

Abstract
Zebrafish (Danio rerio) is an important model for vertebrate development, genomics, physiology, behavior, toxicology, and disease. Additionally, work on numerous Danio species is elucidating evolutionary mechanisms for morphological development. Yet, the relationships of zebrafish and its closest relatives remain unclear possibly due to incomplete lineage sorting, speciation with gene flow, and interspecies hybridization. To clarify these relationships, we first constructed phylogenomic data sets from 30,801 restriction-associated DNA (RAD)-tag loci (483,026 variable positions) with clear orthology to a single location in the sequenced zebrafish genome. We then inferred a well-supported species tree for Danio and tested for gene flow during the diversification of the genus. An approach independent of the sequenced zebrafish genome verified all inferred relationships. Although identification of the sister taxon to zebrafish has been contentious, multiple RAD-tag data sets and several analytical methods provided strong evidence for Danio aesculapii as the most closely related extant zebrafish relative studied to date. Data also displayed patterns consistent with gene flow during speciation and postspeciation introgression in the lineage leading to zebrafish. The incorporation of biogeographic data with phylogenomic analyses put these relationships in a phylogeographic context and supplied additional support for D. aesculapii as the sister species to D. rerio. The clear resolution of this study establishes a framework for investigating the evolutionary biology of Danio and the heterogeneity of genome evolution in the recent history of a model organism within an emerging model genus for genetics, development, and evolution.
Keywords: phylogenomics, restriction-site associated DNA sequencing, next generation sequencing, Cyprinidae, Danio, zebrafish
Introduction
The zebrafish, Danio rerio (Hamilton 1822), is an important model for understanding vertebrate developmental mechanisms (Kinkel and Prince 2009), genome evolution (Postlethwait et al. 2004), physiology (Lohr and Hammerschmidt 2011), behavior (Norton and Bally-Cuif 2010), toxicology (Peterson and MacRae 2012), and disease (Lieschke and Currie 2007; Santoriello and Zon 2012). Furthermore, along with mouse and human, zebrafish has the best genome assembly and gene annotation among vertebrates (Howe et al. 2013).
In addition to zebrafish, the genus Danio (sensu Fang 2003) contains several other species (hereafter referred to as danios) that differ from zebrafish in size, pigment patterns, skeletal morphologies, growth control, and behaviors (Fang 2003; Rosenthal and Ryan 2005; Froelich, Fowler, et al. 2013). Phenotypic differences in species closely related to zebrafish expedite the investigation of the molecular biology of evolution through comparative studies among related species coupled with functional tests in zebrafish. Several developmental studies using various danios showed that seemingly homologous features can develop by different cellular, molecular, and genetic mechanisms: For example, striped pigment patterns derive from different primary cell populations in different species (Quigley et al. 2004); certain molecular pathways are crucial for pattern formation in some species but not others (Quigley et al. 2005; McMenamin et al. 2014); and mutations in the same gene can reduce stripe formation in one species but increase stripe formation in another species (Mills et al. 2007). Other recent work studied the development of keratinized breeding tubercles (a synapomorphy of Danio) and used gain-of-function and loss-of-function mutants in zebrafish to recapitulate the range of morphological variation seen in these structures in other danios (Rodriguez 2013). Another study used functional tests in zebrafish embryos to determine when regulatory modules arose in the lineage leading to zebrafish (Camp et al. 2012).
A feature of genus Danio that facilitates evolutionary analysis is that zebrafish can form hybrids with its congeners and even more distantly related relatives (Parichy and Johnson 2001; Wong et al. 2011). Particularly informative studies identified genes involved in the evolution of species-specific pigment patterns by the strategy of mating zebrafish pigmentation pattern mutants to other danios to test for complementation of phenotypes (Parichy and Johnson 2001). Similar strategies using more distantly related species elucidated the mechanisms that led to the evolution of different patterns of muscle growth (Froelich, Fowler, et al. 2013; Froelich, Galt, et al. 2013). A clear understanding of the historical relationships among species used in such experiments is necessary to interpret the results in an evolutionary context (Meyer, et al. 1993; Conway, et al. 2008). Only with a well-supported phylogeny can we confidently infer ancestral states, distinguish synapomorphic traits from homoplasic traits, and determine the order of events in evolution.
Recent phylogenetic studies involving Danio addressed the placement of the genus in relation to other groups within the incredibly diverse order Cypriniformes (Mayden et al. 2007; Fang et al. 2009; Tang et al. 2010). These studies used numerous taxa to accommodate the diversity of species within Cypriniformes, but used sequences from relatively few loci. As such, these data sets were well suited to resolving relationships at the genus level and above, but relationships below the genus level, particularly between closely related species, were often unresolved. These studies agree on the placement of several groups of species within Danio (fig. 1), although not all species within those groups are represented in every study. First, the large danios—D. feegradei (yoma danio) and D. dangila (moustached danio), which is the type species of the genus—are consistently recovered basal to all other danios. Second, three species—D. choprae (glowlight danio), D. erythromicron (emerald dwarf danio), and D. margaritatus (celestial pearl danio)—are recovered as a monophyletic clade hereafter referred to as the D. choprae species group. Third, four taxa—D. albolineatus (pearl danio), D. roseus (rose danio), D. kerri (Kerr’s danio), and D. sp “hikari”—form a clade hereafter referred to as the D. albolineatus species subgroup. Fourth, a clade within Danio that excludes the large, basal danios and the D. choprae species group has high support in all three studies; we refer to this clade as the “D. rerio species group” because it includes D. rerio and all species recovered as members of its sister group in one or more of these three studies—D. aesculapii (panther danio), D. kyathit (orange-finned danio), D. nigrofasciatus (dwarf danio), and the D. albolineatus species subgroup. The D. rerio species group likely includes three other recently described species—D. jaintianensis (Sen 2007), D. quagga (Kullander et al. 2009), and D. tinwini (Kullander and Fang 2009b)—that were not included in previous molecular phylogenetic studies.
Fig. 1.

Recent Danio phylogenies disagree on the sister group to zebrafish and other relationships. (A) Topology from Mayden et al. (2007) recovered by parsimony and ML from two nuclear and four mitochondrial genes that involved 6,921 positions. (B) Topology from Fang et al. (2009) recovered by ML from Rhodopsin and Cytb sequences that involved 1,542 positions. (C) Topology from Tang et al. (2010) recovered by parsimony based on two nuclear and two mitochondrial genes that involved 4,117 positions. (D) Topology from Tang et al. (2010) recovered by ML based on the same data as in (C). To simplify comparisons across studies, all topologies are presented as phylograms and show support only for the sister group to Danio rerio. In each phylogeny, tips labeled as D. albolineatus and D. choprae species groups include two or more taxa included in these groups.
Despite the emergence of the Danio genus as a model system for understanding the molecular genetics of functional evolution, relationships within the D. rerio species group remain contested, particularly the identity of the sister group to D. rerio (fig. 1). The aforementioned recent phylogenetic studies of Danio and related taxa (Mayden et al. 2007; Fang et al. 2009; Tang et al. 2010) inferred four different species or clades to be the sister group to zebrafish, with each relationship having only limited support. As previous phylogenetic studies were unable to clearly resolve the relationships of species closely related to zebrafish, we performed a phylogenomic study of genus Danio focused on the D. rerio species group. Phylogenomic data sets, which consist of sequences from hundreds or thousands of loci from throughout the genome, can offer several advantages over phylogenetic data sets, which use sequences from only a handful of loci. Phylogenomic approaches are particularly important in clades with short internal branches (Pollard et al. 2006), clades with a history of hybridization (Cui et al. 2013), and clades with introgressed genomic regions (Martin et al. 2013) because under these conditions different regions of the genome have different histories.
The low cost of short-read sequencing makes representational sequencing that samples a large subset of the genome an attractive option for obtaining a genome-wide analysis of informative characters for phylogenomics. A particularly promising method is RAD-seq (restriction-associated DNA sequencing), in which short DNA sequences adjacent to restriction enzyme cutting sites (i.e., RAD-tags) provide phylogenetic signal through polymorphisms within the DNA tag (Baird et al. 2008). The nature of RAD-seq, however, presents two main challenges for phylogenomics. First, RAD-tag loci are shorter than some sequences used for phylogenomic inferences, such as contigs generated from transcriptome data (Cui et al. 2013) and ultraconserved elements (Faircloth et al. 2013). The relatively short length of individual RAD-tag loci makes it a challenge to correctly identify orthologous sequences and to distinguish among alleles, orthologs, and paralogs, especially ohnologs (Wolfe 2001). Moreover, because they are not restricted to coding genes (as with transcriptome-based methods) or predefined loci (as with ultraconserved elements), RAD-tags include sequences from all regions of the genome with the appropriate restriction enzyme cut site including repetitive elements, transposons, and low-complexity regions. To address this challenge, previous phylogenomic studies using RAD-seq employed clustering methods (Emerson et al. 2010; Hohenlohe et al. 2011; Eaton and Ree 2013; Jones et al. 2013; Hipp et al. 2014), BLASTx searches (Wang et al. 2013), or alignments to expressed sequence tag-based transcriptome sequences (Andrew et al. 2013) to infer orthology. Here, we introduce the use of a reference genome and annotations of repetitive elements to define orthology across species.
The second hurdle to using RAD-seq for phylogenomics is that the restriction enzyme cut site must be conserved across taxa to sequence the adjacent genomic DNA and obtain orthologous sequences. The turnover of restriction enzyme recognition sites through evolutionary time results in large amounts of “missing” data as the distance between taxa increases. The long, 8-bp recognition sequences we generally employ exacerbate this problem because, although they have the advantage that they cut only about 25,000 times per genome, they suffer the disadvantage of incurring mutations faster than enzymes with 4- or 6-bp recognition sequences. These issues have been addressed at length for population genomic studies within species (Baird et al. 2008; Emerson et al. 2010; Amores et al. 2011; Catchen et al. 2011; Hohenlohe et al. 2011), but theoretical and modeling work in addition to that performed on sequenced genomes (Rubin et al. 2012; Cariou et al. 2013) would improve the utility of RAD-seq data across taxa separated by longer timescales.
In recent years, RAD-seq has emerged as a common tool for population genomic studies within species, but exploration of its utility for phylogenomics is only beginning. RAD-seq has been used for phylogenomics in instances of recent radiations: Mosquitos diverged less than 22,000 years (Emerson et al. 2010), cichlids diverged less than 15,000 years (Keller et al. 2013; Wagner et al. 2013), pupfishes diverged less than 10,000 years (Martin and Feinstein 2014), and a clade of flowering plants (Eaton and Ree 2013). The utility of RAD-seq for answering phylogenetic questions on longer timescales was verified in silico for Drosophila (crown age 60 My) (Rubin et al. 2012; Cariou et al. 2013), but empirical studies outside of recent radiations are rare. A RAD-seq phylogenomic approach investigating relationships among Xiphophorus fishes (crown age 2.44 My) (Jones et al. 2013) recovered a topology nearly identical to the topology obtained in an independent phylogenomic study based on transcriptomic data (Cui et al. 2013). Other groups (Cruaud et al. 2014) used RAD-seq to infer relationships of ground beetles with a crown age of 17 My and American oaks with a crown age of 23–33 My (Hipp et al. 2014). Several aforementioned studies (Eaton and Ree 2013; Jones et al. 2013; Keller et al. 2013; Hipp et al. 2014) as well as a recent study in sunflowers (Andrew et al. 2013) also used RAD-seq to test for hybridization and gene flow in the diversification of their respective study species.
