

Abstract
White matter tracts are highly vulnerable to damage from impact-acceleration forces of traumatic brain injury (TBI). Mild TBI is characterized by a low density of traumatic axonal injury, whereas associated myelin pathology is relatively unexplored. We examined the progression of white matter pathology in mice after mild TBI with traumatic axonal injury localized in the corpus callosum. Adult mice received a closed-skull impact and were analyzed from 3 days to 6 weeks post-TBI/sham surgery. At all times post-TBI, electron microscopy revealed degenerating axons distributed among intact fibers in the corpus callosum. Intact axons exhibited significant demyelination at 3 days followed by evidence of remyelination at 1 week. Accordingly, bromodeoxyuridine pulse-chase labeling demonstrated the generation of new oligodendrocytes, identified by myelin proteolipid protein messenger RNA expression, at 3 days post-TBI. Overall oligodendrocyte populations, identified by immunohistochemical staining for CC1 and/or glutathione S-transferase pi, were similar between TBI and sham mice by 2 weeks. Excessively long myelin figures, similar to redundant myelin sheaths, were a significant feature at all post-TBI time points. At 6 weeks post-TBI, microglial activation and astrogliosis were localized to areas of axon and myelin pathology. These studies show that demyelination, remyelination, and excessive myelin are components of white matter degeneration and recovery in mild TBI with traumatic axonal injury.
Key Words: Axon damage, Demyelination, Neuroinflammation, Oligodendrocyte, Redundant myelin, Remyelination, Traumatic axonal injury
INTRODUCTION
Traumatic brain injury (TBI) affects millions of people worldwide, many of whom experience a broad range of neurologic deficits (1). Traumatic brain injury often results from impact-acceleration forces and is most commonly classified as mild TBI. However, even mild TBIs leave approximately 33% of patients below full functional status at 3 months postinjury; these deficits will continue through at least 1 year in 22% of patients (2). Traumatic brain injury resulting from impact-acceleration forces leads to traumatic axonal injury (TAI) within white matter tracts. Long axonal projections that traverse the brain in white matter tracts are damaged from forces of torsion, tension, and compression in impact-acceleration injuries to the head (3). The corpus callosum is a common site of high strain from impact, and frequent abnormalities are identified by diffusion tensor imaging even in mild TBI (4–7). The progression of white matter pathologic features that underlie prolonged deficits experienced after mild TBI is poorly understood.
Although myelin is a major component of white matter, the significance of myelin pathology in mild TBI has been relatively unexplored. Studies using a mild fluid percussion model have shown that unmyelinated fibers are particularly vulnerable to damage (8, 9). However, most preclinical TBI studies of oligodendrocyte populations relative to myelin pathology have used models that involve focal axon loss caused by white matter hemorrhage or neuron cell body death with axon loss caused by cortical cavitation (10–12). In these models, focal axon loss causes myelin degeneration because of lack of viable axons to be ensheathed. This overt loss of tissue, involving axons and myelin, is distinct from demyelination of intact axons. In a model with TAI in the corpus callosum, demyelination not correlated with axon degeneration was observed, indicating separate pathologic processes after TBI (13). Demyelination of viable axons impairs saltatory conduction and renders the axons more vulnerable to further damage. Slow conduction along demyelinated axons desynchronizes neural circuitry, which may contribute to deficits in information-processing speed after TBI (14, 15). Damage to myelin may also cause myelin debris that can stimulate microglial activation (16). This progression could contribute to chronic neuroinflammation localized in white matter tracts, which is observed in models of single and repetitive mild TBI with dispersed TAI (13, 17).
Subsequent to demyelination, remyelination of viable axons may be particularly relevant to recovery after mild TBI but has not yet been sufficiently studied. As with demyelination, analysis of remyelination requires models of TBI that do not incur extensive focal tissue loss from cavitation or hemorrhage. Studies in demyelinating disease models have shown that remyelination can both enable recovery of function and protect denuded axons from progressive degeneration and disconnection, which results in permanent loss of function (18, 19). Furthermore, viable axons may exhibit myelin remodeling, which is a form of adult plasticity that can be modified by cortical activity and experience (20, 21).
A marked feature of myelin pathology in our initial study of TAI in the corpus callosum was abnormal myelin sheaths with excessive myelin folding back onto itself (13). These excessive myelin figures with double-layered sheaths resembled “redundant” myelin observed during central nervous system (CNS) development (22). Recent 3-dimensional reconstruction analysis revealed excess myelin outfoldings as part of normal myelin sheath formation in the developing CNS (23). In the context of TBI, further evaluation is needed to determine whether excessive myelin figures are a component of white matter damage and/or myelin remodeling.
The purpose of the current study was to determine the progression of myelin pathology and repair in a model of mild TBI that exhibits localized TAI in the corpus callosum without tissue loss from hemorrhage or cavitation. We examine the corpus callosum pathology across a time course from 3 days to 6 weeks post-TBI. Electron microscopy captures the ultrastructural details required to quantify the spectrum of pathologic features indicative of degenerating axons relative to myelin changes, including demyelination, excessive myelin figures, and remyelination. Interpretation of these ultrastructural studies is further supported by analysis of cellular responses associated with the specific myelin findings. These experiments clarify the contribution of both axon and myelin components of white matter to the progression of pathology after mild TBI.
MATERIALS AND METHODS
Traumatic Brain Injury
All procedures were performed on 8-week-old male C57BL/6 mice (Jackson Laboratories, Bar Harbor, ME). Mice were housed in pairs according to the guidelines of the National Institutes of Health and the Institutional Animal Care and Use Committee of the Uniformed Services University of the Health Sciences.
Mice received a mild TBI, using a previously characterized model of impact onto the closed skull, to produce TAI in the corpus callosum over the lateral ventricles (13, 24). Impact to the skull was centered at bregma using an Impact One™ Stereotaxic Impactor (Leica, Buffalo Grove, IL) device with a 3-mm-diameter flat tip set to a depth of 1.5 mm, a velocity of 4 m/second, and a dwell time of 100 milliseconds. Isoflurane was administered continuously at 2% via a nose cone until immediately after impact. Video analysis of the impact velocity and depth verified an average impact velocity of 4.3 ± 0.2 m/second (mean ± SE) and an average depth of 1.8 ± 0.1 mm. Sham mice received anesthesia and a scalp incision. Naive mice did not receive either anesthesia or an incision. These impact parameters resulted in postsurgical apnea of 17.93 ± 3.35 seconds. Immediate postimpact visual inspection of the skull revealed a minimal extent of typically linear fracture near bregma in approximately 62% of mice. Mice with unexpectedly severe skull fracture, including depressed fractures, were excluded from further analysis. Postsurgery, mice exhibited an average righting reflex time of 6.65 ± 0.48 minutes in TBI mice and 1.27 ± 0.34 minutes in sham mice. Mice killed 2 hours after injury did not exhibit blood-brain barrier leakage based on immunohistochemistry to detect mouse IgG (data not shown). At the time of brain dissection (i.e. 3, 7, 14, or 42 days postsurgery), there was no visible evidence of fractures on the skull or contusion on the surface of the brain.
Tissue Preparation for Electron Microscopy
For electron microscopy, mice were anesthesized with ketamine/xylazine before transventricular cardiac perfusion with 2% paraformaldehyde and 3% glutaraldehyde at 3 days, 1 week, 2 weeks, or 6 weeks after surgery and stored in fixative overnight before dissection. Dissected brains were postfixed in fixative overnight. Brains were hemisected along the midline. One hemisphere was sectioned (40 μm) in coronal slices and the other was sectioned in sagittal slices using a Leica vibrating VT-1000 vibratome (Leica). Sections were selected for further processing based on localization at the coronal level of the anterior commissure and at parasagittal levels to include the corpus callosum over the lateral ventricles. Samples were immersed in 2% osmium tetroxide in PBS (OsO4; Electron Microscopy Sciences, Hatfield, PA) for 1 hour. Samples were then washed again in PBS, dehydrated in ethanol, and infiltrated with Spurr epoxy resin (Electron Microscopy Sciences). Samples were then polymerized at 70 °C for 11 hours, and thin sections (∼70 nm) were cut on an Ultracut UC6 ultramicrotome (Leica). Copper grids containing thin sections were poststained for 20 minutes in 2% aqueous uranyl acetate (Electron Microscopy Sciences) and for 5 minutes in Reynolds lead citrate (25). Grids were reviewed on a JEOL JEM-1011 transmission electron microscope (JEOL USA Inc, Peabody, MA). Images were captured on an AMT XR50S-A digital camera (Advanced Microscopy Techniques, Woburn, MA).
Tissue Preparation for Light Microscopy
Mice used for histopathology, immunohistochemistry, or in situ hybridization were anesthesized with ketamine/xylazine before transventricular cardiac perfusion with 3% paraformaldehyde. Brains were postfixed overnight, infiltrated with sucrose, and embedded in optimal cutting temperature (OCT) medium. Coronal cryostat sections (14 μm) were selected at the level of the anterior commissure.
