

Abstract
To date, no truly efficacious drugs for Alzheimer’s disease (AD) have been developed; moreover, all new anti-AD drugs developed since 2003 have failed. To succeed where previous ones have failed in drug development, new approaches for AD therapy are needed. Here we discuss the potential application of network medicine as a new approach to AD treatment. Unlike traditional approaches focused on a single target/pathway, network medicine targets and restores disease-disrupted networks through simultaneous modulation of numerous proteins (targets)/pathways involved in AD pathogenesis. We consider several drug candidates under development for AD therapy, including Keap1–Nrf2 regulators, endogenous neurogenic agents, and hypoxia-inducible factor 1 (HIF-1) activators. These drug candidates are multi-target ligands with the potential to further develop as network medicines, since they act as master regulators to initiate a broad range of cellular defense mechanisms/cytoprotective genes that exert their efficacy in a holistic way. We also explore their diverse mechanisms of action and potential disease-modifying effects, which may have profound implications for drug discovery.
Keywords: Keap1–Nrf2 regulator, neurogenic agents, HIF-1 activator, AChE-MAO-A/B inhibitor, M30, Alzheimer’s disease
Introduction
Alzheimer’s disease (AD) is a fatal, complex, neurodegenerative disease affecting over 5 million Americans as of 2014, with one new AD case being reported every 67 seconds in the United States.1 The annual cost for AD treatment is estimated to be approximately 172 billion dollars in USA and anticipated to soar to 1.1 trillion dollars by 2050.1 Unfortunately, to date, no truly effective drugs have been developed for AD treatment. And moreover, all new anti-AD drugs developed since 2003 have failed.2 To succeed where previous ones have failed in drug discovery, new therapeutic approaches must be developed. Recently, network medicine has attracted increasing interest as a promising alternative for effectively treating complex diseases like AD and cancer.3,4 The advances in network medicine have greatly increased our knowledge of multi-pathway interactions in complex diseases and may revolutionize our approaches to next-generation drug design and development. From the network medicine perspective, diseases are the results of system breakdown of biological networks within the body because of suppression or activation of certain stages, leading to an unbalance of input–output in the biological networks.5 The goal of therapy is to restore the disturbed disease networks by simultaneously targeting key components in disease networks.6 However, most disturbed disease networks are difficult to restore through intervention of a single node (protein/signaling pathway) in the network, because there exist robust and redundant cell mechanisms in biological systems.7 These may be the reasons that conventional approaches (one drug, one target) have yet to yield effective drugs for treating complex diseases, no matter how strongly these drugs bind to their respective targets.8
Complex diseases are network-related diseases, which are rarely caused by a single gene (or single signaling pathway) dysfunction. Instead, these diseases are likely a result of disturbed disease networks, involving dysfunction of numerous genes, proteins, and signaling pathways.6 System biology suggests that effective treatment of complex diseases – like AD and cancer – needs to restore disrupted disease networks, which often requires simultaneous (or even dynamically simultaneous) modulation of multiple proteins (targets)/pathways.4 In AD patients, many brain networks are disrupted, including default mode network (DMN), sensory–motor network (SMN), dorsal attention network (DAN), salience network (SAL), and control network (CON).9 A recent study that included 510 adults (average age 77 years) demonstrated that brain networks, like DMN, SMN, DAN, SAL, and COM, exhibited reduced correlations in mild AD patients. DMN, a network of brain regions that becomes active when the mind is at rest, was disrupted in individuals with very mild to mild AD. Cross-network relations were consistently lost with increasing AD severity.9 In addition, widespread loss of both intra-network and inter-network connections was also seen with AD progression.10 Other studies found that brain networks were decayed and disrupted in early AD, which occurred at about the same time that Aβ levels began to rise and tau levels started to drop in spinal fluid.11 Moreover, DMN disruption, which emerges during the presymptomatic phase and intensifies with AD progression, was linked to core memory and visuospatial deficits.12 Studies also found that there was obvious loss in parietal cortex functional connectivity in AD.13 The loss of communication between different functional brain regions reflected cognitive decline in AD patients.13
The fundamental principles of network medicine have been used intentionally or unintentionally in the development of multi-target and cocktail drugs. Multi-target and cocktail drugs may not be considered as network medicines, but they could be further developed into network medicines. In fact, clinical studies have demonstrated that multi-target and cocktail drugs are more efficacious than single-target drugs in treating complex diseases such as cancer, HIV, and depression.14–16 In AD treatment, cocktail drugs (eg, memantine plus donepezil) produce substantially reduced rates of clinical worsening, demonstrate good safety and tolerability, and offer greater improvement in cognition, function, and global status in patients with moderate and severe AD.17
In this review, we examine several drug candidates under development for AD therapy, including Kelch like-ECH-associated protein 1 (Keap1)–nuclear factor (erythroid-derived 2)-like 2 (Nrf2) regulators, endogenous neurogenic agents, and hypoxia-inducible factor 1 (HIF-1) activators. These candidates are multi-target ligands that may be viewed as network medicines, since they act as master regulators of cellular defense mechanisms or cytoprotective genes that may function synergistically to exert their efficacy in a holistic way. We also explore their underlying mechanisms of action and potential disease-modifying effects, which may have profound implications for drug discovery.
