

Background: Riboswitches regulate purine biosynthesis and transport in many bacteria.
Results: Mutagenic analysis of an adenine-responsive riboswitch revealed features important for efficient co-transcriptional regulation.
Conclusion: Adenine binding to the riboswitch results in a local barrier to strand exchange by the transcriptional terminator, whose formation is facilitated by a stem-loop element.
Significance: This is the first study comprehensively analyzing an RNA structural switch in a cellular context.
Keywords: Fluorescence, Mutagenesis, Purine, RNA Structure, Transcriptional Regulation, Riboswitch
Abstract
Riboswitches are a broadly distributed form of RNA-based gene regulation in Bacteria and, more rarely, Archaea and Eukarya. Most often found in the 5′-leader sequence of bacterial mRNAs, they are generally composed of two functional domains: a receptor (aptamer) domain that binds an effector molecule and a regulatory domain (or expression platform) that instructs the expression machinery. One of the most studied riboswitches is the Bacillus subtilis adenine-responsive pbuE riboswitch, which regulates gene expression at the transcriptional level, up-regulating expression in response to increased intracellular effector concentrations. In this work, we analyzed sequence and structural elements that contribute to efficient ligand-dependent regulatory activity in a co-transcriptional and cellular context. Unexpectedly, we found that the P1 helix, which acts as the antitermination element of the switch in this RNA, supported ligand-dependent activation of a reporter gene over a broad spectrum of lengths from 3 to 10 bp. This same trend was also observed using a minimal in vitro single-turnover transcription assay, revealing that this behavior is intrinsic to the RNA sequence. We also found that the sequences at the distal tip of the terminator not directly involved in alternative secondary structure formation are highly important for efficient regulation. These data strongly support a model in which the switch is highly localized to the P1 helix adjacent to the ligand-binding pocket that likely presents a local kinetic block to invasion of the aptamer by the terminator.
Introduction
Since their discovery and structural characterization, purine-responsive riboswitches have emerged as an important model system for exploring small molecule-RNA interactions, RNA structure, in vitro and in vivo folding, and conformational dynamics (reviewed in Ref. 1). Purine riboswitches present themselves as an ideal model system to explore various aspects of RNA chemistry and biology because of their small size and simple architecture of the ligand-binding domain, very well behaved folding properties, and multiple activities (small-molecule recognition and regulation of gene expression). However, the majority of studies of riboswitches have focused solely upon the ligand-binding (aptamer) domain in isolation using in vitro biochemical, biophysical and structural approaches. Thus, the wealth of information about the structure and function of the aptamer domain is poorly linked to an understanding of regulatory activity, as is the case for most classes of riboswitches. The lack of structural and mechanistic insights into their regulatory activity makes a comprehensive understanding of riboswitches elusive.
The Bacillus subtilis pbuE adenine riboswitch, a transcriptional “ON” switch, embodies the simplest arrangement of a riboswitch acting through a secondary structural switch comprising two mutually exclusive and competing structures (2). At high intracellular concentrations of adenine, binding of adenine stabilizes the aptamer domain of the riboswitch, including the P1 helix that serves as the antitermination element (Fig. 1A). This directly results in up-regulation of expression of the downstream pbuE gene, which codes for a purine efflux pump. When intracellular adenine concentrations are low, the terminator element forms at the expense of the P1 and P3 helices (Fig. 1B). Together, the P1 and terminator (P4a) helices constitute the ligand-dependent secondary structural switch.
FIGURE 1.

Sequence and secondary structure of the B. subtilis pbuE adenine riboswitch. A, proposed secondary structure of the adenine-bound state, in which the switching sequence (outlined sequence, nucleotides 79–94) is incorporated into the aptamer. The adenine nucleobase interacts with nucleotide 89 via Watson-Crick base pairing. Within the expression platform, only P4b likely forms. B, proposed structure of the terminated state, in which P1 and P3 are disrupted by pairing of the switching sequence with a downstream sequence (outlined) to form P4a, which is part of the intrinsic terminator element.
The folding and binding properties of the pbuE aptamer domain, along with several other representative purine riboswitch aptamers, have been studied extensively using a broad set of structural and biophysical techniques (1). Notably, the first comprehensive analysis of co-transcriptional folding of an RNA by single-molecule techniques was recently reported by Frieda and Block (3) using the pbuE riboswitch. In this study, it was observed that the long-range L2-L3 tertiary interaction, a conserved feature of all purine riboswitch aptamer domains, forms independently of ligand, consistent with previous force extension studies of the isolated aptamer domain (4) and NMR studies (5). This leads to an adenine-binding competent state in which the three-way junction (the adenine-binding pocket) is likely preorganized, but the P1 helix is predominantly unformed (3). Adenine binding is required for further structure formation (e.g. the P1 helix), leading to a decrease in transcriptional termination from 85 to 5%. Finally, it was noted that adenine binding only temporarily stalls formation of the terminator element (P4a/b), but this delay is hypothesized to be sufficient for RNA polymerase (RNAP)2 to escape past the polyuridine tract of the regulatory element and continue to synthesize the entire mRNA.
It must be emphasized that these experiments were performed under non-physiological solution conditions and with a minimal Escherichia coli RNAP holoenzyme (3). Thus, the conclusions of this pioneering study may not fully reflect the behavior of the RNA in the cellular context. Indeed, recent studies have pointed to differences between in vitro and in vivo folding (6–8). For example, using SHAPE chemical probing, it was shown that the cellular environment stabilizes the pbuE aptamer domain structure such that even elevated magnesium ion concentrations in vitro (10 mm) fail to fully reflect the structure in E. coli (6). Furthermore, there is some disagreement as to whether RNAP contains a programmed pause in the expression platform that would significantly alter the regulatory properties of the RNA (9). Although no pausing was observed in the single-molecule force extension study (3), another study by Lafontaine and co-workers (10) using single-turnover transcription assays found significant pausing of the polymerase around nucleotides 114–117 for both E. coli and B. subtilis RNAPs. Finally, exchange between alternative structures has been proposed to follow a significantly lower energy pathway in the cell than under standard in vitro conditions for reasons that are not fully known (11). Thus, it is critically important to interrogate key features of biophysically derived observations and resultant hypotheses regarding the relationship between RNA structure and regulatory activity of riboswitches using co-transcriptional and cell-based assays.
Toward this end, we performed a mutational analysis of the B. subtilis pbuE riboswitch using both in vitro and cell-based approaches to determine the sequence and structural features of the riboswitch contributing to efficient switching. We focused primarily upon features of the expression platform and determined the effect of sequence and structure variation on regulatory activity via the riboswitch's ability to activate gfpuv expression after the addition of 2-aminopurine (2-AP), an adenine analog, to the cell culture medium. First, we demonstrate that although the studies of binding affinity for mutants of the isolated aptamer roughly correlate with cellular regulatory activity, there are differences that may reflect deficiencies in tertiary structure acquisition. Second, the riboswitch is highly tolerant to a distribution of P1 lengths spanning from 3 to 10 bp. This reveals that the relative stability of the two competing secondary structures is not requisite for efficient gene regulation. In particular, a very minimal P1 helix supports regulatory activity, indicating that the “switch” is a very localized event around the effector-binding site. Finally, we show that a small stem-loop at the tip of the terminator that is not part of the secondary structural switch (P4b) is essential for efficient regulation, revealing that terminator helix nucleation is a critical feature of the switch. Together, the in vivo data presented in this study are consistent with a model of regulation derived from in vitro studies of the isolated aptamer that predict the secondary structural switch is dictated by a series of kinetic parameters affecting the rate of ligand binding/P1 formation and terminator element formation.