Cryosections were processed for Prussian blue histologic stain for hemosiderin combined with pararosaniline nuclear stain according to the manufacturer’s instructions (Sigma, St Louis, MO). Cryosections were immunostained with the following primary antibodies as cell type specific markers: astrocytes: glial fibrillary acidi protein (GFAP; rabbit polyclonal, 1:500; DAKO, Carpinteria, CA), microglia: CD11b (rat monoclonal, 1:100; AbCam, Cambridge, MA), oligodendrocytes: glutathione S-transferase pi (GSTpi; rabbit polyclonal, 1:200; MBL International, Woburn, MA) and CC1 (mouse monoclonal, 1:200; Millipore, Billerica, MA). Secondary antibodies included goat anti-rabbit IgG conjugated with Alexa Fluor 488 (1:200; Life Technologies, Grand Island, NY) to detect GFAP and GSTpi, goat anti-rat IgG conjugated with Alexa Fluor 555 (1:200; Life Technologies) to detect CD11b, and donkey anti-mouse IgG conjugated with Cy3 to detect CC1. Sections were counterstained with DAPI (Sigma) before being mounted with Vectashield (Vector Laboratories, Burlingame, CA).
In situ hybridization was performed, using a previously described riboprobe, to hybridize proteolipid protein (PLP) messenger RNA (mRNA) transcripts as a marker of oligodendrocytes (26). After hybridization, labeling was detected with alkaline phosphatase–conjugated sheep anti-digoxigenin and reaction with substrate solution (nitroblue tetrazolium chloride/5-bromo-4-chloro-3-indolylphosphate; DAKO). After completion of the in situ hybridization procedure, we performed terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining, using ApopTag Plus Peroxidase In Situ Apoptosis Detection Kit (S7101 EMD; Millipore Corporation, Temecula, CA), to detect apoptotic cells. To examine the conversion of progenitor cells into oligodendrocytes, we prelabeled progenitor cells with a pulse of bromodeoxyuridine (BrdU). Bromodeoxyuridine administration before TBI/sham surgery labels endogenous cycling cells and minimizes postinjury concerns of labeling proliferating astrocytes or microglia, or cells with DNA damage. Mice received intraperitoneal injections of 200 mg/kg BrdU (Sigma) at 24 and 22 hours before surgery and were subsequently perfused on Day 3 after TBI or sham surgery. After completion of in situ hybridization, sections were treated with 2 N HCl and incubated overnight with anti-BrdU antibody conjugated to fluorescein (Roche Diagnostics, Indianapolis, IN).
Quantification of Immunohistochemistry and In Situ Hybridization
Digital images were captured on an Olympus IX70 microscope using a Spot CCD camera (Diagnostic Instruments, Sterling Heights, MI). Areas of the corpus callosum were measured using Spot Advanced software. The area within the corpus callosum immunostained for GFAP or CD11b was estimated based on a cutoff for pixel intensity values that were above background levels using the Metamorph software thresholding function (Molecular Devices, Downingtown, PA). Immunostaining intensity across sections was controlled for by comparison of regions outside the corpus callosum. CD11b expressing cells within the corpus callosum were also counted individually to differentiate activation stage as resting (fine processes), activated (hypertrophic with thickened processes), or amoeboid (retracted processes), as previously characterized (13). All other cell phenotypes were counted manually within the area of the corpus callosum. Cells expressing CC1 and/or GSTpi were counted as single- or double-labeled phenotypes. Cells with cytoplasmic PLP mRNA hybridization signal were counted based on nuclear 3,3′ diaminobenzidine chromogen reaction for either BrdU or TUNEL. Under all conditions, quantification in the corpus callosum included at least 3 sections per mouse and included 24 mice (BrdU and TUNEL with PLP: n = 3 naive, n = 3 TBI; CC1/GSTpi: n = 5 sham, n = 3 TBI; CD11b/GFAP: n = 5 sham, n = 5 TBI).
Corpus Callosum Width
Corpus callosum widths were measured in osmicated coronal sections (40 μm) at the level of the anterior commissure. Measurements were taken in 4 locations: the midline, the peak of the cingulum, the lateral edge of the ventricle, and 2 μm lateral to the midline. Quantification included 19 mice (3 days: n = 3 sham, n = 4 TBI; 1 week: n = 3 TBI; 2 weeks: n = 5 TBI; 6 weeks: n = 4 sham, n = 4 TBI).
Electron Microscopy Image Quantification
Images of sagittal sections through the corpus callosum were acquired at 5,000× magnification for measurements of axon diameter and myelin thickness along with manual counting of degenerating axons, demyelinated axons, and excessive myelin figures. Analysis included 3 to 5 images per animal (at least 120 axons per image); 3 to 6 animals per condition were used. Degenerating axons were defined as axons with cytoskeletal changes (microtubule and neurofilament density extremely low or high with irregular spacing), axons filled with vesicles, or axons with swollen mitochondria (filling >50% of the axon cross-section). Demyelinated axons were manually counted based on a lack of detectable compact myelin and a diameter larger than 0.3 μm. Axons smaller than 0.3 μm were excluded from analysis as potentially unmyelinated fibers (27). An excessive myelin figure was identified as a myelin sheath that was not tightly enwrapping an axon but instead folded back on itself (i.e. redundant myelin) (13, 22). These figures can be seen surrounding and extending from an intact axon or a degenerating axon or remaining without an axon. Within each counting frame, all apparently intact axons were measured to determine axon diameter and myelin thickness using Metamorph software (Molecular Devices), as previously detailed (28, 29). These values were also used to calculate the g-ratio (axon diameter divided by myelinated fiber diameter) as an indicator of remyelination. Excessive myelin figures were measured along the lumen of the myelin sheaths, and that length was doubled to determine the length of each excessive myelin figure. Additional images were taken at 10,000× for illustration of pathology. Quantification included 26 mice (3 days: n = 3 sham, n = 4 TBI; 1 week: n = 5 TBI; 2 weeks: n = 5 TBI; 6 weeks: n = 4 sham, n = 5 TBI).
Electron Microscopy in the Cuprizone Model of Corpus Callosum Demyelination
Male C57BL/6 mice administered cuprizone have been previously characterized by electron microscopy during the acute and chronic stages of demyelination (28). In addition, electron microscopy has shown that the absence of fibroblast growth factor 2 (Fgf2) dramatically increases remyelination in Fgf2-null versus wild-type mice after chronic cuprizone demyelination (29). Images from each of these prior cuprizone studies were analyzed, as described previously, to quantify excessive myelin figures during specific stages of demyelination versus remyelination to inform interpretation of excessive myelin figures in the TBI model. Quantification included 15 C57 mice (no cuprizone, n = 5; 4-week cuprizone, acute demyelination, n = 3; 12-week cuprizone, chronic demyelination, n = 7) and 18 Fgf2 mice (n = 3 for each of the 3 time points for both null and wild-type genotypes).
Statistics
Prism 6.0 (GraphPad Software) was used for statistical analyses and graphing of quantitative data. One-way analysis of variance was performed to determine significant differences across multiple postinjury time points compared to each sham time point, with post hoc multiple comparisons using Dunnett test or Kruskall-Wallis test if D’Agostino-Pearson normality test failed. Two-way analysis of variance was used to compare Fgf2 and wild-type genotypes across time points and to compare oligodendrocyte populations (ie, cells expressing CC1 and/or GSTpi) between sham and TBI conditions. Significant differences between 2 groups for a given time point or condition were determined using Student t-test. Slopes were calculated using linear regression and analyzed using Pearson correlation. Statistical significance was determined as p < 0.05.
RESULTS
Overview of Corpus Callosum Pathology in a Mild TBI Model With TAI
The current studies of corpus callosum pathology take advantage of our previously characterized TBI model of impact onto the skull at bregma, which focuses on TAI over the lateral ventricles at the coronal level of the anterior commissure (13). Brains were examined at multiple points after injury for evaluation at the gross level for overall pathology and by electron microscopy for subcellular ultrastructure (Fig. 1). The current impact protocol produced an injury categorized as mild TBI based on the absence of overt pathology such as contusion with cavitation in the cortex or corpus callosum hemorrhage (Figs. 1A–C, E, F). Corpus callosum thinning, along with ventricular enlargement, was observed in some TBI mice (Fig. 1C), but this was not significantly different from sham mice (Fig. 1D). Cortical regions under the impact site maintained appropriate cytoarchitecture, with evidence of prior bleeding observed only rarely along the cortical surface (Figs. 1E, F). The injury did not produce hemorrhage or cavitation within the corpus callosum (data not shown) (13). Ultrastructural examination of the corpus callosum demonstrated axon and myelin pathologic features across all post-TBI time points (Figs. 1H–M). Degenerating axons are dispersed among intact axons, consistent with a pattern of TAI. Myelin abnormalities include demyelination, collapsed myelin, and excessive myelin figures that are not tightly enwrapping an axon and so are similar to “redundant myelin.” These features and the associated cellular responses were further examined to evaluate the progression of myelin pathology in this model of TAI.
FIGURE 1.