Here, we used RAD-tag sequences flanking cut sites for the restriction enzyme SbfI to resolve relationships in the genus Danio. We investigated 12 danios and 7 outgroup taxa and analyzed data either by aligning reads to the D. rerio reference genome or by clustering RAD-tags into de novo loci based on sequence similarity independent of the reference genome. Using concatenated data sets, which were orders of magnitude larger than previous data sets and originated from thousands of loci across the genome, both approaches recovered the same topology with D. aesculapii as the sister taxon to D. rerio using both maximum likelihood (ML) and Bayesian inference. The topology inferred using maximum parsimony (MP) differed only in the placement of the two basal danios. A third analysis using a multilocus approach and Patterson’s D-statistic revealed complicated historical relationships consistent with rapid speciation and introgression during the diversification of genus Danio, particularly in the lineage leading to zebrafish. We found that the biogeographical distribution of danio species compared with recovered historical relationships among D. rerio and its closest congeners is consistent with species distributions across geographically distinct hydrological basins. Our findings suggest that previous phylogenetic studies obtained limited support for the relationships of zebrafish and its closest relatives due to a lack of phylogenetic signal on the internal branches near the base of the D. rerio species group. Furthermore, our findings provide evidence that the discordance among previous studies could be due to different gene trees underlying the limited number of loci investigated in each study. The new, more detailed understanding of the history of Danio given by this RAD-seq study provides a better framework for understanding the molecular biology and evolution of a preeminent vertebrate “model species” and an emerging “model genus.”
Results
The Number of Total RAD-Tags Varies Widely across Species due to Expansion of Repeats and Gene Families
Using the restriction enzyme SbfI, we digested genomic DNA and prepared RAD-tag libraries from 41 individuals representing 12 species within Danio and 7 outgroup species (Baird et al. 2008). Sequencing the libraries provided 1.0–3.8 million quality-filtered reads per sample (supplementary table S1, Supplementary Material online). This sequencing depth is equivalent to 38 × to 152 × average coverage of the 58,720 RAD-tags we identified on the 25 chromosomes of the zebrafish reference genome (Zv9 version 72). We then used Stacks (Catchen et al. 2011) to create RAD-tag loci from highly similar sequences with sufficient sequencing depth. The total number of RAD-tags per individual ranged from 26,132 to 58,750 (fig. 2A). This variation was significantly associated with species (F[18,22] = 17.81, P < 0.001) and independent of sequencing depth (t[39] = 3.70, P = 0.062, R2 = 0.087), suggesting that the observed variation is not due to undersequencing, but rather to a fluctuation in the total number of RAD-tags across species.
Fig. 2.

Analysis of RAD-tag alignments to the zebrafish genome and description of constructed data sets. (A) RAD-tags across taxa. Top bars represent in silico RAD-tags from the Zv9 reference genome aligned back to the genome in the same manner as the RAD-tags from the biological samples. Species are ordered according to their relatedness to Danio rerio (middle column) as recovered by the ML phylogeny of Tang et al. (2010) such that species near the top of the graph are more closely related to D. rerio. Error bars represent the range of RAD-tags obtained for each species except for D. rerio, where error bars represent the standard error of the mean for the seven zebrafish individuals. The left bar graph (gray tones) shows the number of RAD-tags that were excluded from phylogenetic analyses due to questionable orthology across taxa, including tags that were repetitive (pale gray), were unmapped (dark gray), or that mapped to multiple locations (medium gray). The right bar graph (color) shows the number of RAD-tag loci (i.e., RAD-tags with clear orthology to a single, nonrepetitive, locus in the zebrafish genome). Use of a reference genome identifies a large number of RAD-tag loci with well-supported orthology in species closely related to the reference genome. (B) Comparison of data sets used for phylogenetic inference in this study. The three graphics represent all RAD-tag loci from (B1) chromosome 1 (chr-1) for the Min. Taxa data set, (B2) chr-1 of the Dre Group data set, and (B3) chr-1 of the All Danios data set. Each narrow row of blocks represents one of the 19 species included in this study (listed in the order shown in [A]). Each narrow column of blocks represents a RAD-tag locus (1,318 in Min. Taxa, 133 in Dre Group, and 66 in All Danios). Note that the Min. Taxa data set is broken into four segments. Taxa within Danio are surrounded by a black rectangle. Loci at splice acceptors are denoted by red blocks over the loci. The horizontal scale and the color code for each block denoting the genomic feature of the locus (B4) apply to (B) and (C). The darkness of each block denotes whether that locus was sequenced in all samples (dark), some samples (midtone), or no samples (white) for each taxon. (C) Data sets used for phylogenetic inference in previous studies. The data sets of (C1) Mayden et al. (2007), (C2) Fang et al. (2009), and (C3) Tang et al. (2010) are shown with taxa restricted to the genera included in this study. (D) Density of RAD-tag loci from the Min. Taxa, Dre Group, and All Danios data sets mapped across the Zebrafish Genome. Alternating light and dark vertical bars represent chromosomes with centromeres denoted as black dots below each column. Colored lines represent the average number of loci in a 10-Mb sliding window for Min. Taxa data set (green), Dre Group data set (blue), and All Danios data set (fuschia). See text for explanation of the anomalously low number of tags on the right arm of chr-4.
Unlike previous phylogenomic RAD-seq studies, we could use the high-quality reference genome and annotated repeat information for one of our species to provide protection against incorrect orthology assumptions. Using these resources, we performed stringent quality filtering. We removed RAD-tags if they matched annotated repeats, failed to align to the zebrafish genome with high support, or aligned to more than one location with equal support. Using these filters, we limited the RAD-tags in our phylogenomic data sets to those that aligned with high support to a single, nonrepetitive location in the zebrafish genome. We therefore refer to these sets of orthologous RAD-tags mapped to the same genomic location as “RAD-tag loci,” or simply “loci” to distinguish them from RAD-tags excluded from our data sets due to unclear orthology. To evaluate how well these filtering steps worked, we also applied them in silico to the 58,720 RAD-tags flanking SbfI cut sites on the 25 chromosomes in the zebrafish reference genome (Zv9). We include these in silico results for comparison to the actual results obtained for our seven zebrafish samples.
The percentage of total RAD-tags removed from further analyses because of similarity to zebrafish repetitive elements varied greatly across species (fig. 2A, light gray bars) from 9.0% in Danionella translucida to 55.9% in D. tinwini. Compared with species more distantly related to zebrafish, members of the D. rerio species group had on average more than twice as many RAD-tags with high similarity to annotated repetitive elements. DNA transposons, which make up 39% of the entire zebrafish genome (Howe et al. 2013), comprised 26.2% of the RAD-tags generated in silico from the zebrafish genome and 26.9% of the RAD-tags from the seven sequenced zebrafish specimens, the highest of any species in the study. DNA transposons made up 14.2–26.0% of the RAD-tags for the other species in the D. rerio species group, 5.6–8.8% in danios outside of the D. rerio species group, and 2.2–6.8% in outgroups. This trend suggests that the 39% of the zebrafish genome made up of DNA transposons is the result of a recent expansion of DNA transposons in the lineage leading to zebrafish. Satellite repeats, which constitute 0.9% of the total zebrafish reference genome (Howe et al. 2013), made up 6.3% of our in silico RAD-tag data set based on the reference sequence and 7.2% of the RAD-tags from our zebrafish samples. Other species in the D. rerio species group had similar levels of satellite repeats (4.1–13.8%) with the exception of D. tinwini, in which satellite repeats accounted for a staggering 32.4% of RAD-tags, suggesting a significant recent expansion of these elements in D. tinwini. Satellite repeats were less frequent in the more basally diverging danios (0.3–2.7% of RAD-tags) and in outgroups (0.3–0.9% of RAD-tags). This difference in DNA transposon and satellite content explains much of the variation in the number of total RAD-tags across species and suggests that the high repeat content of the zebrafish genome (52.2%, the highest reported repeat content of any sequenced vertebrate) (Howe et al. 2013) is a relatively recent occurrence. For comparison, the closest relative to zebrafish with a sequenced genome is currently the common carp, Cyprinus carpio, the genome of which is estimated to contain just 11.7–28.0% repeats (Henkel et al. 2012). We note, however, that the nature of the present RAD-seq experiment limits the repeat families that we can assay to those with SbfI sites. Further investigations into the historical origin of repeats in the zebrafish genome will require other forms of data, such as whole-genome sequences from related species.
After removing RAD-tags related to repetitive elements, we found that 0.8% of the total RAD-tags from the D. rerio samples mapped to genomic scaffolds not incorporated into the 25 chromosomes and 1.6% of RAD-tags failed to align anywhere in the zebrafish reference genome (fig. 2A, dark gray). Some of these unmappable RAD-tags may represent sequencing artifacts, but many unmapped RAD-tags appeared independently in several different zebrafish samples suggesting that they belong to elements absent from the TU-strain-based zebrafish reference genome. RAD-tags from species other than zebrafish that failed to map to the D. rerio genome (15.8–29.1% per danio sample, 49.4–76.8% in the outgroups) likely represent sequences from three sources: Regions orthologous to unassembled zebrafish genomic scaffolds, loci orthologous to locations on the 25 zebrafish chromosomes but diverged beyond recognition, and sequences with no orthologous locus in the zebrafish genome.
Across species, a small proportion of RAD-tags mapped to multiple locations on chromosomes in the zebrafish reference genome with equal support (fig. 2A, medium gray). Among RAD-tags with multiple mapping locations were 884 RAD-tags (1.7% of the total) generated in silico from chromosomes in the zebrafish reference genome. As we knew the genomic locations of these markers, we could identify the genetic elements from which they came. The long (right) arm of chromosome 4 (chr-4R), which has been noted for its elevated GC-content and highly duplicated gene families (Howe et al. 2013), contained 232 multiple-mapping RAD-tags with an average of 11.1 mapping locations in chr-4R alone. Many multiply mapped RAD-tags occurred within members of duplicated gene families in this region including a subfamily of NOD-like receptors, which have been reported as unique to teleosts (Laing et al. 2008). Other genomic elements that generated RAD-tags with multiple best mapping positions included centromeres, lncRNAs, and some tandemly duplicated genes (e.g., ints12 at Chr-1: 25414014–25419172 and 1: 25321186–25326343). In other sampled taxa, many of the 0.6–4.7% of RAD-tags that mapped to multiple locations appeared to come from similar elements, but may also represent sequences from other paralogous gene families, unannotated repeats, or constrained regions of ohnologs tracing back to genome duplications at the base of the teleost or the vertebrate radiations.
