

Abstract
The brains of teleost fish show extensive adult neurogenesis and neuronal regeneration. The patterns of gene regulation during fish brain aging are unknown. The short-lived teleost fish Nothobranchius furzeri shows markers of brain aging including reduced learning performances, gliosis, and reduced adult neurogenesis. We used RNA-seq to quantify genome-wide transcript regulation and sampled five different time points to characterize whole-genome transcript regulation during brain aging of N. furzeri. Comparison with human datasets revealed conserved up-regulation of ribosome, lysosome, and complement activation and conserved down-regulation of synapse, mitochondrion, proteasome, and spliceosome. Down-regulated genes differ in their temporal profiles: neurogenesis and extracellular matrix genes showed rapid decay, synaptic and axonal genes a progressive decay. A substantial proportion of differentially expressed genes (∼40%) showed inversion of their temporal profiles in the last time point: spliceosome and proteasome showed initial down-regulation and stress-response genes initial up-regulation. Extensive regulation was detected for chromatin remodelers of the DNMT and CBX families as well as members of the polycomb complex and was mirrored by an up-regulation of the H3K27me3 epigenetic mark. Network analysis showed extensive coregulation of cell cycle/DNA synthesis genes with the uncharacterized zinc-finger protein ZNF367 as central hub. In situ hybridization showed that ZNF367 is expressed in neuronal stem cell niches of both embryonic zebrafish and adult N. furzeri. Other genes down-regulated with age, not previously associated with adult neurogenesis and with similar patterns of expression are AGR2, DNMT3A, KRCP, MEX3A, SCML4, and CBX1. CBX7, on the other hand, was up-regulated with age.
Keywords: animal model, brain aging, epigenetics, gene expression, neural stem cells, neurogenesis, teleost, transcriptomics
Introduction
Several studies analyzed genome-wide transcript regulation during aging of the primate and rodent brain (Lee et al., 2000; Loerch et al., 2008; Somel et al., 2010) including analysis of human samples across the entire life stages (Colantuoni et al., 2011). Some conserved features linked to brain aging are as follows: (i) down-regulation of genes coding for synaptic and axonal proteins (Loerch et al., 2008), (ii) increased activity of the polycomb repressive complex (Horvath et al., 2012), and (iii) reduced adult neurogenesis (Kempermann, 2011). In primates, where analyses along the entire postnatal life are available, gene regulation observed during brain aging is either the continuation, or the inversion, of a process that started during development. (Somel et al., 2010; Colantuoni et al., 2011).
The brain of teleost fish, as opposed to mammals, shows widespread adult neurogenesis and remarkable regenerative properties (Kempermann, 2011). The process of brain aging in teleost has received very little attention so far (Terzibasi Tozzini et al., 2012; Edelmann et al., 2013). It is therefore of interest to investigate whether the patterns of gene regulation identified in mammals are observed also in teleosts.
The short-lived fish Nothobranchius furzeri is a novel model organism for aging research. N. furzeri inhabits ephemeral habitats that last on average 75 days (Terzibasi Tozzini et al., 2013), and their captive lifespan is the shortest ever recorded for a vertebrate (Genade et al., 2005). As an adaptation to this environment, N. furzeri shows explosive growth (Blazek et al., 2013) and rapid expression of aging phenotypes at the behavioral, histological, and molecular levels (Valenzano et al., 2006a; Di Cicco et al., 2011; Hartmann et al., 2011) including age-dependent gliosis and rapid decay of adult neurogenesis (Terzibasi Tozzini et al., 2012). The N. furzeri transcriptome is available, and an initial study of RNA-seq, based on pool sequencing and two age steps, has confirmed a prominent decay of mitotic activity during brain aging (Petzold et al., 2013). A striking characteristic of N. furzeri is the existence of large differences in longevity across strains (Terzibasi et al., 2008) that has a polygenic origin (Kirschner et al., 2012). Here, we used the strain MZM-04/10 that has a median lifespan of ∼ 30 weeks and was characterized in previous studies (Terzibasi et al., 2008; Terzibasi Tozzini et al., 2012).
The aim of this study was to perform a systematic analysis of genome-wide transcript regulation during brain aging, using biological replicates and multiple time points. In particular, we concentrated our analysis on the following aspects: (i) characterization of the temporal profiles of expression, (ii) comparison with human data, (iii) characterization of networks of coregulated genes, and iv) localization of cells expressing differentially expressed genes in the brain of both adult N. furzeri and zebrafish embryos.
Results
Identification of differentially expressed genes and global analysis of gene expression
To describe age-dependent transcriptome regulation, we used RNA-seq (Illumina) and a reference N. furzeri transcriptome containing 19 812 annotated transcript contigs. The data cover male animals, 5 age-groups (5, 12, 20, 27, and 39 weeks), and five biological replicates for each age. These ages correspond to sexual maturity, young adult, adult (as defined by a decrease in growth rate), median lifespan, and old (∼30% survivorship) (Figs S1, S2). Differentially expressed genes (DEGs) were defined by the intersection of three independent statistical tests without cutoff for effect size (see Supplementary Material and Methods) in at least one of all pairwise comparisons between the age-groups, and 4104 DEGs were detected (Table S1). The highest number of DEGs between adjacent time points was detected in the comparison between 5 and 12 weeks and the minimum between 20 and 27 weeks (Fig. S3).
To validate the findings obtained by RNA-seq, we performed qPCR on 20 DEGs (Table S2). The fold-changes measured using the two techniques are highly correlated (Pearson's R2 = 0.70; Fig. S4).
To visualize the impact of age on global transcript regulation, we applied multidimensional scaling (MDS) (Fig.1). Using this approach, the 5-week-, 12-week-, and 39-week-old samples can be separated in nonoverlapping regions in the MDS plot. However, the largest separation is clearly between the 5-week-old samples and the other samples in dimension 1 (see also Fig. S5), while in dimension 2, there is a progression from the 12-week-old samples to 20- and 27-week-old to 39-week-old samples.
Figure 1.
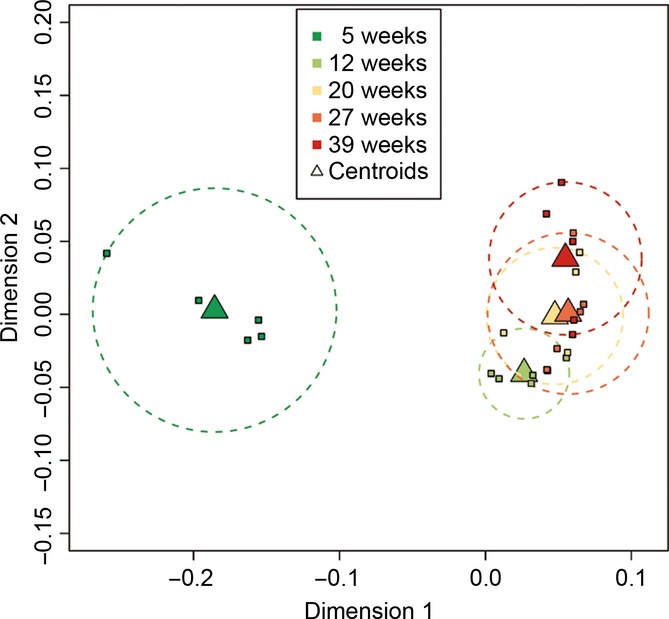
Multidimensional scaling. Age of samples is color-coded. Individual samples are represented by small squares, the centroids by triangles, and the confidence interval by dotted circles.