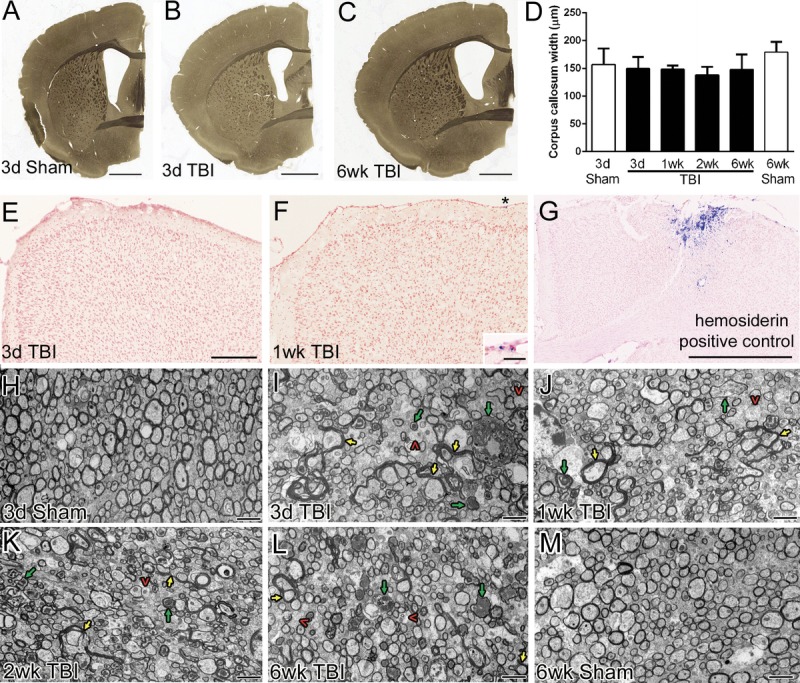
Overview of pattern of axon and myelin pathology in the corpus callosum. (A–C) Osmicated 40-μm sections show little overt pathology at the coronal level of the anterior commissure where the injury was centered. In some injured animals, there seems to be thinning of the corpus callosum with ventricular enlargement (C). (D) The widths of the corpus callosum do not differ significantly across injury conditions. (E–G) Prussian blue histologic stain to detect hemosiderin-laden macrophages indicating areas of prior blood extravasation. In mice representative of those included in this study (E, F), the impact site shows appropriate cortical cytoarchitecture (pink pararosaniline nuclear counterstain) without cavitation. Rare hemosiderin deposits (blue) were located superficially (F; inset shows area below asterisk). In contrast, a mouse excluded from the study because of atypical skull fracture is shown as a positive control for the Prussian blue detection of hemosiderin (G). (H–M) Sagittal sections (axons cut in cross-section) were examined by electron microscopy at multiple time points in sham (H, M) and TBI (I, J, K, L) mice. Degenerating axons (green arrows), demyelinated axons (red caret symbols), and excessive myelin figures (yellow arrows) are each prominent pathologic findings in TBI mice. These features are dispersed among a majority of normal-appearing axons. Ultrastructural changes are shown in more detail in subsequent figures. Scale bars = (A–C) 1 mm; (E–G) 250 μm; (F, inset) 25 μm; (H–M) 2 μm. d, days; wk, weeks.
Persistent Axon Degeneration After TAI
A distributed pattern of degenerating axons among intact fibers within white matter tracts is characteristic of TAI (30–33). In our previous characterization of this model, we detected axon damage that was highest early during the first week post-TBI, using immunohistochemistry for β-amyloid precursor protein (βAPP) (13, 24). We now examined axon degeneration at the ultrastructural level to detect the full range of pathologic features and thus avoid the technical caveats of histologic stains and immunohistochemical markers that often detect specific stages of axon damage within the postinjury time course. By electron microscopy, degenerating axons exhibited breakdown of the cytoskeleton with abnormally high cytoplasmic density and vesicle accumulation (51.1%–59.9%) or with abnormally low cytoplasmic density (23.7%–33.1%) (Figs. 2A–I). Mitochondrial swelling was also noted (13.8%–25.2% of degenerating axons) as a subtler indicator of axon damage. At each time point examined, degenerating axons were significantly increased in TBI mice, with a 2.4- to 3.8-fold increase from sham values (Fig. 2J). The 2.9-fold increase in degenerating axons at 6 weeks post-TBI indicates a prolonged pathologic response to injury.
FIGURE 2.
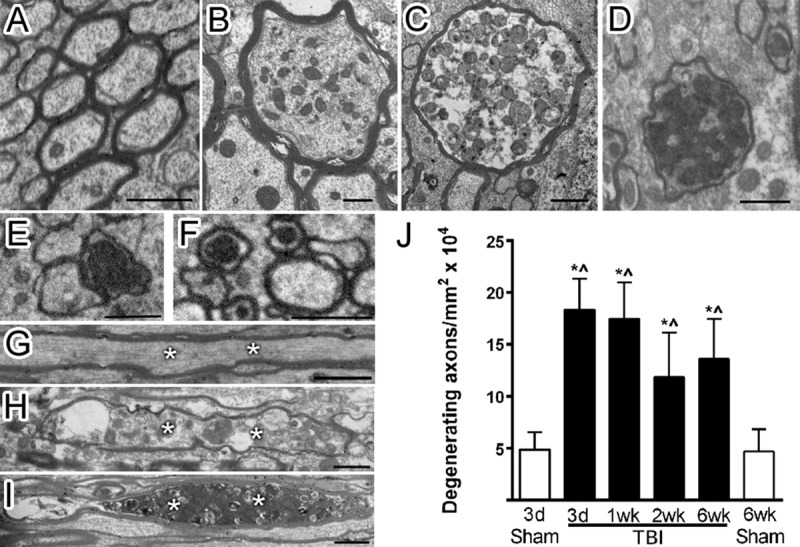
Multiple stages of degenerating axons observed postinjury. (A–I) After TBI, corpus callosum axons exhibit multiple stages of degeneration, as shown in cross-section (A, sham; B–F, TBI) and longitudinal sections (G, sham; H, I, TBI). Sham axons (A) have a uniform cytoskeletal structure. In TBI animals, axon degeneration involves swelling and varying densities of accumulated vesicles (B–D). Axons with very dense cytoplasm (E) and swollen mitochondria (F) were also scored as degenerating. Longitudinal views also demonstrate uniform cytoskeletal structure in sham axons (G, asterisks) and accumulation of vesicles in degenerating axons after TBI (H, I, asterisks). (J) Degenerating axons are increased across all time points in TBI mice. * p ≤ 0.0001 compared to 3-day sham. ^p ≤ 0.0001 compared to 6-week sham. Image time points: (A, G) 3-day sham; (B, C, E, F, I) 3-day TBI; (D, H) 1-week TBI. Scale bar = 1 μm. d, days; wk, weeks.
Prominent Excessive Myelin Figures
After loss of an axon, the associated myelin sheath may collapse to a double-layered myelin figure before degradation. Also, during myelin biogenesis, myelin can extend out beyond the axon circumference and fold back onto itself. For analysis across these potential categories of myelin structures, double-layered myelin sheaths extending out from an axon or lacking a visible axon were categorized as excessive myelin figures. We quantified excessive myelin figures relative to axon pathology across a range of postinjury time points. Excessive myelin figures were associated with intact axons (Fig. 3A) or degenerating axons (Fig. 3B), and were also observed without an associated axon (Fig. 3C). Among the excessive myelin figures observed, the frequency across time points ranged from 3.0% to 4.2% for myelin figures surrounding intact axons, from 29.0% to 35.5% for myelin figures associated with degenerating axons, and from 60.3% to 67.8% for myelin figures without an axon. All 3 configurations were present at similar frequencies across time points. Excessive myelin figures were significantly increased at 3 days (62.6-fold), 1 week (45.9-fold), and 2 weeks (23.2-fold) compared to sham mice (Fig. 3D). The presence of excessive myelin figures correlated positively with the increasing frequency of degenerating axons in TBI mice (Fig. 3E). Thus, mild TBI pathology in the corpus callosum results in both axon damage and abnormal myelin sheaths.
FIGURE 3.
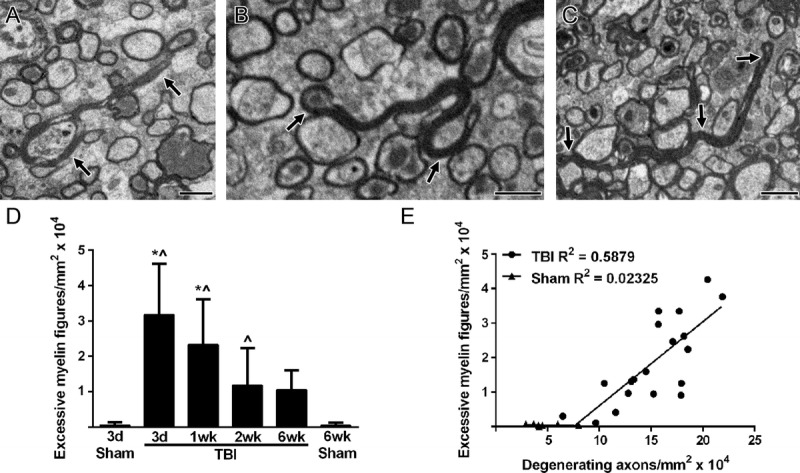
Excessive myelin figures correlating with degenerating axons. (A–C) Sagittal sections of the corpus callosum show examples of excessively long myelin folding back onto itself rather than tightly wrapping around an axon. Excessive myelin figures (black arrows) are associated with intact axons (A) and degenerating axons (B) or remain without an axon (C). (D) Excessive myelin figures are prevalent at 3 days post-TBI and continue to be elevated through at least 2 weeks after injury. * p ≤ 0.0001 compared to 3-day sham. ^p ≤ 0.0001 compared to 6-week sham for TBI at 3 days and 1 week but p ≤ 0.05 for 2-week TBI. (E) Excessive myelin figure values are graphed relative to degenerating axons. Each value represents a single mouse. All time points (3 days to 6 weeks) are included. There is a strong correlation within individual animals (Pearson r = 0.7667, p < 0.0001). Image time points: (A) 6-week TBI; (B) 1-week TBI; (C) 3-day TBI. Scale bar = 1 μm. d, days; wk, weeks.