Across all 41 samples, 648,326 RAD-tags aligned to a single location on one of the 25 chromosomes in the zebrafish reference genome. These mapping locations corresponded to 89,593 loci with an RAD-tag alignment from at least one sample (table 1) with 30,801 loci having RAD-tags from four or more samples. Most loci that occurred in fewer than four samples were species-specific and therefore of little use for phylogenetic inference. We expected to recover 26,476 RAD-tag loci in our zebrafish samples based on our in silico analysis of the zebrafish reference genome. The mean number of loci identified across our D. rerio samples (23,770 loci) was lower than this genomic estimate, but substantially higher than the average of all species in the D. rerio species group (18,339 loci), the average across all danio species (17,734 loci), and the average for outgroup taxa (7,148 loci). This decrease in the number of RAD-tag loci with increasing phylogenetic distance from the reference genome mirrors the increase in unmappable RAD-tags mentioned previously. Interestingly, in contrast to the high variability in repetitive RAD-tags across species, the total number of nonrepetitive RAD-tags (mapped RAD-tag loci and unmappable RAD-tags) was relatively constant across the phylogenetic distance investigated in this study with the mean across Danio (25,301 RAD-tags) being only slightly higher than the mean across outgroup species (24,953 RAD-tags).
Table 1.
Data Set Characteristics.
| Data Set | Loci | Repeat Filtering | Total Positions | Parsimony Positions | % Loci |
Species per Locus | |||
|---|---|---|---|---|---|---|---|---|---|
| Exons | Introns | UTRs | Intergenic | ||||||
| All mapped loci | 89,593 | RepBase | 7,884,184 | 401,097 | 25.41 | 39.97 | 3.83 | 30.79 | 2.96 |
| Min. Taxa | 30,801 | RepBase | 2,710,488 | 401,097 | 26.56 | 40.42 | 3.86 | 29.17 | 6.31 |
| Dre Group | 3,406 | RepBase | 299,728 | 62,805 | 39.12 | 31.53 | 4.84 | 24.52 | 13.77 |
| All Danios | 1,720 | RepBase | 151,360 | 27,799 | 38.38 | 30.89 | 3.99 | 26.73 | 15.25 |
| pyRAD | 60,216 | None | 5,296,122 | 258,690 | NA | NA | NA | NA | 3.47 |
Note.—NA, not available.
Based on alignment locations, we inferred RAD-tags from different samples to represent orthologous loci if they mapped to the same unique location in the zebrafish genome. We constructed three data sets based on the degree of conservation across species (table 1 and fig. 2B). Compared with the data sets used by previous phylogenetic studies (fig. 2C), the resulting phylogenomic data sets were orders of magnitude larger in terms of total alignment positions and had similar or greater taxon sampling within Danio. The “minimum taxa” data set (Min. Taxa) contains all 30,801 loci with RAD-tags from four or more samples including 5,370 loci with RAD-tags exclusive to zebrafish samples. The “D. rerio species group” data set (Dre Group) consists of the 3,406 loci present in all 20 individuals sampled from seven species in the D. rerio species group. The “all danios” data set (All Danios) is restricted to the 1,720 loci that are conserved in all 28 sequenced samples of 12 species in genus Danio.
Most RAD-based phylogenomic studies have not had the luxury of a sequenced genome for any of the investigated taxa and have therefore inferred orthology based solely on sequence similarity. To complement our genome-assisted analyses and to better compare analyses across studies, we therefore also employed a genome-independent approach to infer RAD-tag orthology based only on sequence similarity. We generated the resulting data set (“pyRAD Data set” in table 1) using the pyRAD package (Eaton and Ree 2013), which was designed explicitly for phylogenomic analysis of RAD-seq data. This data set contained 60,216 loci with RAD-tags from four or more samples. By comparing results from our genome-assisted approach and a genome-independent approach, we were able to infer the benefit or detriment of using available genomic resources.
Genomic Distribution of RAD-Tag Loci
Having identified orthologous loci, we used the annotation of the D. rerio reference genome to determine the distribution and degree of conservation of RAD-tag loci in our data sets. In all three genome-assisted data sets, the left arm of chr-4 had the greatest density of RAD-tag loci per megabase in the genome, whereas the highly repetitive, heterochromatic right arm of chr-4 (chr-4R), the putative sex chromosome of D. rerio (Anderson et al. 2012), had the fewest loci per megabase (fig. 2D). As mentioned previously, chr-4R contains a number of highly duplicated gene families, which limits our ability to make well-supported orthology inferences for RAD-tags within those loci. Thus, the paucity of chr-4R loci included in our data sets is partly due to excluding loci mapping to these gene families and does not reflect perfectly the distribution of RAD-seq loci across the zebrafish genome. In addition to chr-4R, other regions of the genome, particularly regions surrounding the centromeres of several chromosomes, also have fewer loci than the genome-wide average.
To find the proportion of SbfI-based RAD-tags occurring in exons, introns, untranslated regions (UTRs), and intergenic regions, we compared the sequences of RAD-seq loci with sequences of genes annotated in Ensembl Zv9 (table 1). Results showed that RAD-tags in our data sets were more than 4-fold enriched within and around coding sequences. Annotated transcripts from Zv9 (Ensembl version 75) contained 278 RAD-tag alignments per megabase compared with the average of 66 RAD-tag alignments per megabase in nonrepetitive regions of the genome. Because SbfI recognizes a GC-rich octamer (CCTGCAGG; fig. 3A), the enrichment of loci around coding sequences is likely due in part to the high GC content of zebrafish transcripts (47.6% GC) relative to the AT-rich genome-wide average (38.6% GC). This enrichment for GC content continued outside of the SbfI sites in the RAD-tags themselves with observed GC content ranging from 47.6% to 49.3% across taxa. Conserved regions within coding sequences also affect the enrichment of SbfI sites around coding sequence. For instance, SbfI sites are enriched in the NOD-like receptor gene family, which includes 135 annotated paralogs on chr-4R and 190 annotated paralogs elsewhere in the genome. In particular, genes in subfamily C of the NOD-like receptors have repetitive exons with a highly conserved leucine-rich region that often contains an SbfI site (e.g., see fig. 3B).
Fig. 3.
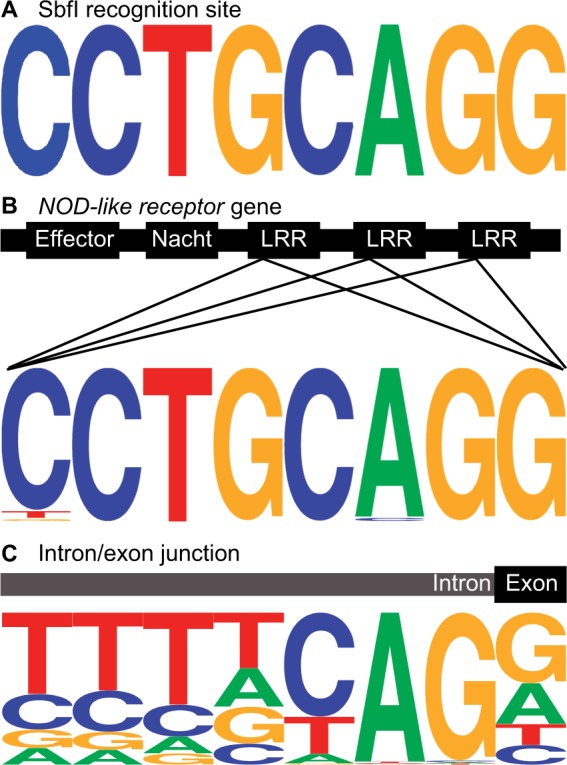
Enrichment of SbfI sites in the zebrafish genome. (A) The SbfI restriction cut site. (B) SbfI sites often occur in NOD-like receptor genes (subfamily C) due to the consensus sequence of Leucine-rich repeat (LRR) domains. (C) SbfI sites are enriched at splice acceptors due to the zebrafish consensus splice acceptor sequence. Base heights represent frequency of each base at each position.
We noticed that a striking number of RAD-tag loci were located at splice acceptor sites. In silico analysis of the zebrafish reference genome showed that SbfI sites are enriched about 1,600-fold at splice acceptor sites (738 observed vs. <0.5 expected assuming genome-wide nucleotide levels). We surmise that this is due to the similarity of the SbfI recognition site to the zebrafish splice acceptor motif (fig. 3C). Because each SbfI cut site has two associated RAD-tags, a cut site at a splice junction has one RAD-tag extending into the exon and its directly adjacent sister RAD-tag extending in the opposite direction into the intron. Realizing this situation allowed us to ask how well our approach recovered RAD-tag loci in exons compared with introns. Of the nearly 90,000 loci across all samples that had orthology to the zebrafish genome, 1,404 loci began at splice acceptors. Most of these loci (66.4% of 1,404 loci) occurred as pairs with both the exonic and intronic loci recovered in our data set. When our data set contained only the exonic or only the intronic locus, the exonic RAD-tag was obtained more often (25.9% of loci) than the intronic RAD-tag (7.7% of loci). We presume that this asymmetry arises because the higher rate of sequence conservation in exons than introns provided more frequent alignment of the exon partner of the RAD-tag pair to the zebrafish reference genome. In support of this hypothesis, the number of RAD-tags at splice acceptors with a confidently mapped intronic locus but no exonic locus generally occurred for duplicated genes. For example, of the 16 paralogs of ms4a17a in a 500-kb section of chr-4, only ms4a17a.11 had a confidently mapped intronic RAD-tag at a splice acceptor. Recovery of the intronic rather than the exonic RAD-tag likely occurred due to our exclusion of exonic reads that mapped to multiple paralogs, whereas the corresponding intronic reads retained sufficient paralog-specific sequence to align to a unique spot in the reference genome.
ML Phylogenetic Inference Based on Concatenated Data Sets
Having identified orthologous loci, we constructed ML trees from all three genome-assisted data sets and the genome-independent pyRAD data set. All four data sets supported the same topology with most nodes having full bootstrap support (fig. 4). For nodes with less support, data sets containing more RAD-tags (Min. Taxa and pyRAD) had higher support than did data sets with fewer sites. All currently recognized species and genera were recovered as monophyletic clades across data sets and bootstraps. All four data sets support the same relationships for the outgroup species used in this study, demonstrating the potential for using RAD-seq on longer timescales. Consistent with previous studies, analysis placed the Danio species with the largest body size, D. dangila and D. feegradei, basally within the genus across all ML analyses. These two species formed a clade to the exclusion of all other danios in all bootstrap replicates of the Min. Taxa and pyRAD data sets as well as in the majority of bootstrap replicates of the other two data sets. The D. choprae species group was recovered basal to the D. rerio species group with full support across all data sets. Within the D. rerio species group, the D. albolineatus species subgroup had full support and was the most basally diverging. Because all data sets fully supported the monophyly of the remaining species in the D. rerio species group to the exclusion of the D. albolineatus species group, we conclude that the D. albolineatus species group is unlikely to constitute the sister group to D. rerio in contrast to previous findings (Fang et al. 2009). RAD-tag analysis recovered the formerly unplaced D. tinwini as a close relative of D. nigrofasciatus, suggesting that reduced adult body size is a synapomorphy of these two species and evolved independently of reduced body size in D. margaritatus and D. erythromicron within the D. choprae species group.
Fig. 4.

ML phylogeny of the Danio genus based on RAD-tag sequences. Unlabeled nodes have 100% bootstrap support across all data sets. Labeled nodes give the bootstrap support for the node in (from top to bottom) the Min. Taxa data set, the Dre Group data set, the All Danios data set, and the pyRAD data set. Branch lengths are based on the ML analysis of Min. Taxa data set, the largest genome-based data set. The inset shows the support across data sets for the relationships within a subclade of the D. rerio species group. White scale bars in photos are 0.5 cm in length.
