

Background: The Ski oncoprotein and tumor suppressor is a negative regulator of the antimitotic TGF-β/Smad pathway.
Results: The Ski protein is localized in the nucleus and cytoplasm of hepatocytes. Ski protein stability is controlled differentially by actin cytoskeleton dynamics.
Conclusion: TGF-β/Smads and GPCR/actin cytoskeleton-dynamic signals regulate Ski protein stability via the proteasome in hepatocytes.
Significance: Stabilization of Ski protein may favor the proliferation of regenerating hepatocyte.
Keywords: Actin, Cytoskeleton, G Protein-coupled Receptor (GPCR), Proteasome, SMAD Transcription Factor, Transcription Corepressor, TGF-β
Abstract
TGF-β-induced antimitotic signals are highly regulated during cell proliferation under normal and pathological conditions, such as liver regeneration and cancer. Up-regulation of the transcriptional cofactors Ski and SnoN during liver regeneration may favor hepatocyte proliferation by inhibiting TGF-β signals. In this study, we found a novel mechanism that regulates Ski protein stability through TGF-β and G protein-coupled receptor (GPCR) signaling. Ski protein is distributed between the nucleus and cytoplasm of normal hepatocytes, and the molecular mechanisms controlling Ski protein stability involve the participation of actin cytoskeleton dynamics. Cytoplasmic Ski is partially associated with actin and localized in cholesterol-rich vesicles. Ski protein stability is decreased by TGF-β/Smads, GPCR/Rho signals, and actin polymerization, whereas GPCR/cAMP signals and actin depolymerization promote Ski protein stability. In conclusion, TGF-β and GPCR signals differentially regulate Ski protein stability and sorting in hepatocytes, and this cross-talk may occur during liver regeneration.
Introduction
Cell proliferation is positively regulated by a myriad of signaling pathways downstream of growth factor receptors and G protein-coupled receptors (GPCRs),4 among others, and it is negatively regulated by homeostatic signals such as TGF-β. Liver development and regeneration are excellent models for studying cell proliferation in a physiological context that is distinct from cancer (1–3). The liver regenerates upon injury, and all hepatic cells coordinately go through the cell cycle to restore the original mass and function of the organ. Interestingly, the liver grows despite the presence of TGF-β, a potent antiproliferative cytokine for epithelial cells. The TGF-β pathway is a major modulator of hepatocyte proliferation and death. However, it is unclear how hepatocytes become insensitive to TGF-β antiproliferative effects under conditions such as liver regeneration or hepatocarcinoma development (4–8).
Ligands of the TGF-β family regulate gene expression through the activation of Ser/Thr kinase receptors and the phosphorylation of receptor-regulated Smads (R-Smads) (9–11). This canonical pathway is tightly regulated by negative feedback loops generated by the up-regulation of inhibitors such as Smad7 and SnoN, which are encoded by immediate-early genes regulated by the TGF-β/Smad pathway (12). Any dysregulation of these negative feedback loops may contribute to the development of some diseases. TGF-β also regulates the levels of the Ski and SnoN corepressors by inducing their degradation via the ubiquitin-proteasome system. The phosphorylated R-Smads act as adaptors to recruit the E3 ubiquitin ligases that catalyze the polyubiquitination of the Ski and SnoN proteins (12, 13). This process facilitates TGF-β/Smad-induced transcriptional regulation of target genes. Therefore, any disruption of Ski and SnoN protein down-regulation might increase their protein levels and inhibit TGF-β signals.
The Ski corepressor is a negative regulator of TGF-β/Smad signaling that may also act as a transcriptional cofactor for some nuclear receptors and other transcriptional factors (14–17). Ski functions as a nuclear transcriptional corepressor for the Smad transcriptional factors by recruiting other corepressors, such as nuclear receptor co-repressor (NCoR) and Sin3A, and different histone deacetylases to repress TGF-β target genes such as smad7 and skil (12, 14, 18). However, the Ski protein is also localized in the cytoplasm of some cell types, where its function is less clear. It has been reported that overexpressed cytoplasmic Ski may escape from the down-regulation exerted by TGF-β and that it may sequester the Smad proteins to block TGF-β signals (17). To date, the TGF-β-independent functions of Ski are poorly studied, particularly the function of cytoplasmic Ski (19, 20).
A major cross-talk among diverse signaling pathways occurs during liver regeneration, where the antiproliferative actions of TGF-β may be under tight control exerted by different pathways, mainly those implicated in cell cycle promotion, such as the pathways downstream of growth factors receptors and GPCRs, among others (2). One of the mechanisms used by cells to prevent TGF-β-induced antimitotic actions, which have been observed in normal and some cancer cells, is the up-regulation of negative modulators of the canonical pathway, such as the Ski and SnoN corepressors (12). We previously reported the up-regulation of Ski and SnoN during liver regeneration, mainly in proliferating hepatocytes, and we also suggested that the inhibitory actions of Ski and SnoN against the TGF-β/Smad signals might explain why hepatocytes escape from TGF-β-induced antiproliferative control during regeneration (21).
In this study, we demonstrate that Ski protein stability is regulated differentially by TGF-β and GPCR signals in hepatocytes and that the molecular mechanisms involved are influenced by the dynamics of the actin cytoskeleton. Furthermore, we show that Ski protein stability is increased during liver regeneration, where it may facilitate hepatocyte proliferation by controlling TGF-β signaling.
EXPERIMENTAL PROCEDURES
Materials
Recombinant hTGF-β1 (TGF-β) was obtained from PeproTech. Methyl-β-cyclodextrin (MβCD), CHAPS, sphingosine 1-phosphate (S1P), lysophosphatidic acid (LPA), 3-isobutyl-1-methylxanthine (IBMX) and forskolin (F) were obtained from Sigma. Latrunculin B (LatB) and jasplakinolide (Jasp) compounds were obtained from Calbiochem. MG132 (a proteasome inhibitor), SB431542 (an ALK5 receptor inhibitor), and Y27632 (a Rho-associated protein kinase (ROCK) inhibitor) were obtained from Tocris Bioscience. Culture reagents and media were obtained from Invitrogen. Anti-FLAG M2 and anti-β-tubulin mouse monoclonal antibodies were obtained from Sigma. The following antibodies were obtained from Santa Cruz Biotechnology: anti-SnoN (catalog no. H-317), anti-Ski (catalog no. H-329), and anti-hepatocyte growth factor-regulated tyrosine kinase substrate (HRS) (catalog no. V-20) rabbit polyclonal antibodies; anti-Smad2/3 (catalog no. N-19) and anti-Smad4 (catalog no. C-20) goat-polyclonal antibodies; and anti-Ski (catalog no. G8) and anti-flotillin-2 (catalog no. B-6) mouse monoclonal antibodies. Anti-Ski (catalog no. 07-060), anti-Smad4, and anti-phospho-Smad2 rabbit polyclonal antibodies were from Millipore. Anti-EEA1 and anti-GM130 mouse monoclonal antibodies were obtained from BD Transduction Laboratories. Anti-Smad2 and anti-Yes-associated protein/transcriptional co-activator with PDZ-binding motif (YAP/TAZ) rabbit polyclonal antibodies were obtained from Cell Signaling Technology. Secondary anti-rabbit IgG and anti-rabbit IgG (light chain) HRP-coupled antibodies were from Zymed Laboratories Inc. and Jackson ImmunoResearch Laboratories, respectively. Secondary anti-mouse IgG HRP-coupled antibody was from Santa Cruz Biotechnology. Alexa Fluor 488 (anti-rabbit IgG) and Alexa Fluor 594 (anti-mouse IgG) secondary antibodies were from Molecular Probes. Alexa Fluor 594 anti-goat IgG secondary antibody was from Jackson ImmunoResearch Laboratories.