Demyelination and Remyelination of Intact Axons
Given the extent of myelin abnormalities associated with degenerating axons, we next explored whether TBI may also result in myelin remodeling along intact axons through demyelination and remyelination. Demyelinated axons were significantly increased at 3 days (1.8-fold) and 6 weeks (1.5-fold) post-TBI but composed only a small proportion (<5%) of total axons (Figs. 4A–C). The frequency of demyelinated intact axons did not correlate with the presence of degenerating axons (Fig. 4D).
FIGURE 4.
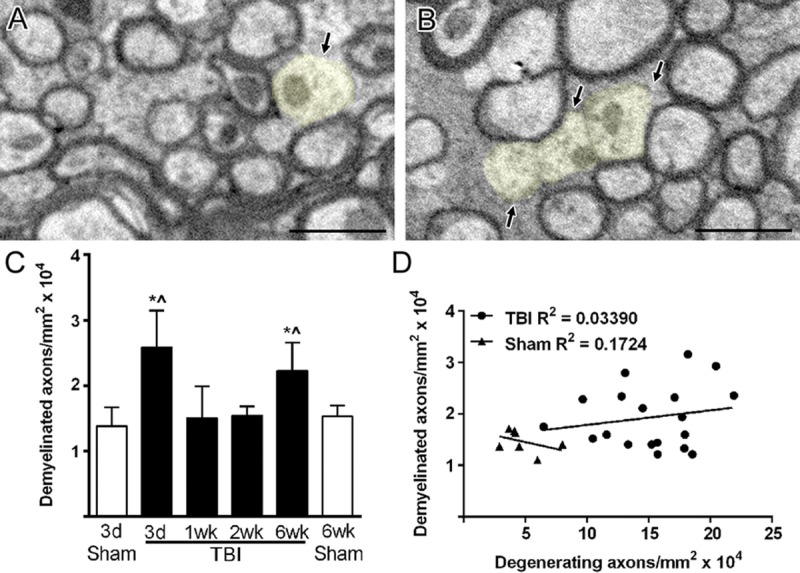
Intact axons undergoing demyelination after TBI. (A, B) Demyelinated axons (light yellow) have normal mitochondria and cytoskeletal structure but lack a myelin sheath. Only axons with a diameter greater than 0.3 μm were counted as demyelinated to avoid potential inclusion of unmyelinated axons. (C) Demyelinated axons are significantly increased after TBI but only at 3 days and 6 weeks. * p ≤ 0.01 compared to 3-day sham. ^p ≤ 0.05 compared to 6-week sham. (D) Values for demyelinated axons do not correlate with values for degenerating axons within individual animals across the post-TBI time course (3 days to 6 weeks). Image time points: (A) 3-day TBI; (B) 6-week TBI. Scale bar = 1 μm. d, days; wk, weeks.
Remyelination is indicated by myelin that is thinner than normal for a given diameter of axon (34–36). Among TBI mice, the thickness of myelin among intact fibers was reduced at 1, 2, and 6 weeks in comparison with sham time points (Fig. 5A). Comparison of sham and TBI mice across the full range of axon diameters showed a shift in the overall distribution (Figs. 5B, C) and a significant reduction of the mean axon diameter in TBI mice at both 3 days (10.2%) and 6 weeks (16.2%). The g-ratio (calculated as axon diameter divided by myelinated fiber diameter [36]) indicates remyelination more specifically than myelin thickness by accounting for axon size. The slope of the g-ratio relative to axon diameter is increased for TBI animals, indicating that myelin is thinner for a given axon diameter (Figs. 5D–G). Starting at 1 week, remyelination among the population of intact fibers is indicated by both thinner myelin (Fig. 5A) and an increased slope in the g-ratio plots (Fig. 5E). Furthermore, demyelination that was significant at 3 days returned to normal levels by 1 week (Fig. 4C), which is consistent with remyelination progressing during this interval.
FIGURE 5.
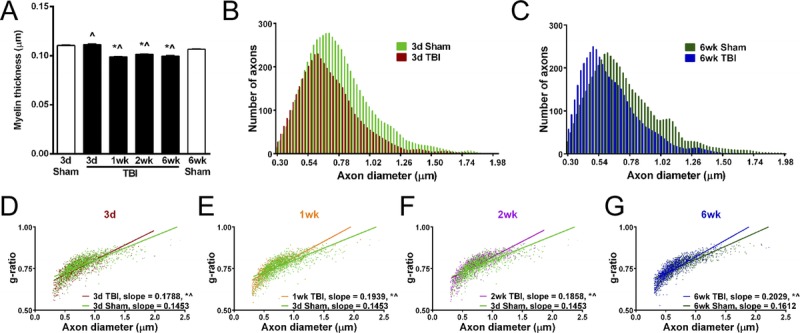
Reduction in myelin thickness after TBI. Quantitative analysis of axon diameter and myelin sheath thickness from sagittal electron micrographs of the corpus callosum. (A) Myelin thickness is significantly reduced by 1 week post-TBI and continues to be thinner through 6 weeks. (B, C) Among the total myelinated fiber populations examined, axon diameters are reduced across the range of fiber sizes at both early (B) (3 days) and late (C) (6 weeks) time points. The mean axon diameter is significantly reduced at 3 days (B) (sham, 0.78 ± 0.26; TBI, 0.70 ± 0.23; p ≤ 0.001) and 6 weeks (C) (sham, 0.74 ± 0.28; TBI, 0.62 ± 0.22; p ≤ 0.001). (D–G) Analysis of the g-ratio (axon diameter divided by myelinated fiber diameter) facilitates a more specific analysis of myelin thickness relative to a given axon size. Consistent with remyelination, the g-ratio slope is increased at each time point post-TBI. * p ≤ 0.0001 compared to 3-day sham. p ≤ 0.0005 compared to 6-week sham. d, days; wk, weeks.
Oligodendrocyte Response to Injury
Remyelination is carried out by newly generated oligodendrocytes. We prelabeled cycling progenitor cells with BrdU 1 day before injury and perfused the mice at 3 days post-TBI to examine oligodendrogenesis. Bromodeoxyuridine detection was combined with in situ hybridization for PLP to identify newly generated oligodendrocytes. Both naive and TBI mice showed BrdU incorporation in the subventricular zone, a germinal zone that serves as an internal positive control (Figs. 6A, B). Within the corpus callosum, newly generated oligodendrocytes expressing PLP and BrdU (PLP+ BrdU+) were readily detected by PLP mRNA transcript expression in the cytoplasm and by BrdU incorporation in the nucleus (Figs. 6C–E). Proteolipid protein+, BrdU+ cells were relatively rare but were significantly more prevalent in TBI versus naive mice (Fig. 6F). This increase in newly generated oligodendrocytes at 3 days post-TBI supports the potential for subsequent remyelination of demyelinated axons between 3 days and 1 week post-TBI.
FIGURE 6.
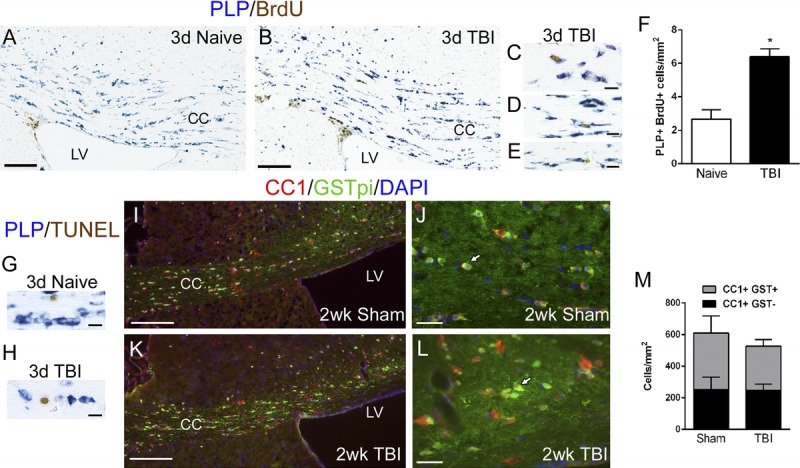
Characterization of oligodendrocyte populations after TBI. (A–E) The generation of new oligodendrocytes was examined by labeling endogenous cycling cells with a pulse of BrdU 1 day before TBI followed by a chase period through 3 days post-TBI to allow time for cell differentiation. Naive mice were run in parallel. Oligodendrocytes were identified by in situ hybridization for myelin PLP mRNA combined with immunostaining to detect nuclei with BrdU incorporation. (F) Cells double labeled for PLP and BrdU (i.e. newly differentiated oligodendrocytes) were significantly increased in the corpus callosum after TBI (* p = 0.0068). (G, H) To examine apoptosis of oligodendrocytes in the corpus callosum, we combined in situ hybridization for PLP mRNA with TUNEL staining at 3 days post-TBI or in age-matched naive mice. Terminal deoxynucleotidyl transferase dUTP nick end labeling–labeled cells were rare (1–2 cells per corpus callosum), and the labeled cells did not express PLP. (I–L) The overall population of mature oligodendrocytes was identified by double immunostaining for CC1 and GSTpi. Glutathione S-transferase is expressed in a subset of CC1-labeled oligodendrocytes (white arrows). (M) The CC1-immunolabeled populations, with or without GSTpi, are not significantly different between sham and TBI mice. Scale bars = (A, B) 250 μm; (I, K) 100 μm; (J, L) 50 μm; (C–E, G, H) 5 μm. CC, corpus callosum; d, days; LV, lateral ventricle; wk, weeks.