All four data sets recovered D. aesculapii as the sister species of D. rerio. This finding was surprising because, to our knowledge, this relationship had not been recovered in previous phylogenetic studies that included these species (Mayden et al. 2007; Fang et al. 2009; Tang et al. 2010). Moreover, the adult pigmentation pattern of the two species is markedly different (see fig. 4).
Within D. rerio, we sequenced two lab strains (AB and Tübingen) and two wild strains (Nadia and WIK). Other groups have investigated the relatedness and population structure of zebrafish strains (Guryev et al. 2006; Whiteley et al. 2011), but the strains used in each study overlapped relatively little across studies. The relationships we recovered do, however, confirm the previous finding that AB and Tubingen are more closely related to each other than either is to WIK (Guryev et al. 2006).
The remaining member of the D. rerio species group used in this study, D. kyathit, was originally described as having morphs with different pigment patterns—spotted or continuous stripes—with the holotype being conspicuously spotted (Fang 1998). This study included a pair of spotted D. kyathit individuals and a pair of striped individuals. We refer to the striped D. kyathit specimens as D. aff. kyathit to denote the difference in pigmentation pattern relative to the holotype while recognizing that the type series of the species included one striped individual. Branch lengths separating striped D. aff. kyathit from spotted D. kyathit were greater than branch lengths separating any other conspecific samples, including the four D. rerio strains, but they are shorter than branch lengths separating any other two species in this study. The intermediate branch lengths separating D. kyathit morphs offer no solid answer as to whether spotted and striped D. kyathit pigment morphs are different species. The relatedness and species status of the spotted and striped D. kyathit morphs and taxa from other studies—D. aff. kyathit (Quigley et al. 2004), D. kyathit “spotted” (Tang et al. 2010), and Danio sp. “Ozelot” (Fang et al. 2009; Tang et al. 2010)—remain unclear and warrant further investigation by the examination of morphologies, vigor, and fertility of hybrids.
An additional issue that makes D. kyathit problematic is its variable phylogenetic location within the D. rerio species group in two of our four data sets. Analyses of the Min. Taxa and the pyRAD data sets placed D. kyathit as sister to the (D. nigrofasciatus, D. tinwini) clade with full support. The Dre Group and All Danios data sets also recovered this topology in the majority of bootstrap replicates, but without full support. Variant minority placements of D. kyathit in the Dre Group and All Danios data sets did not corroborate previous phylogenetic studies (fig. 4, inset). In our bootstrap replicates where D. kyathit did not fall with (D. nigrofasciatus, D. tinwini), it usually fell basal to the (D. rerio, D. aesculapii) clade (86% of the replicates) and infrequently as sister to D. rerio (14% of the replicates) but never as sister to D. aesculapii. The asymmetrical placement of D. kyathit with respect to D. rerio and D. aesculapii is striking and is investigated further in later subsections.
Bayesian Inference and MP Analyses of Concatenated Data Sets
Bayesian analysis of the Dre Group and the All Danios data sets supported the same topology as ML, with equivalent support for most terminal nodes (supplementary fig. S1, Supplementary Material online). Notably, Bayesian analyses of the Dre Group data set and the All Danios data set placed D. kyathit as sister to the (D. nigrofasciatus, D. tinwini) clade with posterior probabilities of 0.95 and 0.94, respectively. This result seems to contrast to the relatively weak support for this relationship obtained from these two data sets using ML (75% bootstrap support in the Dre Group data set; 59% in the All Danios data set). Close inspection of topologies sampled from the posterior distribution in these analyses showed that the reduced support for several deeper nodes, including even the base of genus Danio, is due to the erratic behavior of one of the outgroups, Danion. translucida. The only member of its genus included in this study, this species was separated from all others by a long branch, consistent with previous studies, which inferred high mutation rates for Danionella species (Mayden et al. 2007; Ruber et al. 2007; Fang et al. 2009; Tang et al. 2010). The two Danion. translucida samples also had the highest proportion of unmappable RAD-tags and the fewest mapped loci of any taxon across all four data sets. Thus, the erratic nature of Danion. translucida in this study is likely due to its failure to clear the two hurdles that RAD-seq faces for application to phylogenomics: Sufficient sequence similarity to infer orthology across taxa and conservation of the restriction enzyme cut sites. The behavior of Danion. translucida reiterates the importance of thorough taxon sampling and the impact of long branches when using RAD-seq for phylogenomics. Bayesian analyses of the larger data sets (Min. Taxa data set and the pyRAD data set) were not completed because the chains failed to converge after 2 weeks of running on mpi nodes and were estimated by MrBayes to take several months to complete.
Topologies obtained through MP analyses of the three genome-assisted data sets were nearly identical to the topology obtained by ML across all three data sets (supplementary fig. S2, Supplementary Material online). Under ML, D. feegradei and D. dangila formed a monophyletic clade; in contrast, using MP, D. feegradei consistently fell basal to D. dangila. This minor difference between the findings of the two methods is not surprising given that the topology inferred using parsimony was recovered in some of the likelihood-based bootstrap analyses and because of the relatively long branches leading to D. feegradei and D. dangila, which can be particularly susceptible to long branch attraction, especially when using parsimony (Felsenstein 1978).
The few nodes lacking full bootstrap support using MP were the same nodes that lacked full support using ML. For instance, among the four sampled strains within D. rerio, MP and ML recovered WIK as the most basally diverging strain in most, but not all, bootstrap replicates of the Dre Group data set and the All Danios data set. The other two data sets, which each contained more than 250,000 parsimony-informative characters, unambiguously supported WIK as the most basally diverging D. rerio strain. Similarly, D. kyathit formed a clade with (D. nigrofasciatus, D. tinwini) in all four data sets, but this relationship lacked full support in the Dre group data set (80% bootstrap support with MP; 75% with ML) and the All Danios data set (67% bootstrap support with MP; 59% with ML). Consistent with our findings using ML, the variant topologies supported a (D. kyathit, (D. rerio, D. aesculapii)) clade.
Introgression Testing with pyRAD
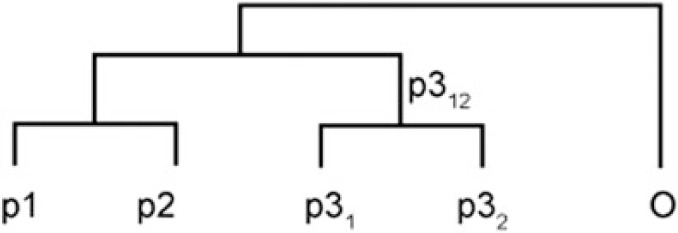
A possible explanation for variability and asymmetry in the placement of D. kyathit in our analyses of concatenated data sets is the process of introgression within the D. rerio species group. To test for past gene flow between the various taxa within the D. rerio species group, we used a partitioned Patterson’s D-statistic on the topology recovered with ML, MP, and Bayesian inference. This approach, which tests for an imbalance of character state frequencies on a hypothesized five-taxon topology with an outgroup and two pairs of sister taxa, has been used with RAD-tag sequences to measure unidirectional gene flow from outcrossing flowers into closely related incrossing species (Eaton and Ree 2013) and to test for past introgression in American oaks (Hipp et al. 2014). To avoid biasing our analysis by testing for signs of gene flow on only a subset of topologies supported by our prior analyses, we tested all five-taxon topologies consistent with the topology recovered by ML and Bayesian analysis. Because a partitioned Patterson’s D-statistic can only be determined with four ingroup taxa, we sequentially excluded two of the six members of the D. rerio species group with variable placement in ML bootstrap replicates (D. rerio, D. aesculapii, D. kyathit, D. aff. kyathit, D. nigrofasciatus, and D. tinwini) while retaining the other four taxa as the ingroup taxa. The outgroup alternated between all more basally diverging danios (table 2).
Table 2.
Partitioned Patterson’s D-Statistic Tests.
 |
Tests (Significant/Total) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| p3→p1 | p3→p2 | p31→p1 | p31→p2 | p32→p1 | p32→p2 | |||||
| Dre | Dae | Dni | Dti | Various | 0/7 | 0/7 | 0/7 | 0/7 | 0/7 | 0/7 |
| Dre | Dae | Dky | Dni | Various | 0/14 | 0/14 | 8/14 | 0/14 | 0/14 | 0/14 |
| Dre | Dae | Dky | Dti | Various | 0/14 | 0/14 | 12/14 | 0/14 | 0/14 | 0/14 |
| Dni | Dti | Dre | Dae | Various | 0/7 | 0/7 | 0/7 | 0/7 | 0/7 | 0/7 |
| Dky | Dni | Dre | Dae | Various | 14/14 | 0/7 | 0/14 | 0/7 | 0/7 | 0/7 |
| Dky | Dti | Dre | Dae | Various | 14/14 | 0/7 | 0/14 | 0/7 | 0/7 | 0/7 |
| Dre | Dae | Dky | Dky aff. | Various | 1/7 | 0/7 | 0/7 | 0/7 | 0/7 | 0/7 |
| Dky | Dky aff. | Dae | Dre | Various | 0/7 | 0/7 | 0/7 | 0/7 | 0/7 | 0/7 |
| Dky | Dky aff. | Dni | Dti | Various | 0/7 | 0/7 | 0/7 | 0/7 | 0/7 | 0/7 |
| Dni | Dti | Dky | Dky aff. | Various | 0/7 | 0/7 | 0/7 | 0/7 | 0/7 | 0/7 |
The observed partitioned Patterson’s D-statistics provide strong support for two instances of past introgression in the recent history of the D. rerio species group. Tests for introgression into D. rerio or D. aesculapii (rows 1–5 in table 2) provided evidence for introgression of alleles present in D. kyathit into D. rerio after its divergence from D. aesculapii. Tests for introgression of alleles originating in D. rerio, D. aesculapii, or their common ancestor (rows 6–10 in table 2) revealed strong evidence for introgression of alleles predating the divergence of the two species into D. kyathit. Analysis provided no evidence, however, for introgression between D. kyathit and D. rerio or D. aesculapii since the divergence of D. kyathit and D. aff. kyathit (rows 11–14 in table 2). Taken together, these tests support a model in which alleles present in a common ancestor of D. rerio and D. aesculapii were introduced into the common ancestor of D. kyathit and D. aff. kyathit after they diverged from D. nigrofasciatus and D. tinwini. Subsequently, after it diverged from D. aesculapii, the D. rerio lineage acquired alleles from an ancestor of D. kyathit and D. aff. kyathit. These instances of inferred introgression are consistent with the asymmetric results we observed previously in which D. kyathit occasionally appeared as sister to D. rerio or (D. rerio, D. aesculapii) but never as sister to D. aesculapii in the ML and MP analyses of the Dre Group data set and the All Danios data set.