EXPERIMENTAL PROCEDURES
Generation of Riboswitches in a Reporter Vector
Each riboswitch was cloned into a parental reporter vector upstream of gfpuv to regulate its expression in a ligand-dependent fashion derived from a low-copy pBR327 plasmid (12). The parental vector contains a moderately strong synthetic insulated transcriptional promoter, “proD” (13), downstream of the strong rrnB terminator to limit transcriptional read-through from upstream genes. Construction of each plasmid vector was accomplished using standard molecular cloning methods (14) and sequence-verified. Table 1 gives the full and annotated sequence for each riboswitch, starting from the transcription initiation site (+1) to the translation initiation codon.
TABLE 1.
Sequences of riboswitches used in this study
| Riboswitch | Sequence |
|---|---|
| Wild-typea | TTTACGGGCATGCATAAGGCTCGTATAATATATTCGGAAACGAATCAATTAAATAGCTATTATCACTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTTATTTATCAAAACATTTAAGTAAAGGAGTTTGTTATG |
| U66Cb | GGAAACGAATCAATTAAATAGCTATTATCACTTGTATAACCTCAATAATATGGTTTGAGGGTGTCCACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| U89A | GGAAACGAATCAATTAAATAGCTATTATCACTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGAATACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| 5′-Δ3 | AACGAATCAATTAAATAGCTATTATCACTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| 5′-Δ6 | GAATCAATTAAATAGCTATTATCACTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| 5′-Δ9 | TCAATTAAATAGCTATTATCACTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| 5′-Δ11 | AATTAAATAGCTATTATCACTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| 5′-Δ27 | CTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-A48U | AATTAAATAGCTATTATCACTTGTATAACCTCAATATTATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-U49A | AATTAAATAGCTATTATCACTTGTATAACCTCAATAAAATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-A50U | AATTAAATAGCTATTATCACTTGTATAACCTCAATAATTTGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-U51G | AATTAAATAGCTATTATCACTTGTATAACCTCAATAATAGGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-G52C,G53C | AATTAAATAGCTATTATCACTTGTATAACCTCAATAATATCCTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-(P1−4) | AATTAAATAGCTATTATCACAACAATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-(P1−3) | AATTAAATAGCTATTATCACAACTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-(P1−2) | AATTAAATAGCTATTATCACAAGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-(P1−1) | AATTAAATAGCTATTATCACATGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-(P1+1) | AATTAAATAGCTATTATCATTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-(P1+2) | AATTAAATAGCTATTATCTTTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-(P1+3) | AATTAAATAGCTATTATATTTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-(P1+4) | AATTAAATAGCTATTCAATTTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-(P1+5) | AATTAAATAGCTATTAAATTTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-(P1+6) | AATTAAATAGCTATCAAATTTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-(P1+8) | AATTAAATAGCTAACAAATTTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-(P1+10) | AATTAAATAGCAAACAAATTTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-G100A,C108U | AATTAAATAGCTATTATCACTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTATTTATGATATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-G100C | AATTAAATAGCTATTATCACTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTCTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-G100C,C108G | AATTAAATAGCTATTATCACTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAATTTCTTTATGAGATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-(ΔP4b) | AATTAAATAGCTATTATCACTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAAAATATTTTTTTGTAATCAGGATTTTTTTT |
| Δ11-U97A | AATTAAATAGCTATTATCACTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAAATTGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-U97A,U98A | AATTAAATAGCTATTATCACTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAAAATGTTTATGACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-U97A,U98A,L4-GAAA | AATTAAATAGCTATTATCACTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAAAATGTCGAAAGACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-(P1+8),NUCL | AATTAAATAGCTAACAAATTTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAAATGTTTGTTTATGACAGACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-(P1+8),NUCL/GC | AATTAAATAGCTAACAAATTTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAAATGTTGGGTTATGCCCAACATTTTTTGTAATCAGGATTTTTTTT |
| Δ11-(P1+8),NUCL/GAAA | AATTAAATAGCTAACAAATTTTGTATAACCTCAATAATATGGTTTGAGGGTGTCTACCAGGAACCGTAAAATCCTGATTACAAAAATGTTTCTCGAAAGAGAAACATTTTTTGTAATCAGGATTTTTTTT |
a The sequence of the promoter, full-length leader sequence, and ATG codon are shown. The promoter and start codon are italic and underlined. For all other sequences, only the leader sequence from the transcription start site to the last nucleotide of the polyuridine tract of the terminator (U133) are shown.
b Point mutations or other sequence alterations are boldface and underlined.
Cell-based Fluorescence Assays
All experiments were performed using E. coli K12 strain BW25113 (parental strain of the Keio knock-out collection (15)). Cells were transformed with a reporter plasmid containing the wild-type pbuE riboswitch, or mutant variants thereof, using standard molecular biological protocols (14). Transformants were grown on LB plates containing 100 μg/ml carbenicillin for selection for the bla resistance marker. Three individual colonies were picked and used to inoculate individual cultures in 5 ml of CSB growth medium (a rich chemically defined medium (12)) containing 100 μg/ml ampicillin and incubated overnight at 37 °C under constant agitation. Saturated cultures were used to inoculate fresh CSB medium as a 1:100 dilution to a final A600 nm of 0.05. At this stage, effector ligand was added to the medium at a defined concentration, and the cells were incubated at 37 °C for 6 h, at which time the cells were in late-log phase growth.
Ligand-induced expression was determined by measuring the A600 nm and fluorescence intensity of the culture using an Infinite M200 PRO plate reader (Tecan). For all fluorescence measurements, the cells were excited at 395 nm, and fluorescence was read at 510 nm. The raw fluorescence values were divided by the A600 nm to calculate the normalized fluorescence per cell density. The background fluorescence of cells was subtracted from this value, calculated as the normalized fluorescence of cells carrying the parental pBR327 vector grown under the same conditions.
In Vitro Transcription Assays
DNA transcription templates were amplified by PCR from the appropriate plasmid constructed to test activity of the riboswitch in vivo (see above). The two primers used were 5′-GTGGTTGCTGGATAACTTTACGGGC and 5′-CCCGGGGATCCTCTAGAGTCGAC at 1 μm in a standard PCR amplification. These DNA templates were transcribed as described previously (12, 16, 17). Polymerase was bound to the promoter in a first reaction step by incubating 50 ng of DNA template at 37 °C for 10 min in 12.5 μl of 2× transcription buffer (140 mm Tris-Cl (pH 8.0), 140 mm NaCl, 0.2 mm EDTA (pH 8.0), 28 mm β-mercaptoethanol, and 70 μg/ml BSA), 2.5 μl of 25 mm MgCl2, 0.5 μCi of [α-32P]ATP, and 0.25 units of E. coli RNAP σ70 holoenzyme (Epicenter). Transcription was initiated by the addition of 7.5 μl of NTP mixture (75 μm final concentration of each NTP, 0.2 mg/ml heparin, and 3.3 mm 2-AP), followed by incubation for an additional 20 min at 37 °C. Transcription was quenched by the addition of 25 μl of RNA loading buffer (95% (v/v) deionized formamide, 10 mm EDTA (pH 8.0), 0.25% (w/v) bromphenol blue, and 0.25% xylene cyanol) and incubation at 65 °C for 3 min. The RNA products were electrophoretically separated on a denaturing 29:1 acrylamide/bisacrylamide gel. Quantification of the intensities for the bands was performed with ImageJ (http://imagej.nih.gov/ij/).