To further explore the basis of demyelination observed at 3 days post-TBI, we examined oligodendrocyte apoptosis using TUNEL detection combined with PLP in situ hybridization. Terminal deoxynucleotidyl transferase dUTP nick end labeling–labeled cells were readily detected but rare in the corpus callosum (Figs. 6G, H). Cells double labeled for PLP and TUNEL were even less frequent, with only 1 to 2 cells in the corpus callosum per section from either naive mice (2 ± 0 cells) or TBI mice (1 day: 1.0 ± 0.3; 3 days: 1.4 ± 0.9). Although TUNEL may not detect the full extent of apoptosis at a given point in time, this low level of TUNEL labeling during and before demyelination indicates that demyelination is not likely due to oligodendrocyte loss in this mild TBI model.
To examine the overall oligodendrocyte population more broadly, we quantified mature oligodendrocytes based on immunolabeling with 2 markers, CC1 and GSTpi (Figs. 6I–M). In both sham and TBI mice, more cells were immunolabeled by CC1 than by GSTpi. In addition, all cells labeled for GSTpi were double labeled for CC1. We observed no significant difference in the total population of oligodendrocytes labeled with CC1 or in the subpopulation labeled by GSTpi after injury (Fig. 6M). Therefore, this mild TBI does not produce a prolonged detrimental effect on the oligodendrocyte population in the corpus callosum.
Excessive Myelin Figure Frequency in a Model of Widespread Demyelination and Remyelination
The excessive myelin figures in TBI are difficult to interpret relative to the processes of demyelination and remyelination. Therefore, we compared the current TBI results with cuprizone ingestion, which exhibits distinct stages of demyelination and remyelination (Fig. 7). Short-term cuprizone treatment (4 weeks) causes death of oligodendrocytes with strong microglial activation and prevalent axon beading (28). This cuprizone time point had the highest frequency of excessive myelin figures, but the levels were only approximately one tenth of the levels observed in mice with mild TBI (Figs. 3, 7A). Prolonged cuprizone treatment (12 weeks) induced chronic demyelination throughout the corpus callosum, which resulted in lower levels of excessive myelin figures (Fig. 7A). Excessive outfoldings of myelin can occur during myelin formation in CNS development (23). Therefore, we examined excessive myelin figures during the spontaneous remyelination that occurs after removal of cuprizone from the diet (Fig. 7B). This analysis was performed with chronic cuprizone demyelination followed by a recovery period on normal chow (12 + 6 weeks) during which partial spontaneous remyelination occurs. To specifically test whether the frequency of excessive myelin figures corresponds with the extent of remyelination, we compared wild-type mice with Fgf2-null mice that undergo significantly more remyelination (29, 37). Excessive myelin figures were not more frequent in Fgf2-null mice versus wild-type mice (Fig. 7B). Notably, in comparison with the mild TBI model, excessive myelin figures were observed at much lower frequencies in the cuprizone study and were typically associated with intact axons (Figs. 7C–F).
FIGURE 7.
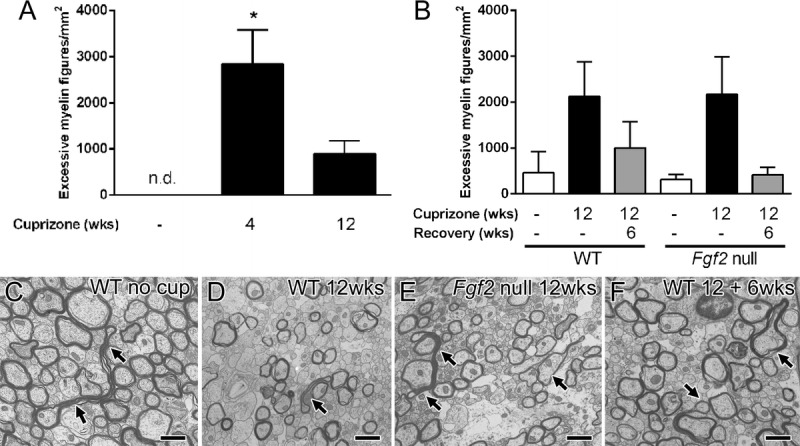
Excessive myelin figures during cuprizone-induced demyelination and remyelination. (A) In C57BL/6 mice, cuprizone ingestion produces widespread demyelination of the corpus callosum. Cuprizone treatment for 4 weeks results in oligodendrocyte loss, initial demyelination, and axon damage. Prolonged cuprizone administration through 12 weeks produces chronic demyelination. Excessive myelin figures are not detected (n.d.) in 8-week-old mice not taking cuprizone but are significantly increased during the period of initial demyelination (* p = 0.0009). (B–F) Excessive myelin figures were also analyzed relative to the extent of remyelination. Removal of cuprizone from the diet allows for endogenous progenitor cells to attempt to generate new oligodendrocytes and to initiate remyelination. Remyelination is more extensive in Fgf2-null mice in comparison with wild-type (WT) mice (29). Excessive myelin figures are similar between WT and Fgf2-null mice without cuprizone (26 weeks of age controls), during chronic demyelination (12 weeks), and after a subsequent 6-week period on a normal diet to allow for remyelination (12 + 6 weeks). (C–F) Arrows indicate examples of excessive myelin figures present in all genotypes and time points. Scale bar = 1 μm. wks, weeks.
Myelin Load
Additional characterization of excessive myelin figures was performed to interpret the relevance of this pathologic feature to axon degeneration and collapse of the remaining myelin sheath. We measured the length of each excessive myelin figure and analyzed the frequencies at each time point (Fig. 8). The average length of myelin sheaths enwrapping intact axons was calculated from data in Figure 5 and is marked by a dotted line (Fig. 8). Abnormal myelin figures shorter than this dotted line are likely myelin that collapsed and folded onto itself because of loss of the associated axon. Most abnormal myelin figures are longer than the average length predicted from simple axon collapse and many exceed the length (6.12 μm) predicted for ensheathing the largest axon quantified. The overall distribution of myelin sheath lengths among abnormal myelin figures can be compared from the area under the curve (Fig. 8, table). This calculation cannot accurately estimate the volume of excessive myelin but can serve as a surrogate measure of myelin load. This comparison indicates that the myelin load from excessive myelin figures is highest early after injury (up to 34.8-fold increased) and is still elevated 18.9-fold at 6 weeks post-TBI.
FIGURE 8.
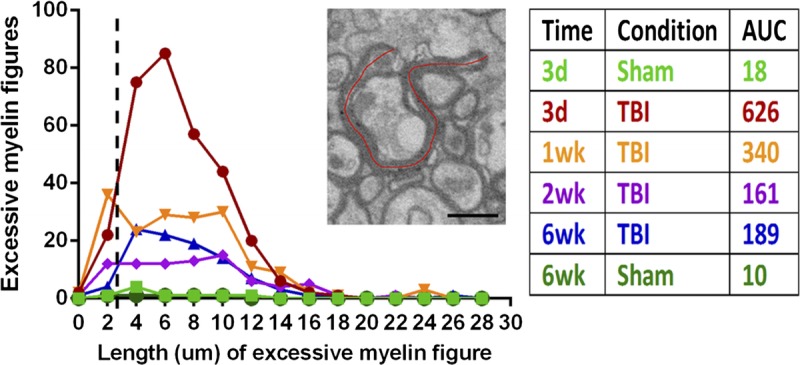
Quantification of myelin associated with excessive myelin figures. Excessive myelin figures were measured along the lumen of the myelin sheaths as shown on the micrograph (red line) and then doubled to determine the length of each excessive myelin figure. The frequency of excessive myelin figures is plotted relative to the length at each sham or TBI time point. The dotted line shows the expected average length of a myelin sheath as calculated from the average myelin thickness and axon circumference. Thus, myelin figures to the left of the dotted line are of the length expected from simple axon loss and collapse of the remaining myelin sheath. Myelin figures longer than expected (i.e. to the right of the dotted line) are present at each post-TBI time point and markedly increased in length and frequency at 3 day post-TBI. The calculated area under the curve (AUC; table) is a relative measure of myelin associated with excessive myelin figures. Scale bar = 1 μm. d, days; wk, weeks.
Persistent Neuroinflammation
Myelin debris can stimulate microglial activation (16). Therefore, excessive myelin figures could contribute to persistent neuroinflammation in this model of mild TBI. Previous characterization of this model through 1 week post-TBI showed significantly increased astrogliosis and microglial activation at 3 days and 1 week postinjury that were localized within the corpus callosum (13). We now demonstrate both ultrastructural and immunohistochemical evidence of neuroinflammation persisting in the corpus callosum through at least 6 weeks post-TBI (Fig. 9). With electron microscopy, a normal astrocyte and a microglia cell are shown in a sham mouse (Fig. 9A). In TBI mice, astrocytes exhibited cytoplasmic changes indicative of cytogenic edema (Fig. 9B), whereas microglia often contained debris indicative of phagocytic activity (Fig. 9C). Immunostaining for GFAP (astrocytes) and CD11b (microglia) detected neuroinflammation more broadly than is possible with electron microscopy sampling and demonstrated localization to the corpus callosum (Figs. 9D, E). Astrocytes and microglia undergo morphologic changes indicative of reactive and activated stages (Figs. 9F–H). Evidence of astrogliosis or microglial activation was not observed in cortical regions (data not shown). Quantification of immunostaining confirmed that GFAP staining is significantly increased in the corpus callosum at 6 weeks post-TBI (Fig. 9I). Similarly, there is also an increase in CD11b immunofluorescence in the corpus callosum of TBI mice (Fig. 9J) caused by an increase in activated microglia (Fig. 9K). Thus, reactive neuroinflammatory cells persist in the corpus callosum for at least 6 weeks after injury in this model of mild TBI.