Clustering temporal profiles of gene expression
To group DEGs with similar temporal profiles, fuzzy c-means (FCM) clustering was used. The number of optimal clusters was estimated by the vote of different cluster validity indices, which capture different aspects of a clustering structure (Guthke et al., 2005). The temporal transcript profiles were grouped into 6 clusters ordered by the number of DEGs included (Fig.2 and Table S3). Three clusters exhibit a monotonic behavior: Cluster 2 shows a gradual increase, while clusters 1 and 5 show rapid and gradual decreases, respectively. The remaining clusters include more complex temporal profiles: Cluster 3 shows a U shape with a minimum at 27 weeks; clusters 4 and 6 include a peak at 27 and 20 weeks, respectively. In summary, 60.9% of DEGs were assigned to clusters with monotonous age regulation, and 39.1% were assigned to clusters with inversion in the initial direction of age regulation. 59.6% of the DEGs showed an initial decrease and 40.4% an initial increase in their expression.
Figure 2.
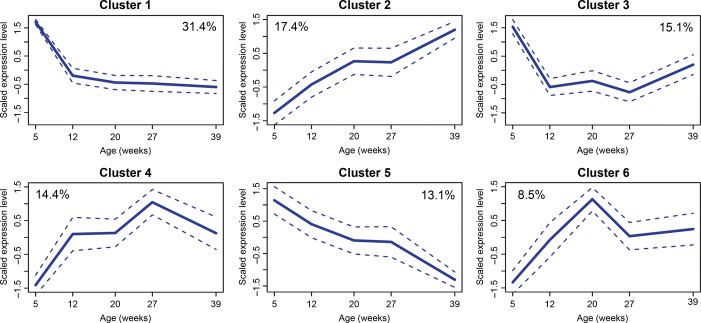
Fuzzy c-means clustering of 4014 differentially expressed genes (DEGs) according to temporal profiles. Each individual DEG profile was centered to mean and scaled to variance. The solid line represents the mean value of the cluster and the dashed lines 95% confidence intervals. For each cluster, the percentage of DEGs assigned to it is reported. Clusters are ordered according total number of members.
As an independent assessment for inversions in the temporal profile of gene expression, we identified genes showing U- or bell-shaped trajectories by quadratic fitting (P < 0.05) of gene expression levels as function of age. The distributions of individual estimated peaks were plotted separately for genes showing U shape (n = 887) or bell shape (n = 2504). In both cases, the large majority of the peaks were in the 20-to 30-week range with median around 25 weeks (Fig. S6).
Gene enrichment analysis
In order to assess whether different temporal profiles correspond to different cellular components or biological processes, we determined for the N. furzeri genes their human orthologs and performed gene enrichment analysis based on annotations of the human genes corresponding to each of the temporal clusters. In a first step, we analyzed KEGG pathways, and we report for each cluster up to the top five annotations (Fig.3) a complete list with percentage of up- and down-regulated genes (Table S4) and a mapping of fold-changes (Appendix S1). To reduce the redundancy of the large numbers of enriched gene ontology (GO) terms (Table1), we used ClueGO to cluster terms that are sharing significant number of genes and report here the results for cellular components only (Fig.3). Cluster 1 (rapid decay) was enriched for genes coding for proteins that are in nuclear components (chromosome, spindle, and telomeres) as well as extracellular matrix and microtubule-organizing center. Consistently, the most enriched KEGG pathways were ‘cell cycle’ and ‘ECM–receptor interaction’. It is of note that 10 genes of the Notch pathway are assigned to cluster 1 (Table S5). Cluster 2 (linear increase) was enriched for genes coding for proteins that are assigned to ‘cytosolic ribosome’ and ‘vacuole’. KEGG analysis showed an extensive enrichment of genes belonging to the pathways ‘ribosome’, ‘lysosome’, and ‘complement and coagulation cascades’. Averaged expression of all genes encoding cytosolic ribosomal proteins (RPs) increased linearly as function of age with 47% increase in expression between 5 and 39 weeks, while mitochondrial ribosomal proteins showed U-shaped profile (Fig.4A). The term ‘GO: 0006413 translational initiation’ is enriched as well (4.4-fold enrichment, Fisher's exact test, P = 0.0023). Cluster 3 (U shaped, minimum at 27 weeks) was enriched for genes coding for nuclear proteins (nuclear pore, chromosome, nucleoplasm, nucleus periphery) as well as ribonucleoprotein and proteasome complexes. KEGG analysis showed highly significant enrichment of ‘proteasome’ and ‘spliceosome’. Cluster 4 (bell shaped, peak at 27 weeks) was enriched for genes coding for proteins in cytoplasm as well as contractile fiber and vacuole. KEGG analysis showed highly significant enrichment of genes belonging to the ‘circadian rhythm’ and ‘MAPK-pathways’ and in addition ‘P53-’, ‘mTOR-’, and ‘neurotrophin-signaling-pathway’. Notably, key negative regulators of the cell cycle such as the cyclin-dependent kinase inhibitor-1 (CDKN1A, P21), members of growth arrest and DNA damage gene family (GADD45), and the TP53 target inhibitory cyclin G1 (CCNG1) (Okamoto & Beach, 1994), are assigned to cluster 4. Cluster 5 (linear decrease) showed significant enrichment in proteins present in neuronal processes: Interestingly, the most significantly enriched GO term for cellular components is synapse (GO:0045202, 23 genes, 3.2-fold enrichment, Fisher's exact test, P = 10−6), and further categories are cell periphery, intracellular, and cytoskeletal part. Top enriched KEGG pathways are ‘axon guidance’ and ‘ECM–receptor interaction’ (Fig.3). It is of note that three key genes of the sonic hedgehog (SHH) signaling pathway, a pathway known to be essential for adult neurogenesis (Favaro et al., 2009), are assigned to this cluster (Table S5). Cluster 6 (bell shaped, peak at 20 weeks) was enriched for genes belonging to KEGG pathways related to innate immunity.
Figure 3.

Gene enrichment analysis of the individual fuzzy c-means (FCM) clusters. The pie charts correspond to clustering of enriched cellular component terms as generated by ClueGO. The dimension of the wedges is proportional to the number of clustered terms, and the term with the highest number of differentially expressed genes was used for annotation. Gray wedges indicate individual terms that could not be clustered. The text below the pie charts reports the five most enriched KEGG pathways for each of the FCM clusters.
