergo regulated proteolytic processing.
The sequestration of GLUT4 in GSVs is thought to stabilize GLUT4 protein, and GLUT4 is degraded more rapidly in 3T3-L1 cells in which TUG is depleted using RNAi compared with controls (16). If SIRT2 deletion enhances the action of TUG to trap GLUT4 in GSVs, then GLUT4 abundance may be increased in SIRT2 KO mice. Consistent with this hypothesis, we observed a ∼20% increase in GLUT4 abundance in SIRT2 KO quadriceps, compared with WT controls (Fig. 7F). The combination of enhanced TUG proteolytic processing, together with increased GLUT4 abundance, argues strongly that GLUT4 is more efficiently retained in insulin-responsive GSVs in SIRT2 KO mice, compared with controls.
We predicted that glucose uptake would be enhanced by SIRT2 deletion. To test this idea, SIRT2 KO and WT control mice were fasted for 6 h and then studied using an intraperitoneal glucose tolerance test. As shown in Fig. 7G, SIRT2 KO mice cleared glucose more rapidly from the circulation, compared with WT controls. This effect was observed both at individual time points between 30 and 120 min after the glucose load and as a cumulative effect in the area under the curve (Fig. 7H). Insulin concentrations were measured in each sample and were not different between WT and KO mice (Fig. 7, I and J). Thus, the pronounced effects on glucose homeostasis were due to enhanced insulin action and did not result from effects on insulin secretion. Of note, body composition analyses revealed no differences in the weight, lean mass, and fat mass of SIRT2 KO mice, compared with WT controls, and cannot account for the increased insulin sensitivity in the SIRT2 KO animals. Together, the data support the idea that TUG acetylation is regulated by SIRT2 in vivo and that deletion of SIRT2 results in increased TUG acetylation, an enlarged pool of insulin-responsive GSVs, and greater insulin-stimulated whole body glucose uptake in mice.
DISCUSSION
Glucose transport across the plasma membrane is the main, rate-controlling step for overall glucose utilization in fat and muscle, which is mediated primarily by GLUT4 and is impaired in insulin-resistant states (1, 39, 40). By translocating GLUT4, insulin controls glucose transport on a moment-to-moment basis. GLUT4 translocation comprises several steps; it has been debated whether there is one main insulin-regulated step and, if so, what step this may be (41). Most data support the idea that insulin acts primarily to mobilize a distinct pool of insulin-responsive GSVs, but the molecular regulation of these vesicles has remained uncertain. Together with previous data, results reported here support a model in which the retention of GLUT4 in insulin-responsive GSVs is mediated by acetylated TUG.
Our data further define the anchoring site to which TUG links GLUT4 in unstimulated cells. Previous results imply that TUG retains GSVs by linking them to the Golgi proteins, Golgin-160 and PIST (17, 18, 27). Binding of IRAP to p115, another Golgi matrix protein that colocalizes with Golgin-160, may also contribute to GSV intracellular retention in unstimulated cells (27, 42). We observe an interaction between the TUG C-terminal peptide and ACBD3 (also called GCP60), a protein that associates with Golgin-160 in a redox-sensitive manner (33, 34). Similar to TUG (12, 16) and Golgin-160 (27), ACBD3 was required for full intracellular retention of GSV cargos within unstimulated 3T3-L1 adipocytes. Because ACBD3 bound preferentially to unacetylated TUG, which our data suggest does not undergo endoproteolytic cleavage, it may act to retain GLUT4 and IRAP in non-insulin-responsive intracellular vesicles. In this case, TUG acetylation may reconfigure its interaction among members of a protein complex, shifting it away from ACBD3 and toward Golgin-160 or another protein, to enhance insulin-responsive trafficking.