FIGURE 9.
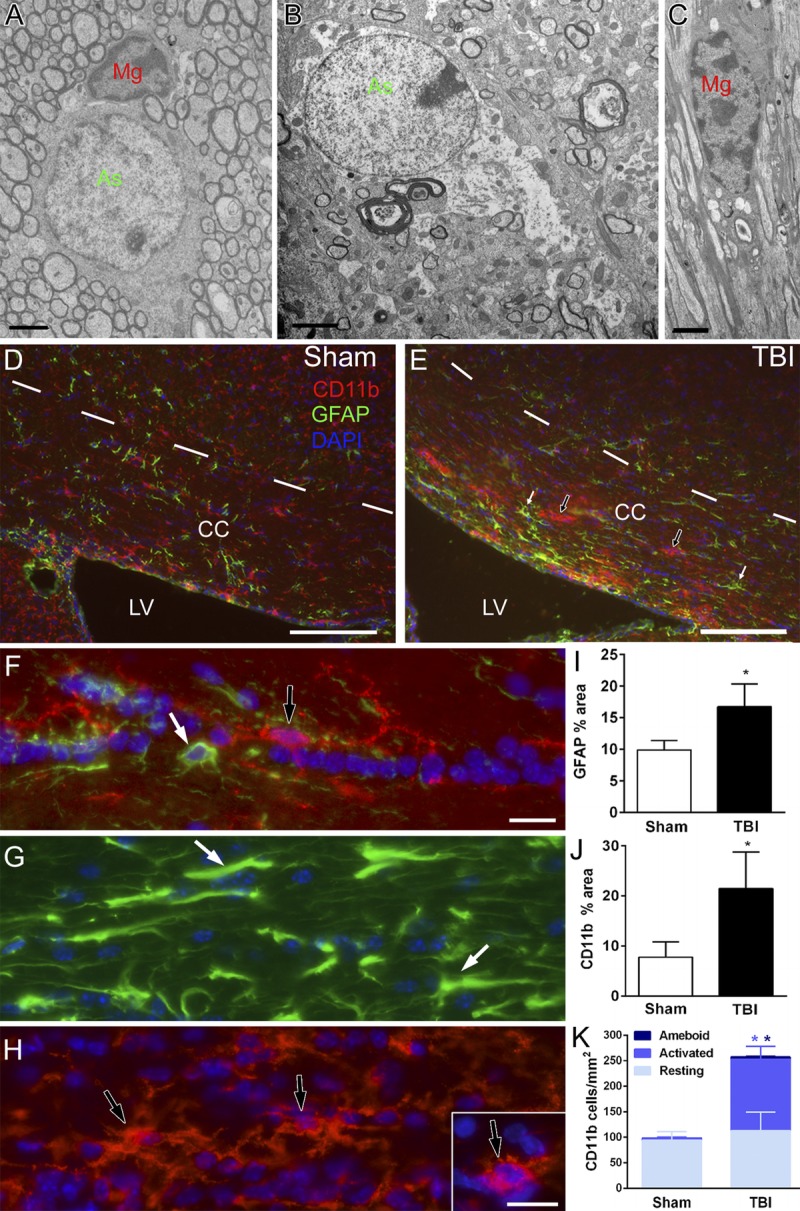
Mild TBI with axonal injury (TAI) resulting in prolonged neuroinflammation. (A–C) Electron microscopy of the corpus callosum (CC) shows normal astrocytes (As; green) and microglia (Mg; red) in sham mice (A) in contrast with images acquired in TBI mice, which exhibited hypertrophic astrocyte (B, green) and microglia (C, red) that often contained debris. (D–H) Immunohistochemistry for GFAP detects reactive astrocyte (E, white arrows) and CD11b shows activated microglia (E, black arrows) indicative of neuroinflammation localized to the CC after TBI (E) (6-week TBI) compared to normal adult phenotypes in sham mice (D) (6-week sham). The superior border of the CC is marked by dashes. The lateral ventricle (LV) is along the inferior border. (F, G) Higher magnification of GFAP immunolabeling of normal (F, white arrow) and reactive (G, white arrows) astrocyte (F, H) Higher magnification of CD11b immunolabeling of normal (F, black arrow), activated (H, black arrows), and amoeboid (H, inset) microglia. (I) At 6 weeks, astrogliosis persists in TBI animals (* p = 0.0040). (J) CD11b staining of microglia is also increased at 6weeks (* p = 0.0286). (K) Resting microglia are similar between sham and TBI mice at 6 weeks. However, activated microglia and amoeboid microglia are significantly increased (activated, * p = 0.0015; amoeboid, * p = 0.0026). Image time points: (A) 3-day sham; (B, C) 3-day TBI; (D) 6-week sham; (E) 6-week TBI. Scale bars = (A–C) 2 μm; (D, E) 100 μm; (F, G, H, and inset) 25 μm.
DISCUSSION
The current study demonstrates distinct features of myelin pathology and plasticity that influence white matter integrity after mild TBI. Closed-skull impact in adult mice resulted in damaged axons dispersed among intact fibers of the corpus callosum. Regions that exhibited this pattern of TAI also showed significant demyelination among the population of intact axons. We provide evidence of oligodendrogenesis and subsequent remyelination among the population of intact axons. Degenerating axons continued to be observed through 6 weeks post-TBI and were strongly correlated with the presence of excessive myelin sheaths. Neuroinflammation was also observed at 6 weeks postinjury and specifically localized to the corpus callosum, indicating that these features of axon and myelin pathology may lead to a long-term neurodegenerative environment.
Reduced white matter integrity is a common component of TBI, including blast exposures and mild TBI (38, 39). Axon damage is the major pathologic feature associated with reduced white matter integrity after TBI (4). However, demyelination of intact axons is a potential factor underlying desynchronized or slowed neural circuitry caused by impaired saltatory conduction. Even in the normal adult CNS, gaps in myelination along cortical projection axons modify circuit function (40). Abnormal gaps in myelination can slow information-processing speed, which is a common deficit in patients with TBI (14, 15). Social isolation has also been shown to alter myelination in the prefrontal cortex of adult mice (20). Importantly, optogenetic modulation of neuron activity in mouse cortex can modulate myelination of the associated axons and gait of the associated limb (21). These studies have led to the recognition of myelin remodeling as a form of adult plasticity. Therefore, myelination may be a neurobiologic substrate of specific neurologic deficits after TBI that could potentially be modified to improve the function of neural circuits.
The closed-skull impact model used in the current study did not cause overt gross brain pathology or evidence of contusion or hemorrhage, which is appropriate for categorization as mild TBI among preclinical models. The impact results in pathology localized in the corpus callosum over the lateral ventricles. In contrast to our initial studies in this model (13), the current experiments minimized skull fractures. Previously, axon damage was detected at intervals throughout the first week post-TBI using immunohistochemistry for βAPP (13). Although βAPP immunostaining detects impaired fast axonal transport associated with early-phase axon damage, it is not sensitive to later-stage axon pathology and neurofilament compaction (41–43). Electron microscopy can detect axon damage based on mitochondrial swelling, accumulation of vesicles, and disruption of the cytoskeleton. Using these parameters, our ultrastructural analysis demonstrated significant axon damage at 3 days postinjury that remained through 1, 2, and 6 weeks (Fig. 2). Degenerating axons were dispersed among intact fibers, as is characteristic of TAI (3, 4, 30). Mechanical injury and secondary insults result in Wallerian degeneration of axons, which involves mitochondrial dysfunction and a conserved axon death signaling pathway (44, 45). Stretch injury causes rapid dislocation of myelin paranodal junctions that may contribute to calcium changes associated with initial TAI and possibly continued axon damage (46). Our data cannot accurately quantify axons damaged from the initial injury relative to continued axon degeneration from secondary processes because Wallerian degeneration can be a prolonged process in the CNS (4, 47). Furthermore, stretch injury of the optic nerve shows that axons continue to initiate degeneration days to weeks after injury (48).
Our ultrastructural analysis demonstrates demyelination of intact axons that is not correlated with axon degeneration, indicating distinct pathologic mechanisms (Fig. 4). At 3 days post-TBI, demyelination was observed without ongoing apoptosis of oligodendrocytes (Fig. 6). These findings of mild TBI in mice contrast with studies of the corpus callosum after moderate TBI in rats. Moderate fluid percussion injury resulted in a loss of oligodendrocytes (identified by CC1) by 3 days and a marked increase in TUNEL-labeled cells in white matter by 12 hours, which continued to increase through 1 week postinjury (11, 49). Myelin loss and oligodendrocyte apoptosis, detected by CC1 combined with activated caspase-3, were significantly increased at 2 to 21 days after fluid percussion injury with hemorrhage in the corpus callosum (12). In the current study, electron microscopy revealed intact axons without myelin that have not been previously characterized in TBI. These demyelinated axons were dispersed among adjacent myelinated fibers and were not readily quantified at the light level using histologic or immunohistochemical techniques ([13] and the current study) (Fig. 1). Additional approaches will be needed to determine the mechanism of demyelination of intact axons after TBI. Distributed demyelinated axons were also observed at 6 weeks post-TBI, which may be attributable to the detrimental effects of persistent neuroinflammation and/or a lack of sufficient remyelination to compensate for continued demyelination.
