

ABSTRACT
The process of osteoclastic bone resorption is complex and regulated at multiple levels. The role of osteoclast (OCL) fusion and motility in bone resorption are unclear, with the movement of OCL on bone largely unexplored. RANKL (also known as TNFSF11) is a potent stimulator of murine osteoclastogenesis, and activin A (ActA) enhances that stimulation in whole bone marrow. ActA treatment does not induce osteoclastogenesis in stroma-free murine bone marrow macrophage cultures (BMM), but rather inhibits RANKL-induced osteoclastogenesis. We hypothesized that ActA and RANKL differentially regulate osteoclastogenesis by modulating OCL precursors and mature OCL migration. Time-lapse video microscopy measured ActA and RANKL effects on BMM and OCL motility and function. ActA completely inhibited RANKL-stimulated OCL motility, differentiation and bone resorption, through a mechanism mediated by ActA-dependent changes in SMAD2, AKT1 and inhibitor of nuclear factor κB (IκB) signaling. The potent and dominant inhibitory effect of ActA was associated with decreased OCL lifespan because ActA significantly increased activated caspase-3 in mature OCL and OCL precursors. Collectively, these data demonstrate a dual action for ActA on murine OCLs.
KEY WORDS: Osteoclast, Motility, Activin A, INHBA
INTRODUCTION
Activins, first identified as proteins originating in the gonads and capable of stimulating the secretion of follicle-stimulating hormone from pituitary cell culture (Vale et al., 2004), have been established as key regulators of many fundamental biological processes, including development, homeostasis, inflammation, and tissue remodeling. Activin A (ActA), a homodimer composed of βAβA subunits (also known as INHBA), exerts stimulatory effects on several hematopoietic cell lineages, including erythroid (Yu et al., 1987), megakaryocyte (Fujimoto et al., 1991; Okafuji et al., 1995) and granulocyte-macrophage cells (Broxmeyer et al., 1988; Moore et al., 1991) as well as on cells of the monocyte and macrophage lineage (Fuller et al., 2000; Gaddy-Kurten et al., 2002; Perrien et al., 2006; Yamada et al., 1992) that are responsible for the differentiation of bone-resorbing osteoclasts (OCLs). Indeed, we and others have demonstrated that Activin βA subunit mRNA (Yu et al., 1994) and protein (Gaddy-Kurten et al., 2002) is locally produced in bone marrow, and, like TGFβ (Bonewald and Mundy, 1990) and bone morphogenetic proteins (BMPs) (Wozney, 1992), ActA is abundantly localized in the bone matrix where it has the capacity to be released during the process of bone resorption (Ogawa et al., 1992).
In addition to storage in the bone matrix, ActA is an endogenous growth factor produced during ex vivo bone marrow cell osteoblast development, where it has the ability to enhance the formation of both bone forming osteoblasts and bone resorbing OCLs (Fuller et al., 2000; Gaddy-Kurten et al., 2002; Nicks et al., 2009; Silbermann et al., 2014). This appears to be the case in humans, and several investigators have suggested an important role for ActA in the fundamental process of bone remodeling (Pearsall et al., 2008; Teitelbaum and Ross, 2003), which is responsible for the maintenance of skeletal integrity (Suva et al., 2011).
OCLs are giant multinucleated bone-resorbing cells derived from mononuclear monocyte and macrophage precursors that require two essential cytokines for survival and differentiation; receptor activator of nuclear factor κB (NF-κB) ligand (RANKL, also known as TNFSF11) and macrophage colony-stimulating factor (MCSF) (Teitelbaum, 1993; Teitelbaum and Ross, 2003). Both cytokines are produced by resident bone cells of stromal origin such as osteoblasts and osteocytes (Nakashima et al., 2011; Xiong et al., 2011). In addition to RANKL and MCSF, cell movement is essential for OCL precursor cell fusion, OCL formation and eventual bone resorption (Faccio et al., 2003). In fact, multi-nucleation is considered to be crucial for the unique ability of OCLs to resorb bone matrix; yet the mechanism(s) of OCL motility and fusion are poorly defined (Novack and Teitelbaum, 2008; Sun et al., 2007; Teitelbaum and Ross, 2003).
However, whether ActA effects on OCL differentiation and bone resorbing activity occur through direct or indirect actions on OCL precursors have not yet been fully elucidated. OCL differentiation has been shown to be regulated by ActA treatment, although these actions are reported as being either stimulatory or inhibitory, depending on the source of osteoclast precursors (Gaddy-Kurten et al., 2002; Sugatani et al., 2003). The ambiguity became even more perplexing with the somewhat surprising observation that a soluble activin receptor type IIA fusion protein (ACE-011), which effectively abolishes all ActA signaling, increases bone mass through both anabolic and antiresorptive effects in vivo. This finding suggested an even more crucial role for ActA signaling in bone resorption and bone remodeling (Lotinun et al., 2010).
Given that the mechanism(s) by which ActA functions during RANKL-mediated OCL formation are unclear, we hypothesized that ActA impacts OCL development at multiple stages. In this study, we determined the effects of ActA on OCL fusion, formation, motility and lifespan, using isolated and expanded murine bone marrow macrophages (mBMMs) as OCL precursors. These studies demonstrated that ActA significantly suppressed the fusion, differentiation and motility of these cells through activation of SMAD2, AKT1 and inhibitor of NF-κB (IκB) signaling. In addition, treatment of mature RANKL-differentiated OCLs with ActA significantly decreased osteoclast multi-nucleation and lifespan. The decreased lifespan was the result of an ActA-mediated stimulation of apoptosis that contributed to the significantly decreased bone resorption observed. These findings implicate ActA as a potent local regulator of OCL development and activity that is synthesized and stored in the bone microenvironment and is dominant over the action of RANKL, thereby providing a level of fine control to balance rates of bone resorption and remodeling.
RESULTS
ActA exerts distinct effects on osteoclast differentiation in whole bone marrow cultures and enriched osteoclast progenitor cultures
Our previous findings have demonstrated that ActA enhances 1,25(OH)2VitaminD3-stimulated OCL differentiation in whole murine bone marrow cultures (Gaddy-Kurten et al., 2002). To determine whether the pro-osteoclastogenic ActA effect was independent of stromal cells, the effects of ActA on whole murine bone marrow cultures were compared with ActA effects on an enriched population of murine stroma-free bone-marrow-derived macrophages (mBMMs) from the same mice. In ex vivo bone marrow cultures the effects of ActA alone, and in combination with RANKL, on tartrate-resistant acid-phosphatase-positive (TRAP+) multinucleated cell formation were determined (Fig. 1A). Similar to what we and others have previously reported, exogenous ActA, alone and in combination with RANKL (50 ng/ml), stimulated osteoclastogenesis in whole bone marrow cultures (Fuller et al., 2000; Gaddy-Kurten et al., 2002; Sugatani et al., 2003). However, when stroma-free mBMMs selected and expanded from the same population were treated with ActA, alone and in combination with RANKL, a significant and robust decrease in osteoclastogenesis was observed after 5 days in culture (Fig. 1B). The suppressive effect of ActA was not due to decreased proliferation, as there were no significant differences in proliferation of cells treated with RANKL with or without ActA on days 1–4 (Fig. 1C). The striking differences in ActA effects in whole bone marrow and stroma-free mBMM cultures suggested that the stimulatory effects of ActA in whole bone marrow cultures (Gaddy-Kurten et al., 2002) and in other stroma-containing BMM cultures (Fuller et al., 2000; Sugatani et al., 2003) are indirect and likely mediated by other, non-macrophage cells. Therefore, mBMMs were utilized for the subsequent experiments to determine the effects of ActA on isolated osteoclast precursor cells and differentiation down the osteoclast lineage.
Fig. 1.

ActA exerts opposing effects on osteoclast differentiation in whole bone marrow cultures and enriched osteoclast progenitor cultures. (A) Murine bone marrow cells were cultured towards osteoclasts with MCSF alone (20 ng/ml) or in the presence of RANKL (50 ng/ml), ActA (50 ng/ml), or RANKL plus ActA. Cultures were harvested on day 10 and stained for TRAP. The graph shows the mean±s.d. number of TRAP+ MNCs per well from at least three triplicate wells per treatment and two independent experiments. Different letters indicate significantly different groups (P<0.05). Representative 40× photomicrographs indicate red TRAP-stained MNCs in each given treatment. (B) Stroma-free murine bone marrow macrophages (BMMs) were isolated. Cells were cultured toward osteoclasts at 2×104 cells/well as above for 5 days. The graph shows the mean±s.d. number of TRAP+ MNCs per well from at least three triplicate wells per treatment and two independent experiments. Different letters indicate significantly different groups (P<0.05). Representative 20× photomicrographs indicate red TRAP-stained MNCs in each given treatment. (C) mBMMs were expanded in culture for 3 days prior to plating in the presence of treatments as above for 1–4 days. Cell proliferation was determined by an MTT assay. Each data point represents normalized mean value ± s.d. in triplicate wells from duplicate experiments. Proliferation was normalized to the daily basal control level (MCSF alone). Different letters indicate significantly different groups (P<0.05).