2-AP Fluorescence Binding Assay
RNA aptamers encompassing nucleotides 32–94 from the pbuE riboswitch with two guanine residues appended to the 5′-end for efficient transcription were generated by T7 in vitro transcription and purified by denaturing PAGE (18). 2-AP binding assays were performed in 50 mm HEPES (pH 7.5), 135 mm KCl, 15 mm NaCl, 10 mm MgCl2, and 50 nm 2-AP with a protocol used previously to monitor 2-AP binding to the purine riboswitch (19). Increasing concentrations of RNA were added to the reactions and incubated for 10 min prior taking fluorescence measurements in the Infinite M200 PRO plate reader. Excitation of 2-AP was performed at 300 nm, and emission was collected from 360 to 370 nm. The change in fluorescence produced by RNA binding to 2-AP was fit to the single-site binding equation (Equation 1),
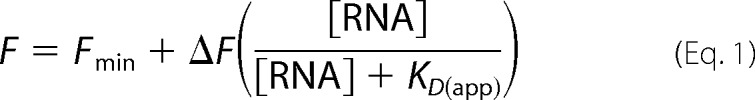 |
where F is the observed fluorescence, Fmin is the fluorescence in the absence of RNA, ΔF is the fluorescence change, and KD(app) is the apparent dissociation constant. The error in the fit values represents the S.E. for three independent titrations.
RESULTS
Validation of Function of an Adenine-responsive Reporter in E. coli
Despite extensive investigation of the isolated aptamer domain of the B. subtilis pbuE adenine riboswitch along with other members of the purine riboswitch family, there have been few studies of its activity in the cellular environment. In one study, the pbuE gene, including its leader sequence, from Bacillus amyloliquefaciens was cloned into E. coli and shown to be induced by the addition of adenine to the medium or by the disruption of the terminator structure by a point mutation (20). This behavior was in agreement with observations of the pbuE riboswitch (previously annotated as ydhL) in its native context (2). The only other published analysis of purine riboswitch function in vivo was a survey of the activity of three guanine riboswitches in B. subtilis (21). In each case, regulatory activity was verified, but no further analysis of function of these riboswitches in a cellular context was pursued.
To interrogate sequence requirements for efficient regulation by the B. subtilis pbuE riboswitch in the cellular environment, we established a reporter system in E. coli. The pbuE riboswitch (spanning sequence from the experimentally verified transcription start site (10) to the initiating codon) (Table 1) was cloned into the leader sequence of a gene encoding a fluorescent protein reporter (gfpuv). This transcription unit was placed under the control of a moderately strong synthetic promoter with an engineered upstream sequence that does not influence initiation by RNAP (13). To monitor riboswitch activity, this plasmid-borne reporter was transformed into E. coli BW25113, the parental strain of the Keio knock-out collection (15). Expression of gfpuv was measured after 6 h of growth (late-log phase) in a rich chemically defined medium by measuring the absorbance-corrected fluorescence of the cells with a background correction using cells with empty vector that does not contain the gfpuv gene.
To validate that this reporter system directly monitors ligand-dependent activation of gene expression, we examined regulation in several different ways. Expression of the reporter was measured as a function of different effector ligands in the growth medium. It is well established that adenine riboswitches productively bind adenine, as well as its analogs 2-AP and 2,6-diaminopurine (2,6-DAP) (2). Conversely, these RNAs have little or no affinity for guanine or hypoxanthine (2). Titration of adenine and its analogs into the growth medium had the expected stimulation of gene expression (Fig. 2A), whereas guanine did not activate gene expression when added to the growth medium at 1 mm (Fig. 2B). Consistent with its higher affinity for the aptamer domain (2, 22), 2,6-DAP began to stimulate expression at lower concentrations compared with adenine or 2-AP, but toxicity effects of the ligand at higher concentrations (>500 μm ligand added to the medium) (Fig. 2A, inset) yielded inflated induction levels (-fold induction) above this concentration. 2-AP and adenine had no toxicity effects up to 1 mm and had clear detectable increases in GFPuv fluorescence. At a moderate effector concentration (500 μm), 2-AP and 2,6-DAP yielded an ∼15-fold increase in fluorescence, whereas adenine was consistently lower (∼10-fold increase). This lower level of response for adenine may be the result of adenine catabolism in the cell or export by the endogenous PbuE efflux pump because adenine and 2-AP have similar properties of binding to the aptamer domain (10, 22, 23). In addition to the increased signal in vivo, several previous reports use 2-AP as a fluorescent probe of ligand binding to facilitate biochemical analysis (19, 23, 24). For these reasons, we conducted the mutagenic analysis of the pbuE riboswitch using 2-AP as a proxy for adenine-dependent binding and regulatory activity.
FIGURE 2.

Ligand-dependent regulatory activity of the pbuE riboswitch in E. coli. A, titration of E. coli cultured in a rich defined medium with varying concentrations of adenine, 2-AP, and 2,6-DAP. At each concentration, the bar height is the cell fluorescence corrected for background fluorescence from cells containing a null reporter vector (no gfpuv) and normalized to measured absorbance at 600 nm. The inset shows the measured absorbance for select ligand concentrations. B, normalized fluorescence for the wild type and select mutants that disrupt the ligand-binding site (U89A and U66C) or the L2-L3 interaction (G53C,G54C) in the absence and presence of 2-AP. As an additional control, the effect of adding 1 mm guanine to the medium was also tested and showed no stimulation of growth. The -fold induction (FI) values are given below the graph. C, effect of deleting nucleotides at the extreme 5′-end of the mRNA on regulatory activity in the presence and absence of the ligand. Error bars represent the S.E. of three separate experiments. A.U., arbitrary units.
To ensure that the observed effector-dependent stimulation of fluorescence is directly due to riboswitch function, we investigated a set of point mutations in the RNA. Conservative mutations in the three-way junction disrupting ligand binding without affecting aptamer structure (U66C) or ablating a tertiary interaction in the aptamer domain (G53C,G54C) in various purine riboswitches (23, 25, 26) were tested for regulatory activity. As expected, both of these mutants had little 2-AP-dependent stimulation of fluorescent protein expression in the presence of 1 mm ligand added to the growth medium (Fig. 2B). Another mutation was made within the aptamer that disrupts both ligand binding and terminator formation (U89A). In this case, the disruption to the terminator stem-loop was expected to be dominant, and indeed, this riboswitch showed increased expression of GFP in the absence of the ligand (Fig. 2B). Thus, these data strongly support the conclusion that this reporter system monitors riboswitch function. It should be noted that our research group has previously used this reporter system to investigate artificial riboswitches, and similar controls validated the assay in each case (12, 27).