Several findings indicate that demyelinated axons can remain viable and undergo remyelination between 3 days and 1 week post-TBI. Demyelination was observed at 3 days, but not at 1 week, postinjury (Fig. 4). Among the population of intact axons evaluated after TBI, remyelination was indicated by higher g-ratio values in conjunction with overall reduction in myelin thickness between 3 days and 1 week postinjury (Fig. 5). Studies of demyelinating disease models, such as cuprizone toxicity in the mouse corpus callosum, have shown that remyelination requires oligodendrocyte progenitor cells to proliferate and differentiate into new oligodendrocytes, which form the new myelin sheaths (19, 50). The cellular responses required for remyelination were found in the corpus callosum after TBI. Newly generated oligodendrocytes, colabeled for PLP and BrdU, were increased in the corpus callosum of TBI mice at 3 days (Fig. 6). Furthermore, our prior studies showed that proliferation of oligodendrocyte progenitors was significantly increased in the corpus callosum by 3 days post-TBI and continued to be elevated at 1 week (13). During this first week postinjury, oligodendrocytes also exhibited a shift to a more active transcription of PLP, which is the most abundant myelin protein (13).
Excessive myelin sheaths were prominent in the corpus callosum at all post-TBI time points and correlated with the presence of degenerating axons (Fig. 3). However, most excessive myelin figures were much longer than would be expected from a simple collapse of myelin sheaths around degenerating axons (Fig. 8). These excessive myelin figures may form from processes associated with axon loss and/or myelin synthesis. In the developing CNS, double-layered myelin figures have been referred to as “myelin outfoldings” or redundant myelin (22, 23). During myelin biogenesis, excess myelin membrane extends outward radially from the growing axon as loose layers that progressively expand around the axon and laterally along the axon (23). These extensions of excess myelin generate double-layered myelin figures. Therefore, after TBI, excessive myelin may reflect productive myelin synthesis to remyelinate viable axons and/or aberrant myelin synthesis around degenerating axons caused by dysregulation of myelin-axon signaling. The current study is the first to characterize excessive myelin figures in TBI or relative to demyelination and remyelination in a demyelinating disease model. Further studies are needed to interpret the processes leading to the generation of these excessive myelin figures in adults and the pathologic implications.
Dorsal rhizotomy studies of ascending spinal cord tracts also support a role for myelin synthesis in the formation of excessively long myelin figures. Adult cats with dorsal rhizotomy exhibit myelin figures of the lengths expected for collapsed myelin sheaths that remain after the axon degenerates (47, 51). In contrast, after dorsal rhizotomy in kittens, tracts with degenerating axons exhibit double-layered myelin figures that are much longer than in adult cats (52). In addition, excessively long double-layered myelin figures without an associated axon were observed extending around cell bodies in the lesioned tracts of kittens (52). In our adult TBI model, the excessively long myelin figures exhibit similarity to structures observed after rhizotomy in the developing CNS. Adult white matter may exhibit more excessive myelin figures only when axon damage is dispersed, as in our TBI model. Consistent with this hypothesis, similar excessive myelin figures have been illustrated in the corpus callosum associated with relatively subtle axon damage in mouse models of repetitive mild closed-skull TBI (53, 54).
Specific for mild TBI, the dispersed white matter axons damaged by TAI may be a subset of only 1 or 2 axons among a cohort of 20 or more axons ensheathed by a surviving oligodendrocyte. This mismatch of axon integrity among the cohort may dysregulate myelin maintenance signals to the oligodendrocyte, resulting in aberrant myelin synthesis. Comparison of the TBI data with the cuprizone data of corpus callosum demyelination (Fig. 7) argues against productive remyelination accounting for most excessive myelin figures observed after TBI. Cuprizone demonstrates similar excessive myelin figures that are present at a much lower frequency (Fig. 7) even though remyelination is much more extensive (29, 37). Similarly, the cuprizone example argues against the frequency of axon damage solely accounting for the abundance of excessive myelin figures after TBI. During acute cuprizone demyelination, axon damage as detected by βAPP or electron microscopy is higher than in this model of TBI (13, 28, 55), yet excessive myelin figures are much fewer (Fig. 7). However, cuprizone elicits strong microglial phagocytosis of damaged myelin, and most cases of axon damage do not progress to transection (29, 56). Interestingly, axon damage is consistently observed with cuprizone demyelination, whereas axon damage is dramatically reduced with genetic manipulations that enhance remyelination (29, 57). Further studies would be important to more specifically demonstrate the extent to which demyelination after TBI may render axons more vulnerable to further damage from secondary processes, such as neuroinflammation, and to what extent remyelination may be neuroprotective.
In this model of TBI, we have shown that neuroinflammation localized within the corpus callosum during the first week after injury (13) and after a prolonged 6-week survival period (Fig. 9). Localization of microglial activation in the corpus callosum has been observed in a spectrum of TBI from repetitive mild TBI models in mice (17, 53) to chronic cases of single moderate to severe TBI in humans (58). Myelin changes, including excessive or redundant myelin, are increased in aged nonhuman primate white matter and potentially related to microglial senescence and reduced efficiency of clearance of myelin debris with aging (59, 60). The relationship between excessive myelin figures and neuroinflammation will be important to delineate in future studies. Activated microglia and reactive astrocytes synthesize increased levels of growth factors and cytokines that regulate oligodendrocyte progenitor proliferation, differentiation, and myelin synthesis (61, 62). On the other hand, myelin debris can stimulate microglial activation (16). Therefore, in addition to axon damage, myelin degradation may be a key element stimulating neuroinflammation and propagating chronic neurodegeneration in white matter tracts.
Our findings highlight the need to evaluate changes in both axon integrity and myelination throughout the postinjury time course after TBI. Magnetic resonance imaging studies in patients with mild TBI demonstrated that diffusion tensor imaging of white matter is a significant predictor of outcome (39). Characteristics of the injury and host factors, including the immune response, may contribute to the progression of white matter damage and repair after TBI. These complex interactions warrant further attention to identify strategies for preventing white matter degeneration and enhancing recovery.
ACKNOWLEDGMENTS
We thank Tuan Le and Dr Fengshan Yu for technical assistance. We thank laboratory members, Dr Kimberly Byrnes, and Dr Aviva Symes for providing critical comments on the manuscript.
Footnotes
This work was supported by the US Department of Defense in the Center for Neuroscience and Regenerative Medicine.
REFERENCES
- 1. Roozenbeek B, Maas AI, Menon DK. Changing patterns in the epidemiology of traumatic brain injury. Nat Rev Neurol 2013; 9: 231– 36 [DOI] [PubMed] [Google Scholar]
- 2. McMahon P, Hricik A, Yue JK, et al. Symptomatology and functional outcome in mild traumatic brain injury: Results from the prospective TRACK-TBI study. J Neurotrauma 2014; 31: 26– 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Smith DH, Hicks R, Povlishock JT. Therapy development for diffuse axonal injury. J Neurotrauma 2013; 30: 307– 23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bigler ED, Maxwell WL. Neuropathology of mild traumatic brain injury: Relationship to neuroimaging findings. Brain Imaging Behav 2012; 6: 108– 36 [DOI] [PubMed] [Google Scholar]
- 5. Bigler ED, Maxwell WL. Neuroimaging and neuropathology of TBI. NeuroRehabilitation 2011; 28: 63– 74 [DOI] [PubMed] [Google Scholar]
- 6. McAllister TW, Ford JC, Ji S, et al. Maximum principal strain and strain rate associated with concussion diagnosis correlates with changes in corpus callosum white matter indices. Ann Biomed Eng 2012; 40: 127– 40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Eierud C, Craddock RC, Fletcher S, et al. Neuroimaging after mild traumatic brain injury: Review and meta-analysis. Neuroimage Clin 2014; 4: 283– 94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reeves TM, Phillips LL, Povlishock JT. Myelinated and unmyelinated axons of the corpus callosum differ in vulnerability and functional recovery following traumatic brain injury. Exp Neurol 2005; 196: 126– 37 [DOI] [PubMed] [Google Scholar]
- 9. Reeves TM, Smith TL, Williamson JC, et al. Unmyelinated axons show selective rostrocaudal pathology in the corpus callosum after traumatic brain injury. J Neuropathol Exp Neurol 2012; 71: 198– 210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Abdel Baki SG, Schwab B, Haber M, Fenton AA, Bergold PJ. Minocycline synergizes with N-acetylcysteine and improves cognition and memory following traumatic brain injury in rats. PLoS One 2010; 5: e12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lotocki G, de Rivero Vaccari JP, Alonso O, et al. Oligodendrocyte vulnerability following traumatic brain injury in rats. Neurosci Lett 2011; 499: 143– 48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Flygt J, Djupsjo A, Lenne F, et al. Myelin loss and oligodendrocyte pathology in white matter tracts following traumatic brain injury in the rat. Eur J Neurosci 2013; 38: 2153– 65 [DOI] [PubMed] [Google Scholar]