Activin A decreases the motility of mBMM precursors
ActA and other TGFβ family members have been shown to modulate the motility and migration of various cell types including mast cell progenitors and dendritic cells (Pilkington et al., 2001; Salogni et al., 2009). Indeed, osteoclast precursor motility is required for cell–cell interaction, leading to subsequent differentiation and fusion (Fujita et al., 2012; Oursler, 2010). We therefore investigated whether ActA altered pre-osteoclast motility using time-lapse video microscopy of OCL precursors plated on both plastic and bovine bone. The 4-hour movements of ten individual motile pre-OCLs plated on plastic were tracked and analyzed throughout the course of OCL differentiation. The tracks of ten individual cells in one field on day 2 are shown in Fig. 2A, and the ten cell tracks superimposed on a Cartesian plane with the origin normalized to zero in Fig. 2B. From the quantitative analysis of the tracks from four independent experiments, it is evident that RANKL significantly stimulated the cumulative motility of pre-OCLs. This robust effect appeared to peak on days 2 and 3 of culture (Fig. 3A). Interestingly, ActA treatment significantly decreased the motility of pre-osteoclasts throughout differentiation compared to cells on plastic stimulated with MCSF or both MCSF and RANKL (Fig. 3A).
Fig. 2.
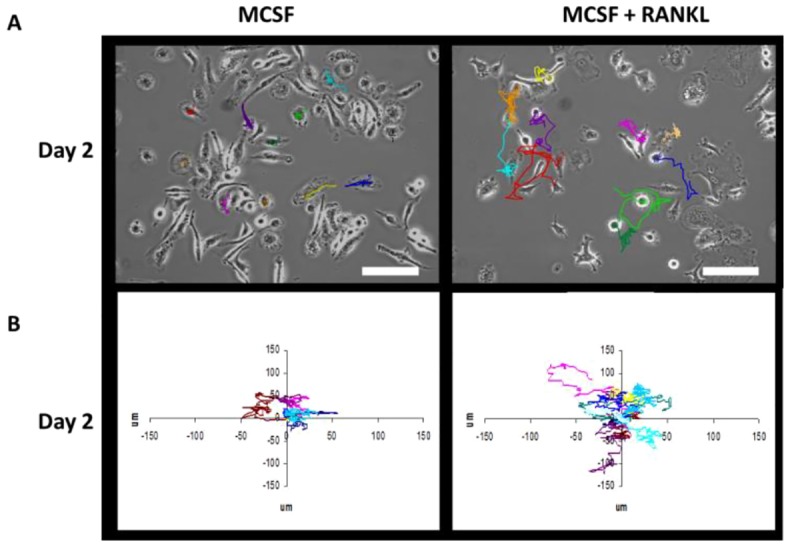
Motility tracking from 10 individual cells on day 2 of mBMM culture. mBMMs were treated with MCSF alone (left panels) or MCSF plus RANKL (right panels) and imaged by time-lapse video microscopy. (A) Representative phase-contrast images at time 0 (10×objective) are shown. The colored lines depict the movements of individual d2 pre-osteoclasts from the point of origin to the end of the imaging time. Tracking of individual cells showed increased cell migration with RANKL treatment. (B) Individual motility tracks (Cartesian plots; µm) from ten d2 pre-osteoclasts normalized to the point of origin at 0.0. Increased movement stimulated by RANKL treatment is apparent. Scale bars: 100 µm.
Fig. 3.
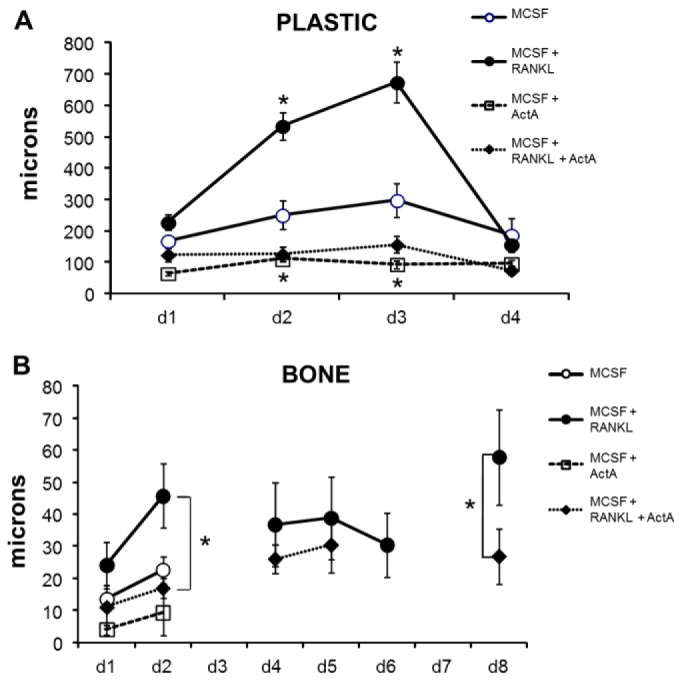
ActA decreases mBMM precursor motility. mBMMs were plated on plastic (A) or bone (B) in the presence of 20 ng/ml MCSF alone or 50 ng/ml ActA, 50 ng/ml RANKL or RANKL plus ActA. (A) 1.5×105 BMMs were plated onto 35-mm culture dishes. Cells were imaged at 37°C and at 5% CO2 in air perfusion under phase-contrast using a 10× objective, and images were collected at 60 images/hour on days 1–4. At each time point, the mean cumulative track length (microns) was measured for 10 individual cells per treatment, over 5 independent experiments, 50 cells total. Error bars are s.d. (B) 1.5×105 GFP–actin-transduced mBMM cells were plated onto 35-mm culture dishes containing bovine cortical bone slices and imaged over 8 days. Cells were imaged as in panel A but on days 1–2 (mononucleated OCL precursors), days 4–6 (mono-, bi-, and multinucleated cells) and day 8 (multinucleated mature OCL). Prior to cell treatment, maintenance medium was removed and replaced with medium containing 0.5% serum for 3 hours before time-lapse video microscopy. At each time point, the average cumulative track length was measured (µm) for ten individual cells per treatment, over two independent experiments (20 cells total). Error bars are s.d. *P<0.05 compared with MCSF alone.
To confirm these observations on the physiologic substrate, mBMMs were transduced with GFP–actin and seeded onto bovine cortical bone slices. As others have shown, transduced cells plated on bone were delayed in their differentiation, requiring ∼8 days to become mature TRAP+ multinucleated cells (MNCs) (Zhou et al., 2006). As expected, RANKL treatment significantly increased motility throughout the time course of OCL maturation (Fig. 3B). In addition, the inhibitory effect of ActA over the RANKL stimulation of motility was also observed (Fig. 3B). These data demonstrate a dominant effect of ActA over RANKL stimulation of pre-OCL motility.
Activin A suppresses mature osteoclasts
Previous reports have demonstrated effects of ActA on the differentiation of OCLs from a variety of precursor populations (Fuller et al., 2000; Murase et al., 2001; Sugatani et al., 2003); however, the effect of ActA on mature OCLs has not been reported. To determine whether ActA exerted effects on mature OCLs, we expanded and differentiated mBMMs to multinucleated TRAP+ cell maturity with both MCSF and RANKL (20 ng/ml + 50 ng/ml) for 5 days, prior to treatment with MCSF and ActA (50 ng/ml) alone or in the presence of RANKL (50 ng/ml) for an additional 24–48 hours (Fig. 4A). After treatment, cells were fixed and TRAP stained, and the number of TRAP+ MNCs with 3–5 nuclei, 6–10 nuclei or >10 nuclei per cell were counted. After 24 hours, ActA, alone or in combination with RANKL, significantly decreased the total number of TRAP+ MNCs and the number of large (10+ nuclei) OCLs (Fig. 4B,D). Although the total number of OCLs decreased significantly after 48 hours of treatment with RANKL, the ActA-mediated decrease was more rapid than with RANKL alone (Fig. 4C,D). These data demonstrate that ActA treatment of mature OCLs significantly decreased OCL number as well as the number of nuclei per OCL, and that the ActA action is dominant over RANKL.
Fig. 4.

ActA suppresses mature osteoclasts. (A) Schematic of expansion, differentiation and maturation of murine OCLs. Murine bone marrow cells were expanded for 3 days in the presence of 100 ng/ml MCSF, followed by trypsinization and plating in the presence of 20 ng/ml MCSF plus 50 ng/ml RANKL for another 5 days. On day 8, medium containing MCSF plus RANKL was aspirated and the cells treated with either MCSF alone or in combination with RANKL and 50 ng/ml ActA for 24 hours (B) or 48 hours (C). After 24 hours (B) or 48 hours (C) treatment, medium was aspirated and cells were stained for TRAP. TRAP+ MNCs and the number of nuclei per cell were counted (3–5 nuclei, white bars, 6–10 nuclei, gray bars; greater than 10 nuclei, black bars). Bars represent the mean±s.d. number of cells from triplicate wells in multiple experiments. (D) TRAP+ MNCs from day 8 MCSF plus RANKL cultures were harvested (t = 0), and treated with MCSF alone, or in the presence of ActA, RANKL or both for 24 or 48 hours. Cells were TRAP-stained, and the total number of TRAP+ MNCs containing at least three nuclei was counted in triplicate wells. *P<0.05 compared with MCSF alone. Analysis is representative of experiments performed in triplicate.