Animals and Partial Hepatectomy Model
Studies were performed on male Wistar rats ∼200–250 g of weight. Animals were maintained on an ad libitum diet and used according to institutional guidelines (Instituto de Fisiología Celular, Universidad Nacional Autónoma de México (UNAM)) for animal experimentation. Anesthetized rats were subjected to a ventral laparotomy, and the anterior two-thirds of the liver were removed. Animals were sacrificed 0, 2, 48, and 120 h after the 70% partial hepatectomy. The livers were harvested, and the nuclear extracts were obtained for protein analysis as described previously (21).
Cell Lines, Hepatocyte Isolation, and Primary Culture
The C9 (rat hepatocytes) and HepG2 (human hepatoma) cell lines were maintained in DMEM supplemented with 10% FBS plus antibiotics (penicillin/streptomycin). Rat hepatocytes were isolated using the collagenase perfusion method adapted from the protocol of Snorri Thorgeirsson as described previously (21). For primary culture, hepatocytes were seeded on plastic Petri dishes coated with 1% rat tail collagen type 1 (BD Biosciences), and cells were cultured for 4 h at 37 °C in attachment medium with 10% FBS. Then the medium was changed to feeding medium (FBS-free), and hepatocytes were cultured for 24 h for further studies (22). Cells were serum-starved for 12 h before treatments. C9 cells were transiently transfected with the pcDNA3/mCherry-FLAG-Ski, pmCherry-C2/hAlix, or pCR3.1/CD63-mCherry constructs using Lipofectamine 2000 (Invitrogen). The pcDNA3/FLAG-Ski WT construct was a gift from Dr. Céline Prunier (INSERM Bat Kourilsky, Hôspital Saint-Antoine, Paris, France). The pCR3.1/CD63-mCherry construct was a gift from Dr. Paul D. Bieniasz (The Aaron Diamond AIDS Research Center, New York, NY). Addgene plasmid no. 21504 (pmCherry-C2/hAlix) was obtained from Dr. James Hurley (23).
Immunoprecipitations and Western Blot Analyses
To obtain whole cell protein extracts (total cell lysates), cells were lysed in SDS lysis buffer (10 mm Tris-HCl (pH 7.5), 150 mm NaCl, 0.5 mm EDTA, 1 mm EGTA, and 1% SDS plus protease and phosphatase inhibitors) and then boiled for 10 min at 100 °C, or cells were either lysed for 30 min with TNTE buffer (50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 0.5% Triton X-100, and 1 mm EDTA plus protease and phosphatase inhibitors) or lysed for 1 h with modified RIPA buffer (50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1 mm EDTA, 0.5% Nonidet-P40, 0.5% sodium deoxycholate, and 0.1% SDS plus protease and phosphatase inhibitors). From whole cell lysates, 50–75 μg of protein/lane was separated by SDS-PAGE and assayed by immunoblotting using specific antibodies, whereas ∼1.5–2 mg of protein was used for each immunoprecipitation (IP). Proteins were detected by either Immobilon Western (Millipore) or SuperSignal West Pico chemiluminescent substrates (Thermo). Densitometric analysis was carried out with ImageJ 1.47 software (National Institutes of Health).
Subcellular Fractionation and Detergent-resistant Membrane (DRM) and non-DRM Fraction Preparation
Nuclear and cytoplasmic fractions were prepared according to a modified protocol described previously (24). C9 cells were harvested with homogenization buffer (250 mm sucrose and 3 mm imidazole (pH 7.4) plus protease and phosphatase inhibitors) and passed 10 times through a 22-gauge needle. The homogenate was centrifuged for 15 min at 3400 rpm at 4 °C. The supernatant contained the cytoplasmic fraction, and the nuclear fraction remained in the pellet. The nuclear fraction was lysed with modified RIPA buffer for 1 h at 4 °C and centrifuged at 13,200 rpm for 5 min. Modified RIPA buffer 5× was added to the cytoplasmic fraction to get 1× final concentration and then incubated for 1 h at 4 °C. β-Tubulin protein was used as a loading control. In addition, DRM (lipid raft-rich) and non-DRM were isolated from confluent cells as described previously with some modifications (25). In brief, cells were harvested with 1.5 ml of TNTE buffer (20 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1 mm EDTA, and 1% Triton X-100 plus protease and phosphatase inhibitors) and passed five times through a 25-gauge needle. Cell lysates were adjusted to 45% final concentration of sucrose (4 ml) and ultracentrifuged through a discontinuous sucrose gradient, formed previously by sequentially layering 35% sucrose (4 ml) and 5% sucrose (4 ml) at 38,000 rpm for 20 h in a Beckman SW40Ti rotor at 4 °C. Twelve fractions of 1 ml were harvested from the top of the gradient. Each fraction was solubilized for 12 h with 1% of CHAPS, and proteins were analyzed by IP and Western blot (WB). Subcellular markers such as flotillin-2 (lipid raft-associated membranes), GM130 (Golgi), and EEA1 (early endosome) proteins were used.
Immunofluorescence Assays
C9 cells were seeded on poly-l-lysine-coated glass coverslips in 24-well tissue culture plates, and immunofluorescence assays were performed as described previously (26). Briefly, cells were fixed for 15 min with 4% paraformaldehyde in PBS at 37 °C. Cells were permeabilized with 0.1% Triton X-100 in PBS for 10 min at 4 °C. Slides were incubated with blocking solution containing either 1% BSA, 10% goat serum, or 10% horse serum in PBS for 1 h at room temperature prior to incubation for 12 h at 4 °C with specific primary antibodies (diluted 1:50 or 1:100 in blocking solution). Slides were incubated with secondary antibodies (diluted 1:500 or 1:750) for 1 h at room temperature in the dark. Slides were mounted on glass coverslips using Vectashield with DAPI (Vector Laboratories). Confocal laser-scanning microscopes used were Fluoview FV1000 equipped with an oil immersion objective (Olympus PlanApoN ×60, numerical aperture = 1.45) and Fluoview FV10i equipped with a water immersion objective (Olympus ×60, numerical aperture = 1.2). FV10ASW software (Olympus) was used for images acquisition, processing, and analysis. The degree of colocalization was examined using the Colocalization Finder plug-in in ImageJ (http://rsb.info.nih.gov/ij/plugins/colocalization-finder.html).
Cell Proliferation
C9 cells seeded at low density (5 × 105 cells) in 12-well tissue culture dishes were serum-starved for 24 h. Cell proliferation was stimulated with 10% FBS, and cell viability and proliferation were evaluated 24–48 h post-treatment. Medium was replaced with 2 ml of fresh medium plus 0.25 mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Sigma) per well (6-well plate), and cells were incubated for 4 h at 37 °C. Then 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide formazan crystals were solubilized with 500 μl of dimethyl sulfoxide for 30 min at room temperature and quantified at 595 nm in an ELx800 absorbance microplate reader.
Protease Protection Assay
Protease protection assays were performed as described previously (27) with some modifications. In brief, liver tissue was homogenized in 4 ml of buffer (250 mm sucrose, 3 mm imidazole (pH 7.4), plus phosphatase inhibitors) without protease inhibitors. Disrupted tissue was centrifuged at 2000 rpm for 10 min at 4 °C. The liver cytoplasmic fraction (30 mg of protein) was incubated for 20 min at room temperature with 0.5 μg/ml Proteinase K (Ambion) in the absence or presence of 0.5% Triton X-100. We added 20 mm PMSF to stop the reaction, and samples were incubated for 2 h at 4 °C by adding 5× modified RIPA. Supernatants were used for IP assays, and proteins were analyzed by WB.
Statistical Analysis
Student's t test was used to calculate statistical significance, and p < 0.05 was considered to be significant.
RESULTS
Ski Protein Is Transiently Up-regulated during Liver Regeneration
The process of liver regeneration exhibits a spatiotemporal synchronization of many signaling pathways positively regulating cell proliferation. Paradoxically, some antimitotic signals, such as TGF-β, are increased during liver regeneration. We reported previously that the up-regulation of TGF-β pathway inhibitors such as Ski and SnoN may counteract TGF-β-induced antiproliferative actions during mouse liver regeneration, allowing for other TGF-β functions, such as extracellular matrix remodeling (21). At that time, we also proposed that the increase in Ski and SnoN protein levels and their association with the Smads might, in part, explain the resistance of hepatocytes to TGF-β signals (21). Since then, we have been interested in discovering the mechanisms whereby hepatocytes become refractory to TGF-β-induced antiproliferative signals, particularly in liver regeneration.