Table 1.
GO categories common between Nothobranchius furzeri and human brain aging
| Category | Human | Intersection | N. furzeri | Top categories | FDR |
|---|---|---|---|---|---|
| Overrepresented terms among up-regulated genes | |||||
| GO: biological process | 194 | 56 (40%) | 142 | Translational elongation | 1.6e-41 |
| Protein targeting to ER | 1.4e-37 | ||||
| Nonsense-mediated decay | 1.5e-35 | ||||
| GO: cellular compartment | 72 | 20 (77%) | 26 | Ribosome | 1.4e-41 |
| Lysosome | 6.3e-6 | ||||
| GO: molecular function | 22 | 4 (80%) | 5 | Structural constituent of ribosome | 1.7e-25 |
| KEGG pathway | 2 | 1 (50%) | 2 | Ribosome | 1.4e-38 |
| Overrepresented terms among down-regulated genes | |||||
| GO: biological process | 349 | 116 (32%) | 364 | Cell cycle | 2.6e-59 |
| DNA damage | 1.7e-22 | ||||
| Cellular response to stress | 5.8e-15 | ||||
| Protein folding | 2.9e-06 | ||||
| RNA processing | 6.2e-06 | ||||
| GO: cellular compartment | 201 | 59 (41%) | 143 | Nucleus | 3.9e-39 |
| Organelle | 6.8e-39 | ||||
| Microtubule cytoskeleton | 3e-16 | ||||
| Mitochondrion | 1.1e-10 | ||||
| GO: molecular function | 99 | 27 (33%) | 82 | ATPase activity | 4.6e-7 |
| RNA binding | 4.9e-05 | ||||
| Threonine-type peptidase activity | 0.00016 | ||||
| Unfolded protein binding | 0.00044 | ||||
| KEGG pathways | 25 | 5 (25%) | 20 | Proteasome | 2.3e-5 |
| RNA transport | 0.00017 | ||||
| Spliceosome | 0.00019 | ||||
| Nucleotide excision repair | 0.00038 | ||||
| Protein processing in endoplasmic reticulum | 0.0006 | ||||
The numbers reported refer to categories significant with hypergeometric test and FDR < 0.05. Human and N. furzeri figures refer to the total number of categories in each gene set, respectively. Percentage in intersection column refers to percentage of N. furzeri categories. The top categories are defined as nonoverlapping categories ranked on FDR-corrected P-value for the N. furzeri dataset whose values are reported in the last column.
Figure 4.
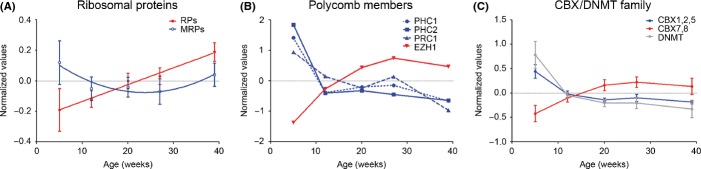
Age-dependent transcript levels of selected differentially expressed genes. Expression values at each age were centered and scaled to the mean. (A) The average of all 87 ribosomal genes (RP) and mitochondrial ribosomal genes (MRP) are shown. Error bars represent standard deviations. (B) Members of the polycomb complexes 1 and 2. (C) Averaged expression of all DNMT genes and members of the CBX genes family. CBX members in cluster 1 are shown in blue, and CBX members in clusters 2 and 6 are shown in red.
Chromatin-remodeling genes
Age-dependent epigenetic remodeling is prominent in the human brain (Horvath et al., 2012; Numata et al., 2012; Watson et al., 2012), and chromosome is a predominant GO term in cluster 1 (Fig.3). We examined the chromatin-remodeling DEGs and found that several members of the DNA methyl-transferase gene family were down-regulated and belong to cluster 1 (DNMT1/3B/3AA/3AB, Fig.4C). Particularly evident was the regulation of members of the polycomb repressive complex 1: protein regulator of cytokinesis (PRC1), polyhomeotic-like 1 and 3 (PHC1 and PHC 2; cluster 1), and enhancer of zeste homolog 1 (EZH1; cluster 4), that is part of polycomb complex 2 (Fig.4B). Similarly, members of the chromobox homolog (CBX) gene family showed opposite trajectories of expression (Fig.4C) with CBX1A/B,CBX2, and CBX5 in cluster 1 and CBX7A/B and CBX8A/B in clusters 2 and 6.
Comparison with other datasets
We compared the list of DEGs obtained in N. furzeri with a public dataset of 269 samples from human prefrontal cortex from fetal to old age (Colantuoni et al., 2011). DEGs from N. furzeri are regulated during human ontogeny, and FCM clustering separates them into three groups: (i) rapid decrease during fetal life, (ii) peak during childhood followed by a decrease, and (iii) increase from fetal life to childhood (Fig. S7). To specifically compare aging-related DEGs from human and N. furzeri, we used a dataset of 13 young (age 25–40) and 15 old subjects (age 70–95) (Loerch et al., 2008) and performed gene enrichment analysis on human and N. furzeri DEGs separately. For both down- and up-regulated GO terms, we detected an intersection that included between 25% and 80% of the N. furzeri terms and that was particularly extensive for up-regulated DEGs (Table1). For a broad-level comparison, we used a meta-analysis of human brain, kidney, and muscle microarray data (Zahn et al., 2006) and a meta-analysis of different tissues from human, primate, and rodent tissues (de Magalhaes et al., 2009). Four up-regulated terms (‘cytosolic ribosome’, ‘lysosome’, ‘negative regulation of apoptosis’, and ‘complement activation’) were shared between N. furzeri, and at least one of these analysis and two down-regulated terms (‘mitochondrion’ and ‘collagen’) were shared between de Magalhaes et al. (2009) and N. furzeri (Table2). In addition, 16% of the up-regulated genes and 21% of the down-regulated genes identified by de Magalhaes et al. (2009) were differentially expressed with concordant direction in N. furzeri (Table S9 and Fig. S12).
Table 2.
Comparison of the meta-analysis of de Magalhaes et al. (2009) and Zahn et al. (2006) with genes differentially expressed in Nothobranchius furzeri brain
| Meta-analysis by de Magalhaes et al. (2009) | Meta-analysis by Zahn et al. (2006) | This study |
|---|---|---|
| Overrepresented terms among down-regulated genes | ||
| Mitochondrion | ND | GO:0005739 mitochondrion, P = 2.5 × 10−5 |
| Oxidative phosphorylation | Oxidative phosphorylation | ND |
| Hydroxylisine, hydroxylation, collagen | ND | GO:0005581 collagen, P = 1.5 × 10−5 |
| Overrepresented terms among up-regulated genes | ||
| Immune response, complement activation | Complement activation | KEGG hsa04610 complement and coagulation cascades, P = 0.035 |
| ND | Cytosolic ribosome | KEGG hsa03010 ribosome, P = 8.4 × 10−49 |
| Lysosome | ND | KEGG hsa04142 lysosome, P = 0.0028 |
| Negative regulation of apoptosis | ND | GO:0043066 negative regulation of apoptosis, P = 0.0053 |
The terms reported for meta-analysis are obtained from Table I de Magalhaes et al. (2009) and represent the broadest of the top annotation of functional clusters obtained by DAVID. The terms reported for Zahn et al. (2006) were detected by GSEA using correlation of gene expression with age. N. furzeri DEGs were analyzed by DAVID using standard setting and the ‘FAT’ level for GO terms. Significance values refer to Fisher's exact test.