Activin A in combination with RANKL induces cleavage of caspase-3 in mature OCLs and precursors
Given that mature OCLs were not maintained by ActA treatment (Fig. 4), and in combination with RANKL, ActA decreased the total number of OCLs (Fig. 4D), we next examined whether ActA stimulated the apoptosis of mature OCLs. Previous reports have suggested that TGFβ and ActA are capable of inducing apoptosis in cells of the hematopoietic lineage (Valderrama-Carvajal et al., 2002), so the effect of ActA treatment on the apoptosis of mature OCL was determined. During the initiation of apoptosis, caspase-3 must be cleaved (Hayashi et al., 2011). Therefore, an antibody specific for cleaved caspase-3 was used for the immunofluorescence analysis of mature OCLs treated with ActA, RANKL or both for 24 hours and the extent of OCL apoptosis determined (Fig. 5). Specific caspase-3 immunofluorescence (green stain in Fig. 5A) demonstrated that 24 hours of RANKL plus ActA treatment caused a significant increase in the number of mature multinucleated OCLs that were positive for cleaved caspase-3 (Fig. 5B) compared to treatment with RANKL alone. Phalloidin staining of the same cells showed that there was substantial actin ring assembly in the RANKL-treated cells (red stain in Fig. 5A). However, when mature OCLs were treated with ActA (50 ng/ml) for 24 hours, the cells appeared disorganized and had few, if any, assembled actin rings (Fig. 5A). In addition, mononuclear cells in the cultures were also positive for cleaved caspase-3 (Fig. 5C). Interestingly, ActA significantly increased the number of mononuclear cells positive for cleaved caspase-3 (Fig. 5A, green), which was further increased in the presence of combined treatment with ActA plus RANKL (Fig. 5C). Taken together, these data demonstrate that ActA significantly enhances apoptosis of both mature multinucleated OCL, and their mononuclear OCL precursors, as measured by cleaved caspase-3. These data suggest that ActA treatment diminishes the mononuclear precursor population prior to fusion, providing an additional mechanism whereby ActA can further suppress the formation of mature bone-resorbing OCLs.
Fig. 5.

ActA effects mononuclear and multinucleated cell apoptosis. (A) mBMMs were grown in the presence of MCSF and RANKL for 5 days before a 24-hour treatment with MCSF alone (20 ng/ml) or RANKL (50 ng/ml), ActA (50 ng/ml), or RANKL plus ActA to assess apoptosis. Left panels, DAPI (blue) and cleaved caspase-3 (green). Right panels show the overlay of DAPI, cleaved caspase-3, and Texas-Red-conjugated Phalloidin (red). Closed arrowheads, multinucleated cells positive for cleaved caspase-3; open arrowheads, F-actin phalloidin-stained ring; closed arrows, mononucleated cells positive for cleaved caspase-3. Scale bars: 100 µm. (B) Total number of multinucleated cells positive for cleaved caspase-3 after 24 hours. (C) Total number of mononucleated apoptotic cells positive for cleaved caspase-3 cells after 24 hours. Each bar represents the mean±s.d. from triplicate wells from two independent experiments. Different letters indicate significantly different groups (P<0.05).
Activin A inhibits OCL resorption pit size and number, as well as Cathepsin K (CatK) secretion
To determine whether ActA treatment decreased mature OCL-mediated bone resorption, functional measurements of OCL activity were employed (Cremasco et al., 2012). First, mature OCLs were treated with combinations of RANKL and ActA in the presence of bovine bone powder. After 24, 48, and 72 hours, conditioned medium was collected and total cell lysate isolated from each treatment. Protein content was quantified and immunoblotted to determine the amount of cathepsin K (CatK) expression. CatK is an osteoclast-specific enzyme absolutely required for the enzymatic digestion of bone matrix (Rieman et al., 2001). Secreted CatK (CM CatK) and the total cell lysate CatK (TCL CatK) were measured (Fig. 6A,B). The secretion and accumulation of CatK in conditioned medium was significantly increased after 48 and 72 hours of RANKL treatment (Fig. 6A,B). ActA in combination with RANKL significantly suppressed the RANKL-stimulated accumulation of CatK in conditioned medium, suggesting that ActA suppressed the ability of mature OCLs to actively secrete CatK in the presence of bone matrix. To determine whether ActA suppressed bone resorption per se, mBMMs were cultured and differentiated into OCL on dentine discs for 5 days in MCSF plus RANKL, and then treated for 24 hours with MCSF plus RANKL in the presence or absence of ActA before fixation and TRAP staining and quantification of the TRAP+ MNCs. ActA significantly suppressed TRAP+ MNC formation within 24 hours (Fig. 7A), as it did when cells were grown on plastic (Fig. 1B). Dentine slices were cleaned of cells and resorption pit area determined. Indeed, ActA treatment significantly decreased the RANKL-stimulated resorption pit area (Fig. 7B) and the number of RANKL-stimulated resorption trails (Fig. 7C). Taken together, these data suggest that the inhibitory effect of ActA is dominant over the pro-resorptive activity of RANKL. In addition, ActA significantly impairs the ability of mature OCL to secrete the enzymes required for matrix degradation, leading to decreased resorptive capacity.
Fig. 6.

ActA decreases CatK release from osteoclasts. Mature murine osteoclasts (day 10) were treated with MCSF alone (20 ng/ml) or in combination with RANKL (50 ng/ml) and ActA (50 ng/ml) in the presence of bovine bone powder. Conditioned medium (CM) and total cell lysates (TCL) were collected at 24, 48 and 72 hours post-treatment initiation. (A) Western blot of 50 µg of total protein from conditioned medium and TCL identifying CatK and β-tubulin, which was used to normalize and account for differences in lane loading. Western blots were visualized using the LI-COR Odyssey Quantitative western blotting system. (B) Ratio of conditioned medium CatK to total CatK, first normalized to the TCL β-tubulin levels to account for differences in lane loading. Values are mean±s.d. calculated from two independent experiments. Different letter designations indicate significantly different groups within each time point (P<0.05).
Fig. 7.

ActA inhibits osteoclastogenesis and bone resorption. Mature murine osteoclasts (day 10) were cultured on dentine slices in the presence of MCSF alone (20 ng/ml), or in combination with RANKL (50 ng/ml) and ActA (50 ng/ml) for an additional 24 hours. (A) TRAP+ MNCs per treatment on bone slices. Each bar represents the mean±s.d. from triplicate wells from two independent experiments. Different letters indicate significantly different groups (P<0.05). (B) Osteoclast resorption pit area as the percentage resorption area of the total area. Different letters indicate significantly different groups (P<0.05). (C) Representative 20× images of dentine slices on which resorption measurements were taken in panel B for each treatment. Asterisks indicate resorption pits with limited resorption trails. Example resorption trails are outlined in black, with diamonds located within the individual trail. Scale bars: 100 µm.
ActA stimulates SMAD2 phosphorylation, AKT1 activation, and increases IκBα levels in mature osteoclasts
To determine the potential mechanisms through which ActA suppressed RANKL-stimulated osteoclastogenesis, whole-cell lysates were prepared from 2 day OCL precursor cultures or from 5 day mature OCL cultures. BMMs were expanded as described in the Materials and Methods prior to culture in MCSF plus RANKL for 2 days or 5 days. Cells were serum starved prior to a short-term treatment of 5–30 minutes (Fig. 8A,B) with MCSF, MCSF plus RANKL, MCSF plus ActA or all three reagents combined. ActA signals through both SMAD-dependent and SMAD-independent pathways, including the activation of AKT1 (Do et al., 2008). Therefore, immunoblot analysis was performed to determine the effects of ActA on SMAD2 phosphorylation and the activation of AKT1 through phosphorylation of Ser473. As expected, both day 2 OCL precursors and day 5 mature OCL demonstrated ActA-dependent SMAD2 phosphorylation, in the presence or absence of RANKL (Fig. 8A). However, although OCL precursors did not respond to ActA or RANKL treatment with respect to altered AKT1 activation, 50 ng/ml ActA treatment of mature OCL (day 5) increased P-AKT1 at Ser473 (Fig. 8B). Increased phosphorylated AKT1 (p-AKT1) was observed following ActA treatment alone or in the presence of RANKL within 5 minutes, and was maintained with ActA plus RANKL for up to 30 minutes of treatment. Thus, although the suppressive effects of ActA on OCL precursors are independent of AKT1 activation, phosphorylation of AKT1 in mature OCL is likely involved in the suppressive effects of ActA on other end points of OCL action.
Fig. 8.