Initially, we observed that Ski and SnoN proteins were up-regulated in parallel with Smad2 protein phosphorylation in rat livers obtained from 2–120 h after partial hepatectomy but not in sham-operated rats (Fig. 1A). These data suggest that, in this cell context, the TGF-β/Smad pathway seems to be unable to cause Ski and SnoN protein down-regulation. Interestingly, we found that a significant fraction of both proteins was associated with the Triton-insoluble fraction when primary cultured hepatocytes were lysed with TNTE buffer, whereas both proteins were better solubilized in RIPA buffer (data not shown). To gain insight into this outcome, we focused on investigating mainly the regulation of Ski protein stability in hepatocytes and the signals involved. In this study, we observed that the amount of Ski protein immunoprecipitated from RIPA lysates was higher than the amount obtained from TNTE lysates. Moreover, the effect of TGF-β on Ski protein down-regulation was better observed in RIPA lysates (Fig. 1B). Therefore, most of the experiments were carried out with RIPA buffer unless indicated otherwise.
FIGURE 1.

Ski protein levels are transiently up-regulated during liver regeneration after partial hepatectomy. A, nuclear protein extracts from regenerating rat livers after PH at the indicated times (or 2-h sham-operated rats) were used to evaluate Ski, SnoN, SnoN2, phospho-Smad2, and proliferating cell nuclear antigen (PCNA) protein levels by IP/WB (top panel). Bottom panel, densitometric analysis of Ski and SnoN protein levels as -fold change induced by PH. Data are represented as mean ± S.D. of three independent experiments. PCNA protein was used as a cell proliferation marker. B, primary cultured hepatocytes were stimulated for 1 h without or with 0.2 nm TGF-β. Cells were lysed with either RIPA or TNTE buffer to obtain two fractions: SN (Triton-soluble fraction) and pellet (Triton-insoluble fraction resuspended in RIPA). Immunoblotting was performed for the indicated proteins from IP or cell lysates. C, hepatocytes were freshly isolated from livers obtained 0 or 48 h post-PH, stimulated for 1 h with 0.2 nm TGF-β, and lysed with RIPA buffer. Ski and Smad2/3 proteins were immunoprecipitated, and immunoblot analyses were performed to identify the indicated proteins from IP or cell lysates. D, nuclear (Nuc) and cytoplasmic (Cyto) protein extracts from regenerating rat livers after PH at the indicated times were used to evaluate the indicated proteins from IP or cell lysates (top panel). Bottom panel, densitometric analysis of the levels of cytoplasmic Ski-actin complexes expressed as -fold change induced by PH. Data are represented as mean ± S.D. of three independent experiments. E, cytoplasmic protein extracts were obtained from regenerating livers 0 or 48 h after PH, and then 35 mg of protein extracts was treated for 20 min with 0.5 μg/ml proteinase K in the absence or presence of 0.5% Triton X-100. Ski protein was immunoprecipitated, and immunoblot analyses were performed for the indicated proteins from IP or protein extracts. The densitometric analysis of Ski protein levels is shown as -fold change over the control. GM130, tubulin, and flotillin-2 were used as controls for protein degradation (asterisk, GM130 fragment). F, mCherry-FLAG-Ski protein levels were detected by immunoblot analysis of RIPA lysates from transiently transfected C9 cells with the indicated amounts of plasmid (top panel). Transiently transfected C9 cells with 30 μg of plasmid were serum-starved for 24 h and then stimulated with 10% FBS for 24–48 h (bottom panel). Cell proliferation was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay (relative cell number). Data are represented as means ± S.D. from two independent experiments performed in triplicate.
With this approach, we were able to detect a clear TGF-β-induced regulation of Ski and SnoN protein levels in freshly isolated hepatocytes from normal (PH 0 h) and regenerating livers (PH 48 h) (Fig. 1C). Interestingly, we observed that Ski and also Smad2 proteins were associated with actin protein (co-IP) at the proliferation phase analyzed (i.e. 48 h post-hepatectomy) (Fig. 1C). Additionally, Ski has been described previously as a soluble nuclear protein in many cell lines, including the hepatoma cell line HepG2. Therefore, it was unexpected to observe that Ski protein was associated with a Triton-insoluble fraction in normal hepatocytes. When we analyzed subcellular localization of Ski in quiescent and regenerating livers (PH 48 h), the data showed that Ski exhibited mainly a nuclear localization. However, a significant fraction of Ski was cytoplasmic, and, intriguingly, the levels of cytoplasmic Ski protein coimmunoprecipitated with actin were higher 48 h post-PH (Fig. 1D, top panel). Fig. 1D, bottom panel, shows that the levels of cytoplasmic Ski-actin complexes increased >2-fold after 48 h post-PH over the control (0 h).
In hepatocytes, Ski protein is partially localized to a Triton-insoluble fraction, which could be explained by a partial association with the cytoskeleton and/or with lipid raft-rich membranes. This hypothesis was proved to be correct by using a protease protection assay. We observed that proteinase K only partially degraded Ski protein from cytoplasmic fractions obtained from quiescent (PH 0 h) or regenerating hepatocytes (PH 48 h) in the absence or presence of 0.5% Triton X-100 (Fig. 1E). As controls, GM130, a cis-Golgi matrix protein, was degraded, whereas a protein associated with lipid raft domains such as flotillin-2 was resistant to degradation by proteinase K. The data suggest that a fraction of cytoplasmic Ski is protected from down-regulation in hepatocytes during regeneration, probably by recruitment to a lipid raft-rich compartment.
Next we analyzed whether the increase in Ski protein levels observed during liver regeneration may favor specific Ski functions, such as the promotion of cell proliferation. We observed that the proliferation rate of hepatic C9 cells in response to 10% FBS treatment for 24–48 h was increased (3- to 6-fold over the control) when Ski protein was transiently overexpressed (Fig. 1F). This result correlated well with the increase in Ski protein levels observed during the proliferative phase of liver regeneration (Fig. 1A) and in regenerating hepatocytes (Fig. 1C).
TGF-β Induces a Transient Down-regulation of Ski Protein via the Proteasome
We first focused on studying the regulation of Ski protein stability and subcellular localization in normal hepatocytes. We observed that TGF-β induced a transient down-regulation of the Ski protein in a time-dependent manner in primary cultured hepatocytes (Fig. 2A). As an additional cell model, we decided to use the C9 cell line because it exhibits similar features as normal hepatocytes. In C9 cells, TGF-β also transiently decreased the levels of Ski protein and induced Smad2 phosphorylation (Fig. 2B). We observed that MG132 inhibited TGF-β-induced Ski down-regulation in C9 cells (Fig. 2C). Unexpectedly, MG132 treatment caused a differential distribution of the Ski protein and the Ski-associated Smad2/4 proteins (co-IPs) in both the Triton-soluble (SN) and Triton-insoluble (pellet) fractions (Fig. 2C, top panel). Fig. 2C, bottom panel, shows that MG132 increased >4-fold the Ski protein levels over the control in the RIPA lysates. We also observed that both the nuclear and cytoplasmic Ski proteins were evenly degraded via the proteasome after TGF-β stimulus and, intriguingly, that the cytoplasmic Ski fraction is bound to actin (Fig. 2D).
FIGURE 2.