Network analysis
We constructed a correlation-based network of the transcript levels of all expressed genes in the 25 samples. Using Pearson′s r > 0.95 as selection criterion for edges, the resulting network contains 1707 nodes and 18 008 edges (Fig.5A). The node of highest edge value is the putative transcription factor zinc-finger protein 367 gene (ZNF367) in cluster 1 (Fig.5A'). The subnetwork of ZNF367 first neighbors (Fig.5B) contains 192 nodes and 1030 edges and is clearly enriched for genes that regulate the cell cycle (Fig.5C).
Figure 5.
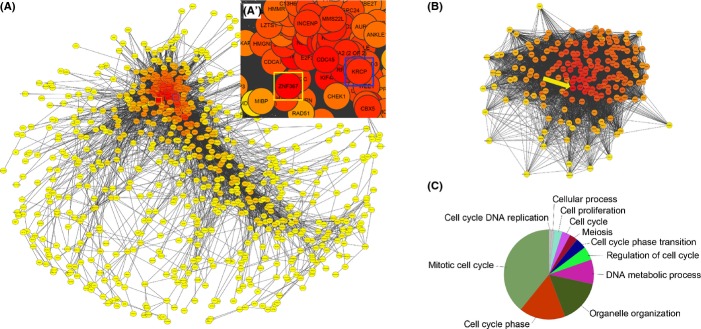
Network analysis. (A) Network of RKPM values of all expressed genes based on Pearson's correlation r ≥ 0.95, spring-embedded layout weighted on correlation. Color of nodes indicates the edge degree value with red assigned to nodes of highest values. Only the cluster with largest number of nodes is shown. (A′) Enlargement of (A): ZNF367, the node of highest edge, is indicated by yellow square, the position of KRCP, a gene of high edge degree value that was further studied by in situ hybridization, is indicated by a blue square. (B) Network of the first neighbors of ZNF367, 192 nodes, 1030 edges. The position of ZNF367 is indicated by a yellow arrow. (C) Gene enrichment analysis of ZNF367 first neighbors.
We further analyzed whether correlation in expression changes as function of age and computed correlation-based networks of the DEGs only across samples of the same age for the five time points separately (Fig. S8). In the 5-week age-group, one single network emerges and contains genes belonging to all above-described FCM clusters. In the last time point, 39 weeks, two distinct clusters emerge that are not connected by edges: one contains mainly genes of FCM clusters one and five (continuous down-regulation) and the second mainly genes of FCM clusters two and three that increase in expression between 27 and 39 weeks.
Expression in the brain and identification of genes related to adult neurogenesis
Cluster 1 is highly enriched for genes related to cell cycle and DNA replication and shows a rapid decay. Adult neurogenesis is known to decrease exponentially with age (Pekcec et al., 2008; Ben Abdallah et al., 2010), and we reasoned that this cluster may contain genes specifically enriched in the niches of neuronal stem cells (NSCs) and not previously characterized in this context. As a first step, we interrogated the ZFIN database and identified 96 genes of cluster 1 for which high-quality in situ hybridization pictures in zebrafish embryos were available. Of these, 86% (83/96) are expressed in the nervous system and 50% (48/96) are expressed specifically in the NSC niches of the zebrafish embryo (Table S6). We selected a short list of genes of putative regulatory function to visualize their expression by in situ hybridization in the central nervous system of zebrafish embryos at age 72 h when niches of NSCs are well delimited (Figs6A and S9). These genes were anterior gradient related 2 (AGR2), previously implicated in regeneration of amphibian limbs (Kumar et al., 2007), the chromatin remodelers CBX1A,CBX7A,DNMT3AA, and sex comb midleg like 4 (Drosophila) (SCML4), and the RNA-binding protein mex3 homolog A (C. elegans) (MEX3A) (Buchet-Poyau et al., 2007). In addition, we analyzed the genes for two putative transcription factors: ZNF367 and the teleost-specific kelch repeat containing protein (KRCP) that both occupy very central positions in the correlation-based network (Fig.5A, A′). The expression of ZNF367, KRCP, and MEX3A was very similar to the expression of the mitotic marker proliferating cell nuclear antigen (PCNA, Fig.6A). Neuronal expression of AGR2 was detected exclusively in the mitotically active olfactory epithelium (Fig. S9). All other genes (Fig. S9) were detected in the ciliary marginal zone that represents the NSC niche of the retina and showed different expression patterns in the other NSC niches. Expression of SCML4 was widespread caudally, but was restricted rostrally to NSC niches in the optic tectum, retina, and olfactory epithelium. CBX1A and DNMT3AA showed more widespread expression in the developing brain. CBX7A was not expressed at detectable levels in zebrafish embryos.
Figure 6.

In situ hybridization and immunohistochemistry. (A) Dorsal view of 72 hpf zebrafish embryos processed by in situ hybridization for ZNF367,KRCP, and MEX3A. Proliferating cell nuclear antigen (PCNA) riboprobe has been used to visualize proliferative region of the zebrafish larval brain in the optic tectum (green arrowhead) and retina (red arrow head). (B) Double-labeling in situ hybridization (ISH) and immunohistochemistry (IHC) on the posterior margin of the optic tectum (OTp) in Nothobranchius furzeri for the following genes: SCML4,MEX3A,KRCP,AGR2,DNMT3AA,ZNF367,CBX1A, and CBX7A – ISH signal was revealed using fluorescent Fast Red and is visualized in red, PCNA IHC is visualized in green to localize the proliferative niche of the OTp. Only merged signals are shown. Young animals were 6 weeks old, and old animals were 25 weeks old. Separated red and green channels are reported in Fig. S10. The images are representative of three replicates. (C) Changes of PRC2 markers with age. Immunohistochemical detection of H3K27me3 in the posterior optic tectum of young (5 weeks) and old (35 weeks) individuals. The images are representative of three replicates. Immunohistochemical signal is shown in red and DNA staining (TOPRO3) in blue. Overlap of signals gives rise to a purple hue.