ActA stimulates SMAD2 phosphorylation, AKT1 activation and increases IκBα levels in mature osteoclasts. (A,B) Stroma-free BMM OCL precursors cultured in the presence of MCSF and RANKL for 2 or 5 days. Cells were serum starved for 6 hours prior to stimulation with MCSF alone (20 ng/ml), or in combination with RANKL (50 ng/ml) and ActA (50 ng/ml) for the time points shown. Whole-cell lysates (50 µg) were analyzed by SDS-PAGE and immunoblotted for P-Smad2 (A) or 60 kDa phosphorylated AKT1 (P-AKT) at Ser473 and compared with total AKT (B). (C). Stroma-free BMMs were cultured on bone powder for 5 days in the presence of MCSF plus RANKL and serum starved for 6 hours (t = 0). Cells were stimulated for 3 hours or 24 hours with MCSF (M), MCSF plus RANKL (M+RL), MCSF plus ActA (M+A), or MCSF, RANKL and ActA (M+R+A) together. Total cell lysates (50 µg) were analyzed by SDS-PAGE and immunoblotted for total IkBα. IkBα was quantified using the LI-COR Odyssey system prior to stripping and reprobing the blot with antibody against β-tubulin. Quantification of β-tubulin was used to normalize for lane loading and to determine changes in total IkBα protein content in each treatment.
Finally, to determine the involvement of the NF-κB–IκBα pathway, a more sustained time-course experiment was performed. The activation of NF-κB is well documented for RANKL during OCL differentiation, and ActA has been shown to enhance its activation in more heterogeneous cultures of OCL precursors (Sugatani et al., 2003). Although ActA did not alter RANKL-stimulated NF-κB phosphorylation during the shorter term mature OCL time course in our more stroma-free BMM cultures (data not shown), ActA alone or in the presence of RANKL increased total IκBα protein levels at 3 hours and at 24 hours compared with RANKL treatment alone (Fig. 8C). These data demonstrate that ActA can suppress RANKL-stimulated NF-κB-dependent actions through its stimulation of negative feedback of the NFκB pathway. Increased levels of the NFκB inhibitor IκBα drives reformation of the NFκB–IκBα complex and diminished activity, thereby providing a mechanism by which RANKL-dependent actions on mature OCL motility, resorption and survival might be inhibited by ActA.
DISCUSSION
The current study investigated the effect of ActA on OCL differentiation and motility, and provides the first evidence that ActA regulates the activity and lifespan of both OCL precursors and mature OCLs. Unlike whole marrow cultures, in which ActA is a potent pro-osteoclastogenic agent (Fuller et al., 2000; Gaddy-Kurten et al., 2002; Silbermann et al., 2014), in stroma-free mBMM ActA was not pro-osteoclastogenic. Instead, when ActA and RANKL were added in combination, there was a significant and dominant ActA inhibitory effect over RANKL on day 5.
The motility of bone marrow macrophages is necessary for cell recruitment to bone-remodeling units (BMUs) and is mediated by various chemotactic responses that remain largely unknown (reviewed in Novack and Faccio, 2011). Bone matrix proteins, Ca2+ gradients and chemotaxis have been suggested as being responsible for the movement of OCL precursors out of blood vessels and toward the BMUs. TGFβ family members, including BMPs and ActA are stored in the bone matrix and their release is coupled to OCL movement and bone resorption (Pilkington et al., 2001; Sakai et al., 2000a). Indeed, Fuller and colleagues have demonstrated that TGFβ enhances the osteoclastogenic activity of RANKL, raising interest in the role of other family members in this process (Fuller et al., 2000). Thus, the idea was tested that ActA (stored in bone matrix or released from cells in the bone microenvironment) (Glowacki and Lian, 1987; Gonzalo et al., 2010; Ishii et al., 2010; Malone et al., 1982) regulates the osteoclast forming potential of precursor cells as they are recruited and migrate towards future sites of bone resorption, as demonstrated for TGFβ (Pilkington et al., 2001; Sakai et al., 2000b).
Our findings suggest that the effects of ActA on the OCL might be to regulate cell motility and limit lifespan, thereby providing an additional autocrine or paracrine level of control to limit the extent of OCL differentiation and/or bone resorption. To test this idea, the effect of ActA, alone and in combination with RANKL, on both pre-OCL and OCL motility was evaluated. Stroma-free mBMM were plated either on plastic or bovine bone and induced to differentiate with MCSF and RANKL. Although baseline MCSF-induced cell motility and migration did not change throughout the time-course, the maximal stimulatory effect of RANKL was observed on days 2–3, and decreased thereafter prior to cell fusion and terminal differentiation. These data indicate that the motile responses of OCL precursors are more sensitive to RANKL than mature OCLs, suggesting that increased progenitor motility precedes the induction of cell fusion. Strikingly, ActA, alone and in the presence of effective doses of RANKL, was capable of suppressing both pre-OCL and mature OCL motility, indicative of a previously unknown and dominant-negative effect of ActA over RANKL-stimulated OCL motility.
To confirm these observations on a relevant biological substrate, mBMMs were virally transduced with GFP–actin and seeded on bovine cortical bone slices. As others have shown (Izawa et al., 2012), bone adherent and transduced mBMMs differentiated into mature OCLs after 8 days of RANKL treatment. Although cells seeded on bone slices had significantly diminished motility compared to cells tracked on plastic, the pattern of stimulation with RANKL and the significant dominant suppression by ActA was sustained. We confirmed that transducing the OCL with GFP–actin did not impair OCL motility, as the measured responses were similar to treatment of GFP–actin-transduced cells plated on plastic (data not shown). This suggests that the cells respond similarly to ActA and RANKL independent of substrate, with respect to the regulation of motility.
Studies are ongoing to better elucidate the molecular targets of ActA-induced motility suppression, as well as other ActA end points. Our data suggest that although the effects of ActA might be mediated in part through SMAD2 phosphorylation on day 2 and day 5, activation of AKT1 through phosphorylation at Ser473 contributes to the ActA-dependent suppression of mature OCLs (day 5), but not day 2 precursors. Interestingly, the effect of ActA on later stage OCLs is to increase the levels of the NF-κB inhibitor IκB, which is not observed in cells treated with RANKL and ActA for 2 days. These results lead to the conclusion that ActA is a potent local regulator of OCL development and bone resorption, whose action is mediated, at least in part, by changes in SMAD2, AKT1 and IκB signaling.
The finding that ActA suppressed the motility of OCL precursors suggested that ActA might also suppress cell fusion during the multi-nucleation phase of murine osteoclastogenesis. Therefore, mBMMs were treated with MCSF and RANKL for 5 days, and the mature OCL treated with ActA (with and without RANKL) for an additional 24–48 hours. ActA treatment significantly decreased total OCL number as well as the number of nuclei per OCL, either alone or in combination with RANKL. These data demonstrate that ActA is not an OCL survival factor, but when administered in combination with RANKL, ActA significantly inhibits RANKL-induced OCL maturation, survival and fusion. Given that ActA has been shown to regulate the lifespan of other hematopoietic cell types (Lee and Kim, 2009; Valderrama-Carvajal et al., 2002), the specific effects of ActA on the survival of mature OCL and their precursors was also examined.
Predominantly, two pathways have been shown to contribute to apoptosis in OCLs: (1) activation of membrane death receptors, and (2) a mitochondrial-initiated pathway (Houde et al., 2009; McManus et al., 2012). The initiation of apoptosis involves initiator caspases (e.g. caspase-3) that can be activated through cleavage by either extrinsic extracellular stimulation, or intrinsic mitochondrial cytochrome c release (Cory and Adams, 2002; Jin and El-Deiry, 2005; Saelens et al., 2004). In this study immunofluorescence of cleaved caspase-3 suggested that RANKL or MCSF have little impact on mature OCL survival on day 6. Day 5 treatment with ActA for 24 hours significantly inhibited basal MNC caspase-3 cleavage; however, the combined treatment with ActA plus RANKL caused a significant increase in caspase-3 cleavage, demonstrating activation of apoptosis in mature OCLs.
When the number of cleaved caspase-3 positive mononuclear cells was determined in the same 6-day cultures, MCSF and RANKL had little effect on apoptosis, given that the number of mononuclear OCL precursors was low. However, ActA had pronounced and significant pro-apoptotic effects on OCL precursors, suggesting cell-type specific ActA actions even in mBMM populations. ActA treatment increased the number of mononuclear cells that were positive for cleaved caspase-3 by twofold over MCSF alone, which was the opposite effect of ActA on mature OCLs. However, combined treatment with MCSF, RANKL and ActA for 24 hours induced a striking and significant tenfold increase in the number of mononuclear cells that were positive for caspase-3. Thus, ActA alone diminishes the OCL precursor population, as well as serving to limit continued RANKL-induced differentiation of OCLs and the maintenance of mature multinucleated OCLs. Interestingly, TGFβ and ActA have been shown to induce apoptosis in other cells of hematopoietic origin by regulating the expression of the inositol phosphate SHIP [Src homology 2 (SH2) domain-containing 5′ inositol phosphatase] (Valderrama-Carvajal et al., 2002). It is interesting to speculate that this pathway of phospholipid metabolism might also be linked to the ActA-induced apoptosis observed here.