- 13. Sullivan GM, Mierzwa AJ, Kijpaisalratana N, et al. Oligodendrocyte lineage and subventricular zone response to traumatic axonal injury in the corpus callosum. J Neuropathol Exp Neurol 2013; 72: 1106– 25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dams-O’Connor K, Spielman L, Singh A, et al. The impact of previous traumatic brain injury on health and functioning: A TRACK-TBI study. J Neurotrauma 2013; 30: 2014– 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Donders J, Strong CA. Clinical utility of the Wechsler Adult Intelligence Scale–Fourth Edition after traumatic brain injury. Assessment 2015; 22: 17– 22 [DOI] [PubMed] [Google Scholar]
- 16. Clarner T, Diederichs F, Berger K, et al. Myelin debris regulates inflammatory responses in an experimental demyelination animal model and multiple sclerosis lesions. Glia 2012; 60: 1468– 80 [DOI] [PubMed] [Google Scholar]
- 17. Mouzon BC, Bachmeier C, Ferro A, et al. Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Ann Neurol 2014; 75: 241– 54 [DOI] [PubMed] [Google Scholar]
- 18. Bruce CC, Zhao C, Franklin RJ. Remyelination—an effective means of neuroprotection. Horm Behav 2010; 57: 56– 62 [DOI] [PubMed] [Google Scholar]
- 19. Irvine KA, Blakemore WF. Remyelination protects axons from demyelination-associated axon degeneration. Brain 2008; 131: 1464– 77 [DOI] [PubMed] [Google Scholar]
- 20. Liu J, Dietz K, DeLoyht JM, et al. Impaired adult myelination in the prefrontal cortex of socially isolated mice. Nat Neurosci 2012; 15: 1621– 23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gibson EM, Purger D, Mount CW, et al. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 2014; 344: 1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rosenbluth J. Redundant myelin sheaths and other ultrastructural features of the toad cerebellum. J Cell Biol 1966; 28: 73– 93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Snaidero N, Mobius W, Czopka T, et al. Myelin membrane wrapping of CNS axons by PI(3,4,5)P3-dependent polarized growth at the inner tongue. Cell 2014; 156: 277– 90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mierzwa AJ, Sullivan GM, Beer LA, Armstrong RC. Comparison of cortical and white matter traumatic brain injury models reveals differential effects in the subventricular zone and divergent Sonic hedgehog signaling pathways in neuroblasts and oligodendrocyte progenitors. ASN Neuro 2014; 6: 1– 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reynolds ES. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J Cell Biol 1963; 17: 208– 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Armstrong RC, Le TQ, Frost EE, et al. Absence of fibroblast growth factor 2 promotes oligodendroglial repopulation of demyelinated white matter. J Neurosci 2002; 22: 8574– 85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sturrock RR. Myelination of the mouse corpus callosum. Neuropathol Appl Neurobiol 1980; 6: 415– 20 [DOI] [PubMed] [Google Scholar]
- 28. Xie M, Tobin JE, Budde MD, et al. Rostrocaudal analysis of corpus callosum demyelination and axon damage across disease stages refines diffusion tensor imaging correlations with pathological features. J Neuropathol Exp Neurol 2010; 69: 704– 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tobin JE, Xie M, Le TQ, et al. Reduced axonopathy and enhanced remyelination after chronic demyelination in fibroblast growth factor 2 (Fgf2)–null mice: Differential detection with diffusion tensor imaging. J Neuropathol Exp Neurol 2011; 70: 157– 65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reichard RR, Smith C, Graham DI. The significance of beta-APP immunoreactivity in forensic practice. Neuropathol Appl Neurobiol 2005; 31: 304– 13 [DOI] [PubMed] [Google Scholar]
- 31. Smith DH, Meaney DF, Shull WH. Diffuse axonal injury in head trauma. J Head Trauma Rehabil 2003; 18: 307– 16 [DOI] [PubMed] [Google Scholar]
- 32. Adams JH, Doyle D, Ford I, et al. Diffuse axonal injury in head injury: Definition, diagnosis and grading. Histopathology 1989; 15: 49– 59 [DOI] [PubMed] [Google Scholar]
- 33. Kim JJ, Gean AD. Imaging for the diagnosis and management of traumatic brain injury. Neurotherapeutics 2011; 8: 39– 53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bunge MB, Bunge RP, Ris H. Ultrastructural study of remyelination in an experimental lesion in adult cat spinal cord. J Biophys Biochem Cytol 1961; 10: 67– 94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ludwin SK, Maitland M. Long-term remyelination fails to reconstitute normal thickness of central myelin sheaths. J Neurol Sci 1984; 64: 193– 98 [DOI] [PubMed] [Google Scholar]
- 36. Rushton WA. A theory of the effects of fibre size in medullated nerve. J Physiol 1951; 115: 101– 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Armstrong RC, Le TQ, Flint NC, et al. Endogenous cell repair of chronic demyelination. J Neuropathol Exp Neurol 2006; 65: 245– 56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yeh PH, Wang B, Oakes TR, et al. Postconcussional disorder and PTSD symptoms of military-related traumatic brain injury associated with compromised neurocircuitry. Hum Brain Mapp 2014; 35: 2652– 73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yuh EL, Cooper SR, Mukherjee P, et al. Diffusion tensor imaging for outcome prediction in mild traumatic brain injury: a TRACK-TBI study. J Neurotrauma 2014; 31: 1457– 77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tomassy GS, Berger DR, Chen HH, et al. Distinct profiles of myelin distribution along single axons of pyramidal neurons in the neocortex. Science 2014; 344: 319– 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stone JR, Singleton RH, Povlishock JT. Antibodies to the C-terminus of the beta-amyloid precursor protein (APP): A site specific marker for the detection of traumatic axonal injury. Brain Res 2000; 871: 288– 302 [DOI] [PubMed] [Google Scholar]
- 42. Gallyas F, Pal J, Farkas O, et al. The fate of axons subjected to traumatic ultrastructural (neurofilament) compaction: An electron-microscopic study. Acta Neuropathol 2006; 111: 229– 37 [DOI] [PubMed] [Google Scholar]
- 43. DiLeonardi AM, Huh JW, Raghupathi R. Impaired axonal transport and neurofilament compaction occur in separate populations of injured axons following diffuse brain injury in the immature rat. Brain Res 2009; 1263: 174– 82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Conforti L, Gilley J, Coleman MP. Wallerian degeneration: An emerging axon death pathway linking injury and disease. Nat Rev Neurosci 2014; 15: 394– 409 [DOI] [PubMed] [Google Scholar]
- 45. Villegas R, Martinez NW, Lillo J, et al. Calcium release from intra-axonal endoplasmic reticulum leads to axon degeneration through mitochondrial dysfunction. J Neurosci 2014; 34: 7179– 89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Maxwell WL. Damage to myelin and oligodendrocytes: A role in chronic outcomes following traumatic brain injury? Brain Sci 2013; 3: 1374– 94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vargas ME, Barres BA. Why is Wallerian degeneration in the CNS so slow? Annu Rev Neurosci 2007; 30: 153– 79 [DOI] [PubMed] [Google Scholar]
- 48. Maxwell W, Bartlet E, Morgan H. Wallerian degeneration in the optic nerve stretch-injury model of TBI: A stereological analysis. J Neurotrauma [published online ahead of print October 21, 2014]. doi:10.1089/neu.2014.3369. [DOI] [PubMed] [Google Scholar]
- 49. Conti AC, Raghupathi R, Trojanowski JQ, et al. Experimental brain injury induces regionally distinct apoptosis during the acute and delayed post-traumatic period. J Neurosci 1998; 18: 5663– 72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Murtie JC, Zhou YX, Le TQ, et al. PDGF and FGF2 pathways regulate distinct oligodendrocyte lineage responses in experimental demyelination with spontaneous remyelination. Neurobiol Dis 2005; 19: 171– 82 [DOI] [PubMed] [Google Scholar]
- 51. Franson P, Ronnevi LO. Myelin breakdown and elimination in the posterior funiculus of the adult cat after dorsal rhizotomy: A light and electron microscopic qualitative and quantitative study. J Comp Neurol 1984; 223: 138– 51 [DOI] [PubMed] [Google Scholar]
- 52. Franson P, Ronnevi LO. Myelin breakdown in the posterior funiculus of the kitten after dorsal rhizotomy. A qualitative and quantitative light and electron microscopic study. Anat Embryol (Berl) 1989; 180: 273– 80 [DOI] [PubMed] [Google Scholar]
- 53. Shitaka Y, Tran HT, Bennett RE, et al. Repetitive closed-skull traumatic brain injury in mice causes persistent multifocal axonal injury and microglial reactivity. J Neuropathol Exp Neurol 2011; 70: 551– 67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bennett RE, Brody DL. Acute reduction of microglia does not alter axonal injury in a mouse model of repetitive concussive traumatic brain injury. J Neurotrauma 2014; 31: 1647– 63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Song SK, Yoshino J, Le TQ, et al. Demyelination increases radial diffusivity in corpus callosum of mouse brain. Neuroimage 2005; 26: 132– 40 [DOI] [PubMed] [Google Scholar]
- 56. Hibbits N, Yoshino J, Le TQ, et al. Astrogliosis during acute and chronic cuprizone demyelination and implications for remyelination. ASN Neuro 2012; 4: 393– 408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhou YX, Pannu R, Le TQ, et al. Fibroblast growth factor 1 (FGFR1) modulation regulates repair capacity of oligodendrocyte progenitor cells following chronic demyelination. Neurobiol Dis 2012; 45: 196– 205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Johnson VE, Stewart W, Smith DH. Axonal pathology in traumatic brain injury. Exp Neurol 2013; 246: 35– 43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: An essential link between degeneration and regeneration. Brain 2009; 132: 288– 95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Peters A. The effects of normal aging on myelin and nerve fibers: A review. J Neurocytol 2002; 31: 581– 93 [DOI] [PubMed] [Google Scholar]
- 61. Gallo V, Deneen B. Glial development: The crossroads of regeneration and repair in the CNS. Neuron 2014; 83: 283– 308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nayak D, Roth TL, McGavern DB. Microglia development and function. Annu Rev Immunol 2014; 32: 367– 402 [DOI] [PMC free article] [PubMed] [Google Scholar]