The idea that the TUG C terminus may interact with distinct sites, depending on its acetylation state, also fits with data from cells overexpressing ACBD3. We observed enhanced insulin-regulated trafficking of GSV cargos in 3T3-L1 cells with very modest overexpression of ACBD3. This effect required intact acetylation sites at the TUG C terminus, despite the observation that unacetylated TUG peptide bound preferentially to ACBD3. A possible explanation to encompass these observations is suggested by the observation that ACBD3 participates in recruiting Golgin-160 to Golgi membranes (33). Thus, ACBD3 overexpression may act indirectly to create additional binding sites for acetylated TUG, as well as acting directly to increase the number of potential binding sites for unacetylated TUG. If the availability of binding sites for acetylated TUG is limiting in 3T3-L1 adipocytes, then this effect could increase insulin-responsive trafficking. Similar reasoning suggests that ACBD3 knockdown may act by a combination of direct effects on TUG and indirect effects, through Golgin-160 (or another protein). Finally, data imply that the size of the insulin-responsive pool was reduced in cells containing TUG 5R, yet this protein did not have a pronounced effect to increase cell surface GLUT4/IRAP in unstimulated cells. Therefore, C-terminal regions of TUG other than the acetylated residues contribute to basal intracellular retention of GSV proteins. This may occur through interactions of TUG with Golgin-160, PIST, or other proteins (17, 18).
ACBD3 is a novel component of the intracellular anchoring site to which TUG links GLUT4 in unstimulated cells. The ACBD3 protein contains an acyl-CoA-binding domain near its N terminus and a GOLD (Golgi dynamics) domain near its C terminus (35). As well as Golgin-160, ACBD3 binds other Golgi proteins, including giantin and PI4KIIIβ (phosphatidylinositol-4-kinase IIIβ) (33, 43–45). The latter interaction raises the possibility that PI4KIIIβ, not PI4KIIα, may be the inositol kinase involved in recruiting coat components for GSV formation (2). Other functions are described for ACBD3, which may be cell type-specific. ACBD3 binds proteins present on mitochondria, notably the 18-kDa translocator protein, TSPO, and it recruits protein kinase A regulatory subunit 1α to facilitate the import of cholesterol for steroid synthesis (46). In the brain, ACBD3 binds complexes containing the GTPases Dexras1 and Rhes to regulate divalent metal transporters (47, 48) and to mediate the neurotoxicity associated with mutated huntingtin proteins, respectively (49). During mitosis, ACBD3 is released into the cytosol and functions together with Numb proteins to specify the developmental fate of daughter cells (22). These multiple functions have led to the idea that ACBD3 functions as a signaling hub (35).
The acyl-CoA binding domain is similar to that of other, structurally characterized domains in proteins in this family, which bind to long and medium chain acyl-CoAs (35). We speculate that the formation of a TUG-ACBD3 complex in unstimulated adipocytes may play some role in fatty acid metabolism. Acyl-CoA dehydrogenases have been identified on GLUT4 vesicles and bind IRAP at a site that is critical for GLUT4 intracellular retention (50, 51). The long chain fatty acyl CoA synthetase ACSL1 was also found in association with TUG6 and sortilin (52) and may function in GSV biogenesis or retention. Adipose ACSL1 directs fatty acids toward β-oxidation (53). If ACBD3 is juxtaposed with an acyl-CoA dehydrogenase and possibly ACSL1, by formation of a TUG-ACBD3 complex in unstimulated adipocytes, then these proteins may act together to control fatty acid metabolism. Of note, ACBD3 overexpression in 293T cells inhibits the maturation and activity of SREBP1 and reduces fatty acid synthase abundance and de novo palmitate synthesis (54). Thus, the idea that this protein may also function in fatty acid metabolism in adipocytes is plausible and may be important for metabolic flexibility (55).
Our data show that TUG acetylation promotes GLUT4 accumulation in an intracellular, insulin-responsive compartment. Furthermore, SIRT2 modulates TUG acetylation to control insulin sensitivity, as shown by in vitro deacetylation data, overexpression of SIRT2 in 3T3-L1 adipocytes, and deletion of SIRT2 in mice. In 3T3-L1 adipocytes, effects of SIRT2 overexpression on GLUT4/IRAP targeting were more marked than effects of TUG 5R expression. Possibly, this may be because lysine residues in TUG are still able to cycle between acetylated and unacetylated states in SIRT2-overexpressing cells. Such cycling may be important for the trapping and release of GSVs. If so, then this site may remain partially occupied by TUG 5R proteins. In addition, binding of overexpressed SIRT2 to TUG may displace other TUG-interacting proteins to prevent GSVs from being trapped.