We then performed double-labeling in situ hybridization with PCNA in the N. furzeri brain at 5 and 25 weeks of age (Figs6B and S10) focusing attention on the posterior margin of the optic tectum that represents one of the most active NSC niches in N. furzeri brain (Terzibasi Tozzini et al., 2012). Mitotically active cells were identified by double labeling with antibodies against PCNA. All these genes were expressed in the NSCs in an age-dependent manner (Fig.6B): CBX7A was not detected in young animals and showed a widespread expression in old animals including both the postmitotic neurons and the stem cell niche. This was opposite to the expression of CBX1A that was widespread in young animals and down-regulated in old animals with expression concentrated in the NSC niche. DNMT3AA,KRCP,MEX3A,SCML4, and ZNF367 all were associated with the NSC niche in young animals, and their expression was down-regulated as a function of age. To obtain an independent confirmation for differential activity of polycomb members, we performed immunohistochemistry for tri-methylation of lysine 27 of histone H3 (H3K27me3) that is a known repressive modification linked to PRC2 activity (Simon & Kingston, 2013). Analysis of optic tectum revealed a clear up-regulation of H3K27me3 signal in old fish (Fig.6C).
To investigate the regulation of these genes in humans, we interrogated the ‘Braincloud’ database (Colantuoni et al., 2011) that reports expression in the prefrontal cortex in 269 human subjects from fetal stage to 80 years. Five of these eight genes show a pattern of age-dependent regulation concordant to N. furzeri:CBX7 is strongly up-regulated starting after birth, CBX1 and ZNF367 are strongly down-regulated between fetal ages and newborn, MEX3A and DNMT3A show more progressive down-regulation, and SCML4 is up-regulated during fetal life and down-regulated during postnatal life (Fig. S11).
Discussion
In the present paper, we analyzed age-dependent genome-wide transcript regulation in the short-lived fish N. furzeri by means of RNA-seq. Analysis of aging-dependent gene regulation requires the use of multiple time points, as different genes may follow different trajectories and reach their peaks or plateaus at different times. In this study, we sampled five time points during adult life starting from sexual maturity and including individuals in the ‘old’ class (around 30% survivorship) to resolve different age-dependent expression profiles. In addition, we compared our results with public data on gene regulation in the aging human cortex and detected conservation in the regulated pathways and GO groups.
Temporal inversions
We observed that almost 40% of the genes showed U or bell shape mostly with inversion at 27 weeks and a change of gene coregulatory networks in the oldest group. This could be the result of heterogeneity in the rate of aging and selection for individuals with retarded aging at the oldest group. This cluster of bell-shaped genes contains GADD45 family members that are typically activated by a variety of stressors and induce apoptosis or senescence via the MAPK and TP53 pathways (Liebermann et al., 2011) and the cell cycle inhibitors CDKN1A and CCNG1. The U-shaped cluster is enriched for cell cycle and DNA replication genes (Table S4), suggesting that the oldest group on average shows lower levels of cellular stress than the animals sampled at median lifespan, possibly because they delayed or escaped age-dependent dysfunctions, as it is the case for human centenarians (Evert et al., 2003). Inversions in the direction of age-dependent gene expression are prominent in the postnatal primate cortex (Somel et al., 2010; Colantuoni et al., 2011) and also observed in the rat brain (Wood et al., 2013). One of these turning points is observed around the age of 60 years in humans (Colantuoni et al., 2011). Our observation that in N. furzeri, the frequency of turning points peaks shortly before median lifespan suggests that this may be a general phenomenon in vertebrates.
Up-regulation of ribosomal genes and protein homeostasis
A surprising observation of the present study is a concerted and progressive up-regulation of genes coding for ribosomal proteins accompanied by up-regulation of genes involved in initiation of translation. Up-regulation of ribosomal protein genes was detected also by a meta-analysis of human brain, kidney, and muscle datasets (Zahn et al., 2006). Enrichment of lysosome-related terms, such as cathepsins, is observed in the same cluster and appears as a conserved signature of aging in a meta-analysis of microarray experiments (de Magalhaes et al., 2009). Aging is linked to disrupted protein homeostasis and accumulation of misfolded proteins (David et al., 2010) that is also a unifying mechanism of progressive neurodegenerative diseases (Ross & Poirier, 2004). In parallel, accumulation of lysosomal aggregates (lipofuscin) is a well-characterized histopathological hallmark of neuronal aging (Jonker et al., 2013; Terzibasi Tozzini et al., 2013). Finally, down-regulation of proteasome is observed both in N. furzeri and human (Table1), and the network of coregulated genes at the oldest age shows enrichment for ER stress response and protein folding (Fig. S8). All these data indicate that impaired protein homeostasis is a conserved hallmark of brain aging.
Different profiles of down-regulation
Clustering down-regulated genes according to temporal profiles revealed two different kinetics of down-regulation: progressive and rapid. Both clusters are enriched for ECM terms clearly showing that brain aging in N. furzeri is associated with major remodeling of the extracellular matrix, as already shown for the skin by an initial study of RNA-seq based on pool sequencing (Petzold et al., 2013) and this is a conserved hallmark of aging across several tissues (de Magalhaes et al., 2009). However, several other GO terms and KEGG pathways are differentially represented in the two clusters. Of particular interest is the down-regulation of synaptic proteins that is also a feature of the aging primate brain (Loerch et al., 2008; Somel et al., 2010) and indicates a progressive deterioration of neuronal function/plasticity during aging that underlies the age-related learning impairments observed in Nothobranchius (Valenzano et al., 2006b).
Rapid decay of gene expression (i.e. large differences between 5 and 12 weeks) can be partly related to the continuation of somatic growth past sexual development that is a typical trait of teleost fish. However, a fraction of DEGs were clearly associated with age-dependent decrease in neurogenesis in agreement with previous histological and transcriptomic studies (Terzibasi Tozzini et al., 2012; Petzold et al., 2013). As the number of neuronal stem cells (radial glia) does not decrease during aging (Terzibasi Tozzini et al., 2012), this difference should be primarily be due to reduced activity of the stem cells that are physically preserved but become increasingly quiescent.
Genes with initial down-regulation and increase in the last point, on the other hand, were enriched for genes coding for proteins of proteasome and spliceosome, and these terms were enriched also in a dataset of human cortex aging (Loerch et al., 2008; Table1). It is highly interesting that RNA-seq studies have shown that both splicing (Wood et al., 2013; Mazin et al., 2013) and the expression of genes coding for splicing factors (Mazin et al., 2013) show a U-shape profile during mammalian brain ontogeny.
Morphogen pathways and aging
Morphogens are known to regulate the state of adult NSCs, and Notch pathway is important in regulating quiescence in teleost fish (Chapouton et al., 2010). In mammals, Notch signaling is activated in NSCs (Lugert et al., 2010), and Notch activity in the NSC niche is down-regulated during aging of the mouse (Sun et al., 2013). In N. furzeri as well, Notch pathway is down-regulated with age and is enriched in cluster 1 (rapid decay).