Multilocus Phylogeny
Evidence for introgression from the partitioned Patterson’s D-statistics demonstrated that a single bifurcating tree is unlikely to best describe the diversification of the Danio genus. Thus, although analyses of concatenated data sets can approximate underlying phylogenies, a more detailed methodology is required to surpass methods that assume a single underlying bifurcating topology. We therefore supplemented the concatenation-based phylogenomic approaches described above with BUCKy, a method based on posterior probabilities of topologies from multiple loci (Larget et al. 2010). Given topologies sampled by Bayesian inference from the posterior distribution of topologies across all loci, BUCKy determines concordance factors (CFs) and confidence intervals (CI) for bipartitions in a topology based on the proportion of topologies that include each bipartition. We restricted our analysis to the D. rerio species group to reduce the effects of missing data while still maintaining sufficient loci for analysis and addressing the relationships of the species that interest us the most. All of our previous analyses concluded that the D. albolineatus species subgroup diverged basal to all other species in the D. rerio species group, so we therefore used D. albolineatus and the closely related D. kerri to root the tree.
The BUCKy analysis recovered topologies across loci that are largely consistent with the phylogeny obtained from analyzing the concatenated data sets. Rather than supporting a single species tree, however, results of this multilocus analysis supported a complex history for D. rerio and its closest relatives (fig. 5). Most individual RAD-tag loci had enough phylogenetic information to separate out closely related taxa as shown by the high CFs for the bipartition basal to the two members of the D. albolineatus species group (CF = 0.978, 95% CI = [0.970,0.985]) and the bipartition basal to the two D. kyathit color morphs (CF = 0.956, 95% CI = [0.943,0.967]). The bipartitions corresponding to the short internal nodes of the ML topology, however, had far less support than the long branches basal to the two D. kyathit color morphs and basal to the D. albolineatus species group, as would be expected under an instance of rapid speciation. When analyzing individual loci, D. kyathit fell as sister to the (D. rerio, D. aesculapii) clade (CF = 0.246, 95% CI = [0.225,0.268]) slightly more often than the (D. nigrofasciatus, D. tinwini) clade (CF = 0.233, 95% CI = [0.211,0.258]) consistent with introgression from the ancestor of D. rerio and D. aesculapii into D. kyathit as inferred by the partitioned Patterson’s D-statistics. Because BUCKy determines CFs for all bifurcations in the topologies sampled from the posterior distribution, we were also able to measure support for relationships that were not in concordance with the topology that was inferred using ML, MP, and Bayesian inference. Of particular note, D. kyathit was found to be sister to D. rerio (CF = 0.207, 95% CI = [0.184,0.229]) significantly more often than D. kyathit was found to be sister to D. aesculapii (CF = 0.144, 95% CI = [0.125,0.166]). The introgression of alleles from D. kyathit into D. rerio explains this asymmetry, as mentioned previously, and recapitulates results from our ML analyses in which D. kyathit grouped with D. rerio in a small percentage of bootstrap replicates (5% in Dre Group data set and 4% in All Danios data set), but never with D. aesculapii. Due in part to the short length of RAD-tag loci and in part to recent divergence times of species within the D. rerio species group, many RAD-tag loci for these closely related taxa lacked strong phylogenetic signal and often supported equally two or more topologies. It remains unclear how often these topologies are the result of synapomorphic changes that support sister group relationships and how many are due to homoplastic changes, incomplete lineage sorting, or interspecies hybridization.
Fig. 5.
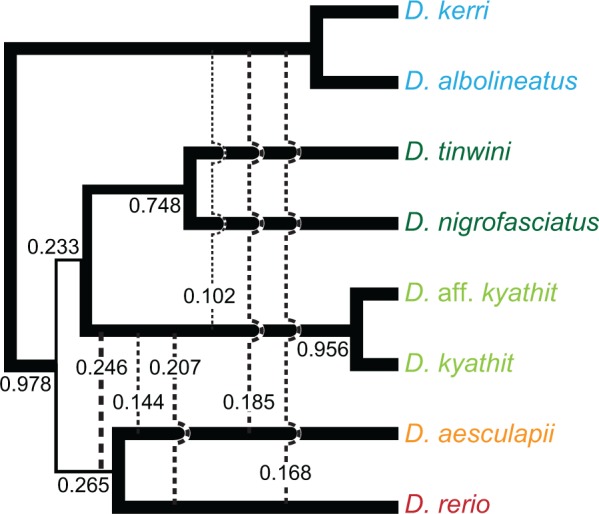
Bipartition frequencies of phylogenies inferred from individual RAD-tag loci. Values represent Bayesian CFs for bipartitions. Solid lines show terminal branches and splits in the topology recovered by analysis of the concatenated data sets. Dashed lines that connect lineages show splits with CFs larger than 0.10 but not concordant with the topology recovered by analysis of the concatenated data sets. Bipartition widths are proportional to their CFs.
Phylogeography of the Danio Genus
A more satisfying appreciation of Danio history can incorporate the group’s phylogeny into its biogeography. Danio species occur across Southeast Asia, from northern India to Malaysia, with the majority of species being endemic to one or two major hydrological basins (Fang et al. 2009 and our fig. 6). Danio rerio and D. albolineatus, however, have considerably wider ranges, distributed across a larger area than all other members of the genus combined, and yet the distributions of D. rerio and D. albolineatus do not overlap. Danio rerio occurs in several hydrological basins across the Indian plateau as well as in the Ganges/Brahmaputra basin at elevations ranging from sea level to well over 1,000 m (Engeszer et al. 2007). Danio albolineatus covers a similarly large range across several major hydrological basins from central Myanmar to southern Thailand. On the border of India and Myanmar, the Arakan Mountains separate the Ganges/Brahmaputra basin from the Irrawaddy basin and delimit the native ranges of a number of Danio species; no Danio species described east of the Arakan Mountains has been collected on the west of the Arakan Mountains and vice versa. Two apparent exceptions appear in the literature, but species designations for the relevant samples have since been amended (Fang 2000; Kullander and Fang 2009a). Hora (1937) designated several individuals collected from the west coast of Myanmar as D. choprae, but these individuals were later assigned to D. aesculapii in the first formal description of the latter species (Kullander and Fang 2009a). Similarly, specimens collected from the northwestern Irrawaddy basin were initially deemed to be D. rerio (Chen et al. 1988), but were later identified as D. albolineatus (Fang 2000).
Fig. 6.

Phylogeography of Danio species. The Arakan Mountains of Myanmar separate D. rerio, D. aesculapii, and the large, basal Danio species from all other danios in this study. Danio rerio occurs in several basins to the West of the area shown. Danio albolineatus occurs in several basins to the East of the area shown.
Discussion
The Phylogeny of Danio
RAD-tag analysis of 12 species of Danio and seven outgroups recovered well-supported species relationships and suggested an explanation for the disparate results of previous phylogenetic studies of this group. Relationships among major clades within Danio (the basal danios, the D. choprae species group, and the D. rerio species group) are consistent across previous studies and the present work. Recovered associations within the D. rerio species group, however, vary across studies and involve nodes with low support both in previous studies and in our most restrictive data set. Variable topologies and weak support for relationships within the D. rerio species group can be explained by two phenomena apparent in our RAD-tag data: 1) Different trees for different loci helps explain variable topologies across studies, and 2) short branches at the base of the D. rerio species group explain weak support for relationships within the group.
The three recent molecular phylogenetic studies of Danio (Mayden et al. 2007; Fang et al. 2009; Tang et al. 2010) varied not only in the topologies they inferred but also in the numbers and types of loci investigated. Mayden et al. (2007) used sequences from six genes but only the four mitochondrial genes, which are maternally inherited and segregate as a single locus, were sampled in more than one Danio. Fang et al. (2009) used one mitochondrial gene and one nuclear gene. Tang et al. (2010) used two mitochondrial genes and two nuclear genes. The RAD-tag data presented in this study are solely of nuclear origin. Thus, the results of Mayden et al. (2007) reflect historical relationships of Danio mitochondria and the results of Fang et al. (2009) and Tang et al. (2010) are based on both mitochondrial and nuclear relationships; in contrast the RAD-tags results presented here reflect only the history of Danio nuclear genomes. Given the evidence for introgression and multiple gene trees uncovered in our data, it is not surprising that inferred topologies within Danio using small data sets are variable across studies because the gene trees from which the species trees were inferred may well have been different for each study.
The topology we recover is consistent with the distribution of species in various hydrological basins of Southeast Asia (fig. 6). All danios sampled in this study can be found in the same major hydrological basin as their closest known relative. Even D. choprae, which appears to be an exception to this pattern, has a close relative native to the same basin—D. flagrans the most recently described danio (Kullander 2012), which has yet to become widely available and was not included in our study. The range of D. rerio encompasses most of India and Bangladesh, extending as far east as the upper Indus basin and as far west as the Brahmaputra basin south of the Himalaya. The large, basal Danio species used in this study (D. feegradei and D. dangila) also occur in the eastern part of this range as well as in adjacent hydrological basins along the western coast of Myanmar in which D. aesculapii is also found (Kullander and Fang 2009a). Interestingly, an uncharacterized specimen referred to as Danio sp. “Bangladesh” appears closely related to D. aesculapii, but was collected in Bangladesh, where D. rerio also occurs (Tang et al. 2010). Whether this individual represents a subspecies of D. aesculapii, a new species altogether, a hybrid between D. rerio and D. aescualapii, or an introduction remains unknown, but warrants further investigation. Two other Danio species—D. jaintianensis (Sen 2007) and D. meghalayensis (Sen and Dey 1985)—have been described from the eastern extent of the D. rerio range, but little is known about how they are related to other species and whether or not they can form fertile hybrids with any of their congeners.
Potential Biases in RAD-Seq Phylogenomics
Various groups have warned of potential biases that can occur when using phylogenomic data sets and RAD-seq data in particular for building phylogenies and inferring introgression. Recently, Roure et al. (2013) showed that larger concatenated data sets with substantial amounts of missing data are more susceptible to phylogenetic artifacts than are smaller more complete data sets, particularly when using an inadequate model of sequence evolution (Roure et al. 2013). To address this potential issue, we used ModelTest to identify the best model of sequence evolution for our RAD-tag data and used data sets with varying degrees of missing data (Min. Taxa, Dre Group, All Danios, and pyRAD data sets). We recovered the same topology for all of these data sets, although analyses based on the larger data sets provided more support for a few nodes (most notably at the base of the D. rerio species group). This increase in support with increasing data set size would be expected if the topology for the D. rerio species group falls in the “Anomaly Zone” (Rosenberg and Tao 2008; Rosenberg 2013), another potential bias that can plague phylogenomic analyses using concatenated data sets. The results of our analysis of individual loci, however, would not be expected if the Danio species tree we recover was, in fact, anomalous. Instead, partitioned Patterson’s D-statistic tests for gene flow between taxa suggest that the lack of resolution within the D. rerio species group by concatenation-based methods is due to instances of introgression, which violate the assumption of a single underlying tree. The study originally describing the partitioned Patterson’s D-statistic test, however, suggested caution when using D-statistics for RAD-seq data due to the limited scope of the simulation studies upon which the test was based (Eaton and Ree 2013). Rather than using RAD-seq data, a recent phylogenomic study showing ancient introgression among fish species relied on contigs generated from transcriptome data (Cui et al. 2013). Although loci used for RAD-seq analysis tend to be considerably shorter than contigs assembled from transcriptome sequences and most RAD-tag loci do not fall in coding sequence, they have higher polymorphism rates than exons in transcripts and thus provide substantial rates of phylogenetically informative characters. Moreover, because D-statistic tests are based on the frequency of character states, the length of individual loci does not matter as long as orthology is accurately assigned. Because the inferred instances of introgression were highly significant and were recovered using multiple outgroup taxa, they are unlikely to be artifacts of incorrect orthology assignment. Thus, we conclude that our results are unlikely to be affected by two known sources of bias affecting phylogenomic data sets.