In SIRT2 KO mice, compared with WT controls, the proteolytic processing of TUG was markedly increased in muscle, both during fasting and after acute insulin stimulation. The increased TUG processing observed during fasting was unexpected. However, fasting plasma insulin concentrations were not reduced in SIRT2 KO mice, compared with WT controls and may have been sufficient to trigger TUG cleavage if insulin sensitivity was greatly enhanced. The increased insulin sensitivity was indeed pronounced, as assessed in glucose tolerance tests, and enhanced glucose uptake may have resulted from increased GLUT4 abundance as well as from increased translocation. Both of these effects are predicted to result from enhanced TUG acetylation and GLUT4 sequestration in insulin-responsive GSVs, according to the model we propose.
Acetylation is well known to act in metabolic sensing and regulation and is regulated in a NAD+-dependent manner by sirtuin proteins (28, 29). NAD+ levels increase during caloric restriction, prolonged fasting, or after exercise, whereas the NAD+/NADH ratio decreases during overnutrition (56–59). Our data predict that during energy stress, increased NAD+ may act through SIRT2 to mobilize GLUT4 to the cell surface in an insulin-independent manner. Conversely, when the NAD+/NADH ratio is reduced, TUG acetylation will be increased, and sequestration of GLUT4 in GSVs will be greater. Whether this mechanism contributes to insulin resistance is not known and may depend on whether an insulin signal is able to stimulate TUG cleavage. Effects of overnutrition are not limited to changes in the NAD+/NADH ratio, and substantial data implicate altered lipid metabolism in insulin resistance (60). Additionally, NAD+ modulates the activities of multiple sirtuin isoforms as well as other proteins (61). Finally, NAD+ concentrations may vary in different cellular compartments and as a result of local NAD+-consuming enzymes, such as poly-ADP-ribose polymerases (62). In this regard, it is notable that tankyrase is a poly-ADP-ribose polymerase that binds IRAP and controls the accumulation of GLUT4 in an insulin-responsive pool (63–66). Based on our results, a possible mechanism for this effect of tankyrase involves NAD+ consumption, reduced SIRT2 activity, and increased TUG acetylation.
Only a handful of targets have been identified for SIRT2, and the identification of TUG as a novel substrate is important (67). Other targets of SIRT2 include FoxO1 and possibly PGC-1α, and it may act through these proteins to influence adipocyte differentiation (68–71). We did not observe marked impairment of differentiation in SIRT2-overexpressing 3T3-L1 cells or altered weight or body composition in SIRT2 KO mice. Recent data show that SIRT2 binds Akt and facilitates Akt phosphorylation (72). However, the increased insulin sensitivity we observed in vivo in intraperitoneal glucose tolerance tests of SIRT2 KO mice, compared with WT controls, is consistent with previous results showing that insulin action on TUG is independent of Akt (12, 14, 16, 17). Thus, the data support a model in which SIRT2 knock-out mice have increased TUG acetylation, which facilitates trapping of GLUT4 in an insulin-responsive pool and which increases insulin sensitivity.
The mechanism we propose may contribute to alterations in insulin sensitivity that occur independent of insulin signaling effects on IRS-1, Akt, and/or AS160/Tbc1D4 (73–75). The data also suggest how insulin resistance may result from altered subcellular GLUT4 localization in fat or muscle from fasting individuals or in unstimulated cells (5, 7, 8). Action through NAD+ and SIRT2 may provide a mechanism for insulin-independent GLUT4 translocation and glucose uptake, which may act together with AMPK-dependent mechanisms (76–78). Finally, our results provide an improved understanding of how GLUT4 is sequestered in insulin-responsive GSVs, with implications for ongoing work to understand metabolic regulation and how it is affected during aging and in metabolic disease.
Acknowledgments
We thank Brad Rubin, Charisse Orme, Ali Nasiri, Jie Liu, Toren Finkel, Weimin Zhong, Carolyn Machamer, Frederick Alt, Eric Verdin, Thomas Melia, and Fred Gorelick for assistance, reagents, and advice. This work used the Cell Biology Core of the Yale Diabetes Endocrinology Research Center (supported by National Institutes of Health Grant P30DK45735) as well as services provided by the W. M. Keck Foundation Biotechnology Resource Laboratory at Yale University.