The results described above were obtained in the context of the full-length leader sequence as described recently by Lafontaine and co-workers (10), where the transcription start site was determined by reverse transcriptase sequencing of the native mRNA. This sequence includes an additional 11 nucleotides of sequence preceding the 5′-side of the P1 helix of the aptamer domain that were not part of the original annotation of the pbuE riboswitch (Fig. 1B, asterisk) (2). To determine whether this additional sequence affects regulatory activity, we examined a set of deletion variants spanning this region (Fig. 2C). Removal of the first 11 nucleotides (referred to as Δ11) had no significant effect on activation of gene expression at 1 mm 2-AP, indicating that this sequence does not play a role in the structure or function of this riboswitch. Further erosion of the leader by 27 nucleotides such that the mRNA began adjacent to the 5′-side of the P1 helix yielded higher levels of fluorescence in both the absence and presence of the ligand, but with similar -fold induction as Δ11. This unambiguously defines the functional boundaries of the pbuE riboswitch as spanning from the beginning of the P1 helix to the polyuridine tract of the terminator. All further experiments in this study were performed in the context of the Δ11 mutation, corresponding to a start site at position +12 of the wild-type RNA, which has the most reproducible behavior in the cell-based assay.
Tertiary Interactions Mediate the Regulatory Activity of the Riboswitch beyond Ligand Binding
All purine riboswitches contain a conserved loop-loop interaction (L2-L3) (Fig. 3A) that orients P2 and P3 adjacent to one another and serves to organize the ligand-binding pocket in the three-way junction (2). For representative guanine and adenine aptamers, this interaction contributes ∼4 kcal/mol of binding energy to the ligand-RNA interaction in vitro (26, 28). Furthermore, as shown above, ablating this interaction by disrupting two universally conserved Watson-Crick interactions in the purine riboswitch family (G52-C75 and G53-C76) results in complete loss of regulatory activity (Fig. 2B), reinforcing the essential nature of this interaction as observed in biochemical analyses for the individual G52C and G53C mutations (26). Detailed mutagenic analysis of the L2-L3 interaction in the context of the B. subtilis pbuE (26) and guanine-sensing xpt-pbuX (25) riboswitches indicated that these 2 central bp and a few other directly interacting nucleotides are critical for ligand binding. The identity of other nucleotides in the loops, which are phylogenetically conserved (Fig. 3A), however, contributes moderately to negligibly with respect to ligand binding affinity (26).
FIGURE 3.
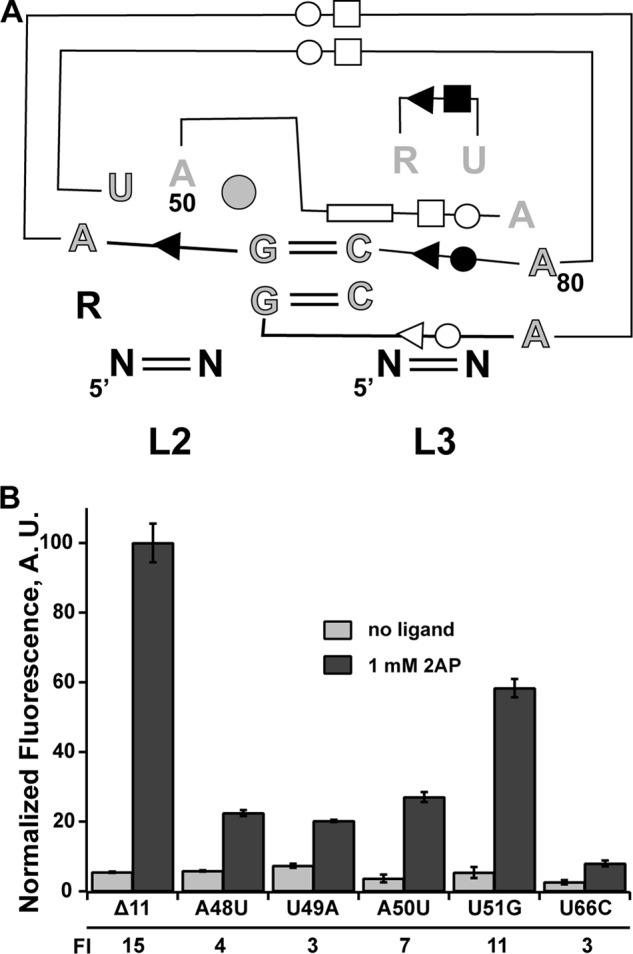
Effect of mutations in loop 2 of the pbuE receptor on binding and regulatory activity. A, schematic of the loop-loop interaction highlighting base-base interactions and conservation patterns. Outlined and gray nucleotides are >97% conserved, gray nucleotides are >90% conserved, and black nucleotides are 75% conserved. The circle at position 51 indicates that there is a nucleotide present at this position in >97% of sequences. The symbols denoting base-base interaction are the Leontis and Westhof notation (42), reflecting Watson-Crick pairing (double lines) and non-canonical base-base interaction (single lines with symbol denoting the edge of the base used in the interaction: circle, Watson-Crick; square, Hoogsteen; triangle, sugar edge). B, effect of point mutations in L2 on 2-AP-dependent regulatory activity. The -fold induction (FI) values are given below the graph. Error bars represent the S.E. of three separate experiments. A.U., arbitrary units.
To assess the impact of these mutations on regulatory activity, we first determined their effect on the affinity of 2-AP using an established in vitro fluorescence quenching assay (26, 28). Binding of 2-AP to the RNA results in significant quenching of its fluorescence, which is used to observe the ligand-RNA interaction (24). These experiments were performed at 37 °C in a buffered solution containing 50 mm HEPES (pH 7.5), 135 mm KCl, 15 mm NaCl, and 10 mm MgCl2, reflecting more physiological conditions than previous experiments (25 °C; Tris-Cl (pH 8.0), 100 mm KCl, and 10 mm MgCl2) (23). We determined the KD(app) values of the wild-type aptamer and four destabilizing loop mutants (Table 2) and found that their values were consistent with previously reported studies of this riboswitch (26). Note that we tested mutants only in L2 because mutations in L3 would also affect the secondary structural switch (Fig. 1). Mutants U49A, A50U, and U51G displayed a 2–3-fold decrease in affinity, whereas mutant A48U displayed a drastic change of ∼10-fold. The larger effect of A48U is consistent with its role as part of one of the critical base quartets, the central feature of the L2-L3 interaction (29, 30).
TABLE 2.
Affinities of 2-AP for wild-type and mutant pbuE aptamer domains
| RNA | KD(app) (nm) | KD(rel)a |
|---|---|---|
| Wild-type | 300 ± 30 | 1.0 |
| U66C | 200,000 ± 10,000 | 660 |
| G52C,G53C | ND | |
| A48U | 2700 ± 300 | 9.2 |
| U49A | 880 ± 90 | 3.0 |
| A50U | 720 ± 120 | 2.4 |
| U51G | 870 ± 90 | 2.9 |
a KD(rel) = KD(app)(mutant)/KD(app)(WT); ND, not detectable.
The effects of these mutations on regulation did not completely correlate with their impact on binding affinity. At 1 mm 2-AP, mutants A48U, U49A and A50U exhibited an ∼5-fold reduction in activity at saturating 2-AP compared with the wild-type riboswitch, despite significant differences in their affinities (Fig. 3B). Consistent with the small impact on the binding affinity of the U51G mutant, the low phylogenetic conservation of this nucleotide, and its being flipped out into the solvent in all purine riboswitch structures (29, 30), this mutant retained substantial ligand-dependent antitermination, but was still reduced relative to the wild type. The differences between equilibrium binding measurements and cell-based assays most likely reflect the role of conserved nucleotides in co-transcriptional folding of the RNA, a critical aspect of riboswitch activity. This limited data set reinforces the need to examine aptamer binding properties in a regulatory context rather than solely using equilibrium binding to the isolated aptamer to fully assess how RNA structural features contribute to its activity.