The time course of OCL fusion, differentiation and apoptosis is extremely interesting and worthy of continued study. Indeed, during the course of the studies described herein, imaging of living OCLs using video microscopy revealed several characteristics of the life and death of these cells (supplementary material Movie 1). OCLs were imaged after 5 days of RANKL plus MCSF differentiation and at 10-minute intervals for 24 hours using a 10× objective. Several notable dynamic cellular structures were apparent in both multinucleated and mononucleated cells. The MNCs show distinct morphologies, with nuclei at either the cell periphery or condensed with centralized nuclei. Within 3 hours, ongoing fusion events were observed between mononuclear and multinuclear cells. In addition, some multinucleated cells begin to shrink at 6 hours and appeared apoptotic between 9 and 12 hours with condensing nuclei and loss of cellular contents. The dramatic changes in the membrane of the cells throughout the imaging time course visually support a potentially critical role of phospholipid metabolism that we are currently pursuing.
Finally, the effect of ActA on the ability of the mature OCLs to perform their primary function, namely bone resorption, was investigated. Consistent with the dominant inhibitory effects of ActA on RANKL-induced OCL motility, formation and apoptosis, ActA significantly inhibited CatK secretion. Furthermore, ActA significantly decreased OCL-mediated bone resorption as measured by pit formation. Taken together, these data suggest that following ActA treatment, OCLs become largely disorganized, reorganize their substantial membrane and undergo apoptosis, culminating in functional impairment and decreased bone resorption. In addition, the data demonstrate the dominant-negative activity of ActA over all reported RANKL-mediated pro-OCL activity.
More than a decade ago, we suggested that ActA could stimulate osteoclastogenesis in ex vivo murine bone marrow cultures (Gaddy-Kurten et al., 2002). Several reports, using a variety of culture models, have confirmed that ActA can not only stimulate and enhance, (Murase et al., 2001; Sugatani et al., 2003) but might be an essential co-factor for osteoclastogenesis (Fuller et al., 2000). In the current study, although these observations in bone marrow cultures were replicated, a very different scenario was observed in isolated stroma-free mBMM cultures. In these cells, ActA potently inhibited RANKL-induced OCL formation, motility, activity and lifespan. In total, these data suggest that in mBMM ActA is a potent negative regulator of OCL development, lifespan, fusion and bone resorption. These findings are in contrast to work of Sugatani et al. (Sugatani et al., 2003) and Murase et al. (Murase et al., 2001), who demonstrated that ActA enhanced RANKL-induced osteoclastogenesis in heterogeneous cultures of mouse bone marrow cells (Murase et al., 2001). There are multiple potential explanations for this apparent discrepancy, but the two most likely possibilities are either differences in the expansion and/or enrichment purity of pre-osteoclast populations, or differences in the concentrations of reagents used or both. In our case, the isolated BMM cell population is stroma-free and absolutely required exogenous RANKL for OCL formation, as indicated by the complete lack of TRAP+ MNCs in the absence of RANKL. Thus, our starting population of mBMMs is comprised almost entirely of RANK-expressing monocyte OCL precursors, with little or no contributions from any contaminating stromal cells. As demonstrated here, and as reported by others (Sugatani et al., 2003), culture expansion parameters appear to be extremely important (Arai et al., 2012).
Perhaps most importantly, our data provide a compelling argument for continued studies into the mechanism of action of ActA on OCLs, as well as suggesting that the pool and phenotype of the available osteoclast precursors in murine bone marrow might be even larger than first thought (Jacquin et al., 2006; Lorenzo, 2003). With the clinical development of activin receptor antagonists ongoing (Lotinun et al., 2012), these results directly question current thinking regarding ActA action in the skeleton, and provide overwhelming evidence that ActA suppresses a variety of fundamental OCL functions, suggesting a promising avenue for the treatment of pathological bone loss. Given that the machinery of OCL fusion and motility is inhibited by anti-catabolic drugs, a more complete understanding of this complex process might produce even more effective treatment options.
MATERIALS AND METHODS
Animals
All animal handling and experimentation was performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and approved under institutional guidelines. 6–10-week-old Swiss Webster mice were purchased from Charles River (Wilmington, MA) and housed (five per cage) in temperature (22°C) and humidity (50%) controlled rooms having a 12 h light and12 h dark cycle with food and water ad libitum.
Materials
Recombinant human ActA, mouse MCSF and mouse RANKL were purchased from R&D Systems (Minneapolis, MN). CMG 14-12 culture medium (equivalent to 100 ng/ml MCSF), the lentiviral construct for expression of recombinant MCSF (Takeshita et al., 2000) and a GFP–actin lentiviral construct (Zou et al., 2010) were generous gifts from Steven Teitelbaum (Washington University, St. Louis, MO). Fetal bovine serum was purchased from Hy-Clone Laboratories, Inc. (Logan, UT), and culture medium (alpha-MEM) and additives were purchased from Gibco Life Technologies (Carlsbad, CA). Antibodies were against CatK (Abcam, Cambridge, MA # ab19027), cleaved caspase-3, P-SMAD2, P-AKT1, total AKT1, total IκBα (Cell Signaling Technology, Danvers, MA, catalog # 9664, 3101, 9271, 9272, 4812, respectively) and β-tubulin (Thermo Scientific, Rockford, IL, clone #TBN06). Secondary antibodies and Odyssey blocking buffer were purchased from LI-COR Biosciences (Lincoln, NE). Texas-Red Phalloidin and Prolong Gold Antifade with DAPI were purchased from Life Technologies (Carlsbad, CA). All non-tissue culture and tissue culture plates and chamber slides were purchased from Thermo Fisher Scientific (Pittsburgh PA).
Murine bone marrow macrophages
Bone marrow and marrow-derived macrophages were derived as previously described (Gaddy-Kurten et al., 2002; Ye et al., 2011). Briefly, the marrow cavities from femurs and tibias were flushed from mice with α-MEM containing 10% fetal bovine serum, antibiotics, and antimycotics (α-10 medium). Marrow cells from all mice were combined and homogenized gently with a 20 gauge needle fitted to a 10 ml syringe, then passed through a 70 µm cell strainer. Cells were then centrifuged and treated with red blood cell lysis buffer (eBioscience, San Diego, CA) for 5 minutes at room temperature, washed and centrifuged. Cells were re-suspended in fresh α-10 medium containing a 1/10th volume of CMG 14-12 culture medium (equivalent to 100 ng/ml MCSF) (Takeshita et al., 2000) and plated in 10 cm Petri-dishes at 1×106 cells/dish. Cells were incubated at 37°C in 5% CO2, 95% air for 3 days. Thereafter, the dishes were washed with PBS, and the adherent enriched population of mBMM cells were trypsinized and plated in BD Falcon tissue culture dishes to generate pre-OCLs and OCLs with 1/50th volume of CMG 14-12 culture supernatant and 50 ng/ml of recombinant mouse RANKL as previously described (Ye et al., 2011).
MTT proliferation assay
mBMMs were expanded in culture for 3 days prior to plating in 96-well culture dishes and cultured in either basal medium containing a 1/50th volume of CMG 14-12 culture supernatant to provide 20 ng/ml MCSF alone (Control), or also in the presence of RANKL (50 ng/ml), ActA (50 ng/ml) or RANKL plus ActA, and cells were cultured for 1–4 days. Cell proliferation was assessed using a Promega CellTiter-96 Non-Radioactive MTT-based Cell proliferation assay (Thermo Fisher Scientific) according to the manufacturer's instructions. The colored Formazen product was detected at a wavelength of 570 nm in a 96-well plate spectrophotometer. For each day, the average absorbance from at least triplicate wells from duplicate experiments was normalized to the daily basal control absorbance value. Each data point represents the mean normalized value ± s.d.
Time-lapse video imaging of pre-osteoclasts and osteoclasts on plastic
Live cell time-lapse video microscopy was performed as described previously (Ye et al., 2011). Briefly, 1.5×105 mBMMs were plated onto 35-mm culture dishes with CMG 14-12 culture medium (20 ng/ml MCSF) plus RANKL (100 ng/ml). The cells were imaged under phase contrast with a 10× objective lens with an Axiovert 100 M microscope fitted with a Zeiss Axiocam ICM1 camera, and images collected using Axiovision 4.8 imaging software at 60 images/hour with culture dishes stable at 37°C and 5% CO2 in air perfusion in a Live Cell Pathology incubator (Westminster, MD). To determine cell trajectories in phase-contrast images, the images were analyzed by NIH ImageJ software (Version 1.46f) with the MTrackJ Plugin. The center of each mononuclear cell nucleus was used as the point of tracking. For each treatment, the average cumulative track length was measured for ten individual cells per treatment, over five independent experiments (50 individual cells total). Overall distance traveled was measured by setting the starting point for each cell at 0,0 and measuring the distance in microns to the final position at the end of the video-microscopy period. The motility rate over the time of tracking was determined by measuring the distance traveled by each cell per minute over the entire video-microscopy period.
Viral transduction and time-lapse video imaging of pre-osteoclasts and osteoclasts on bone slices
Expanded populations of adherent mBMMs were transduced on day 3 in 10-cm Petri dishes with GFP–actin as previously described (Zhou et al., 2006). Plat-E packaging cells were transiently transfected by calcium phosphate precipitation. Virus was collected 24 hours post-transfection and used to transduce mBMM for 24 hours in the presence of 100 ng/ml MCSF and 4 µg/ml polybrene (Sigma). Cells were selected for blastocidin resistance for 3 days and collected by trypsinization, concentrated in basal medium, and seeded onto sterile 1-mm thick bovine cortical slices (prepared as described below) in a 50 µl droplet containing 20,000 cells, and allowed to attach at 37°C for 45 minutes.