The sonic hedgehog (SHH) pathway, in combination with SOX2, is also known to regulate adult NSC activity in mammals (Favaro et al., 2009). SHH pathway is down-regulated with age but with a more progressive time course as opposed to Notch. This suggests that during the course of aging, there is an imbalance between the activities of these two pathways.
Circadian rhythm genes
Circadian rhythm genes are highly enriched in cluster 4 (peak at 27 weeks). All animals in the study were sampled at the same time and in fasted state, and it can be excluded that this difference is due to sampling artifacts. In adult zebrafish, genes of the circadian clock are highly expressed in the NSC niches (Weger et al., 2013), and their regulation might be a consequence of age-dependent decay of adult neurogenesis.
Chromatin remodeling and brain aging
Aging of primate brain is associated with prominent remodeling of chromatin (Horvath et al., 2012). In N. furzeri, histone core proteins of the H2A and H3 family are rapidly down-regulated with age. Chromatin-remodeling genes are also age-regulated, in particular DNMT and polycomb genes. DNMTs decrease rapidly with age and are known to regulate neurogenesis and neuronal differentiation by promoting expression of neurogenic genes via antagonization of polycomb repression (Wu et al., 2010). In human brain and peripheral tissues, polycomb-mediated methylation increases with age (Horvath et al., 2012). Polycomb members are dynamically regulated in N. furzeri. EZH1, the catalytic element of polycomb repressive complex 2 (PRC2) that catalyzes the tri-methylation of K27 on H3 (H3K27me3) (Simon & Kingston, 2013), is up-regulated with age and so is H3K27me3. As for CBX family, CBX7, whose activity is dependent on PRC2 (Morey et al., 2012), is up-regulated. On the other hand, CBX family members that are antagonistic to CBX7 (Morey et al., 2012) are down-regulated and so are components of PRC1: PRC1, PHC1, and PHC2. These data suggest that brain aging in N. furzeri is associated with imbalance in the activity of PRC1 and PRC2.
Identification of novel genes associated with neuronal stem cell niches
Adult neurogenesis is sustained by adult NSC (aNSCs). Mammalian aNSCs are restricted to defined regions, and transcriptome analysis of aNSCs in mammals requires their purification by fluorescence-assisted cell sorting (Beckervordersandforth et al., 2010). Teleost brains, on the other hand, contain widespread aNSCs and no astroglia. Activity of aNSCs decreases exponentially with age (Kempermann, 2011) and is also rapidly down-regulated in N. furzeri between 5 and 12 weeks and further decreased at 25 weeks (Terzibasi Tozzini et al., 2012). Due to the large number of aNSCs, decreased neurogenesis generates in N. furzeri a signal detectable in whole-brain transcriptome. We identified seven genes of possible regulatory function specifically associated with aNSCs in N. furzeri that are also expressed in at least one neurogenic region of the zebrafish embryo. Five of these genes are down-regulated during ontogeny of the human prefrontal cortex (Colantuoni et al., 2011). Some of the identified genes are of unknown function in the nervous system. MEX3A codes for an RNA-binding protein that, in intestinal cells, regulates MSI1 (Pereira et al., 2013), a neurogenic gene expressed in N. furzeri NSCs (Terzibasi Tozzini et al., 2012). ZNF367 and KRCP are putative transcription factors that act as central nodes in gene coregulation networks, and their function in the nervous system is unknown. SCML4 is a putative members of the polycomb repressive complex, DNMT and CBX family members are known to regulate the self-renewal capacity of embryonic stem cells in an interplay that is dependent on activity of the polycomb repressive complex (Wu et al., 2010). Our data strongly suggest that regulation of chromatin-remodeling genes is associated with aging of aNSCs in vivo and that these transcripts are regulated during human ontogeny.
Materials and methods
Materials and Methods are available in the Supplements.
Funding
No funding information provided.
Acknowledgments
This work was partially supported by the German Ministry for Education and Research (JenAge; BMBF, support codes: 0315581A and C) and by intramural grant of Scuola Normale Superiore. We thank Sabine Matz for excellent technical support.
Author′s contribution
A.C., M.P, and R.G. designed the research; M.B., M.G., A.S., G.T., R.R., and F.S. performed the research; M.B., M.G., S.P., A.S., A.D., M.O., C.G., E.T.T., A.C., R.G., and M.P. analyzed and interpreted the data; A.C., M.B., S.P., and M.P. wrote the paper.
Data deposition
Data of this experiment were deposited in gene expression omnibus (GEO), Accession No. GSE52462.
Conflict of interest
The authors declare no competing financial interests.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site.
Fig. S1 Growth curve of N. furzeri MZM-04/10.
Fig. S2 Survivorship of N. furzeri MZM-04/10.
Fig. S3 Number of DEGs in pairwise comparisons.
Fig. S4 Validation by qPCR.
Fig. S5 Sample clustering.
Fig. S6 Distribution of inversion points.
Fig. S7 Clustering of human orthologs of N. furzeri DEGs.
Fig. S8 Age-dependent networks.
Fig. S9In situ hybridization in zebra fish embryos.
Fig. S10In situ hybridization in N. furzeri with separated channels.
Fig. S11 Expression plots of selected genes from Braincloud.
Fig. S12 Overlap of DEGs between this study and de Magalhaes et al.
RPKMs of all genes and list of differentially expressed genes.
List of genes analyzed by qPCR.
Cluster membership of DEGs.
Enriched KEGG pathways.
List of down-regulated genes related to morphogen pathways.
Genes of cluster 1 with expression present in ZFIN.
Primers used to generate zebra fish ISH probes.
Primers and sequences used to generate N. furzeri ISH probes.
Intersection of DEGs with de Magalhaes et al.
Graphical representation of regulated KEGG pathways with rendering of the fold-changes.