TGF-β induces a transient down-regulation of Ski protein via the proteasome in both the nucleus and cytoplasm of C9 cells. A, primary cultures of rat hepatocytes were stimulated with 0.2 nm TGF-β for the indicated times, and Ski protein levels were detected by IP/WB from RIPA lysates. The densitometric analysis of Ski protein levels is shown as -fold change induced by TGF-β. B, C9 cells were stimulated with 0.2 nm TGF-β for the indicated times, and immunoblotting was performed for the indicated proteins from SDS buffer lysates (top panel). Bottom panel, densitometric analysis of Ski protein levels as -fold change induced by TGF-β. Data are represented as mean ± S.D. of three independent experiments. C, C9 cells were pretreated for 4 h without (control) or with 25 μm MG132 and then stimulated for 1 h without or with 0.2 nm TGF-β. Cells were lysed with either RIPA or TNTE buffer to obtain two fractions: SN (Triton-soluble fraction) and pellet (Triton-insoluble fraction resuspended in RIPA). Immunoblotting was performed for the indicated proteins from co-IP with Ski or cell lysates (top panel). Bottom panel, densitometric analysis of Ski protein levels as -fold change over basal. Data are represented as mean ± S.D. of four independent experiments. D, C9 cells were pretreated for 4 h without (control) or with 25 μm MG132, stimulated for 1 h with 0.2 nm TGF-β, and then the nuclear (Nuc) and cytoplasmic (Cyto) fractions were obtained. Ski protein was immunoprecipitated, and immunoblotting was performed for the indicated proteins from co-IP or RIPA-lysates (left panel). Right panel, densitometric analysis of Ski protein levels as a percentage of total levels in the control (100%). Data are represented as mean ± S.D. of five independent experiments.
TGF-β Controls Ski Protein Stability and Subcellular Localization
Knowing that Ski and SnoN proteins mainly exhibit a nuclear localization in many cancer cell lines, including hepatoma cells, we analyzed Ski subcellular distribution in hepatic C9 cells by immunofluorescence. We observed that the Ski protein was localized to both the nucleus and cytoplasm. Surprisingly, control C9 cells exhibited punctate areas of Ski staining in the cytoplasm (Fig. 3).
FIGURE 3.
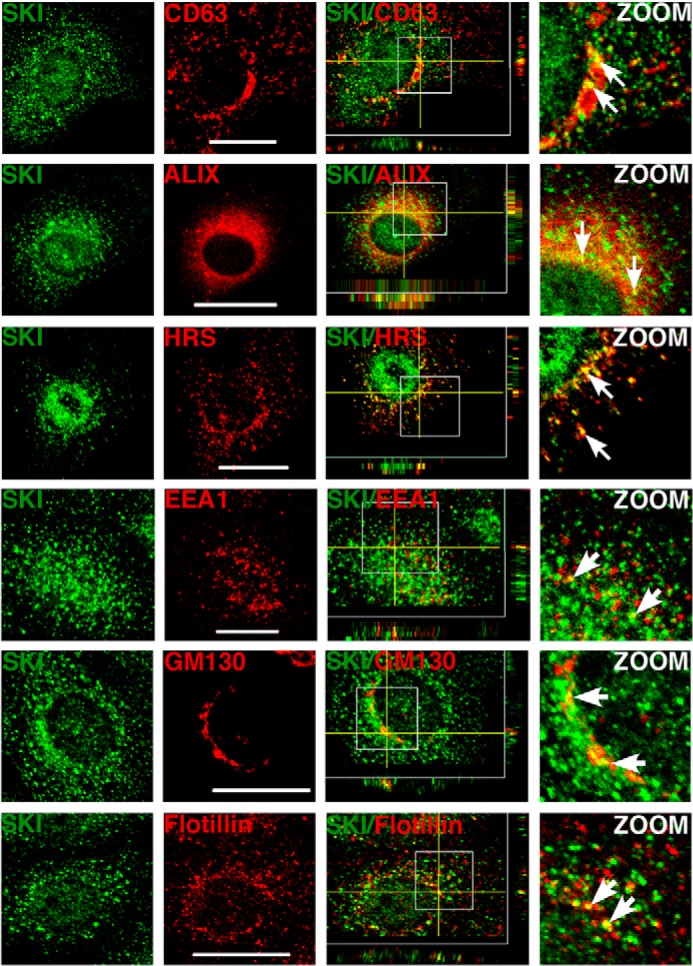
Cytoplasmic Ski protein is partially localized at lipid raft-rich vesicles. Immunofluorescence was performed for Ski protein in C9 cells. Ski protein exhibited a partial colocalization (arrows) with overexpressed CD63-mCherry and mCherry-Alix (MVE markers), endogenous HRS (an endosome marker), EEA1 (an early endosome marker), GM130 (a Golgi marker), and flotillin-2 (a lipid raft marker) in C9 cells. Endogenous proteins were detected by immunofluorescence. Scale bars = 20 μm. Z stack images are shown.
Our data show that cytoplasmic Ski exhibits a punctate pattern of localization, which is typical of the endosomal compartment. To further characterize the subcellular localization of cytoplasmic Ski, we performed colocalization studies of Ski protein with different markers of multivesicular endosomes (MVE) and other organelles. Vesicles containing cholesterol-rich lipid rafts are commonly implicated in vesicular trafficking and also contribute to the formation of MVEs, which are relevant in different cellular processes such as protein degradation via lysosomes and protein export via exosomes. These MVEs may also serve as organelles able to temporally sequester or store signaling proteins, such as GSK3 (27). As shown in Fig. 3, endogenous Ski protein partially colocalized with the overexpressed MVE markers CD63-mCherry and mCherry-Alix and endogenous HRS. Additionally, Ski colocalized with flotillin-2, a lipid raft marker, and, to a lesser extent, with the early endosome marker EEA1 and with the Golgi marker GM130 (Fig. 3, Z stacks in merged images).
Furthermore, TGF-β treatment for 1 h caused a down-regulation of Ski protein in both the nucleus and cytoplasm, which correlated with Smad2 nuclear localization (Fig. 4A). Intriguingly, TGF-β also promoted a partial redistribution of cytoplasmic Ski protein to the perinuclear region after 3 h of treatment (Fig. 4A, magnified images). Additionally, TGF-β treatment enhanced Ski protein colocalization with CD63-mCherry, a late endocytic MVE and exosome marker (Fig. 4B, left panel). The index of colocalization of Ski-CD63 corresponds to 679 ± 136 counts (control) and 1818 ± 291 counts (TGF-β) (p < 0.004, n = 12), as shown in Fig. 4B, right panel. These data indicate that TGF-β treatment for 3 h promoted Ski-CD63 colocalization (2.7-fold over the control) in C9 cells. Together, our findings suggest that, in normal hepatocytes, cytoplasmic Ski protein exhibits different subcellular localizations, including its association with endosomes resembling MVEs, and it is also clear that Ski protein stability and its subcellular localization are regulated by TGF-β.
FIGURE 4.

TGF-β promotes a transient localization of Ski protein at lipid raft-rich vesicles. A, C9 cells were stimulated without (CONTROL) or with 0.2 nm TGF-β for 1 and 3 h. Subcellular localization of the indicated proteins was analyzed by immunofluorescence and confocal microscopy (arrowheads). Nuclei were stained with DAPI. Scale bars = 20 μm. Magnified images are shown (ZOOM). B, C9 cells transiently overexpressing the MVE marker CD63-mcherry were incubated for 3 h without (CONTROL) or with 0.2 nm TGF-β. Subcellular colocalization (arrows) of the indicated proteins was analyzed by immunofluorescence and confocal microscopy. Scale bars = 20 μm. Right panel, quantitative analysis of Ski-CD63 protein colocalization expressed as counts (number of pixels) induced by TGF-β. Data are represented as mean ± S.D. of three independent experiments.