Bovine cortical bone slices were prepared using a diamond-coated blade macro band saw from Exakt Technologies (Oklahoma City, OK) to generate 1-mm thick cross-sectional slices. Each slice was trimmed to an area of 4×4 mm, vigorously rinsed in PBS, followed by 100% ethanol prior to drying under UV light, and subsequently cultured with cells in 35-mm culture dishes. After cell attachment to the prepared bone slices, medium containing 1/50th volume of CMG 14-12 culture supernatant (20 ng/ml MCSF) and 50 ng/ml RANKL was added and cells were maintained at 37°C in a 5% CO2, 95% air atmosphere for 8 days to generate multinucleated OCLs. Bovine cortical bone slices with GFP–actin transduced cells in 35-mm dishes were imaged over 8 days. Prior to treatment initiation, maintenance medium was aspirated and medium containing 0.5% serum was added 3 hours prior to treatment and initiation of time-lapse video microscopy. Culture dishes were maintained at 37°C in 5% CO2, 95% air, in a Live Cell Pathology incubator. Images were taken every minute for 2 hours. For each treatment, the average cumulative track length was measured using ImageJ for 10 individual cells per treatment, over two independent experiments (20 cells total). The overall distance traveled and motility rate were determined as described for cells on plastic.
Immunocytochemistry
Immunocytochemistry was used to determine the expression of cleaved caspase-3 in mBMM cells cultured on 8-well chamber slides for 5 days. After treatment, cells were washed with PBS, fixed with fresh 4% paraformaldehyde for 15 minutes at 4°C then rinsed three times with PBS. Cells were then permeabilized with PBS with 0.02% Triton X-100 for 15 minutes prior to a 30-minute primary antibody rabbit serum block (1% in PBS), followed by addition of a rabbit anti-mouse cleaved caspase-3 (Asp175) primary antibody used at 1∶400 in PBS. Cells were incubated with primary antibody in a humidified container at 4°C overnight, followed by washing three times in PBS, incubation with a secondary antibody goat serum block (1%), and incubation for 30 minutes with a fluorescein goat anti-rabbit-IgG antibody used at 1∶10,000 in PBS (Vector Labs), together with Texas-Red-conjugated phalloidin (1 µM in PBS) (Molecular Probes). Following incubation, cells were washed three times in PBS, and placed under coverslips using Prolong Gold Antifade with DAPI (Life Technologies). Cells were imaged using an EVOS-FL microscope [Advanced Microscopy Group (AMG) Bothell, WA] fitted with light cubes for GFP, DAPI and Texas Red. Each fluorochrome image was collected separately and then stacked using EVOS software. For quantification, light intensity settings were kept at a fixed value while manually counting the number of cells positive for cleaved caspase-3, or the number of multi-nucleated or mononuclear cells per well, from at least triplicate wells in three independent experiments.
CatK assay of osteoclasts cultured on bone powder
Pre-OCLs (adherent mBMMs resulting from 3 days expansion culture in Petri dishes) were seeded into 24-well tissue culture plates at a density of 4×104 cells/well for 5 days with 1/50th volume of CMG 14-12 culture supernatant (20 ng/ml MCSF) and 50 ng/ml RANKL to stimulate differentiation into mature OCLs. On day 5, cells were stimulated with various treatments. Bovine bone powder generated at the bench top as a by-product of cutting 1 mm cortical bone slices (described above) was collected, measured by weight, washed extensively with 70% ethanol, dried and resuspended with basal medium containing various treatments as indicated. After the indicated time points, conditioned medium was collected from each well and total cell protein extracts prepared by cell lysis, for 45 min at 4°C, in modified RIPA buffer, followed by centrifugation at 13,000 g. Supernatants (50–200 µg) were fractionated on 8% SDS-PAGE and electrophoretically transferred onto nitrocellulose membranes. Membranes were blocked using Odyssey blocking buffer from LI-COR Biosciences (Lincoln, NE) containing 0.1% Tween 20 for 30 mins and subsequently incubated with primary anti-CatK antibody at 1∶250 in Odyssey blocking buffer overnight at 4°C. Blots were washed extensively with PBS, and incubated for 1 hour at room temperature with goat anti-rabbit-IgG conjugated to IRDye 800 CW from LI-COR Biosciences (Lincoln, NE) (1∶40,000 dilution) in 5% milk in PBS. Blots were washed four times with PBS and imaged and quantified using the Odyssey LI-COR System according to the manufacturer's instructions. Blots were subsequently probed with an antibody against mouse β-tubulin (1∶1000). Total cell lysate and conditioned medium CatK were specifically normalized to the amount of β-tubulin per lane.
Resorption pit analysis
mBMMs were cultured for 5 days on dentine slices (Immunodiagnostic systems, Scottsdale, AZ) at a concentration of 1×104 cells per well in a 96-well plate in αMEM in the presence of 1/50th volume of CMG 14-12 culture supernatant (20 ng/ml MCSF) and 50 ng/ml RANKL. Cells were treated on day 5 with MCSF alone or in combination with 50 ng/ml RANKL, 50 ng/ml ActA or both, and incubated for a further 24 hours before fixation with fresh 4% paraformaldehyde in PBS. Dentine slices were then stained for tartrate resistant acid phosphatase (TRAP) [Acid Phosphatase, Leukocyte (TRAP) Kit (Sigma)] and the number of TRAP+ multinucleated cells (TRAP+ MNCs) enumerated on each entire slice under light microscopy using a Nikon Eclipse E400. Cells were then removed with fresh 10% bleach solution in PBS incubation for 30 minutes with 1 minute of sonication (Elma Transsonic 570, 35 Hz). Bone resorption pit number and pit area per slice were analyzed using a light microscope (Nixon Eclipse E400) and quantified using Osteomeasure software (Osteometrics, Atlanta, GA).
Immunoblot analysis of P-SMAD2, AKT1 and IκBα
Whole-cell protein extracts were prepared from day 2 BMM OCL precursors or day 5 BMM mature OCL using RIPA buffer, followed by centrifugation at 14,000 g. Supernatants (50 µg) were fractionated on 8% SDS-PAGE and electrophoretically transferred onto nitrocellulose membranes. Membranes were blocked using Odyssey blocking buffer from LI-COR Biosciences containing 0.1% Tween 20 for 30 minutes and subsequently incubated with primary antibody against phospho-SMAD2, total AKT1 or phospho-AKT1 (Ser473) as well as antibody against IkBα at 1∶1000 in Odyssey blocking buffer overnight at 4°C. Blots were washed extensively with PBS, and incubated for 1 hour at room temperature with goat anti rabbit IRDye 800 CW from LI-COR Biosciences (1∶40,000 dilution) in 5% milk in PBS. Blots were washed four times with PBS and imaged and quantified using the Odyssey LI-COR System according to the manufacturer's instructions. IkBα blots were subsequently probed with an antibody against mouse β-tubulin (1∶1000) as a lysate loading control for normalization of IkBα.
Statistics
Data were analyzed by one-way ANOVA to determine significant differences between means of multiple groups in given experiments. Post-hoc analyses with Tukey's were applied as indicated to determine which groups were significantly different from each other. P<0.05 was considered significant and is reported as such.
Supplementary Material
Acknowledgments
The authors acknowledge the use of the institutional Confocal Microscopy Core and thank Steven Teitelbaum and his many colleagues at Washington University, St Louis for their willingness to share reagents, expertise and time in the performance of these studies.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
T.W.F. was involved in all aspects of the manuscript. This includes experimental design, animal handling to isolate cells for culture, cell culture and live cell microscopy, as well as development of live cell motility analysis parameters, as well as osteoclast differentiation, function and protein analysis. T.W.F. also participated in writing and preparing the manuscript text and figures. A.K. participated in animal handling to isolate cells for culture, cell culture and live cell microscopy, live cell motility analysis, as well as osteoclast differentiation and function assays. N.S.A. was responsible for animal husbandry, involved in isolation of cells for culture, and participated in osteoclast resorption assays. R.C.K. designed the video-microscopy instrumentation and methodology that was used for the bulk of the study, and trained T.W.F. in its use and analytical methods. L.J.S. and D.G. designed and supervised all experiments, evaluated all primary data and directed the development of analytical end points to be measured, were involved in figure preparation, as well as writing and editing of the manuscript.