This work was supported, in whole or in part, by National Institutes of Health Grants R21AG041383 and R01DK092661 (to J. S. B.) and U24 DK059635 (to G. I. S.). This work was also supported by American Diabetes Association Grant 1-12-BS-16 (to J. S. B).
J. P. Belman and J. S. Bogan, unpublished data.
- GSV
- GLUT4 storage vesicle
- AcK
- acetyl-lysine
- IRAP
- insulin-regulated aminopeptidase
- LM
- light microsome
- PM
- plasma membrane
- SIRT2
- sirtuin 2
- bis-tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- TUG 5R
- TUG in which Lys-533, -538, -541, -544, and -549 are mutated to arginine
- TfR
- transferrin receptor.
REFERENCES
- 1. Bogan J. S. (2012) Regulation of glucose transporter translocation in health and diabetes. Annu. Rev. Biochem. 81, 507–532 [DOI] [PubMed] [Google Scholar]
- 2. Bogan J. S., Kandror K. V. (2010) Biogenesis and regulation of insulin-responsive vesicles containing GLUT4. Curr. Opin. Cell Biol. 22, 506–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kandror K. V., Pilch P. F. (2011) The sugar is sIRVed: sorting Glut4 and its fellow travelers. Traffic 12, 665–671 [DOI] [PubMed] [Google Scholar]
- 4. Ma J., Nakagawa Y., Kojima I., Shibata H. (2014) Prolonged insulin stimulation down-regulates GLUT4 through oxidative stress-mediated retromer inhibition by a protein kinase CK2-dependent mechanism in 3T3-L1 adipocytes. J. Biol. Chem. 289, 133–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vassilopoulos S., Esk C., Hoshino S., Funke B. H., Chen C. Y., Plocik A. M., Wright W. E., Kucherlapati R., Brodsky F. M. (2009) A role for the CHC22 clathrin heavy-chain isoform in human glucose metabolism. Science 324, 1192–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xu Z., Kandror K. V. (2002) Translocation of small preformed vesicles is responsible for the insulin activation of glucose transport in adipose cells: evidence from the in vitro reconstitution assay. J. Biol. Chem. 277, 47972–47975 [DOI] [PubMed] [Google Scholar]
- 7. Garvey W. T., Maianu L., Zhu J. H., Brechtel-Hook G., Wallace P., Baron A. D. (1998) Evidence for defects in the trafficking and translocation of GLUT4 glucose transporters in skeletal muscle as a cause of human insulin resistance. J. Clin. Invest. 101, 2377–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maianu L., Keller S. R., Garvey W. T. (2001) Adipocytes exhibit abnormal subcellular distribution and translocation of vesicles containing glucose transporter 4 and insulin-regulated aminopeptidase in type 2 diabetes mellitus: implications regarding defects in vesicle trafficking. J. Clin. Endocrinol. Metab. 86, 5450–5456 [DOI] [PubMed] [Google Scholar]
- 9. Bogan J. S., McKee A. E., Lodish H. F. (2001) Insulin-responsive compartments containing GLUT4 in 3T3-L1 and CHO cells: regulation by amino acid concentrations. Mol. Cell. Biol. 21, 4785–4806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shi J., Kandror K. V. (2005) Sortilin is essential and sufficient for the formation of Glut4 storage vesicles in 3T3-L1 adipocytes. Dev. Cell 9, 99–108 [DOI] [PubMed] [Google Scholar]
- 11. Muretta J. M., Romenskaia I., Mastick C. C. (2008) Insulin releases Glut4 from static storage compartments into cycling endosomes and increases the rate constant for Glut4 exocytosis. J. Biol. Chem. 283, 311–323 [DOI] [PubMed] [Google Scholar]
- 12. Xu Y., Rubin B. R., Orme C. M., Karpikov A., Yu C., Bogan J. S., Toomre D. K. (2011) Dual-mode of insulin action controls GLUT4 vesicle exocytosis. J. Cell Biol. 193, 643–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen Y., Wang Y., Zhang J., Deng Y., Jiang L., Song E., Wu X. S., Hammer J. A., Xu T., Lippincott-Schwartz J. (2012) Rab10 and myosin-Va mediate insulin-stimulated GLUT4 storage vesicle translocation in adipocytes. J. Cell Biol. 198, 545–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Belman J. P., Habtemichael E. N., Bogan J. S. (2014) A proteolytic pathway that controls glucose uptake in fat and muscle. Rev. Endocr. Metab. Disord. 15, 55–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bogan J. S., Hendon N., McKee A. E., Tsao T. S., Lodish H. F. (2003) Functional cloning of TUG as a regulator of GLUT4 glucose transporter trafficking. Nature 425, 727–733 [DOI] [PubMed] [Google Scholar]