Regulatory Activity Is Observed over a Broad Spectrum of P1 Helix Lengths
To define the influence of the length and stability of the P1 helix on regulatory activity, a systematic set of mutations were introduced on the 5′-side of the P1 helix. Mutations were made that added or removed Watson-Crick base pairs from P1 while maintaining their ability to form pairs in P4a, and each mutant was verified for minimal potential to form alternative structure using mfold (31). Interestingly, we observed that a broad range of P1 lengths supported 2-AP-dependent gene expression (Fig. 4A). These results contrast with previous reports demonstrating that some riboswitches lose their ability to regulate transcription upon addition or deletion of only a few base pairs to their P1 helix (12). For example, for the B. subtilis metE riboswitch, which terminates transcription upon binding S-adenosylmethionine, deletion of 2 bp yields a constitutively ON riboswitch, whereas the addition of 1 bp turns transcription off regardless of the presence of effector. For the pbuE riboswitch, the minimal length of P1 necessary to support 2-AP-dependent switching is three nucleotides; further erosion of the helix presumably ablates ligand binding. Two of these base pairs, A36-U90 and U35-A91, are required to form ligand-dependent triples with J2/3, whereas the third is weakly conserved as an R-Y pair with no obvious role in ligand binding (29, 30). This is consistent with observations that a 4-bp P1 helix can support near wild-type affinity (3.7-fold lower), whereas only 2 bp in P1 reduce affinity by ∼50-fold (23). Thus, although a 2-bp P1 helix supports binding, the previous base pair (G34-C93) is important for regulation.
FIGURE 4.

Effect of mutations that change the length of the P1 helix on regulatory activity. A, effect of point mutations on the 5′-side of P1 that either remove (−4 through −1) or add (+1 through +10) Watson-Crick pairs on 2-AP-dependent regulatory activity. A binding-incompetent mutant (U66C) is shown for comparison. The -fold induction (FI) values are given below the graph. Error bars represent the S.E. of three separate experiments. A.U., arbitrary units. B, histogram of the length distributions of the P1 helix found within the purine riboswitch family using all non-redundant sequences deposited in Rfam 11.0 (33). This distribution corresponds to an analysis considering the P1 helix to be strictly a contiguous stretch of canonical base pairs (A-U, G-C, and G·U). C, single-turnover transcription assays of a subset of P1 length variants tested in A. The values for the percentage read-through transcription in the absence and presence of 1 mm 2-AP are given in Table 3.
Sequential stabilization of the P1 helix showed that this element can be extended to 10 bp in length before loss of regulatory activity (Fig. 4A). At the upper end of the length series (P1+4 and P1+5), a small increase in gene expression in the absence of 2-AP was observed, suggesting that these lengths may begin to impair efficient terminator helix formation. Further extension of the P1 helix by a single nucleotide (11 bp total) sharply reduced regulatory activity by ∼5-fold via a significant increase in expression in the absence of 2-AP. These data indicate that the terminator element of pbuE effectively forms in the context of a broad spectrum of P1 lengths and stabilities. In contrast, a recent study by Schwalbe and co-workers (32) using the isolated B. subtilis pbuE aptamer domain suggests that elongation of the P1 helix is deleterious to the riboswitch's binding activity, again emphasizing that observations of in vitro binding activity may not perfectly correlate with in vivo regulatory activity.
The lower and upper boundaries of P1 length supporting riboswitch function in this mutational analysis correlate with the length distribution observed across purine riboswitch phylogeny. The length of the P1 helix in a set of RNAs comprising all non-redundant purine riboswitch sequences (Rfam 11.0, accession number RF00167 (33)) was determined by considering P1 either as the total number of contiguous Watson-Crick base pairs counting back from the universally conserved A-U pair adjacent to the three-way junction (A36-U90 in Fig. 1A) or as the total number of contiguous pairs allowing for a single mismatched pair. A histogram of P1 lengths for the first means of calculating length in shown in Fig. 4B; the more generous method leads to a small increase in sequences in the upper tail of the distribution (mean lengths of 7.1 and 7.3 bp, respectively). Thus, the results of the mutational analysis of the P1 helix length are consistent with observed natural variation within the purine riboswitch family. This mutational analysis also predicts that in natural purine riboswitches there is a low correlation between length and/or stability of the P1 and alternative helices in the structural switch.
The above experiments only indirectly monitored secondary structural switching through expression of the gfpuv reporter. Furthermore, these experiments were performed in a cellular context, and efficient switching may require protein factors or other cellular agents rather than being encoded solely by RNA sequence (9). Other factors influencing the magnitude of expression, such as potential changes in mRNA stability across the series of variants, were also not assessed by this assay. To directly observe intrinsic regulatory activity, we assayed the above set of P1 mutants using an in vitro single-turnover transcription assay (34). This assay is commonly used to determine the effect of small molecules on transcriptional activity using a minimal system comprising a DNA template encoding the riboswitch, E. coli RNAP holoenzyme, and 75 μm each NTP and ligand. To evaluate the effect of the ligand on disruption of the terminator element, the transcribed 32P-labeled terminated and read-through products were separated by denaturing PAGE, and the amount of terminated and read-through transcript was quantified.
The in vitro transcription assay revealed an identical range of P1 lengths supporting regulatory switching as observed in the cellular environment (Fig. 4C and Table 3), which is generally consistent with a more limited set of observations made using this assay by another group (10). The highest dynamic range/-fold induction observed in both assays was over the P1−2 to P1+3 lengths, with a moderate decrease in the P1+4 and P1+5 lengths due to read-through transcription under no-effector conditions. It is important to emphasize that the concentration of nucleotides in the in vitro reaction is substantially lower than in rapidly dividing E. coli (1–2 mm) (35), which influences the rate of transcription (9). Despite this difference, along with the absence of factors (such as NusA) that influence pausing and transcriptional termination (9), the in vitro transcription assay was strongly consistent with the cell-based assay. This directly demonstrates that efficient switching is intrinsic to the RNA sequence in the context of a minimal transcriptional apparatus rather than assisted by components found in the cellular context (see “Discussion”).
TABLE 3.
Transcriptional regulation by P1 length variants of the pbuE riboswitch in vitro
| Mutant | Read-through transcription |
|
|---|---|---|
| No ligand | 1 mm 2-AP | |
| % | ||
| P1−4 | 2 ± 2 | 3 ± 3 |
| P1−3 | 0 ± 2 | 0 ± 2 |
| P1−2 | 5 ± 4 | 87 ± 3 |
| P1−1 | 4 ± 4 | 80 ± 6 |
| WT | 2 ± 2 | 67 ± 4 |
| P1+1 | 2 ± 2 | 64 ± 8 |
| P1+2 | 6 ± 3 | 50 ± 3 |
| P1+3 | 6 ± 3 | 90 ± 2 |
| P1+4 | 47 ± 9 | 86 ± 6 |
| P1+5 | 51 ± 6 | 92 ± 2 |
| P1+6 | 91 ± 2 | 94 ± 2 |
| P1+8 | 92 ± 2 | 91 ± 3 |
| P1+10 | 91 ± 2 | 94 ± 2 |
A “Nucleator” Stem-Loop Is Required for Promoting Efficient Regulatory Activity
An interesting observation from the above data relates to the boundary at which further lengthening the P1 helix eliminates regulatory activity. In this series, the P1+6 mutant corresponds to the formation of a base pair that competes with formation of the G100-C108 pair in P4b (Fig. 1). This suggests that disruption of P4b is deleterious to efficient terminator formation and thereby transcriptional termination in the absence of the ligand. A similar type of stem-loop motif capping an antiterminator element in the B. subtilis lysC riboswitch was previously shown to be required for efficient lysine-dependent regulatory activity in a single-turnover transcription assay (36). Although not universally conserved among all expression platforms, a stem-loop capping the helix competing with P1 that does not directly participate in the secondary structural switch is common and may be an important feature for regulatory activity in many riboswitches.