Methodological Considerations for Application of RAD-Seq to Phylogenomics
RAD-seq has been successfully used to answer biogeographic and phylogenomic questions for short timescales (Emerson et al. 2010; Eaton and Ree 2013; Jones et al. 2013; Keller et al. 2013; Wagner et al. 2013; Martin and Feinstein 2014), but its utility on longer timescales has been questioned (Rubin et al. 2012; Cariou et al. 2013; Jones et al. 2013) and remains largely unexplored. Although a study has yet to explicitly estimate the root age of Danio, investigations estimating the root ages of various Cyprinid clades from relaxed molecular clock methods for the cytB gene placed the origin of genus Danio in the mid Miocene, at least 13 Ma, and the last common ancestor of all taxa included in this study at more than 31 Ma (Ruber et al. 2007). If Danio is indeed a 13-My-old genus, this study represents, to the best of our knowledge, the longest timescale to which RAD-seq has been empirically applied in vertebrates. Despite the antiquity of this divergence time, we were still able to infer orthologous RAD-tags across both ingroup and outgroup taxa through both our genome-assisted method and genome-independent, de novo method. Application of RAD-seq on this timescale empirically demonstrates its utility for answering phylogenomic questions across larger phylogenetic distances than those to which it has previously been applied.
The number of RAD-tags that lacked discernable orthologs in other species increased in outgroup samples, as expected. With RAD-seq, as with all phylogenetic methods, dense taxon sampling is likely to improve results. As seen with our Danion. translucida samples, the lack of a close relative combined with a high mutation rate can result in unusable loci due to mutations in the restriction enzyme site on the branch leading to the divergent taxon, to the creation of new restriction enzyme sites in the divergent taxon, and to the inability to determine the orthology of loci in the divergent taxon to loci in other species due to a high number of mutations within individual loci.
Our study provides several key insights for designing and analyzing future phylogenetic investigations based on RAD-seq. First, we show that our analysis using a reference genome yielded results that are highly consistent with a similar genome-independent approach. Our validation of the reference genome independent approach is an important result given that the majority of organisms lack a close relative with a sequenced genome. Second, we analyzed the genomic positions of RAD-seq loci aligned to a reference genome and showed that mapping locations are not a random sample of the genome. Rather, when using SbfI, RAD-seq loci are enriched in and around exons, particularly at splice acceptor sites, in some repeat elements, and in certain conserved protein domains (e.g., leucine-rich repeat domains). A large proportion of the data, in some samples up to half of the mapped loci, are orthologous to repetitive regions of the zebrafish genome. These loci may warrant exclusion from phylogenomic investigations due to the uncertainty associated with separating alleles from paralogs within a sample and identifying orthologs across samples. We note, however, that the pyRAD analysis, which was agnostic as to the identity of annotated repeats, performed comparable to our largest genome-assisted analysis. This finding suggests that the substantial number of repetitive loci filtered out of the genome-assisted data sets due to similarity to repetitive elements did not ultimately impact the efficacy of the genome-independent pyRAD approach. The extent to which this finding will apply to other taxa, however, warrants further investigation in future RAD-seq studies of taxa closely related to other model species. Third, in our study, concatenated data sets with fewer, more conserved loci (e.g., the All Danios data set) provided less support for certain species relationships than did concatenated data sets with more loci that were less conserved (e.g., Min. Taxa and pyRAD data sets). This increase in support with increased data is only advantageous if the concatenated data sets converge on the true species tree. When a single species tree is not the best descriptor of the group’s history, using more data may result in looking past important historical relationships such as gene flow between species unless additional analyses are performed that do not assume a single species tree.
Conclusions
We provide the first well-supported phylogeny of the cyprinid genus Danio based on more than 30,000 nuclear RAD-tag loci from 4 strains of D. rerio, 11 other danios, and 7 closely related outgroup species. Our analyses of concatenated data sets all provide strong support for D. aesculapii as the sister species to D. rerio, a relationship that has not been recovered in previous phylogenetic studies. Tests for introgression and analysis of phylogenies based on individual loci revealed striking asymmetries that are inconsistent with a single bifurcating species tree, suggesting that the diversification of Danio involved instances of rapid speciation and introgression. The two inferred instances of introgression within the D. rerio species group both involve D. rerio, demonstrating the necessity of understanding the history of the genus as a whole to understand more fully this important model organism. Given the evidence we see for multiple topologies explaining the recent evolution of D. rerio and its closest relatives, we infer that the seemingly incongruent results of previous phylogenetic studies, while likely to be correct gene trees for the loci included in each of those studies, are not accurate representations of the genome-wide species tree for Danio. To better understand the recent evolutionary history of zebrafish, future work will necessarily include: Further phylogenomic inferences involving increased taxon sampling to characterize the mosaic history of its genome; whole-genome sequencing of several other danios to identify recent structural rearrangements; hybrid studies and examination of natural isolates to explore the extent to which these species can and do interbreed; as well as cellular and developmental studies to understand the evolution of pigmentation, size, and other phenotypic differences among these species.
To varying degrees, all species in genus Danio share with zebrafish the biological characteristics that allowed D. rerio to become a preeminent model organism (e.g., small size, high fecundity, externally developing transparent embryos, and ease of laboratory culture) so many tools designed for zebrafish can also be used to study its closest relatives. Because danios can hybridize with zebrafish, the natural genetic variation in evolutionarily important traits that these species possess can be seen as an extension to the genetic resources available for zebrafish including induced null activity alleles in over 45% of its 26,000 genes, more than any other vertebrate (Kettleborough, Busch-Nentwich, et al. 2013, and http://www.sanger.ac.uk/Projects/D_rerio/zmp/, last accessed December 1, 2014). These features, along with the phylogenetic context we provide in this work, promise to make Danio the premier vertebrate “model genus.”
Materials and Methods
Animals
DNAs were collected from caudal fin clips of 41 individuals representing 12 Danio species and 7 outgroup species. Taxa from the University of Oregon Fish Facility included: D. rerio (AB), S#23336; D. rerio (Tu), S#23891; D. rerio (Nad), S#22583; WIK strain, S#23069; D. nigrofasciatus, S#23139; and D. albolineatus, S#23658. Taxa from Eugene Research Aquatics, LLC included: D. aesculapii, S#ERA.Daes.1; D. dangila, S#ERA.Ddan.1; and D. feegradei, S#ERA.Dfee.1. Taxa from the aquarium trade included: D. kyathit, WS24.Dkyp and WS25.Dkyp; D. aff. kyathit, WS22.Dkyt and WS23.Dkyt; D. tinwini, WS02.Dtin; D. kerri, WS08.Dker and WS20.Dker; D. choprae, WS12.Dcho and NT01.Dcho; D. margaritatus, WS05.Dmar and S#23036; D. erythromicron, WS14.Dery and WS15.Dery; Danion. translucida, WS120729.DLAtr; Devario aequipinnatus, NT05.DEVaeq; Devario pathirana, WS120416.DEVpat; Microdevario kubotai, WS120416.MICkub; Sundadanio axelrodi, S#Q12824.SUNaxe; Rasbora espei, WS07.RASesp; and Rasbora maculata, WS10.RASmac. The University of Oregon Animal Care and Use Committee approved all protocols associated with this work. We compared mitochondrial sequences from our animals to COI and CytB sequences published in previous phylogenetic studies (Mayden et al. 2007; Tang et al. 2010) to confirm species identification.
Genomic DNA Extraction and Sequencing
We used the restriction enzyme SbfI-HF (New England Biolabs) to digest genomic DNA and ligated barcoded Illumina sequencing adapters to the four-base overhangs left by the enzyme. We sequenced RAD-tags flanking these sites for all samples on an Illumina HiSeq 2000 with single-end, 100-bp reads. Sequences passed through several filtering steps prior to use for phylogenetic inference. To sort by sample barcode and exclude sequences without an SbfI site, we used “process_radtags.pl” from the Stacks software package (Catchen et al. 2011) with the following parameters: -b barcodes.txt -e sbfI -E phred33 –D. Sequences containing Illumina adapters were excluded from analysis. Remaining sequences were run through condetri v 2.2 (Smeds and Kunstner 2011) to exclude reads with quality less than 20 at any site. After these quality filtering steps, we obtained 1.0–3.8 million reads per sample (supplementary table S1, Supplementary Material online).
From caudal fin clips collected from euthanized or anesthetized adults of each species, we extracted genomic DNA and prepared sequencing libraries as described (Amores et al. 2011), except that adapters had six-nucleotide barcode sequences and were optimized for sequencing on the Illumina HiSeq 2000. All barcodes differed by at least two nucleotides to prevent attribution of sequence to the wrong sample due to sequencing error in the barcode. Samples were sequenced in one lane of an Illumina HiSeq 2000 using single-end 100-nucleotide reads. Three samples had low coverage (fewer than one million sequences) and were resequenced.
Locus Generation and Orthology Inference
For the Min. Taxa, Dre Group, and All Danios data sets, we used the “ustacks” program in the Stacks package (Catchen et al. 2011) to merge RAD-tag alleles into loci within individuals allowing up to two mismatches (-M 2) between alleles. We enforced a minimum stack depth of three reads (-m 3) to account for possible read misattribution from other samples and rare sequencing artifacts. Repetitive and overmerged stacks were accounted for with the parameters (-r -d). For polymorphic loci, the consensus sequence was extracted from the output of Stacks. Following locus generation with ustacks, we removed sequences from repetitive regions of the Zebrafish genome with RepeatMasker v 3.3.0 (Smit et al. 1996–2010) (http://www.repeatmasker.org/RMDownload.html, last accessed December 1, 2014) using the zebrafish repeat database (-species danio). For in silico RAD-seq analysis of the zebrafish reference genome (Zv9 version 72), we extracted sequences flanking SbfI sites and ran them through the ustacks and RepeatMasker steps. In ustacks, the minimum stack depth requirement was removed to accommodate the 1× coverage of the in silico sequences.
For the genome-independent approach, quality-filtered sequences were run through pyRAD v 1.5.1 (Eaton and Ree 2013) with a minimum sample cutoff of four samples, a clustering threshold of 0.90, and a maximum of three heterozygous samples to exclude merging potentially paralogous loci. The 60,216 loci meeting these requirements were concatenated to form the pyRAD data set.
In addition to using a completely de novo approach, we used the available and well-annotated D. rerio reference genome (Zv9 version 72) to define orthology. Quality-filtered RAD-tag loci from each sample were aligned against the zebrafish reference genome using GSNAP (Wu and Watanabe 2005). Several sets of parameters varying minimum percent identity to reference, indel penalty, and mismatch trimming were tested to accommodate the genetic distance of some species from the zebrafish reference genome (data not shown). Ultimately, parameters were chosen that maximized the number of reads across samples that mapped best to a single site in the genome with high support (mapping quality > 30); these parameters were: -m 0.5 –indel-penalty = 1 –trim-mismatch-score = 0 –trim-indel-score = 0 –max-middle-insertions = 20 –max-middle-deletions = 20 –max-end-insertions = 20 –max-end-deletions = 20. Aligned RAD-tag loci were inferred to be orthologous based on genomic location.