- 16. Yu C., Cresswell J., Löffler M. G., Bogan J. S. (2007) The glucose transporter 4-regulating protein TUG is essential for highly insulin-responsive glucose uptake in 3T3-L1 adipocytes. J. Biol. Chem. 282, 7710–7722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bogan J. S., Rubin B. R., Yu C., Löffler M. G., Orme C. M., Belman J. P., McNally L. J., Hao M., Cresswell J. A. (2012) Endoproteolytic cleavage of TUG protein regulates GLUT4 glucose transporter translocation. J. Biol. Chem. 287, 23932–23947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Löffler M. G., Birkenfeld A. L., Philbrick K. M., Belman J. P., Habtemichael E. N., Booth C. J., Castorena C. M., Choi C. S., Jornayvaz F. R., Gassaway B. M., Lee H. Y., Cartee G. D., Philbrick W., Shulman G. I., Samuel V. T., Bogan J. S. (2013) Enhanced fasting glucose turnover in mice with disrupted action of TUG protein in skeletal muscle. J. Biol. Chem. 288, 20135–20150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Orme C. M., Bogan J. S. (2012) The ubiquitin regulatory X (UBX) domain-containing protein TUG regulates the p97 ATPase and resides at the endoplasmic reticulum-Golgi intermediate compartment. J. Biol. Chem. 287, 6679–6692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Govers R., Coster A. C., James D. E. (2004) Insulin increases cell surface GLUT4 levels by dose dependently discharging GLUT4 into a cell surface recycling pathway. Mol. Cell Biol. 24, 6456–6466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Naviaux R. K., Costanzi E., Haas M., Verma I. M. (1996) The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J. Virol. 70, 5701–5705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou Y., Atkins J. B., Rompani S. B., Bancescu D. L., Petersen P. H., Tang H., Zou K., Stewart S. B., Zhong W. (2007) The mammalian Golgi regulates numb signaling in asymmetric cell division by releasing ACBD3 during mitosis. Cell 129, 163–178 [DOI] [PubMed] [Google Scholar]
- 23. North B. J., Marshall B. L., Borra M. T., Denu J. M., Verdin E. (2003) The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 11, 437–444 [DOI] [PubMed] [Google Scholar]
- 24. Liu X., Constantinescu S. N., Sun Y., Bogan J. S., Hirsch D., Weinberg R. A., Lodish H. F. (2000) Generation of mammalian cells stably expressing multiple genes at predetermined levels. Anal. Biochem. 280, 20–28 [DOI] [PubMed] [Google Scholar]
- 25. Huang G., Buckler-Pena D., Nauta T., Singh M., Asmar A., Shi J., Kim J. Y., Kandror K. V. (2013) Insulin responsiveness of glucose transporter 4 in 3T3-L1 cells depends on the presence of sortilin. Mol. Biol. Cell 24, 3115–3122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Camporez J. P., Jornayvaz F. R., Petersen M. C., Pesta D., Guigni B. A., Serr J., Zhang D., Kahn M., Samuel V. T., Jurczak M. J., Shulman G. I. (2013) Cellular mechanisms by which FGF21 improves insulin sensitivity in male mice. Endocrinology 154, 3099–3109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Williams D., Hicks S. W., Machamer C. E., Pessin J. E. (2006) Golgin-160 is required for the Golgi membrane sorting of the insulin-responsive glucose transporter GLUT4 in adipocytes. Mol. Biol. Cell 17, 5346–5355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xiong Y., Guan K. L. (2012) Mechanistic insights into the regulation of metabolic enzymes by acetylation. J. Cell Biol. 198, 155–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Johnson E. S., Kornbluth S. (2012) Life, death, and the metabolically controlled protein acetylome. Curr. Opin. Cell Biol. 24, 876–880 [DOI] [PubMed] [Google Scholar]