References
- Beckervordersandforth R, Tripathi P, Ninkovic J, Bayam E, Lepier A, Stempfhuber B, Kirchhoff F, Hirrlinger J, Haslinger A, Lie DC, Beckers J, Yoder B, Irmler M, Gotz M. In vivo fate mapping and expression analysis reveals molecular hallmarks of prospectively isolated adult neural stem cells. Cell Stem Cell. 2010;7:744–758. doi: 10.1016/j.stem.2010.11.017. [DOI] [PubMed] [Google Scholar]
- Ben Abdallah NM, Slomianka L, Vyssotski AL, Lipp HP. Early age-related changes in adult hippocampal neurogenesis in C57 mice. Neurobiol. Aging. 2010;31:151–161. doi: 10.1016/j.neurobiolaging.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Blazek R, Polacik MJ, Reichard M. Rapid growth, early maturation and short generation time in African annual fishes. EvoDevo. 2013;4:24. doi: 10.1186/2041-9139-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchet-Poyau K, Courchet J, Le Hir H, Seraphin B, Scoazec JY, Duret L, Domon-Dell C, Freund JN, Billaud M. Identification and characterization of human Mex-3 proteins, a novel family of evolutionarily conserved RNA-binding proteins differentially localized to processing bodies. Nucleic Acids Res. 2007;35:1289–1300. doi: 10.1093/nar/gkm016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapouton P, Skupien P, Hesl B, Coolen M, Moore JC, Madelaine R, Kremmer E, Faus-Kessler T, Blader P, Lawson ND, Bally-Cuif L. Notch activity levels control the balance between quiescence and recruitment of adult neural stem cells. J. Neurosci. 2010;30:7961–7974. doi: 10.1523/JNEUROSCI.6170-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT, Colantuoni EA, Elkahloun AG, Herman MM, Weinberger DR, Kleinman JE. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature. 2011;478:519–523. doi: 10.1038/nature10524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David DC, Ollikainen N, Trinidad JC, Cary MP, Burlingame AL, Kenyon C. Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol. 2010;8:e1000450. doi: 10.1371/journal.pbio.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cicco E, Tozzini ET, Rossi G, Cellerino A. The short-lived annual fish Nothobranchius furzeri shows a typical teleost aging process reinforced by high incidence of age-dependent neoplasias. Exp. Gerontol. 2011;46:249–256. doi: 10.1016/j.exger.2010.10.011. [DOI] [PubMed] [Google Scholar]
- Edelmann K, Glashauser L, Sprungala S, Hesl B, Fritschle M, Ninkovic J, Godinho L, Chapouton P. Increased radial glia quiescence, decreased reactivation upon injury and unaltered neuroblast behavior underlie decreased neurogenesis in the aging zebra fish telencephalon. J. Comp. Neurol. 2013;521:3099–3115. doi: 10.1002/cne.23347. [DOI] [PubMed] [Google Scholar]
- Evert J, Lawler E, Bogan H, Perls T. Morbidity profiles of centenarians: survivors, delayers, and escapers. J. Gerontol. A Biol. Sci. Med. Sci. 2003;58:232–237. doi: 10.1093/gerona/58.3.m232. [DOI] [PubMed] [Google Scholar]
- Favaro R, Valotta M, Ferri AL, Latorre E, Mariani J, Giachino C, Lancini C, Tosetti V, Ottolenghi S, Taylor V, Nicolis SK. Hippocampal development and neural stem cell maintenance require Sox2-dependent regulation of Shh. Nat. Neurosci. 2009;12:1248–1256. doi: 10.1038/nn.2397. [DOI] [PubMed] [Google Scholar]
- Genade T, Benedetti M, Terzibasi E, Roncaglia P, Valenzano DR, Cattaneo A, Cellerino A. Annual fishes of the genus Nothobranchius as a model system for aging research. Aging Cell. 2005;4:223–233. doi: 10.1111/j.1474-9726.2005.00165.x. [DOI] [PubMed] [Google Scholar]
- Guthke R, Moller U, Hoffmann M, Thies F, Topfer S. Dynamic network reconstruction from gene expression data applied to immune response during bacterial infection. Bioinformatics. 2005;21:1626–1634. doi: 10.1093/bioinformatics/bti226. [DOI] [PubMed] [Google Scholar]
- Hartmann N, Reichwald K, Wittig I, Drose S, Schmeisser S, Luck C, Hahn C, Graf M, Gausmann U, Terzibasi E, Cellerino A, Ristow M, Brandt U, Platzer M, Englert C. Mitochondrial DNA copy number and function decrease with age in the short-lived fish Nothobranchius furzeri. Aging Cell. 2011;10:824–831. doi: 10.1111/j.1474-9726.2011.00723.x. [DOI] [PubMed] [Google Scholar]
- Horvath S, Zhang Y, Langfelder P, Kahn RS, Boks MP, van Eijk K, van den Berg LH, Ophoff RA. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012;13:R97. doi: 10.1186/gb-2012-13-10-r97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonker MJ, Melis JP, Kuiper RV, van der Hoeven TV, Wackers PF, Robinson J, van der Horst GT, Dolle ME, Vijg J, Breit TM, Hoeijmakers JH, van Steeg H. Life spanning murine gene expression profiles in relation to chronological and pathological aging in multiple organs. Aging Cell. 2013;12:901–909. doi: 10.1111/acel.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempermann G. Adult Neurogenesis 2. Oxford: Oxford University Press; 2011. [Google Scholar]
- Kirschner J, Weber D, Neuschl C, Franke A, Bottger M, Zielke L, Powalsky E, Groth M, Shagin D, Petzold A, Hartmann N, Englert C, Brockmann GA, Platzer M, Cellerino A, Reichwald K. Mapping of quantitative trait loci controlling lifespan in the short-lived fish Nothobranchius furzeri- a new vertebrate model for age research. Aging Cell. 2012;11:252–261. doi: 10.1111/j.1474-9726.2011.00780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Godwin JW, Gates PB, Garza-Garcia AA, Brockes JP. Molecular basis for the nerve dependence of limb regeneration in an adult vertebrate. Science. 2007;318:772–777. doi: 10.1126/science.1147710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CK, Weindruch R, Prolla TA. Gene-expression profile of the ageing brain in mice. Nat. Genet. 2000;25:294–297. doi: 10.1038/77046. [DOI] [PubMed] [Google Scholar]
- Liebermann DA, Tront JS, Sha X, Mukherjee K, Mohamed-Hadley A, Hoffman B. Gadd45 stress sensors in malignancy and leukemia. Crit. Rev. Oncog. 2011;16:129–140. doi: 10.1615/critrevoncog.v16.i1-2.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loerch PM, Lu T, Dakin KA, Vann JM, Isaacs A, Geula C, Wang J, Pan Y, Gabuzda DH, Li C, Prolla TA, Yankner BA. Evolution of the aging brain transcriptome and synaptic regulation. PLoS ONE. 2008;3:e3329. doi: 10.1371/journal.pone.0003329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugert S, Basak O, Knuckles P, Haussler U, Fabel K, Gotz M, Haas CA, Kempermann G, Taylor V, Giachino C. Quiescent and active hippocampal neural stem cells with distinct morphologies respond selectively to physiological and pathological stimuli and aging. Cell Stem Cell. 2010;6:445–456. doi: 10.1016/j.stem.2010.03.017. [DOI] [PubMed] [Google Scholar]