Ski Protein Is Partially Associated with Lipid Rafts
Our findings indicating that a fraction of the whole Ski protein is partially associated with a Triton-insoluble fraction suggest that Ski might be located in cholesterol-rich vesicles. To test this, C9 cells were pretreated with the cholesterol-chelating agents filipin and MβCD, followed by TGF-β treatment for 1 h, and lysed with TNTE or RIPA buffer. Ski protein was detected by IP/WB. As shown in Fig. 5A, MβCD treatment not only enriched Ski protein levels in the TNTE-soluble fraction but also disrupted the Ski-actin protein association in RIPA buffer. Despite the fact that neither agent altered the TGF-β effect on Ski protein stability (Fig. 5A), the ability of MβCD to extract cholesterol from the plasma membrane and intracellular compartments, promoting raft disruption and the solubilization of proteins from rafts (28), could explain the differential results obtained with MβCD and filipin treatments on the Ski-actin protein association. Moreover, MG132 pretreatment of C9 cells partially increased Ski protein levels in the Triton-insoluble fraction (pellet) (25% from total) compared with the soluble fraction (SN) (16% from total), and MβCD pretreatment blocked this effect (pellet) (13% from total) (Fig. 5B). These data suggest that Ski protein is partially associated with lipid rafts that are generally associated with the DRMs (29, 30).
FIGURE 5.

Ski protein is localized in both the DRMs and non-DRMs. A, C9 cells were pretreated for 4 h without (Control) or with 2.5 μm filipin or 2.5 mm MβCD, stimulated for 1 h with 0.2 nm TGF-β, and lysed with TNTE or RIPA buffer. Ski protein was immunoprecipitated, and immunoblotting was performed for the indicated proteins from IP or cell lysates. B, C9 cells were preincubated for 4 h without (Control) or with 2.5 mm MβCD, treated for 2 h without or with 25 μm MG132, and lysed with TNTE buffer to obtain two fractions: SN (Triton-soluble fraction) and pellet (Triton-insoluble fraction resuspended in RIPA). Ski protein was immunoprecipitated, and immunoblotting was performed for the indicated proteins from IP or cell lysates. The partitioning of Ski protein between the SN and pellet after MG132 treatment is shown as a percentage of total Ski protein (100%). C, C9 cells were stimulated in the absence (Control) or presence of 0.2 nm TGF-β for 1 h or incubated for 4 h with 25 μm MG132 or 2.5 mm MβCD. DRMs and non-DRMs were obtained. Ski protein was immunoprecipitated, and immunoblotting was performed for the indicated proteins from IP or cell lysates. Subcellular markers were detected: flotillin-2 (lipid raft-associated), GM130 (Golgi), and EEA1 (early endosomes).
Next we decided to analyze DRMs (cholesterol-rich) by separating DRM and non-DRM fractions from C9 cells. As shown in Fig. 5C, Ski protein was localized in both the DRM and non-DRM fractions from control cells. Notably, Ski protein at non-DRMs was more sensitive to TGF-β-induced down-regulation. It is possible that Ski protein becomes partially sequestered at MVEs to prevent total protein depletion after TGF-β stimulation. MG132 induced an accumulation of Ski protein in both the DRM and non-DRM fractions, whereas MβCD blocked Ski protein association with the DRM fraction (Fig. 5C). Therefore, our findings support the hypothesis that Ski protein is partially associated with lipid raft-rich vesicles in normal hepatocytes.
Actin Cytoskeleton Dynamics Modulate Ski Protein Stability
Our data suggesting that changes in actin cytoskeleton dynamics (actin polymerization or depolymerization) might control Ski protein stability led us to investigate different cellular contexts where the cytoskeleton rearrangement is relevant, such as cell-cell contact controlled by cellular confluence. Therefore, C9 cells were seeded at low (sparse) and high (confluent) density, and Ski protein stability was analyzed. As shown in Fig. 6A, Ski protein is highly stabilized in confluent C9 cells in both the nuclear and cytoplasmic compartments, suggesting that the Ski protein could be sensitive to actin cytoskeleton rearrangements that occur in cells when cell-cell junctions are decreased (sparse) or increased (confluent). Furthermore, TGF-β treatment efficiently down-regulated Ski protein levels in both sparse and confluent cells (Fig. 6B, top panel). Fig. 6B, bottom panel, shows that the levels of Ski protein were 6-fold higher in confluent than in sparse C9 cells.
FIGURE 6.

Actin cytoskeleton dynamics modulate Ski protein stability. A, the nuclear (Nuc) and cytoplasmic (Cyto) fractions were obtained from sparse or confluent C9 cells, Ski protein was immunoprecipitated, and immunoblotting was performed for the indicated proteins from IP or cell lysates. B, sparse or confluent C9 cells were treated for 1 h with TGF-β. Ski and Smad2 proteins were immunoprecipitated, and immunoblotting was performed for the indicated proteins from IP or cell lysates (top panel). Bottom panel, densitometric analysis of Ski protein levels as -fold change in sparse versus confluent C9 cells. C, C9 cells were treated with 1 μm LatB or 0.5 μm Jasp for the indicated times, and immunoblotting was performed for the indicated proteins from SDS buffer lysates. The densitometric analysis of Ski protein levels is shown as -fold change over basal. D, C9 cells were treated for 6 h with 0.5 μm Jasp or 1 μm LatB, and DRMs and non-DRMs were obtained. Ski protein was immunoprecipitated, and immunoblotting was performed for the indicated proteins from co-IP. E, C9 cells were treated for 2 and 6 h with 0.5 μm Jasp or 1 μm LatB. Ski protein was immunoprecipitated, and immunoblotting was performed for the indicated proteins from co-IP (left panel). Right panel, densitometric analysis of Ski-actin complex levels as -fold change induced by Jasp or LatB. Data are represented as mean ± S.D. of three independent experiments.
Next we tested whether pharmacological modulators of actin cytoskeleton dynamics might affect the stability of Ski protein and its association with actin. Strikingly, LatB, an inhibitor of actin polymerization, increased Ski protein stability (Fig. 6C), whereas Jasp, a potent inducer of actin polymerization, decreased Ski protein stability (Fig. 6C). Interestingly, actin cytoskeleton dynamics modulated Ski protein association to DRM and non-DRM fractions. Therefore, Jasp induced an increase in Ski protein levels associated with the DRM fraction, whereas LatB induced an increase in Ski protein levels associated with the non-DRM fraction and impaired the association of Ski protein with the DRM fraction (Fig. 6D). Notably, the Ski-actin interaction (co-IP) was increased in both the DRM and non-DRM fractions by Jasp treatment (Fig. 6D). Jasp treatment increased the levels of Ski-actin complexes 15-fold over the control, whereas LatB decreased such complexes 0.25-fold compared with the control (Fig. 6E). Neither LatB nor Jasp affected basal or TGF-β-induced Smad2 phosphorylation (data not shown). All of these data indicate that actin cytoskeleton dynamics control Ski protein stability and also strongly suggest that a fraction of cytoplasmic Ski is associated, possibly along with actin, to a subset of vesicles containing cholesterol-rich lipid rafts.
GPCR Signaling Pathways Differentially Modulate Ski Protein Stability
Knowing that GPCRs control diverse pathways that are highly active during liver regeneration and that it has been reported recently that these receptors can differentially regulate the dynamics of the actin cytoskeleton to control the Hippo pathway (2, 31, 32), we further analyzed whether Ski protein stability was regulated by either GPCRs controlling cAMP levels, such as the glucagon receptor, or GPCRs coupled to G12/13 and Rho-GTPase activation, such as the S1P and LPA receptors. When C9 cells were treated for different times with LPA or S1P, we observed that both stimuli induced a transient down-regulation of Ski protein (Fig. 7A). As a positive control, we observed that TAZ protein stability was increased (Fig. 7A). Intriguingly, we did not see any significant modulation of Smad2 phosphorylation or its subcellular localization by agents controlling actin cytoskeleton dynamics or the Hippo pathway in hepatocytes (data not shown), even though it has been shown previously that the Hippo pathway controls Smad2 in embryonic cells (33). In addition, Ski protein down-regulation and Ski-actin association (co-IPs) induced by S1P were inhibited by pretreatment with MG132, LatB, and Y27632 (a ROCK inhibitor) (Fig. 7B). By contrast, we observed that stimulators of cAMP accumulation, such as glucagon or a mixture of F/IBMX, induced an increase in Ski protein stability (Fig. 7C) and that F/IBMX pretreatment inhibited the LPA- and S1P-mediated reduction of Ski stability (Fig. 7D, left panel). Fig. 7D, right panel, shows that S1P and LPA increased the levels of Ski-actin complexes 15-fold over basal in control cells. Intriguingly, MβCD treatment prevented Ski down-regulation induced by S1P and also the Ski-actin interaction (Fig. 7E), but it did not change the Ski/Smad4 interaction (Fig. 7, E and F) or Ski subcellular localization (Fig. 7, F and G). Together, these data suggest considering the possibility that the association between Ski and actin may rely on the participation of an adaptor or scaffold protein rather than a direct interaction. However, this remains to be investigated.