- 30. Sanders B. D., Zhao K., Slama J. T., Marmorstein R. (2007) Structural basis for nicotinamide inhibition and base exchange in Sir2 enzymes. Mol. Cell 25, 463–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang X. J., Seto E. (2008) Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol. Cell 31, 449–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hicks S. W., Machamer C. E. (2005) Isoform-specific interaction of golgin-160 with the Golgi-associated protein PIST. J. Biol. Chem. 280, 28944–28951 [DOI] [PubMed] [Google Scholar]
- 33. Sbodio J. I., Hicks S. W., Simon D., Machamer C. E. (2006) GCP60 preferentially interacts with a caspase-generated golgin-160 fragment. J. Biol. Chem. 281, 27924–27931 [DOI] [PubMed] [Google Scholar]
- 34. Sbodio J. I., Machamer C. E. (2007) Identification of a redox-sensitive cysteine in GCP60 that regulates its interaction with golgin-160. J. Biol. Chem. 282, 29874–29881 [DOI] [PubMed] [Google Scholar]
- 35. Fan J., Liu J., Culty M., Papadopoulos V. (2010) Acyl-coenzyme A binding domain containing 3 (ACBD3; PAP7; GCP60): an emerging signaling molecule. Prog. Lipid Res. 49, 218–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chalkiadaki A., Guarente L. (2012) Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat. Rev. Endocrinol. 8, 287–296 [DOI] [PubMed] [Google Scholar]
- 37. Luo J., Nikolaev A. Y., Imai S., Chen D., Su F., Shiloh A., Guarente L., Gu W. (2001) Negative control of p53 by Sir2α promotes cell survival under stress. Cell 107, 137–148 [DOI] [PubMed] [Google Scholar]
- 38. Blander G., Olejnik J., Krzymanska-Olejnik E., McDonagh T., Haigis M., Yaffe M. B., Guarente L. (2005) SIRT1 shows no substrate specificity in vitro. J. Biol. Chem. 280, 9780–9785 [DOI] [PubMed] [Google Scholar]
- 39. Cline G. W., Petersen K. F., Krssak M., Shen J., Hundal R. S., Trajanoski Z., Inzucchi S., Dresner A., Rothman D. L., Shulman G. I. (1999) Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. N. Engl. J. Med. 341, 240–246 [DOI] [PubMed] [Google Scholar]
- 40. Goodpaster B. H., Bertoldo A., Ng J. M., Azuma K., Pencek R. R., Kelley C., Price J. C., Cobelli C., Kelley D. E. (2014) Interactions among glucose delivery, transport, and phosphorylation that underlie skeletal muscle insulin resistance in obesity and type 2 diabetes: studies with dynamic PET imaging. Diabetes 63, 1058–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rubin B. R., Bogan J. S. (2009) Intracellular retention and insulin-stimulated mobilization of GLUT4 glucose transporters. Vitam. Horm. 80, 155–192 [DOI] [PubMed] [Google Scholar]
- 42. Hosaka T., Brooks C. C., Presman E., Kim S. K., Zhang Z., Breen M., Gross D. N., Sztul E., Pilch P. F. (2005) p115 interacts with the GLUT4 vesicle protein, IRAP, and plays a critical role in insulin-stimulated GLUT4 translocation. Mol. Biol. Cell 16, 2882–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sohda M., Misumi Y., Yamamoto A., Yano A., Nakamura N., Ikehara Y. (2001) Identification and characterization of a novel Golgi protein, GCP60, that interacts with the integral membrane protein giantin. J. Biol. Chem. 276, 45298–45306 [DOI] [PubMed] [Google Scholar]
- 44. Sasaki J., Ishikawa K., Arita M., Taniguchi K. (2012) ACBD3-mediated recruitment of PI4KB to picornavirus RNA replication sites. EMBO J. 31, 754–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Greninger A. L., Knudsen G. M., Betegon M., Burlingame A. L., Derisi J. L. (2012) The 3A protein from multiple picornaviruses utilizes the Golgi adaptor protein ACBD3 to recruit PI4KIIIbeta. J. Virol. 86, 3605–3616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Miller W. L. (2013) Steroid hormone synthesis in mitochondria. Mol. Cell. Endocrinol. 379, 62–73 [DOI] [PubMed] [Google Scholar]