For each genomic locus with sequences from four or more samples, sequences were aligned against each other using Muscle v 3.8.31 (Edgar 2004) with default settings. The resulting alignments were trimmed of their SbfI restriction sites and concatenated to give the Min. Taxa, Dre Group, and All Danios data sets based on the number of samples and species possessing each locus. The Min. Taxa data set contained all 30,801 loci present in at least four samples; the Dre Group data set contained the 3,406 loci present in all members of the D. rerio species group; the All Danios data set contained 1,720 loci present in all danio samples.
Phylogenetic Inference
To determine the most appropriate model of sequence evolution for the data sets in this study, we employed ModelTest v 2.1.4 (Guindon and Gascuel 2003; Darriba et al. 2012). Based on these results, ML phylogenies were inferred under a GTR+I+Γ model in RAxML v 7.3.0 (Stamatakis 2006). MP analyses were also run with RAxML v 7.3.0. For Bayesian inference, we used MrBayes v 3.2.1 (Huelsenbeck et al. 2001; Ronquist and Huelsenbeck 2003; Ronquist et al. 2012) to analyze the Dre Group data set and All Danios data set. The Min. Taxa data set and the pyRAD data set were estimated to take several months to complete even using MPI nodes on the University of Oregon’s super computer (http://aciss-computing.uoregon.edu, last accessed December 1, 2014). We allowed 1 million generations for burn-in, then sampled every thousand generations for 10 million generations. For the multilocus analysis, we used MrBayes v 3.2.1 with the same parameters to sample posterior probability distributions for individual loci with sequence for all species in the D. rerio species group. These distributions were combined and analyzed in BUCKy v 1.4.2 (Larget et al. 2010) with α = 1 and default settings. For the analysis of Patterson’s D-statistic tests, we sampled and analyzed character states in the pyRAD data set using the partitioned D-statistic test integrated into the pyRAD v 1.5.1 package (Eaton and Ree 2013).
Analyses of Genomic Features
We downloaded genomic feature files in “bed” format from Ensembl and UCSC using the BioMart tool and UCSC Table Browser, respectively. We created our own “bed” files for genomic alignments using the “bamtobed” tool from the “bedtools” package (Quinlan and Hall 2010). To determine overlap between these various sets of genomic features, we used the “intersect” tool from the bedtools package (Quinlan and Hall 2010).
For analyses of splice acceptor sequences and Nod-like receptors, we downloaded the appropriate reference sequences from UCSC genome browser (splice acceptors) or Ensembl (Nod-like receptors). Nucleotide sequences for Nod-like receptors sequences were aligned with Muscle v 3.8.31 and the region corresponding to an SbfI site in most sequences was extracted. Base frequency graphics were generated using WebLogo (Crooks et al. 2004).
Biogeographic Analyses
We retrieved hydrological basin data from Aquastat, the Food and Agricultural Organization of the United Nations’s global water information system (fao.org/nr/water/aquastat/gis/index.stm) and danio locality information from the Global Biodiversity Information Facility (gbif.org) and imported them into ArcGIS (esri.com/software/arcgis) for visualization and comparison. Some additions and corrections were made to the locality information based on recent publications and redescriptions. Namely, two localities of D. choprae from the Western Ghats were corrected to D. aesculapii according to Kullander and Fang (2009a).
Supplementary Material
Supplementary figures S1 and S2 and table S1 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
This work was supported by the National Institute of Health (NIH) grant R01OD011116 (alias R01 RR020833) and NIH HD22486 for maintenance of the wild-type zebrafish colony. During this work, B.M.M. was supported by NIH Genetics Training Grant 5-T32-GMO7413. Computational resources and support were provided by Robert Lowe and ACISS at the University of Oregon (National Science Foundation grant OCI-0960354). The authors thank Amanda Rapp, Trevor Enright, Tim Mason, and the staff of the University of Oregon Zebrafish Facility for assistance with animals; Allison Fuiten for assistance with biogeographical data; Bill Trevarrow of Eugene Research Aquatics for providing specimens and photographs; Thomas Desvignes for guidance and assistance with photographs; the University of Oregon Genomics Core Facility for sequencing; members of the Postlethwait Lab and B.M.M.’s dissertation advisory committee for meaningful discussion; and two reviewers for useful comments.
References
- Amores A, Catchen J, Ferrara A, Fontenot Q, Postlethwait JH. Genome evolution and meiotic maps by massively parallel DNA sequencing: spotted gar, an outgroup for the teleost genome duplication. Genetics. 2011;188:799–808. doi: 10.1534/genetics.111.127324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JL, Mari AR, Braasch I, Amores A, Hohenlohe P, Batzel P, Postlethwait JH. Multiple sex-associated regions and a putative sex chromosome in zebrafish revealed by RAD mapping and population genomics. PLoS One. 2012;7:e40701. doi: 10.1371/journal.pone.0040701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew RL, Kane NC, Baute GJ, Grassa CJ, Rieseberg LH. Recent nonhybrid origin of sunflower ecotypes in a novel habitat. Mol Ecol. 2013;22:799–813. doi: 10.1111/mec.12038. [DOI] [PubMed] [Google Scholar]
- Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. 2008;3:e3376. doi: 10.1371/journal.pone.0003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp JG, Jazwa AL, Trent CM, Rawls JF. Intronic cis-regulatory modules mediate tissue-specific and microbial control of angptl4/fiaf transcription. PLoS Genet. 2012;8:e1002585. doi: 10.1371/journal.pgen.1002585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cariou M, Duret L, Charlat S. Is RAD-seq suitable for phylogenetic inference? An in silico assessment and optimization. Ecol Evol. 2013;3:846–852. doi: 10.1002/ece3.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen JM, Amores A, Hohenlohe P, Cresko W, Postlethwait JH. Stacks: building and genotyping loci de novo from short-read sequences. 2011 doi: 10.1534/g3.111.000240. G3 1:171-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YR, Cui GH, Shao JJ. Three cyprinid fishes new to Chinese fauna. Zool Res. 1988;9:439–440. [Google Scholar]
- Conway KW, Chen WJ, Mayden RL. The “Celestial Pearl danio” is a miniature Danio (s.s) (Ostariophysi : Cyprinidae): evidence from morphology and molecules. Zootaxa. 2008;1686:1–28. [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruaud A, Gautier M, Galan M, Foucaud J, Saune L, Genson G, Dubois E, Nidelet S, Deuve T, Rasplus JY. Empirical assessment of RAD sequencing for interspecific phylogeny. Mol Biol Evol. 2014;31:1272–1274. doi: 10.1093/molbev/msu063. [DOI] [PubMed] [Google Scholar]
- Cui RF, Schumer M, Kruesi K, Walter R, Andolfatto P, Rosenthal GG. Phylogenomics reveals extensive reticulate evolution in Xiphophorus fishes. Evolution. 2013;67:2166–2179. doi: 10.1111/evo.12099. [DOI] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton DAR, Ree RH. Inferring phylogeny and introgression using RADseq data: an example from flowering plants (Pedicularis: Orobanchaceae) Syst Biol. 2013;62:689–706. doi: 10.1093/sysbio/syt032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson KJ, Merz CR, Catchen JM, Hohenlohe PA, Cresko WA, Bradshaw WE, Holzapfel CM. Resolving postglacial phylogeography using high-throughput sequencing. Proc Natl Acad Sci U S A. 2010;107:16196–16200. doi: 10.1073/pnas.1006538107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engeszer RE, Patterson LB, Rao AA, Parichy DM. Zebrafish in the wild: a review of natural history and new notes from the field. Zebrafish. 2007;4:21–38. doi: 10.1089/zeb.2006.9997. [DOI] [PubMed] [Google Scholar]
- Faircloth BC, Sorenson L, Santini F, Alfaro ME. A phylogenomic perspective on the radiation of ray-finned fishes based upon targeted sequencing of ultraconserved elements (UCEs) PLoS One. 2013;8:e65923. doi: 10.1371/journal.pone.0065923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang F. Danio kyathit, a new species of cyprinid fish from Myitkyina, northern Myanmar. Ichthyol Explor Freshw. 1998;8:273–280. [Google Scholar]
- Fang F. A review of Chinese Danio species (Teleostei:Cyprinidae) Acta Zootaxon Sin. 2000;25:213–227. [Google Scholar]
- Fang F. Phylogenetic analysis of the Asian cyprinid genus Danio (Teleostei, Cyprinidae) Copeia. 2003;2003:714–728. [Google Scholar]
- Fang F, Noren M, Liao TY, Kallersjo M, Kullander SO. Molecular phylogenetic interrelationships of the south Asian cyprinid genera Danio, Devario and Microrasbora (Teleostei, Cyprinidae, Danioninae) Zool Scr. 2009;38:237–256. [Google Scholar]
- Felsenstein J. Cases in which parsimony or compatibility methods will be positively misleading. Syst Zool. 1978;27:401–410. [Google Scholar]
- Froelich JM, Fowler ZG, Galt NJ, Smith DL, Jr, Biga PR. Sarcopenia and piscines: the case for indeterminate-growing fish as unique genetic model organisms in aging and longevity research. Front Genet. 2013;4:159. doi: 10.3389/fgene.2013.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froelich JM, Galt NJ, Charging MJ, Meyer BM, Biga PR. In vitro indeterminate teleost myogenesis appears to be dependent on Pax3. In Vitro Cell Dev Biol Anim. 2013;49:371–385. doi: 10.1007/s11626-013-9616-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Guryev V, Koudijs MJ, Berezikov E, Johnson SL, Plasterk RHA, van Eeden FJM, Cuppen E. Genetic variation in the zebrafish. Genome Res. 2006;16:491–497. doi: 10.1101/gr.4791006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton F. An Account of the Fishes of the River Ganges and its Branches. Edinburgh: Archibald Constable & Co; 1822. [Google Scholar]
- Henkel CV, Dirks RP, Jansen HJ, Forlenza M, Wiegertjes GF, Howe K, van den Thillan GEEJM, Spaink HP. Comparison of the exomes of common carp (Cyprinus carpio) and zebrafish (Danio rerio) Zebrafish. 2012;9:59–67. doi: 10.1089/zeb.2012.0773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipp AL, Eaton DAR, Cavender-Bares J, Fitzek E, Nipper R, Manos PS. A framework phylogeny of the American oak clade based on sequenced RAD data. PLoS One. 2014;9:e93975. doi: 10.1371/journal.pone.0093975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenlohe PA, Amish SJ, Catchen JM, Allendorf FW, Luikart G. Next-generation RAD sequencing identifies thousands of SNPs for assessing hybridization between rainbow and westslope cutthroat trout. Mol Ecol Resour. 2011;11:117–122. doi: 10.1111/j.1755-0998.2010.02967.x. [DOI] [PubMed] [Google Scholar]