FIGURE 7.

GPCR signaling pathways differentially modulate Ski protein stability. A, C9 cells were stimulated with 1 μm LPA or 1 μm S1P for the indicated times. TAZ protein levels indicate LPA or S1P receptor activation. B, C9 cells were pretreated for 4 h without (Control) or with 25 μm MG132, 5 μm LatB, or 30 μm Y27632 and then stimulated for 1.5 h with 1 μm S1P. C, C9 cells were stimulated with a mixture of 1 μm F/100 μm IBMX mixture or with 2 μm glucagon for the indicated times. D, C9 cells were preincubated for 1 h in the absence (Control) or presence of a mixture of 2 μm F/100 μm IBMX and then stimulated for 1.5 h with 1 μm S1P or for 1 h with 1 μm LPA (left panel). Right panel, densitometric analysis of Ski-actin complex levels as -fold change induced by S1P or LPA. Data are represented as mean ± S.D. of three independent experiments. E, C9 cells were pretreated without (Control) or with MβCD for 4 h and stimulated with 1 μm S1P for 1.5 h. F, C9 cells were treated with MβCD for 3 h, and the nuclear (Nuc) and cytoplasmic (Cyto) fractions were obtained. In A–F, Ski protein was immunoprecipitated, and immunoblotting was performed for the indicated proteins from IP or cell lysates. G, C9 cells were treated with MβCD for 3 h, and endogenous Ski protein was detected by immunofluorescence. Nuclei were stained with DAPI. Scale bars = 20 μm.
On the basis of these results, we show a model depicting the proposed mechanism (Fig. 8). Accordingly, TGF-β/Smad pathway and/or the stabilization of actin filaments promoted by the GPCR/G13/Rho signaling axis can decrease Ski protein stability, whereas the GPCR/cAMP pathway disrupts actin filaments and, consequently, increases Ski protein stability in hepatocytes.
FIGURE 8.

Model showing the regulation of Ski protein by TGF-β signaling and actin polymerization in hepatocytes. All Ski protein pools (nuclear and cytoplasmic) are similarly sensitive to down-regulation induced by TGF-β or actin polymerization independently of Ski subcellular localization (left panel). Intriguingly, cytoplasmic Ski protein stability regulated by the actin cytoskeleton dynamics depends on Ski-actin complex association, and the pool of Ski protein localized at lipid raft-rich vesicles seems to be protected from degradation induced by actin polymerization (right panel). (P = phosphate; Act = actin; Ub = ubiquitin).
DISCUSSION
Ski protein is a major transcriptional corepressor for the TGF-β/Smad pathway. Therefore, its levels may determine the TGF-β signaling outcome (e.g. high Ski protein levels are associated with an inhibition of TGF-β-induced signals). The relevance of maintaining appropriate Ski protein levels for homeostasis has been clearly demonstrated. Recently, several in-frame mutations in exon 1 of Ski (the R-Smad binding region) have been identified as the main cause of the human Shprintzen-Goldberg syndrome, and increased TGF-β signaling has been associated with this pathogenesis (34). Furthermore, Ski knockout mice exhibit embryonic lethality, and heterozygous mice show a high sensitivity to developing cancer (35, 36). By contrast, transgenic mice overexpressing Ski protein exhibit increased muscle mass (17). Additionally, high levels of Ski protein have been involved in the switch from glycolysis to oxidative energy metabolism in cancer cells (37).
In many cancer cells, Ski overexpression occurs mainly in the nucleus, where it may recruit histone deacetylases to eventually repress TGF-β target genes. Some reports suggest that cytoplasmic Ski protein may act by sequestering Smad proteins and impairing their translocation to the nucleus. In some cancer cells, TGF-β induces Ski protein down-regulation via the proteasome, whereas, in other cancer cells, Ski protein seems to be resistant to TGF-β-induced degradation. Although the mechanisms underlying this cell-to-cell variability are unclear, it has been suggested that expression of specific E3 ubiquitin ligases may be important (38). Notably, there are very few studies of Ski and SnoN protein regulation and function in normal cells (39).
TGF-β-induced signaling is highly modulated during many physiological processes. One example is its regulation in cells with an elevated proliferation rate, such as those found in liver regeneration and cancer. Hepatic regeneration is an excellent model for studying the regulation of cell proliferation and how hepatic cells can proliferate in the presence of an antimitotic cytokine as TGF-β, which has been an enigma. We have proposed that the up-regulation of Ski and SnoN inhibitors might explain the transient resistance to TGF-β signals exhibited by hepatocytes during liver regeneration (21). Surprisingly, during this study, we found a novel mechanism for the regulation of Ski protein stability in normal hepatocytes involving a convergence of TGF-β and GPCR signals with actin cytoskeleton participation. Therefore, we focused on studying the molecular mechanisms involved in controlling Ski protein stability in normal hepatocytes.
In our model of normal hepatocytes, Ski protein appears to be distributed in both the nucleus and cytoplasm. Cytoplasmic Ski shows a punctate localization and partial association with the DRM fraction. We found that a fraction of cytoplasmic Ski is partially localized to a subset of lipid raft-rich vesicles, some of them containing MVE markers such as CD63 and Alix. We also found that nuclear and cytoplasmic Ski are quite sensitive to proteasome-dependent down-regulation in response to TGF-β or any stimulus that increases the polymerization of actin filaments, such as Jasp, S1P, or LPA. However, Ski protein stability is increased in response to any stimulus that decreases actin filament polymerization, such as LatB or glucagon-stimulated GPCR/cAMP signals (Fig. 7). To our knowledge, this is the first demonstration of Ski protein localization at the MVE in normal hepatocytes, and we also show that its stability is associated with the dynamics of the actin cytoskeleton. We suggest that this novel regulation of Ski protein stability is an event that possibly occurs during liver regeneration, when extracellular matrix remodeling modifies the architecture of the liver, causing hepatocytes to reorganize their cytoskeletons to migrate to new positions within the regenerating liver. Intriguingly, this novel regulation of Ski stability in normal hepatocytes seems to be lost in hepatoma cells.
Many signals are involved in regulating liver regeneration, and, possibly, many of them control the expression of the transcriptional coregulator Ski during this process. Therefore, our work is an initial approach to reveal the molecular mechanisms and the potential consequences of the regulation of Ski protein stability and sorting in hepatocytes. Although TGF-β signaling is present during regeneration, its inhibitory action on Ski protein levels may be counteracted by other signals that modulate hepatocyte polarity, a tightly regulated process that takes place during regeneration (40). However, Ski is a transcriptional coregulator for other signaling pathways distinct from TGF-β, such as Wnt, Shh, retinoic acid, and other pathways involving nuclear receptors (17). Further investigation will be required to determine whether the regulation of Ski stability and sorting in hepatocytes influences those other signaling pathways during the regenerative process.
Notably, there is also evidence suggesting a positive role for Ski during cell cycle progression because Ski seems to promote proliferation of some cell types, such as muscle cells, melanoma cells, and fibroblasts (41–43). The mechanisms are unknown, but they may be related to the inhibition of TGF-β/Smad signaling (44–46) or by the sequestration of the tumor suppressor Rb in the cytoplasm (47). Additionally, Ski has been localized at centrosomes and the mitotic spindle during cell mitosis (48). Here we found that Ski seems to favor the proliferation of normal hepatocytes. However, the mechanisms involved have also not been fully elucidated.