To determine whether P4b influences regulatory activity, a series of mutations in this region expected to have either destabilizing or stabilizing influences on this small stem-loop structure were investigated. Again, this set of mutations was designed to minimize potential for alternative structures in the P1/P4a switch. A mutation in P4b converting the G100-C108 base pair into an A-U pair was predicted to destabilize P4b by 1.9 kcal/mol, and introduction of a C-C mismatch (G100C) was predicted to eliminate this helix by mfold. These mutations retained weak 2-AP-dependent stimulation of reporter expression, but in the absence of ligand, there was significantly elevated expression of GFP (Fig. 5A). Introducing a compensatory mutation to G100C (G100C,C109G) rescued riboswitch activity by allowing the system to terminate in the absence of the ligand. Interestingly, another mutation eliminating P4b and the 2 × 2 internal loop separating P4a and P4b by deletion of nucleotides 97–108 (ΔP4b) (Fig. 5A) showed wild-type regulatory activity. In this RNA, a UAU triloop directly capping P4a was sufficient to promote regulatory activity (note that the terminal 2 bp of this stem do not participate in the regulatory switch). Together, these data reveal that although P4b is not essential for regulatory activity, it positively influences the efficiency of termination.
FIGURE 5.

Effects of mutations that alter that terminator hairpin on regulatory activity. A, effect of point mutations in P4b or the internal loop between P4a and P4b on 2-AP-dependent regulatory activity. To the right is the secondary structure of each mutant tested. The -fold induction (FI) values are given below the graph. B, effect of extending the P4b helix to compensate for its partial disruption by the P1+8 mutation. Error bars represent the S.E. of three separate experiments. A.U., arbitrary units.
Stabilizing mutations in P4b displayed differing levels of regulatory activity through decreased expression of GFP in both the absence and presence of 2-AP compared with the wild-type riboswitch. Stabilization of the RNA around the junction of the P4a/P4b helices by mutating one (U98A) or two (U98A,U99A) uridines in the internal loop to create Watson-Crick pairs supported significant levels of 2-AP-stimulated expression. Furthermore, expression levels in the absence of the ligand were reduced to background levels compared with the empty vector control. Further mutation in which the terminal loop was converted to an ultra-stable GAAA tetraloop (37) along with ablating the internal loop (Fig. 5A) yielded a functional riboswitch, but with strongly decreased expression levels at both low and high 2-AP concentrations compared with the wild type. Notably, all of these riboswitches had greater values for overall -fold induction relative to the wild type due to the increased repression at low ligand concentrations. An alternative explanation for the observed decrease in read-through product is that these mutations may affect transcriptional pausing. One study of the pbuE riboswitch observed pausing of both E. coli and B. subtilis RNAPs at a short polyuridine tract in region 112–117 (10). However, none of these mutations directly altered this tract and thus would not be expected to affect pausing. Furthermore, in the aforementioned study and that of Frieda and Block (3), transcriptional pausing was not observed at high NTP concentrations mimicking the cellular environment. Therefore, these data, especially those in the absence of 2-AP, can be interpreted as P4b promoting efficient formation of the terminator element, likely through rapid nucleation and subsequent propagation of the terminator stem via invasion of the P1 helix.
To further test this hypothesis, we constructed a set of terminator stem-loop variants that aimed to rescue the highly deleterious effect of the P1+8 mutant (13 bp in P1), which was predicted to disrupt formation of P4b (Fig. 5B). To accomplish this, we extended P4b with additional base pairs and/or stabilizing tetraloops. Extension of P4b with a weak stem-loop of the sequence of wild-type P4b did not rescue the loss of termination at low 2-AP concentrations (P1+8,NUCL) (Fig. 5B). However, the addition of stem-loops that either strengthen base pairing or add an ultra-stable C(GAAA)G tetraloop restored efficient 2-AP-dependent regulatory activity ((P1+8),NUCL/GC and (P1+8),NUCL/GAAA) (Fig. 5B). The tetraloop itself is not expected to contribute to the rate of hairpin nucleation, but rather the stability of the closing base pairs (38). The observed rescue by the addition of G-C pairs in both NUCL/GC and NUCL/GAAA that was predicted to increase the rate of stem-loop nucleation suggests that efficient nucleation of the terminator is critical for regulatory activity.
It is important to note that the NUCL/GAAA variant had a 2-AP-dependent regulatory response nearly identical to that of the wild type, with a P1 helix of 13 bp in length. This again contrasts with a recent report suggesting that a pbuE riboswitch variant with an elongated P1 helix (9 bp stabilized with four additional G-C pairs) and stabilizing P1 have a negative impact on RNA folding and ligand affinity (32). We observed that the concentration of 2-AP in the growth medium required to elicit a half-maximal regulatory response was also very similar between the two RNAs (∼300 μm for the wild type and ∼100 μm for (P1+8),NUCL/GAAA), suggesting that P1 stability is not important for regulatory function. This further reinforces views that riboswitches may behave differently in vitro and in the context of transcription and the cellular environment.
DISCUSSION
Over the past decade, isolated riboswitch aptamer domains have been studied extensively using a broad spectrum of in vitro approaches (1). This study has taken the next step by investigating effector-dependent regulation of the B. subtilis pbuE adenine-responsive riboswitch (which contains one of the best characterized aptamers) in the context of transcription and under cellular conditions that more closely match the speed of RNA transcription, salt concentrations, and molecular crowdedness of the native environment of the riboswitch. We must emphasize that this analysis was conducted in a heterologous system with respect to organism and genetic context (promoter strength, copy number, plasmid-borne versus integrated into the bacterial chromosome, etc.) and thus should not be strictly considered “in vivo” but rather as a molecular analysis under cellular conditions. Nonetheless, there is at least in vitro evidence that multiple classes of riboswitches behave similarly when transcribed with B. subtilis or E. coli RNAPs, including pbuE (9, 10, 39). Thus, our observations are likely to strongly approximate the behavior of this riboswitch in its native biological context.
This study represents the first comprehensive analysis of an RNA secondary structural switch in the cellular context, critically important for developing a comprehensive understanding of the mechanism of these regulatory elements. First, we have shown that observations of ligand binding to the isolated aptamer domain do not fully account for patterns of phylogenetic conservation in purine riboswitches. Analysis of mutations of the loop-loop interaction indicates that components of L2 contributing modestly to equilibrium binding affinity are important for regulatory activity in the cellular environment. We interpret these differences as these nucleotides being important for efficient formation of tertiary structure required for ligand binding. This can influence regulatory activity in one of two ways. First, the identity of these nucleotides could be important for high-fidelity folding, i.e. mutations in L2 may result in a fraction of mRNAs that are misfolded. The alternative is that these nucleotides impact the rate of L2-L3 formation. For a temporal (kinetically driven) process, the time for the aptamer to become competent to bind ligand will have a significant effect on the aptamer's occupancy status when RNAP reaches the intrinsic terminator and the mandatory regulatory decision is made. The cell-based data cannot discriminate between these alternatives, as each would manifest itself as a lowering of reporter expression at high 2-AP concentrations, as shown in Fig. 4B. In contrast to several studies indicating that the identity of the nucleotides composing the two terminal base-base interactions in L2 and L3 are not important for in vitro ligand affinity (25, 26), intracellular activity measurements more closely reflect observed conservation of nucleotide identity within L2 in the purine riboswitch family, which likely reflect efficient co-transcriptional folding requirements.