- 47. Cheah J. H., Kim S. F., Hester L. D., Clancy K. W., Patterson S. E., 3rd, Papadopoulos V., Snyder S. H. (2006) NMDA receptor-nitric oxide transmission mediates neuronal iron homeostasis via the GTPase Dexras1. Neuron 51, 431–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen Y., Khan R. S., Cwanger A., Song Y., Steenstra C., Bang S., Cheah J. H., Dunaief J., Shindler K. S., Snyder S. H., Kim S. F. (2013) Dexras1, a small GTPase, is required for glutamate-NMDA neurotoxicity. J. Neurosci. 33, 3582–3587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sbodio J. I., Paul B. D., Machamer C. E., Snyder S. H. (2013) Golgi protein ACBD3 mediates neurotoxicity associated with Huntington's disease. Cell Rep. 4, 890–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Katagiri H., Asano T., Yamada T., Aoyama T., Fukushima Y., Kikuchi M., Kodama T., Oka Y. (2002) Acyl-coenzyme A dehydrogenases are localized on GLUT4-containing vesicles via association with insulin-regulated aminopeptidase in a manner dependent on its dileucine motif. Mol. Endocrinol. 16, 1049–1059 [DOI] [PubMed] [Google Scholar]
- 51. Waters S. B., D'Auria M., Martin S. S., Nguyen C., Kozma L. M., Luskey K. L. (1997) The amino terminus of insulin-responsive aminopeptidase causes Glut4 translocation in 3T3-L1 adipocytes. J. Biol. Chem. 272, 23323–23327 [DOI] [PubMed] [Google Scholar]
- 52. Huang G. (2010) Biogenesis and Insulin Responsiveness of GLUT4 Storage Vesicles in 3T3-L1 Cells, Ph.D. thesis, Boston University [Google Scholar]
- 53. Ellis J. M., Li L. O., Wu P. C., Koves T. R., Ilkayeva O., Stevens R. D., Watkins S. M., Muoio D. M., Coleman R. A. (2010) Adipose acyl-CoA synthetase-1 directs fatty acids toward β-oxidation and is required for cold thermogenesis. Cell Metab. 12, 53–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen Y., Patel V., Bang S., Cohen N., Millar J., Kim S. F. (2012) Maturation and activity of sterol regulatory element binding protein 1 is inhibited by acyl-CoA binding domain containing 3. PLoS One 7, e49906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Galgani J. E., Moro C., Ravussin E. (2008) Metabolic flexibility and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 295, E1009–E1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen D., Bruno J., Easlon E., Lin S. J., Cheng H. L., Alt F. W., Guarente L. (2008) Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 22, 1753–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cantó C., Jiang L. Q., Deshmukh A. S., Mataki C., Coste A., Lagouge M., Zierath J. R., Auwerx J. (2010) Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 11, 213–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kim H. J., Kim J. H., Noh S., Hur H. J., Sung M. J., Hwang J. T., Park J. H., Yang H. J., Kim M. S., Kwon D. Y., Yoon S. H. (2011) Metabolomic analysis of livers and serum from high-fat diet induced obese mice. J. Proteome Res. 10, 722–731 [DOI] [PubMed] [Google Scholar]
- 59. Houtkooper R. H., Auwerx J. (2012) Exploring the therapeutic space around NAD+. J. Cell Biol. 199, 205–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Samuel V. T., Shulman G. I. (2012) Mechanisms for insulin resistance: common threads and missing links. Cell 148, 852–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Imai S. I., Guarente L. (2014) NAD and sirtuins in aging and disease. Trends Cell Biol. 10.1016/j.tcb.2014.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Koch-Nolte F., Fischer S., Haag F., Ziegler M. (2011) Compartmentation of NAD+-dependent signalling. FEBS Lett. 585, 1651–1656 [DOI] [PubMed] [Google Scholar]