- de Magalhaes JP, Curado J, Church GM. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics. 2009;25:875–881. doi: 10.1093/bioinformatics/btp073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazin P, Xiong J, Liu X, Yan Z, Zhang X, Li M, He L, Somel M, Yuan Y, Phoebe Chen YP, Li N, Hu Y, Fu N, Ning Z, Zeng R, Yang H, Chen W, Gelfand M, Khaitovich P. Widespread splicing changes in human brain development and aging. Mol. Syst. Biol. 2013;9:633. doi: 10.1038/msb.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morey L, Pascual G, Cozzuto L, Roma G, Wutz A, Benitah SA, Di Croce L. Nonoverlapping functions of the Polycomb group Cbx family of proteins in embryonic stem cells. Cell Stem Cell. 2012;10:47–62. doi: 10.1016/j.stem.2011.12.006. [DOI] [PubMed] [Google Scholar]
- Numata S, Ye T, Hyde TM, Guitart-Navarro X, Tao R, Wininger M, Colantuoni C, Weinberger DR, Kleinman JE, Lipska BK. DNA methylation signatures in development and aging of the human prefrontal cortex. Am. J. Hum. Genet. 2012;90:260–272. doi: 10.1016/j.ajhg.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Beach D. Cyclin G is a transcriptional target of the p53 tumor suppressor protein. EMBO J. 1994;13:4816–4822. doi: 10.1002/j.1460-2075.1994.tb06807.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekcec A, Baumgartner W, Bankstahl JP, Stein VM, Potschka H. Effect of aging on neurogenesis in the canine brain. Aging Cell. 2008;7:368–374. doi: 10.1111/j.1474-9726.2008.00392.x. [DOI] [PubMed] [Google Scholar]
- Pereira B, Sousa S, Barros R, Carreto L, Oliveira P, Oliveira C, Chartier NT, Plateroti M, Rouault JP, Freund JN, Billaud M, Almeida R. CDX2 regulation by the RNA-binding protein MEX3A: impact on intestinal differentiation and stemness. Nucleic Acids Res. 2013;41:3986–3999. doi: 10.1093/nar/gkt087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petzold A, Reichwald K, Groth M, Taudien S, Hartmann N, Priebe S, Shagin D, Englert C, Platzer M. The transcript catalogue of the short-lived fish Nothobranchius furzeri provides insights into age-dependent changes of mRNA levels. BMC Genomics. 2013;14:185. doi: 10.1186/1471-2164-14-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat. Med. 2004;10(Suppl):S10–S17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- Simon JA, Kingston RE. Occupying chromatin: polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol. Cell. 2013;49:808–824. doi: 10.1016/j.molcel.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somel M, Guo S, Fu N, Yan Z, Hu HY, Xu Y, Yuan Y, Ning Z, Hu Y, Menzel C, Hu H, Lachmann M, Zeng R, Chen W, Khaitovich P. MicroRNA, mRNA, and protein expression link development and aging in human and macaque brain. Genome Res. 2010;20:1207–1218. doi: 10.1101/gr.106849.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F, Mao X, Xie L, Ding M, Shao B, Jin K. Notch1 signaling modulates neuronal progenitor activity in the subventricular zone in response to aging and focal ischemia. Aging Cell. 2013;12:978–987. doi: 10.1111/acel.12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzibasi Tozzini E, Baumgart M, Battistoni G, Cellerino A. Adult neurogenesis in the short-lived teleost Nothobranchius furzeri: localization of neurogenic niches, molecular characterization and effects of aging. Aging Cell. 2012;11:241–251. doi: 10.1111/j.1474-9726.2011.00781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzibasi Tozzini E, Dorn A, Ng'oma E, Polacik M, Blazek R, Reichwald K, Petzold A, Watters B, Reichard M, Cellerino A. Parallel evolution of senescence in annual fishes in response to extrinsic mortality. BMC Evol. Biol. 2013;13:77. doi: 10.1186/1471-2148-13-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzibasi E, Valenzano DR, Benedetti M, Roncaglia P, Cattaneo A, Domenici L, Cellerino A. Large differences in aging phenotype between strains of the short-lived annual fish Nothobranchius furzeri. PLoS ONE. 2008;3:e3866. doi: 10.1371/journal.pone.0003866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzano DR, Terzibasi E, Cattaneo A, Domenici L, Cellerino A. Temperature affects longevity and age-related locomotor and cognitive decay in the short-lived fish Nothobranchius furzeri. Aging Cell. 2006a;5:275–278. doi: 10.1111/j.1474-9726.2006.00212.x. [DOI] [PubMed] [Google Scholar]
- Valenzano DR, Terzibasi E, Genade T, Cattaneo A, Domenici L, Cellerino A. Resveratrol prolongs lifespan and retards the onset of age-related markers in a short-lived vertebrate. Curr. Biol. 2006b;16:296–300. doi: 10.1016/j.cub.2005.12.038. [DOI] [PubMed] [Google Scholar]
- Watson CT, Disanto G, Sandve GK, Breden F, Giovannoni G, Ramagopalan SV. Age-associated hyper-methylated regions in the human brain overlap with bivalent chromatin domains. PLoS ONE. 2012;7:e43840. doi: 10.1371/journal.pone.0043840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weger M, Weger BD, Diotel N, Rastegar S, Hirota T, Kay SA, Strahle U, Dickmeis T. Real-time in vivo monitoring of circadian E-box enhancer activity: a robust and sensitive zebra fish reporter line for developmental, chemical and neural biology of the circadian clock. Dev. Biol. 2013;380:259–273. doi: 10.1016/j.ydbio.2013.04.035. [DOI] [PubMed] [Google Scholar]
- Wood SH, Craig T, Li Y, Merry B, de Magalhaes JP. Whole transcriptome sequencing of the aging rat brain reveals dynamic RNA changes in the dark matter of the genome. Age (Dordr) 2013;35:763–776. doi: 10.1007/s11357-012-9410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Coskun V, Tao J, Xie W, Ge W, Yoshikawa K, Li E, Zhang Y, Sun YE. Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science. 2010;329:444–448. doi: 10.1126/science.1190485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn JM, Sonu R, Vogel H, Crane E, Mazan-Mamczarz K, Rabkin R, Davis RW, Becker KG, Owen AB, Kim SK. Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genet. 2006;2:e115. doi: 10.1371/journal.pgen.0020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Growth curve of N. furzeri MZM-04/10.
Fig. S2 Survivorship of N. furzeri MZM-04/10.
Fig. S3 Number of DEGs in pairwise comparisons.
Fig. S4 Validation by qPCR.
Fig. S5 Sample clustering.
Fig. S6 Distribution of inversion points.
Fig. S7 Clustering of human orthologs of N. furzeri DEGs.
Fig. S8 Age-dependent networks.
Fig. S9In situ hybridization in zebra fish embryos.
Fig. S10In situ hybridization in N. furzeri with separated channels.
Fig. S11 Expression plots of selected genes from Braincloud.
Fig. S12 Overlap of DEGs between this study and de Magalhaes et al.
RPKMs of all genes and list of differentially expressed genes.
List of genes analyzed by qPCR.
Cluster membership of DEGs.
Enriched KEGG pathways.
List of down-regulated genes related to morphogen pathways.
Genes of cluster 1 with expression present in ZFIN.
Primers used to generate zebra fish ISH probes.
Primers and sequences used to generate N. furzeri ISH probes.
Intersection of DEGs with de Magalhaes et al.
Graphical representation of regulated KEGG pathways with rendering of the fold-changes.