Most interestingly, we found that switching in pbuE is tolerant to a spectrum of P1 helical lengths. Erosion of P1 to three nucleotides still enables 2-AP-dependent regulatory activity with cellular normalized fluorescence increased to nearly the same levels as in the wild-type RNA at high effector levels (Fig. 4), indicating that the reduced aptamer is still an effective antiterminator. At the other end of the spectrum, the longest functional P1 helices (P1+4 and P1+5) also regulate expression in a 2-AP-dependent fashion, but exhibit a small but significant increase in expression in the absence of 2-AP. This indicates that the terminator does not form in a fraction of the transcription events even in the absence of ligand, despite the fact that it remains significantly more thermodynamically stable that its P1 counterpart (mfold (31) predicts −26.4 and −13.3 kcal/mol, respectively).
These observations are best understood in the context of a co-transcriptional folding model in which secondary structures fold sequentially but also exchange rapidly through branch migration (40). In this mechanism, the P1 helix forms initially but is disrupted by the 5′-side of the terminator helix. In the riboswitches with the longest functional P1 helices, increased background expression in the absence of 2-AP is likely due to the terminator not being able to fully displace P1 and P3 prior to the polymerase escaping past the polyuridine tract of the intrinsic terminator in a fraction of transcription events. In most events, regardless of P1 length, the terminator invades the less stable P1 helix until it reaches base pairs adjacent to the junction. Further branch migration is stalled by ligand-dependent structure around P1 and J3/1. In particular, ligand-induced formation of base triples between J2 and J3 (U65 and C66), and the two Watson-Crick pairs proximal to the junction (U35-A91 and A36-U90, respectively) (29, 30) likely serve as the primary barrier to strand exchange in the bound state, consistent with interpretations of single-molecule observations of co-transcriptional folding of the pbuE riboswitch by Frieda and Block (3). In this study, it was proposed that the ligand-bound aptamer presents a short kinetic block that is sufficient to stall completion of the terminator element until the RNAP has escaped past the polyuridine tract. Thus, the key switch of the RNA is a localized event involving the progression of branch migration through a few critical nucleotides in the aptamer.
Although the pbuE riboswitch is able to accommodate a spectrum of P1 lengths, there is a clear upper boundary (P1+6) (Fig. 4). One interpretation of this model is that the terminator can no longer branch-migrate through a P1 helix of >10 bp in the timescale of transcription. We hypothesize that the activity loss is instead due to disruption of a small stem-loop (P4b) (Fig. 1) that forms independently of the occupancy status of the aptamer domain and likely serves to promote efficient terminator formation. Mutations that destabilize this element negatively impact transcriptional termination in the absence of ligand, and one of these mutations (G100C) can be rescued to wild-type activity through a compensatory C108G mutation. Conversely, mutations that stabilize P4b substantially reduce reporter expression, regardless of 2-AP. These variants exhibit the largest -fold induction, primarily because the amount of reporter expression in the absence of 2-AP approaches that of the negative control. As direct support that the length of P1 alone is not the cause of loss of regulatory activity, we were able to rescue the regulatory activity of P1+8 (13 bp) by extending the length of the terminator helix and restoring a nucleation site not disrupted by P1. Thus, we propose that another key determinant in the regulatory activity of the pbuE riboswitch is the rate of terminator nucleation, which is related to sequences not directly involved in alternative base pairing. For many riboswitches, the rate of nucleation of the helix alternative to P1 may be a means of tuning the dynamic range to meet the needs of the transcriptional unit (41). Unfortunately, very little quantitative information is available regarding how stem-loop sequence and structure determine the rate at which it forms (38).
It is worth noting that the pbuE riboswitch relies upon the ligand directly blocking branch migration through the aptamer to form the functional terminator element. However, there are a number of riboswitches in which this is not the case. In these RNAs, the minimal sequence required for high-affinity ligand binding is distinct from sequences that directly participate in the secondary structural switch, i.e. in these RNAs, the helix alternative to P1 does not invade into the aptamer domain. These expression platforms and associated regulatory switch can host a variety of distinct aptamers and retain efficient ligand-dependent regulatory activity (12, 27). An interesting difference is that these riboswitches (e.g. the B. subtilis metE SAM-I riboswitch) can be exquisitely sensitive to P1 length and that efficient regulatory activity in one case is restricted to single-P1 length (12). How these observations relate to the observations of this study is not clear and merits further investigation. It is likely that elements of the above model relate to these riboswitches but that the details of how ligand binding prevents formation of the competing helix may differ. Nonetheless, this work reveals new insights into the mechanism of regulation by the pbuE riboswitch, and these findings are likely to be broadly representative of a number of riboswitches controlling transcription or translation, and highlights the importance of investigating riboswitches and, by extension, many other RNAs in a cellular and co-transcriptional context.
Acknowledgments
We thank Chris Wostenberg, Jacob Polaski, Ely Porter, and Jeremy Trausch for critical reading of this manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant GM073850 (to R. T. B.) and Biophysics Training Grant T32 GM065103 (to J. G. M.-V.).
- RNAP
- RNA polymerase
- 2-AP
- 2-aminopurine
- 2,6-DAP
- 2,6-diaminopurine.