Funding
This work was supported by the National Institutes of Health (NIH) [grant numbers R21-DK74024 to D.G. and R01-CA166060 to L.J.S.]; University of Arkansas for Medical Sciences (UAMS) Translational Research Institute (TRI) NIH CTSA [grant number #1 UL1TR000039]; a UAMS Medical Research Foundation grant (to D.G.); the Carl L Nelson Chair in Orthopaedic Creativity (to L.J.S.); and a UAMS Graduate Student Educational Research Fellowship (to T.W.F.). Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.157834/-/DC1
References
- Arai A., Mizoguchi T., Harada S., Kobayashi Y., Nakamichi Y., Yasuda H., Penninger J. M., Yamada K., Udagawa N., Takahashi N. (2012). Fos plays an essential role in the upregulation of RANK expression in osteoclast precursors within the bone microenvironment. J. Cell Sci. 125, 2910–2917 10.1242/jcs.099986 [DOI] [PubMed] [Google Scholar]
- Bonewald L. F., Mundy G. R. (1990). Role of transforming growth factor-beta in bone remodeling. Clin. Orthop. Relat. Res (250), 261–276. [PubMed] [Google Scholar]
- Broxmeyer H. E., Lu L., Cooper S., Schwall R. H., Mason A. J., Nikolics K. (1988). Selective and indirect modulation of human multipotential and erythroid hematopoietic progenitor cell proliferation by recombinant human activin and inhibin. Proc. Natl. Acad. Sci. USA 85, 9052–9056 10.1073/pnas.85.23.9052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cory S., Adams J. M. (2002). The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2, 647–656 10.1038/nrc883 [DOI] [PubMed] [Google Scholar]
- Cremasco V., Decker C. E., Stumpo D., Blackshear P. J., Nakayama K. I., Nakayama K., Lupu T. S., Graham D. B., Novack D. V., Faccio R. (2012). Protein kinase C-delta deficiency perturbs bone homeostasis by selective uncoupling of cathepsin K secretion and ruffled border formation in osteoclasts. J. Bone Miner. Res. 27, 2452–2463 10.1002/jbmr.1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do T. V., Kubba L. A., Antenos M., Rademaker A. W., Sturgis C. D., Woodruff T. K. (2008). The role of activin A and Akt/GSK signaling in ovarian tumor biology. Endocrinology 149, 3809–3816 10.1210/en.2007-1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faccio R., Novack D. V., Zallone A., Ross F. P., Teitelbaum S. L. (2003). Dynamic changes in the osteoclast cytoskeleton in response to growth factors and cell attachment are controlled by beta3 integrin. J. Cell Biol. 162, 499–509 10.1083/jcb.200212082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto K., Kawakita M., Kato K., Yonemura Y., Masuda T., Matsuzaki H., Hirose J., Isaji M., Sasaki H., Inoue T. et al. (1991). Purification of megakaryocyte differentiation activity from a human fibrous histiocytoma cell line: N-terminal sequence homology with activin A. Biochem. Biophys. Res. Commun. 174, 1163–1168 10.1016/0006-291X(91)91543-L [DOI] [PubMed] [Google Scholar]
- Fujita K., Iwasaki M., Ochi H., Fukuda T., Ma C., Miyamoto T., Takitani K., Negishi-Koga T., Sunamura S., Kodama T. et al. (2012). Vitamin E decreases bone mass by stimulating osteoclast fusion. Nat. Med. 18, 589–594 10.1038/nm.2659 [DOI] [PubMed] [Google Scholar]
- Fuller K., Bayley K. E., Chambers T. J. (2000). Activin A is an essential cofactor for osteoclast induction. Biochem. Biophys. Res. Commun. 268, 2–7 10.1006/bbrc.2000.2075 [DOI] [PubMed] [Google Scholar]
- Gaddy-Kurten D., Coker J. K., Abe E., Jilka R. L., Manolagas S. C. (2002). Inhibin suppresses and activin stimulates osteoblastogenesis and osteoclastogenesis in murine bone marrow cultures. Endocrinology 143, 74–83 10.1210/endo.143.1.8580 [DOI] [PubMed] [Google Scholar]
- Glowacki J., Lian J. B. (1987). Impaired recruitment and differentiation of osteoclast progenitors by osteocalcin-deplete bone implants. Cell Differ. 21, 247–254 10.1016/0045-6039(87)90479-9 [DOI] [PubMed] [Google Scholar]
- Gonzalo P., Guadamillas M. C., Hernández-Riquer M. V., Pollán A., Grande-García A., Bartolomé R. A., Vasanji A., Ambrogio C., Chiarle R., Teixidó J. et al. (2010). MT1-MMP is required for myeloid cell fusion via regulation of Rac1 signaling. Dev. Cell 18, 77–89 10.1016/j.devcel.2009.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S., Wang Z., Bryan J., Kobayashi C., Faccio R., Sandell L. J. (2011). The type II collagen N-propeptide, PIIBNP, inhibits cell survival and bone resorption of osteoclasts via integrin-mediated signaling. Bone 49, 644–652 10.1016/j.bone.2011.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houde N., Chamoux E., Bisson M., Roux S. (2009). Transforming growth factor-beta1 (TGF-beta1) induces human osteoclast apoptosis by up-regulating Bim. J. Biol. Chem. 284, 23397–23404 10.1074/jbc.M109.019372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii M., Kikuta J., Shimazu Y., Meier-Schellersheim M., Germain R. N. (2010). Chemorepulsion by blood S1P regulates osteoclast precursor mobilization and bone remodeling in vivo. J. Exp. Med. 207, 2793–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa T., Zou W., Chappel J. C., Ashley J. W., Feng X., Teitelbaum S. L. (2012). c-Src links a RANK/αvβ3 integrin complex to the osteoclast cytoskeleton. Mol. Cell. Biol. 32, 2943–2953 10.1128/MCB.00077-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquin C., Gran D. E., Lee S. K., Lorenzo J. A., Aguila H. L. (2006). Identification of multiple osteoclast precursor populations in murine bone marrow. J. Bone Miner. Res. 21, 67–77 10.1359/JBMR.051007 [DOI] [PubMed] [Google Scholar]
- Jin Z., El-Deiry W. S. (2005). Overview of cell death signaling pathways. Cancer Biol. Ther. 4, 147–171 10.4161/cbt.4.2.1508 [DOI] [PubMed] [Google Scholar]
- Lee H. J., Kim P. H. (2009). Further Characterization of Activin A-induced IgA Response in Murine B Lymphocytes. Immune Netw. 9, 133–137 10.4110/in.2009.9.4.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo J. (2003). Characterization of osteoclast precursor cells in murine bone marrow. J. Musculoskelet. Neuronal Interact. 3, 273–277discussion 292-294 [PubMed] [Google Scholar]
- Lotinun S., Pearsall R. S., Davies M. V., Marvell T. H., Monnell T. E., Ucran J., Fajardo R. J., Kumar R., Underwood K. W., Seehra J. et al. (2010). A soluble activin receptor Type IIA fusion protein (ACE-011) increases bone mass via a dual anabolic-antiresorptive effect in Cynomolgus monkeys. Bone 46, 1082–1088 10.1016/j.bone.2010.01.370 [DOI] [PubMed] [Google Scholar]
- Lotinun S., Pearsall R. S., Horne W. C., Baron R. (2012). Activin receptor signaling: a potential therapeutic target for osteoporosis. Curr. Mol. Pharmacol 5, 195–204 10.2174/1874467211205020195 [DOI] [PubMed] [Google Scholar]
- Malone J. D., Teitelbaum S. L., Griffin G. L., Senior R. M., Kahn A. J. (1982). Recruitment of osteoclast precursors by purified bone matrix constituents. J. Cell Biol. 92, 227–230 10.1083/jcb.92.1.227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus S., Chamoux E., Bisson M., Roux S. (2012). Modulation of tumor necrosis factor related apoptosis-inducing ligand (TRAIL) receptors in a human osteoclast model in vitro. Apoptosis 17, 121–131 10.1007/s10495-011-0662-5 [DOI] [PubMed] [Google Scholar]
- Moore M. A., Gotoh Y., Rafidi K., Gerstenfeld L. C. (1991). Characterization of a cDNA for chicken osteopontin: expression during bone development, osteoblast differentiation, and tissue distribution. Biochemistry 30, 2501–2508 10.1021/bi00223a029 [DOI] [PubMed] [Google Scholar]
- Murase Y., Okahashi N., Koseki T., Itoh K., Udagawa N., Hashimoto O., Sugino H., Noguchi T., Nishihara T. (2001). Possible involvement of protein kinases and Smad2 signaling pathways on osteoclast differentiation enhanced by activin A. J. Cell. Physiol. 188, 236–242 10.1002/jcp.1113 [DOI] [PubMed] [Google Scholar]
- Nakashima T., Hayashi M., Fukunaga T., Kurata K., Oh-Hora M., Feng J. Q., Bonewald L. F., Kodama T., Wutz A., Wagner E. F. et al. (2011). Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 17, 1231–1234 10.1038/nm.2452 [DOI] [PubMed] [Google Scholar]