In conclusion, our work demonstrates a novel mechanism that regulates Ski protein levels in normal hepatocytes through different stimuli (Fig. 8). In these cells, Ski protein is able to sense changes in actin cytoskeleton dynamics that control its own stability and endosomal sorting. Additionally, the regulation of Ski protein stability by GPCR pathways seems to be tightly associated with actin cytoskeleton dynamics.
Acknowledgments
We thank Drs. J. Vázquez-Prado and J. Chimal-Monroy for helpful discussions. We also thank Dr. J. A. García-Sáinz (IFC, UNAM) for the C9 and HepG2 cell lines and all members from Unidad de Imagenología, Bioterio, and Unidad de Cómputo at IFC, UNAM.
This work was supported by PAPIIT/DGAPA/UNAM Grants IN222909 and IN206012) and CONACyT Grant 101826 (to M. M. S.).
- GPCR
- G protein-coupled receptor
- MβCD
- methyl-β-cyclodextrin
- S1P
- sphingosine 1-phosphate
- LPA
- lysophosphatidic acid
- F
- forskolin
- IBMX
- 3-isobutyl-1-methylxanthine
- LatB
- Latrunculin B
- Jasp
- jasplakinolide
- RIPA
- radioimmune precipitation assay
- IP
- immunoprecipitation
- DRM
- detergent-resistant membrane
- WB
- Western blot
- PH
- partial hepatectomy
- MVE
- multivesicular endosome
- SN
- supernatant.
REFERENCES
- 1. Taub R. (2004) Liver regeneration: from myth to mechanism. Nat. Rev. Mol. Cell Biol. 5, 836–847 [DOI] [PubMed] [Google Scholar]
- 2. Michalopoulos G. K. (2007) Liver Regeneration. J. Cell Physiol. 213, 286–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Godoy P., Hewitt N. J., Albrecht U., Andersen M. E., Ansari N., Bhattacharya S., Bode J. G., Bolleyn J., Borner C., Böttger J., Braeuning A., Budinsky R. A., Burkhardt B., Cameron N. R., Camussi G., Cho C. S., Choi Y. J., Craig Rowlands J., Dahmen U., Damm G., Dirsch O., Donato M. T., Dong J., Dooley S., Drasdo D., Eakins R., Ferreira K. S., Fonsato V., Fraczek J., Gebhardt R., Gibson A., Glanemann M., Goldring C. E., Gómez-Lechón M. J., Groothuis G. M., Gustavsson L., Guyot C., Hallifax D., Hammad S., Hayward A., Häussinger D., Hellerbrand C., Hewitt P., Hoehme S., Holzhütter H. G., Houston J. B., Hrach J., Ito K., Jaeschke H., Keitel V., Kelm J. M., Kevin Park B., Kordes C., Kullak-Ublick G. A., LeCluyse E. L., Lu P., Luebke-Wheeler J., Lutz A., Maltman D. J., Matz-Soja M., McMullen P., Merfort I., Messner S., Meyer C., Mwinyi J., Naisbitt D. J., Nussler A. K., Olinga P., Pampaloni F., Pi J., Pluta L., Przyborski S. A., Ramachandran A., Rogiers V., Rowe C., Schelcher C., Schmich K., Schwarz M., Singh B., Stelzer E. H., Stieger B., Stöber R., Sugiyama Y., Tetta C., Thasler W. E., Vanhaecke T., Vinken M., Weiss T. S., Widera A., Woods C. G., Xu J. J., Yarborough K. M., Hengstler J. G. (2013) Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch. Toxicol. 87, 1315–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Herrera B., Alvarez A. M., Beltrán J., Valdés F., Fabregat I., Fernández M. (2004) Resistance to TGF-β-induced apoptosis in regeneration hepatocytes. J. Cell Physiol. 201, 385–392 [DOI] [PubMed] [Google Scholar]
- 5. Romero-Gallo J., Sozmen E. G., Chytil A., Russell W. E., Whitehead R., Parks W. T., Holdren M. S., Her M. F., Gautam S., Magnuson M., Moses H. L., Grady W. M. (2005) Inactivation of TGF-β signaling in hepatocytes results in an increased proliferative response after partial hepatectomy. Oncogene 24, 3028–3041 [DOI] [PubMed] [Google Scholar]
- 6. Coulouarn C., Factor V. M., Thorgeirsson S. S. (2008) Transforming growth factor-β gene expression signature in mouse hepatocytes predicts clinical outcome in human cancer. Hepatology 47, 2059–2067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dooley S., ten Dijke P. (2012) TGF-β in progression of liver disease. Cell Tissue Res. 347, 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mu X., Lin S., Yang J., Chen C., Chen Y., Herzig M. C., Washburn K., Halff G. A., Walter C. A., Sun B., Sun L. Z. (2013) TGF-β signaling is often attenuated during hepatumorigenesis, but is retained for the malignancy of hepatocellular carcinoma cells. PLoS ONE 8, e63436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wrana J. L., Attisano L., Wieser R., Ventura F., Massagué J. (1994) Mechanism of activation of the TGF-β receptor. Nature 370, 341–347 [DOI] [PubMed] [Google Scholar]
- 10. Macías-Silva M., Abdollah S., Hoodless P. A., Pirone R., Attisano L., Wrana J. L. (1996) MADR2 is a substrate of the TGF-β receptor and its phosphorylation is required for nuclear accumulation and signaling. Cell 87, 1215–1224 [DOI] [PubMed] [Google Scholar]
- 11. Massagué J. (2012) TGF-β signaling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Luo K. (2004) Ski and SnoN: negative regulators of TGF-β signaling. Curr. Opin. Genet. Dev. 14, 65–70 [DOI] [PubMed] [Google Scholar]
- 13. Imamura T., Oshima Y., Hikita A. (2013) Regulation of TGF-β family signaling by ubiquitination and deubiquitination. J. Biochem. 154, 481–489 [DOI] [PubMed] [Google Scholar]
- 14. Luo K., Stroschein S. L., Wang W., Chen D., Martens E., Zhou S., Zhou Q. (1999) The Ski oncoprotein interacts with the Smad proteins to repress TGF-β signaling. Genes Dev. 13, 2196–2206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nomura T., Khan M. M., Kaul S. C., Dong H. D., Wadhwa R., Colmenares C., Kohno I., Ishii S. (1999) Ski is a component of the histone deacetylase complex required for transcriptional repression by Mad and Thyroid hormone receptor. Genes Dev. 13, 412–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xu W., Angelis K., Danielpour D., Haddad M. M., Bischof O., Campisi J., Stavnezer E., Medrano E. E. (2000) Ski acts as a co-repressor with Smad2 and Smad3 to regulate the response to type β transforming growth factor. Proc. Natl. Acad. Sci. 97, 5924–5929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bonnon C., Atanasoski S. (2012) c-Ski in health and disease. Cell Tissue Res. 347, 51–64 [DOI] [PubMed] [Google Scholar]
- 18. Tecalco-Cruz A. C., Sosa-Garrocho M., Vázquez-Victorio G., Ortiz-García L., Domínguez-Hüttinger E., Macías-Silva M. (2012) Transforming growth factor-β/SMAD target gene skil is negatively regulated by the transcriptional cofactor complex SNON-SMAD4. J. Biol. Chem. 287, 26764–26776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Atanasoski S., Notterpek L., Lee H. Y., Castagner F., Young P., Ehrengruber M. U., Meijer D., Sommer L., Stavnezer E., Colmenares C., Suter U. (2004) The protooncogene Ski controls Schwann cell proliferation and myelination. Neuron 43, 499–511 [DOI] [PubMed] [Google Scholar]
- 20. Nagata M., Goto K., Ehata S., Kobayashi N., Saitoh M., Miyoshi H., Imamura T., Miyazawa K., Miyazono K. (2006) Nuclear and cytoplasmic c-Ski differently modulates cellular functions. Genes Cells 11, 1267–1280 [DOI] [PubMed] [Google Scholar]
- 21. Macias-Silva M., Li W., Leu J. I., Crissey M. A., Taub R. (2002) Up-regulated transcriptional repressors SnoN and Ski bind Smad proteins to antagonize transforming growth factor-β signals during liver regeneration. J. Biol. Chem. 277, 28483–28490 [DOI] [PubMed] [Google Scholar]