REFERENCES
- 1. Porter E. B., Marcano-Velázquez J. G., Batey R. T. (2014) The purine riboswitch as a model system for exploring RNA biology and chemistry. Biochim. Biophys. Acta 1839, 919–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mandal M., Breaker R. R. (2004) Adenine riboswitches and gene activation by disruption of a transcription terminator. Nat. Struct. Mol. Biol. 11, 29–35 [DOI] [PubMed] [Google Scholar]
- 3. Frieda K. L., Block S. M. (2012) Direct observation of cotranscriptional folding in an adenine riboswitch. Science 338, 397–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Greenleaf W. J., Frieda K. L., Foster D. A., Woodside M. T., Block S. M. (2008) Direct observation of hierarchical folding in single riboswitch aptamers. Science 319, 630–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Noeske J., Buck J., Fürtig B., Nasiri H. R., Schwalbe H., Wöhnert J. (2007) Interplay of ‘induced fit’ and preorganization in the ligand induced folding of the aptamer domain of the guanine binding riboswitch. Nucleic Acids Res. 35, 572–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tyrrell J., McGinnis J. L., Weeks K. M., Pielak G. J. (2013) The cellular environment stabilizes adenine riboswitch RNA structure. Biochemistry 52, 8777–8785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Strulson C. A., Boyer J. A., Whitman E. E., Bevilacqua P. C. (2014) Molecular crowders and cosolutes promote folding cooperativity of RNA under physiological ionic conditions. RNA 20, 331–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Strulson C. A., Yennawar N. H., Rambo R. P., Bevilacqua P. C. (2013) Molecular crowding favors reactivity of a human ribozyme under physiological ionic conditions. Biochemistry 52, 8187–8197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wickiser J. K., Winkler W. C., Breaker R. R., Crothers D. M. (2005) The speed of RNA transcription and metabolite binding kinetics operate an FMN riboswitch. Mol. Cell 18, 49–60 [DOI] [PubMed] [Google Scholar]
- 10. Lemay J. F., Desnoyers G., Blouin S., Heppell B., Bastet L., St-Pierre P., Massé E., Lafontaine D. A. (2011) Comparative study between transcriptionally- and translationally-acting adenine riboswitches reveals key differences in riboswitch regulatory mechanisms. PLoS Genetics 7, e1001278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mahen E. M., Harger J. W., Calderon E. M., Fedor M. J. (2005) Kinetics and thermodynamics make different contributions to RNA folding in vitro and in yeast. Mol. Cell 19, 27–37 [DOI] [PubMed] [Google Scholar]
- 12. Ceres P., Garst A. D., Marcano-Velázquez J. G., Batey R. T. (2013) Modularity of select riboswitch expression platforms enables facile engineering of novel genetic regulatory devices. ACS Synth. Biol. 2, 463–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Davis J. H., Rubin A. J., Sauer R. T. (2011) Design, construction and characterization of a set of insulated bacterial promoters. Nucleic Acids Res. 39, 1131–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sambrook J., Russell D. W. (2001) Molecular Cloning: A Laboratory Manual, 3 Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York [Google Scholar]
- 15. Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K. A., Tomita M., Wanner B. L., Mori H. (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Trausch J. J., Ceres P., Reyes F. E., Batey R. T. (2011) The structure of a tetrahydrofolate-sensing riboswitch reveals two ligand binding sites in a single aptamer. Structure 19, 1413–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Garst A. D., Porter E. B., Batey R. T. (2012) Insights into the regulatory landscape of the lysine riboswitch. J. Mol. Biol. 423, 17–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Edwards A. L., Garst A. D., Batey R. T. (2009) Determining structures of RNA aptamers and riboswitches by x-ray crystallography. Methods Mol. Biol. 535, 135–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stoddard C. D., Widmann J., Trausch J. J., Marcano-Velázquez J. G., Knight R., Batey R. T. (2013) Nucleotides adjacent to the ligand-binding pocket are linked to activity tuning in the purine riboswitch. J. Mol. Biol. 425, 1596–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zakataeva N. P., Gronskiy S. V., Sheremet A. S., Kutukova E. A., Novikova A. E., Livshits V. A. (2007) A new function for the Bacillus PbuE purine base efflux pump: efflux of purine nucleosides. Res. Microbiol. 158, 659–665 [DOI] [PubMed] [Google Scholar]
- 21. Mulhbacher J., Lafontaine D. A. (2007) Ligand recognition determinants of guanine riboswitches. Nucleic Acids Res. 35, 5568–5580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wickiser J. K., Cheah M. T., Breaker R. R., Crothers D. M. (2005) The kinetics of ligand binding by an adenine-sensing riboswitch. Biochemistry 44, 13404–13414 [DOI] [PubMed] [Google Scholar]
- 23. Lemay J. F., Lafontaine D. A. (2007) Core requirements of the adenine riboswitch aptamer for ligand binding. RNA 13, 339–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gilbert S. D., Stoddard C. D., Wise S. J., Batey R. T. (2006) Thermodynamic and kinetic characterization of ligand binding to the purine riboswitch aptamer domain. J. Mol. Biol. 359, 754–768 [DOI] [PubMed] [Google Scholar]
- 25. Gilbert S. D., Love C. E., Edwards A. L., Batey R. T. (2007) Mutational analysis of the purine riboswitch aptamer domain. Biochemistry 46, 13297–13309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lemay J. F., Penedo J. C., Tremblay R., Lilley D. M., Lafontaine D. A. (2006) Folding of the adenine riboswitch. Chem. Biol. 13, 857–868 [DOI] [PubMed] [Google Scholar]
- 27. Ceres P., Trausch J. J., Batey R. T. (2013) Engineering modular ‘ON’ RNA switches using biological components. Nucleic Acids Res. 41, 10449–10461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stoddard C. D., Gilbert S. D., Batey R. T. (2008) Ligand-dependent folding of the three-way junction in the purine riboswitch. RNA 14, 675–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Batey R. T., Gilbert S. D., Montange R. K. (2004) Structure of a natural guanine-responsive riboswitch complexed with the metabolite hypoxanthine. Nature 432, 411–415 [DOI] [PubMed] [Google Scholar]
- 30. Serganov A., Yuan Y. R., Pikovskaya O., Polonskaia A., Malinina L., Phan A. T., Hobartner C., Micura R., Breaker R. R., Patel D. J. (2004) Structural basis for discriminative regulation of gene expression by adenine- and guanine-sensing mRNAs. Chem. Biol. 11, 1729–1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zuker M. (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nozinovic S., Reining A., Kim Y.-B., Noeske J., Schlepckow K., Wöhnert J., Schwalbe H. (2014) The importance of helix P1 stability for structural pre-organization and ligand binding affinity of the adenine riboswitch aptamer domain. RNA Biol. 11, 655–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Griffiths-Jones S., Bateman A., Marshall M., Khanna A., Eddy S. R. (2003) Rfam: an RNA family database. Nucleic Acids Res. 31, 439–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Artsimovitch I., Henkin T. M. (2009) In vitro approaches to analysis of transcription termination. Methods 47, 37–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bennett B. D., Kimball E. H., Gao M., Osterhout R., Van Dien S. J., Rabinowitz J. D. (2009) Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 5, 593–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Blouin S., Chinnappan R., Lafontaine D. A. (2011) Folding of the lysine riboswitch: importance of peripheral elements for transcriptional regulation. Nucleic Acids Res. 39, 3373–3387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Blose J. M., Proctor D. J., Veeraraghavan N., Misra V. K., Bevilacqua P. C. (2009) Contribution of the closing base pair to exceptional stability in RNA tetraloops: roles for molecular mimicry and electrostatic factors. J. Am. Chem. Soc. 131, 8474–8484 [DOI] [PubMed] [Google Scholar]
- 38. Nagel J. H., Flamm C., Hofacker I. L., Franke K., de Smit M. H., Schuster P., Pleij C. W. (2006) Structural parameters affecting the kinetics of RNA hairpin formation. Nucleic Acids Res. 34, 3568–3576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McDaniel B. A., Grundy F. J., Artsimovitch I., Henkin T. M. (2003) Transcription termination control of the S box system: direct measurement of S-adenosylmethionine by the leader RNA. Proc. Natl. Acad. Sci. U.S.A. 100, 3083–3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mahen E. M., Watson P. Y., Cottrell J. W., Fedor M. J. (2010) mRNA secondary structures fold sequentially but exchange rapidly in vivo. PLoS Biol. 8, e1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tomsic J., McDaniel B. A., Grundy F. J., Henkin T. M. (2008) Natural variability in S-adenosylmethionine (SAM)-dependent riboswitches: S-box elements in Bacillus subtilis exhibit differential sensitivity to SAM in vivo and in vitro. J. Bacteriol. 190, 823–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Leontis N. B., Westhof E. (2001) Geometric nomenclature and classification of RNA base pairs. RNA 7, 499–512 [DOI] [PMC free article] [PubMed] [Google Scholar]