- Hora SL. Notes on fishes in the Indian Museum XXXI. On a small collection of fish from Sandoway, Lower Burma. Records of the Indian Museum. 1937;39:323–331. [Google Scholar]
- Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, Collins JE, Humphray S, McLaren K, Matthews L, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013;496:498–503. doi: 10.1038/nature12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F, Nielsen R, Bollback JP. Evolution—Bayesian inference of phylogeny and its impact on evolutionary biology. Science. 2001;294:2310–2314. doi: 10.1126/science.1065889. [DOI] [PubMed] [Google Scholar]
- Jones JC, Fan SH, Franchini P, Schartl M, Meyer A. The evolutionary history of Xiphophorus fish and their sexually selected sword: a genome-wide approach using restriction site-associated DNA sequencing. Mol Ecol. 2013;22:2986–3001. doi: 10.1111/mec.12269. [DOI] [PubMed] [Google Scholar]
- Keller I, Wagner CE, Greuter L, Mwaiko S, Selz OM, Sivasundar A, Wittwer S, Seehausen O. Population genomic signatures of divergent adaptation, gene flow and hybrid speciation in the rapid radiation of Lake Victoria cichlid fishes. Mol Ecol. 2013;22:2848–2863. doi: 10.1111/mec.12083. [DOI] [PubMed] [Google Scholar]
- Kinkel MD, Prince VE. On the diabetic menu: zebrafish as a model for pancreas development and function. Bioessays. 2009;31:139–152. doi: 10.1002/bies.200800123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullander SO. Description of Danio flagrans, and redescription of D. choprae, two closely related species from the Ayeyarwaddy River drainage in northern Myanmar (Teleostei: Cyprinidae) Ichthyol Explor Freshw. 2012;23:245–262. [Google Scholar]
- Kullander SO, Fang F. Danio aesculapii, a new species of danio from south-western Myanmar (Teleostei: Cyprinidae) Zootaxa. 2009a;2164:41–48. [Google Scholar]
- Kullander SO, Fang F. Danio tinwini, a new species of spotted danio from northern Myanmar (Teleostei: Cyprinidae) Ichthyol Explor Freshw. 2009b;20:223–228. [Google Scholar]
- Kullander SO, Liao TY, Fang F. Danio quagga, a new species of striped danio from western Myanmar (Teleostei: Cyprinidae) Ichthyol Explor Freshw. 2009;20:193–199. [Google Scholar]
- Laing KJ, Purcell MK, Winton JR, Hansen JD. A genomic view of the NOD-like receptor family in teleost fish: identification of a novel NLR subfamily in zebrafish. BMC Evol Biol. 2008;8:42. doi: 10.1186/1471-2148-8-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larget BR, Kotha SK, Dewey CN, Ane C. BUCKy: gene tree/species tree reconciliation with Bayesian concordance analysis. Bioinformatics. 2010;26:2910–2911. doi: 10.1093/bioinformatics/btq539. [DOI] [PubMed] [Google Scholar]
- Lieschke GJ, Currie PD. Animal models of human disease: zebrafish swim into view. Nat Rev Genet. 2007;8:353–367. doi: 10.1038/nrg2091. [DOI] [PubMed] [Google Scholar]
- Lohr H, Hammerschmidt M. Zebrafish in endocrine systems: recent advances and implications for human disease. Annu Rev Physiol. 2011;73:183–211. doi: 10.1146/annurev-physiol-012110-142320. [DOI] [PubMed] [Google Scholar]
- Martin CH, Feinstein LC. Novel trophic niches drive variable progress towards ecological speciation within an adaptive radiation of pupfishes. Mol Ecol. 2014;23:1846–1862. doi: 10.1111/mec.12658. [DOI] [PubMed] [Google Scholar]
- Martin SH, Dasmahapatra KK, Nadeau NJ, Salazar C, Walters JR, Simpson F, Blaxter M, Manica A, Mallet J, Jiggins CD. Genome-wide evidence for speciation with gene flow in Heliconius butterflies. Genome Res. 2013;23:1817–1828. doi: 10.1101/gr.159426.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayden RL, Tang KL, Conway KW, Freyhof J, Chamberlain S, Haskins M, Schneider L, Sudkamp M, Wood RM, Agnew M, et al. Phylogenetic relationships of Danio within the order Cypriniformes: a framework for comparative and evolutionary studies of a model species. J Exp Zool B Mol Dev Evol. 2007;308B:642–654. doi: 10.1002/jez.b.21175. [DOI] [PubMed] [Google Scholar]
- McMenamin SK, Bain EJ, McCann AE, Patterson LB, Eom DS, Waller ZP, Hamill JC, Kuhlman JA, Eisen JS, Parichy DM. Thyroid hormone-dependent adult pigment cell lineage and pattern in zebrafish. Science. 2014;345:1358–1361. doi: 10.1126/science.1256251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer A, Biermann CH, Orti G. The phylogenetic position of the zebrafish (Danio rerio), a model system in developmental biology: an invitation to the comparative method. Proc Biol Sci. 1993;252:231–236. doi: 10.1098/rspb.1993.0070. [DOI] [PubMed] [Google Scholar]
- Mills MG, Nuckels RJ, Parichy DM. Deconstructing evolution of adult phenotypes: genetic analyses of kit reveal homology and evolutionary novelty during adult pigment pattern development of Danio fishes. Development. 2007;134:1081–1090. doi: 10.1242/dev.02799. [DOI] [PubMed] [Google Scholar]
- Norton W, Bally-Cuif L. Adult zebrafish as a model organism for behavioural genetics. BMC Neurosci. 2010;11:90. doi: 10.1186/1471-2202-11-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parichy DM, Johnson SL. Zebrafish hybrids suggest genetic mechanisms for pigment pattern diversification in Danio. Dev Genes Evol. 2001;211:319–328. doi: 10.1007/s004270100155. [DOI] [PubMed] [Google Scholar]
- Peterson RT, MacRae CA. Systematic approaches to toxicology in the zebrafish. Annu Rev Pharmacol Toxicol. 2012;52:433–453. doi: 10.1146/annurev-pharmtox-010611-134751. [DOI] [PubMed] [Google Scholar]
- Pollard DA, Iyer VN, Moses AM, Eisen MB. Widespread discordance of gene trees with species tree in Drosophila: evidence for incomplete lineage sorting. PLoS Genet. 2006;2:1634–1647. doi: 10.1371/journal.pgen.0020173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postlethwait J, Amores A, Cresko W, Singer A, Yan YL. Subfunction partitioning, the teleost radiation and the annotation of the human genome. Trends Genet. 2004;20:481–490. doi: 10.1016/j.tig.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Quigley AK, Turner JM, Nuckels RJ, Manuel JL, Budi EH, MacDonald EL, Parichy DM. Pigment pattern evolution by differential deployment of neural crest and post-embryonic melanophore lineages in Danio fishes. Development. 2004;131:6053–6069. doi: 10.1242/dev.01526. [DOI] [PubMed] [Google Scholar]
- Quigley IK, Manuel JL, Roberts RA, Nuckels RJ, Herrington ER, MacDonald EL, Parichy DM. Evolutionary diversification of pigment pattern in Danio fishes: differential fms dependence and stripe loss in D. albolineatus. Development. 2005;132:89–104. doi: 10.1242/dev.01547. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A. The zebrafish as a model for the evolution and development of breeding tubercles in fishes. Boulder (CO): University of Colorado; 2013. [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg NA. Discordance of species trees with their most likely gene trees: a unifying principle. Mol Biol Evol. 2013;30:2709–2713. doi: 10.1093/molbev/mst160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg NA, Tao R. Discordance of species trees with their most likely gene trees: the case of five taxa. Syst Biol. 2008;57:131–140. doi: 10.1080/10635150801905535. [DOI] [PubMed] [Google Scholar]
- Rosenthal GG, Ryan MJ. Assortative preferences for stripes in danios. Anim Behav. 2005;70:1063–1066. [Google Scholar]
- Roure B, Baurain D, Philippe H. Impact of missing data on phylogenies inferred from empirical phylogenomic data sets. Mol Biol Evol. 2013;30:197–214. doi: 10.1093/molbev/mss208. [DOI] [PubMed] [Google Scholar]
- Ruber L, Kottelat M, Tan HH, Ng PKL, Britz R. Evolution of miniaturization and the phylogenetic position of Paedocypris, comprising the world's smallest vertebrate. BMC Evol Biol. 2007;7:38. doi: 10.1186/1471-2148-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin BER, Ree RH, Moreau CS. Inferring phylogenies from RAD sequence data. PLoS One. 2012;7:e33394. doi: 10.1371/journal.pone.0033394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoriello C, Zon LI. Hooked! Modeling human disease in zebrafish. J Clin Invest. 2012;122:2337–2343. doi: 10.1172/JCI60434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N. Description of a new species of Brachydanio Weber and de Beaufort, 1916 (Pisces : Cypriniformes : Cyprinidae) from Meghala, North East India with a note on comparative studies of other known species. Rec Zool Surv India. 2007;107:27–31. [Google Scholar]
- Sen N, Dey SC. Two new fish species of the genus Danio Hamilton (Pisces: Cyprinidae) from Meghalaya, India. Journal Assam Scientific Society. 1985;27:60–68. [Google Scholar]
- Smeds L, Kunstner A. CONDETRI—a content dependent read trimmer for Illumina data. PLoS One. 2011;6:e26314. doi: 10.1371/journal.pone.0026314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. 2010. “RepeatMasker Open-3.0. 1996–2010”.
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Tang KL, Agnew MK, Hirt MV, Sado T, Schneider LM, Freyhof J, Sulaiman Z, Swartz E, Vidthayanon C, Miya M, et al. Systematics of the subfamily Danioninae (Teleostei: Cypriniformes: Cyprinidae) Mol Phylogenet Evol. 2010;57:189–214. doi: 10.1016/j.ympev.2010.05.021. [DOI] [PubMed] [Google Scholar]
- Wagner CE, Keller I, Wittwer S, Selz OM, Mwaiko S, Greuter L, Sivasundar A, Seehausen O. Genome-wide RAD sequence data provide unprecedented resolution of species boundaries and relationships in the Lake Victoria cichlid adaptive radiation. Mol Ecol. 2013;22:787–798. doi: 10.1111/mec.12023. [DOI] [PubMed] [Google Scholar]
- Wang XQ, Zhao L, Eaton DAR, Li DZ, Guo ZH. Identification of SNP markers for inferring phylogeny in temperate bamboos (Poaceae: Bambusoideae) using RAD sequencing. Mol Ecol Resour. 2013;13:938–945. doi: 10.1111/1755-0998.12136. [DOI] [PubMed] [Google Scholar]
- Whiteley AR, Bhat A, Martins EP, Mayden RL, Arunachalam M, Uusi-Heikkila S, Ahmed ATA, Shrestha J, Clark M, Stemple D, et al. Population genomics of wild and laboratory zebrafish (Danio rerio) Mol Ecol. 2011;20:4259–4276. doi: 10.1111/j.1365-294X.2011.05272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe KH. Yesterday’s polyploids and the mystery of diploidization. Nat Rev Genet. 2001;2:333–341. doi: 10.1038/35072009. [DOI] [PubMed] [Google Scholar]
- Wong TT, Saito T, Crodian J, Collodi P. Zebrafish germline chimeras produced by transplantation of ovarian germ cells into sterile host larvae. Biol Reprod. 2011;84:1190–1197. doi: 10.1095/biolreprod.110.088427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.