- 22. Delgado-Coello B., Bravo-Martínez J., Sosa-Garrocho M., Briones-Orta M. A., Macías-Silva M., Mas-Oliva J. (2010) Plasma membrane calcium ATPase isoform 3 expression in single cell types isolated from rat liver. Mol. Cell Biochem. 344, 117–124 [DOI] [PubMed] [Google Scholar]
- 23. Lee H. H., Elia N., Ghirlando R., Lippincott-Schwartz J., Hurley J. H. (2008) Midbody targeting of the ESCRT machinery by a noncanonical coiled coil in CEP55. Science 322, 576–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grewal T., Heeren J., Mewawala D., Schnitgerhans T., Wendt D., Salomon G., Enrich C., Beisiegel U., Jäckle S. (2000) Annexin VI stimulates endocytosis and is involved in the trafficking of low density lipoprotein to the prelysosomal compartment. J. Biol. Chem. 275, 33806–33813 [DOI] [PubMed] [Google Scholar]
- 25. Slimane T. A., Trugnan G., Van Ijzendoorn S. C., Hoekstra D. (2003) Raft-mediated trafficking of apical resident proteins occurs in both direct and transcytotic pathways in polarized hepatic cells: role of distinct lipid microdomains. Mol. Biol. Cell 14, 611–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sancak Y., Bar-Peled L., Zoncu R., Markhard A. L., Nada S., Sabatini D. M. (2010) Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Taelman V. F., Dobrowolski R., Plouhinec J. L., Fuentealba L. C., Vorwald P. P., Gumper I., Sabatini D. D., De Robertis E. M. (2010) Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 143, 1136–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Awasthi-Kalia M., Schnetkamp P. P., Deans J. P. (2001) Differential effects of filipin and methyl-β-cyclodextrin on B cell receptor signaling. Biochem. Biophys. Res. Commun. 287, 77–82 [DOI] [PubMed] [Google Scholar]
- 29. Simons K., Toomre D. (2000) Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1, 31–39 [DOI] [PubMed] [Google Scholar]
- 30. Delgado-Coello B., Briones-Orta M. A., Macías-Silva M., Mas-Oliva J. (2011) Cholesterol: recapitulation of its active role during liver regeneration. Liver Int. 31, 1271–1284 [DOI] [PubMed] [Google Scholar]
- 31. Yu F. X., Zhao B., Panupinthu N., Jewell J. L., Lian I., Wang L. H., Zhao J., Yuan H., Tumaneng K., Li H., Fu X. D., Mills G. B., Guan K. L. (2012) Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 150, 780–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yu F.-X., Zhang Y., Park H. W., Jewell J. L., Chen Q., Deng Y., Pan D., Taylor S. S., Lai Z.-C., Guan K.-L. (2013) Protein kinase A activates the hippo pathway to modulate cell proliferation and differentiation. Genes Dev. 27, 1223–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Varelas X., Samavarchi-Tehrani P., Narimatsu M., Weiss A., Cockburn K., Larsen B. G., Rossant J., Wrana J. L. (2010) The crumbs complex couples cell density sensing to hippo-dependent control of the TGF-β-Smad pathway. Dev. Cell 19, 831–844 [DOI] [PubMed] [Google Scholar]
- 34. Doyle A. J., Doyle J. J., Bessling S. L., Maragh S., Lindsay M. E., Schepers D., Gillis E., Mortier G., Homfray T., Sauls K., Norris R. A., Huso N. D., Leahy D., Mohr D. W., Caulfield M. J., Scott A. F., Destrée A., Hennekam R. C., Arn P. H., Curry C. J., Van Laer L., McCallion A. S., Loeys B. L., Dietz H. C. (2012) Mutations in the TGF-β repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat. Genet. 44, 1249–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Berk M., Desai S. Y., Heyman H. C., Colmenares C. (1997) Mice lacking the ski proto-oncogene have defects in neurulation, craniofacial, patterning, and skeletal muscle development. Genes Dev. 11, 2029–2039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shinagawa T., Nomura T., Colmenares C., Ohira M., Nakagawara A., Ishii S. (2001) Increased susceptibility to tumorigenesis of ski-deficient heterozygous mice. Oncogene 20, 8100–8108 [DOI] [PubMed] [Google Scholar]
- 37. Ye F., Lemieux H., Hoppel C. L., Hanson R. W., Hakimi P., Croniger C. M., Puchowicz M., Anderson V. E., Fujioka H., Stavnezer E. (2011) Peroxisome proliferators-activated receptorγ (PPARγ) mediates a Ski oncogene-induced shift from glycolysis to oxidative energy metabolism. J. Biol. Chem. 286, 40013–40024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Briones-Orta M. A., Levy L., Madsen C. D., Das D., Erker Y., Sahai E., Hill C. S. (2013) Arkadia regulates tumor metastasis by modulation of the TGF-β pathway. Cancer Res. 73, 1800–1810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Krakowski A. R., Laboureau J., Mauviel A., Bissell M. J., Luo K. (2005) Cytoplasmic SnoN in normal tissues and nonmalignant cells antagonizes TGF-β signaling by sequestering of the Smad proteins. Proc. Natl. Acad. Sci. 102, 12437–12442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang L., Boyer J. L. (2004) The maintenance and generation of membrane polarity in hepatocytes. Hepatology 39, 892–899 [DOI] [PubMed] [Google Scholar]
- 41. Soeta C., Suzuki M., Suzuki S., Naito K., Tachi C., Tojo H. (2001) Possible role for the c-ski gene in the proliferation of myogenic cells in regenerating skeletal muscles of rats. Dev. Growth Differ. 43, 155–164 [DOI] [PubMed] [Google Scholar]
- 42. Reed J. A., Lin Q., Chen D., Mian I. S., Medrano E. E. (2005) Ski pathways inducing progression of human melanoma. Cancer Metastasis Rev. 24, 265–272 [DOI] [PubMed] [Google Scholar]
- 43. Liu X., Li P., Liu P., Xiong R., Zhang E., Chen X., Gu D., Zhao Y., Wang Z., Zhou Y. (2008) The essential role for c-Ski in mediating TGF-β1-induced bi-directional effects on skin fibroblast proliferation through a feedback loop. Biochem. J. 409, 289–297 [DOI] [PubMed] [Google Scholar]
- 44. Kokura K., Kim H., Shinagawa T., Khan M. M., Nomura T., Ishii S. (2003) The Ski-binding protein C184M negatively regulates tumor growth factor-β signaling by sequestering the smad proteins in the cytoplasm. J. Biol. Chem. 278, 20133–20139 [DOI] [PubMed] [Google Scholar]
- 45. Prunier C., Pessah M., Ferrand N., Seo S. R., Howe P., Atfi A. (2003) The oncoprotein Ski acts as an antagonist if transforming growth factor-β signaling by suppressing Smad2 phosphorylation. J. Biol. Chem. 278, 26249–26257 [DOI] [PubMed] [Google Scholar]
- 46. Ferrand N., Atfi A., Prunier C. (2010) The oncoprotein c-ski functions as a direct antagonist of the transforming growth factor-β type I receptor. Cancer Res. 70, 8457–8466 [DOI] [PubMed] [Google Scholar]
- 47. Jacob C., Grabner H., Atanasoski S., Suter U. (2008) Expression and localization of Ski determine cell type-specific TGF-β signaling effects on the cell cycle. J. Cell Biol. 182, 519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Marcelain K., Hayman M. J. (2005) The Ski oncoprotein is up-regulated and localized at the centrosomes and mitotic spindle during mitosis. Oncogene 24, 4321–4329 [DOI] [PubMed] [Google Scholar]