- 63. Chi N. W., Lodish H. F. (2000) Tankyrase is a Golgi-associated mitogen-activated protein kinase substrate that interacts with IRAP in GLUT4 vesicles. J. Biol. Chem. 275, 38437–38444 [DOI] [PubMed] [Google Scholar]
- 64. Yeh T. Y., Beiswenger K. K., Li P., Bolin K. E., Lee R. M., Tsao T. S., Murphy A. N., Hevener A. L., Chi N. W. (2009) Hypermetabolism, hyperphagia, and reduced adiposity in tankyrase-deficient mice. Diabetes 58, 2476–2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yeh T. Y., Sbodio J. I., Tsun Z. Y., Luo B., Chi N. W. (2007) Insulin-stimulated exocytosis of GLUT4 is enhanced by IRAP and its partner tankyrase. Biochem. J. 402, 279–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Guo H. L., Zhang C., Liu Q., Li Q., Lian G., Wu D., Li X., Zhang W., Shen Y., Ye Z., Lin S. Y., Lin S. C. (2012) The Axin/TNKS complex interacts with KIF3A and is required for insulin-stimulated GLUT4 translocation. Cell Res. 22, 1246–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. de Oliveira R. M., Sarkander J., Kazantsev A. G., Outeiro T. F. (2012) SIRT2 as a therapeutic target for age-related disorders. Front. Pharmacol. 3, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jing E., Gesta S., Kahn C. R. (2007) SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 6, 105–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang F., Tong Q. (2009) SIRT2 suppresses adipocyte differentiation by deacetylating FOXO1 and enhancing FOXO1's repressive interaction with PPARγ. Mol. Biol. Cell 20, 801–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhao Y., Yang J., Liao W., Liu X., Zhang H., Wang S., Wang D., Feng J., Yu L., Zhu W. G. (2010) Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 12, 665–675 [DOI] [PubMed] [Google Scholar]
- 71. Krishnan J., Danzer C., Simka T., Ukropec J., Walter K. M., Kumpf S., Mirtschink P., Ukropcova B., Gasperikova D., Pedrazzini T., Krek W. (2012) Dietary obesity-associated Hif1α activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes Dev. 26, 259–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ramakrishnan G., Davaakhuu G., Kaplun L., Chung W. C., Rana A., Atfi A., Miele L., Tzivion G. (2014) Sirt2 deacetylase is a novel AKT binding partner critical for AKT activation by insulin. J. Biol. Chem. 289, 6054–6066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Arias E. B., Kim J., Funai K., Cartee G. D. (2007) Prior exercise increases phosphorylation of Akt substrate of 160 kDa (AS160) in rat skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 292, E1191–E1200 [DOI] [PubMed] [Google Scholar]
- 74. Castorena C. M., Arias E. B., Sharma N., Cartee G. D. (2014) Postexercise improvement in insulin-stimulated glucose uptake occurs concomitant with greater AS160 phosphorylation in muscle from normal and insulin-resistant rats. Diabetes 63, 2297–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hoehn K. L., Hohnen-Behrens C., Cederberg A., Wu L. E., Turner N., Yuasa T., Ebina Y., James D. E. (2008) IRS1-independent defects define major nodes of insulin resistance. Cell Metab. 7, 421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rafaeloff-Phail R., Ding L., Conner L., Yeh W. K., McClure D., Guo H., Emerson K., Brooks H. (2004) Biochemical regulation of mammalian AMP-activated protein kinase activity by NAD and NADH. J. Biol. Chem. 279, 52934–52939 [DOI] [PubMed] [Google Scholar]
- 77. Cantó C., Gerhart-Hines Z., Feige J. N., Lagouge M., Noriega L., Milne J. C., Elliott P. J., Puigserver P., Auwerx J. (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Friedrichsen M., Mortensen B., Pehmøller C., Birk J. B., Wojtaszewski J. F. (2013) Exercise-induced AMPK activity in skeletal muscle: role in glucose uptake and insulin sensitivity. Mol. Cell. Endocrinol. 366, 204–214 [DOI] [PubMed] [Google Scholar]