- Nicks K. M., Perrien D. S., Akel N. S., Suva L. J., Gaddy D. (2009). Regulation of osteoblastogenesis and osteoclastogenesis by the other reproductive hormones, Activin and Inhibin. Mol. Cell. Endocrinol. 310, 11–20 10.1016/j.mce.2009.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novack D. V., Faccio R. (2011). Osteoclast motility: putting the brakes on bone resorption. Ageing Res. Rev. 10, 54–61 10.1016/j.arr.2009.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novack D. V., Teitelbaum S. L. (2008). The osteoclast: friend or foe? Annu. Rev. Pathol. 3, 457–484 10.1146/annurev.pathmechdis.3.121806.151431 [DOI] [PubMed] [Google Scholar]
- Ogawa Y., Schmidt D. K., Nathan R. M., Armstrong R. M., Miller K. L., Sawamura S. J., Ziman J. M., Erickson K. L., de Leon E. R., Rosen D. M. et al. (1992). Bovine bone activin enhances bone morphogenetic protein-induced ectopic bone formation. J. Biol. Chem. 267, 14233–14237. [PubMed] [Google Scholar]
- Okafuji K., Kaku K., Seguchi M., Tanaka H., Azuno Y., Kaneko T. (1995). Effects of activin A/erythroid differentiation factor on erythroid and megakaryocytic differentiations of mouse erythroleukemia (Friend) cells: evidence for two distinct modes of cell response. Exp. Hematol. 23, 210–216. [PubMed] [Google Scholar]
- Oursler M. J. (2010). Recent advances in understanding the mechanisms of osteoclast precursor fusion. J. Cell. Biochem. 110, 1058–1062 10.1002/jcb.22640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearsall R. S., Canalis E., Cornwall-Brady M., Underwood K. W., Haigis B., Ucran J., Kumar R., Pobre E., Grinberg A., Werner E. D. et al. (2008). A soluble activin type IIA receptor induces bone formation and improves skeletal integrity. Proc. Natl. Acad. Sci. USA 105, 7082–7087 10.1073/pnas.0711263105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrien D. S., Achenbach S. J., Bledsoe S. E., Walser B., Suva L. J., Khosla S., Gaddy D. (2006). Bone turnover across the menopause transition: correlations with inhibins and follicle-stimulating hormone. J. Clin. Endocrinol. Metab. 91, 1848–1854 10.1210/jc.2005-2423 [DOI] [PubMed] [Google Scholar]
- Pilkington M. F., Sims S. M., Dixon S. J. (2001). Transforming growth factor-beta induces osteoclast ruffling and chemotaxis: potential role in osteoclast recruitment. J. Bone Miner. Res. 16, 1237–1247 10.1359/jbmr.2001.16.7.1237 [DOI] [PubMed] [Google Scholar]
- Rieman D. J., McClung H. A., Dodds R. A., Hwang S. M., Holmes M. W., James I. E., Drake F. H., Gowen M. (2001). Biosynthesis and processing of cathepsin K in cultured human osteoclasts. Bone 28, 282–289 10.1016/S8756-3282(00)00445-2 [DOI] [PubMed] [Google Scholar]
- Saelens X., Festjens N., Vande Walle L., van Gurp M., van Loo G., Vandenabeele P. (2004). Toxic proteins released from mitochondria in cell death. Oncogene 23, 2861–2874 10.1038/sj.onc.1207523 [DOI] [PubMed] [Google Scholar]
- Sakai R., Fujita S., Horie T., Ohyama T., Miwa K., Maki T., Okimoto N., Nakamura T., Eto Y. (2000a). Activin increases bone mass and mechanical strength of lumbar vertebrae in aged ovariectomized rats. Bone 27, 91–96 10.1016/S8756-3282(00)00307-0 [DOI] [PubMed] [Google Scholar]
- Sakai R., Fujita S., Horie T., Ohyama T., Miwa K., Maki T., Okimoto N., Nakamura T., Eto Y. (2000b). Activin increases bone mass and mechanical strength of lumbar vertebrae in aged ovariectomized rats. Bone 27, 91–96 10.1016/S8756-3282(00)00307-0 [DOI] [PubMed] [Google Scholar]
- Salogni L., Musso T., Bosisio D., Mirolo M., Jala V. R., Haribabu B., Locati M., Sozzani S. (2009). Activin A induces dendritic cell migration through the polarized release of CXC chemokine ligands 12 and 14. Blood 113, 5848–5856 10.1182/blood-2008-12-194597 [DOI] [PubMed] [Google Scholar]
- Silbermann R., Bolzoni M., Storti P., Guasco D., Bonomini S., Zhou D., Wu J., Anderson J. L., Windle J. J., Aversa F. et al. (2014). Bone marrow monocyte-/macrophage-derived activin A mediates the osteoclastogenic effect of IL-3 in multiple myeloma. Leukemia 28, 951–954 10.1038/leu.2013.385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugatani T., Alvarez U. M., Hruska K. A. (2003). Activin A stimulates IkappaB-alpha/NFkappaB and RANK expression for osteoclast differentiation, but not AKT survival pathway in osteoclast precursors. J. Cell. Biochem. 90, 59–67 10.1002/jcb.10613 [DOI] [PubMed] [Google Scholar]
- Sun L., Peng Y., Zaidi N., Zhu L. L., Iqbal J., Yamoah K., Wang X., Liu P., Abe E., Moonga B. S. et al. (2007). Evidence that calcineurin is required for the genesis of bone-resorbing osteoclasts. Am. J. Physiol. 292, F285–F291 10.1152/ajprenal.00415.2005 [DOI] [PubMed] [Google Scholar]
- Suva L. J., Washam C., Nicholas R. W., Griffin R. J. (2011). Bone metastasis: mechanisms and therapeutic opportunities. Nat. Rev. Endocrinol. 7, 208–218 10.1038/nrendo.2010.227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita S., Kaji K., Kudo A. (2000). Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J. Bone Miner. Res. 15, 1477–1488 10.1359/jbmr.2000.15.8.1477 [DOI] [PubMed] [Google Scholar]
- Teitelbaum S. L. (1993). Bone remodeling and the osteoclast. J. Bone Miner. Res. 8, Suppl. 2S523–S525 10.1002/jbmr.5650081318 [DOI] [PubMed] [Google Scholar]
- Teitelbaum S. L., Ross F. P. (2003). Genetic regulation of osteoclast development and function. Nat. Rev. Genet. 4, 638–649 10.1038/nrg1122 [DOI] [PubMed] [Google Scholar]
- Valderrama-Carvajal H., Cocolakis E., Lacerte A., Lee E. H., Krystal G., Ali S., Lebrun J. J. (2002). Activin/TGF-beta induce apoptosis through Smad-dependent expression of the lipid phosphatase SHIP. Nat. Cell Biol. 4, 963–969 10.1038/ncb885 [DOI] [PubMed] [Google Scholar]
- Vale W., Wiater E., Gray P., Harrison C., Bilezikjian L., Choe S. (2004). Activins and inhibins and their signaling. Ann. N. Y. Acad. Sci. 1038, 142–147 10.1196/annals.1315.023 [DOI] [PubMed] [Google Scholar]
- Wozney J. M. (1992). The bone morphogenetic protein family and osteogenesis. Mol. Reprod. Dev. 32, 160–167 10.1002/mrd.1080320212 [DOI] [PubMed] [Google Scholar]
- Xiong J., Onal M., Jilka R. L., Weinstein R. S., Manolagas S. C., O'Brien C. A. (2011). Matrix-embedded cells control osteoclast formation. Nat. Med. 17, 1235–1241 10.1038/nm.2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada R., Suzuki T., Hashimoto M., Eto Y., Shiokawa K., Muramatsu M. (1992). Induction of differentiation of the human promyelocytic cell line HL-60 by activin/EDF. Biochem. Biophys. Res. Commun. 187, 79–85 10.1016/S0006-291X(05)81461-5 [DOI] [PubMed] [Google Scholar]
- Ye S., Fowler T. W., Pavlos N. J., Ng P. Y., Liang K., Feng Y., Zheng M., Kurten R., Manolagas S. C., Zhao H. (2011). LIS1 regulates osteoclast formation and function through its interactions with dynein/dynactin and Plekhm1. PLoS ONE 6, e27285 10.1371/journal.pone.0027285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Shao L. E., Lemas V., Yu A. L., Vaughan J., Rivier J., Vale W. (1987). Importance of FSH-releasing protein and inhibin in erythrodifferentiation. Nature 330, 765–767 10.1038/330765a0 [DOI] [PubMed] [Google Scholar]
- Yu A. W., Shao L. E., Frigon N. L., Jr and Yu J. (1994). Detection of functional and dimeric activin A in human marrow microenvironment. Implications for the modulation of erythropoiesis. Ann. N. Y. Acad. Sci. 718, 285–298discussion 298-299 10.1111/j.1749-6632.1994.tb55727.x [DOI] [PubMed] [Google Scholar]
- Zhou P., Kitaura H., Teitelbaum S. L., Krystal G., Ross F. P., Takeshita S. (2006). SHIP1 negatively regulates proliferation of osteoclast precursors via Akt-dependent alterations in D-type cyclins and p27. J. Immunol. 177, 8777–8784 10.4049/jimmunol.177.12.8777 [DOI] [PubMed] [Google Scholar]
- Zou W., Zhu T., Craft C. S., Broekelmann T. J., Mecham R. P., Teitelbaum S. L. (2010). Cytoskeletal dysfunction dominates in DAP12-deficient osteoclasts. J. Cell Sci. 123, 2955–2963 10.1242/jcs.069872 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.