Keap1–Nrf2 Regulators
A large body of evidence has demonstrated that oxidative stress (OS) in the brain plays a pivotal role in AD pathogenesis.18 OS occurs at early stages of AD before the appearance of amyloid plaques and neurofibrillary tangles and acts to exacerbate the disease progression. It induces and activates multiple cell signaling pathways that contribute to neurodegeneration in AD.19 In addition, OS is closely associated with Aβ, in which OS increases Aβ production and subsequently Aβ aggregation that further induces and exacerbates OS. This represents a vicious circle that favors Aβ toxicity and neurodegeneration.19–23 In fact, several reports have demonstrated antioxidants functioning as effective anti-AD drugs.23–25 And a wide variety of antioxidants have shown promising results in culture and animal AD models.23–25 However, large-scale clinical trials on antioxidants for AD therapy so far have been rather controversial and elusive, and none of the investigated antioxidant drugs survive.23 Various factors may contribute to this failure, which may include: (1) the beneficial effects of most antioxidants are tested in animal models, and in most cases, these animal models do not sufficiently reflect disease in humans. (2) The poor bioavailability to targeted site (brain) and low antioxidant capacity to counteract the wide variety of sources and sites of OS. (3) Many antioxidants may also induce OS in high concentrations or under unfavored environmental conditions. The pro-oxidative property of antioxidants may counteract the benefits of antioxidants in the clinic. (4) The complexity of OS network in the human body. OS is a complex cellular process with numerous sources and sites of production, and without specific treatment target or a major metabolic pathway. Treating with only a single exogenous antioxidant may not be sufficient to restore the unbalanced network in body’s defense system.
In 2013, FDA approved Tecfidera for the treatment of multiple sclerosis (MS). MS is a chronic inflammatory disease of the central nervous system (CNS), in which oxidative damage is extensive and widespread in the disease lesions. The oxidative damage is seen predominantly in MS initial stage, which contributes to the underlying neurodegeneration.26 Therefore, antioxidant therapy has been long proposed as a promising treatment for MS. Indeed, a wide variety of antioxidants have been shown to be beneficial in culture and animal models for MS treatment.26 However, prior to the discovery of Tecfidera, no antioxidants demonstrated therapeutic efficacy in MS treatment in phase 3 clinical trials.23,27 Tecfidera or dimethyl fumarate (DMF) is a novel antioxidant that is intrinsically different from those failed antioxidants. In phase 3 trials, DMF (240 mg, three times daily for 2 years) reduced annualized relapse rate (ARR) 51% and the risk of relapse 50% in patients with relapsing-remitting MS, compared with placebo. DMF was generally well tolerated with an acceptable safety profile and similar side effects across treatment groups.28
The recent FDA approval of DMF for MS treatment has profound implications in drug discovery for designing novel antioxidant drugs, which may be more effective than current antioxidant drugs for treating OS-related diseases. It is known that most of the antioxidant drugs that failed in clinical trials are exogenous antioxidants. These antioxidants may act through several mechanisms including scavenging oxidant species, neutralizing radicals, and/or chelating metal ions that catalyze radicals generation. By contrast, DMF works by combined or synergistic effects of diverse endogenous antioxidants produced upon activation of the Keap1–Nrf2 stress response pathway.29 Nrf2 is a transcription factor that plays a key role in cellular stress responses. Under normal conditions, Keap1 binds to Nrf2 in the cytoplasm, promoting Nrf2 ubiquitination, and subsequent degradation by the proteasome. Under OS or in the presence of Nrf2 activators, the binding of Keap1 with Nrf2 is disrupted, which releases Nrf2. The released Nrf2 translocates from the cytoplasm into the nucleus where it forms heterodimers with other transcription factors such as c-Jun and binds to antioxidant response elements (AREs). This ARE–Nrf2 binding leads to activation of the Keap1–Nrf2 pathway, which induces the expression of over 100 cytoprotective genes. The induced genes include (1) the cellular antioxidant and anti-inflammatory defense enzymes such as NAD(P)H quinone oxyreductase, glutathione. (2) Glutathione biosynthesis enzymes, extracellular superoxide dismutase, and glutamate-6-phosphate dehydrogenase. (3) Pro- and anti-inflammatory enzymes such as cyclooxygenase-2 (COX-2), inducible nitric oxide synthase (iNOS), and heme oxygenase-1 (HO-1). In addition, Nrf2 activation also induces the expression of genes that regulate mitochondrial biogenesis and glycolytic metabolism, angiogenesis, cell survival, apoptosis, and other processes that interfere with cell survival.30 Therefore, Nrf2 activators act as master regulators to activate a broad range of cellular defense processes and cytoprotective genes. These activated genes and processes may work together or even synergistically to exert their efficacy in a holistic way. In this regard, Nrf2 activators may be considered as network medicines.
The underlying mechanisms of action of DMF have profound implications for drug discovery. Accumulated evidence has revealed that DMF is a novel antioxidant intrinsically different from those failed antioxidants in the clinic. It has multiple activities, including acting as an Nrf2 activator and working on both antioxidant and immunomodulatory pathways.29 DMF, after oral intake, was rapidly converted into its active metabolite monomethyl fumarate (MMF), and administration of either DMF or MMF before an oxidative challenge prevented free radical-mediated cell death in primary astrocytes and neurons in an Nrf2-dependent manner. Either DMF or MMF treatment was found to increase cellular redox potential, glutathione, ATP levels, and improve mitochondrial function. In addition, studies have shown that DMF, via either Nrf2-dependent or Nrf2-independent pathway, promoted anti-inflammatory activities including (1) attenuation of pro-inflammatory cytokine production31; (2) regulation of NFkB activation32; and (3) reduced activation of macrophages, microglia, and astrocytes.33 DMF also acted as an immunomodulator to regulate immune homeostasis and reduce CNS infiltration of immune cells. Other activities of DMF include: (1) regulation of OS-induced intracellular Ca2+ accumulation; (2) modulation of IL-12 and IL-23 production to ameliorate inflammatory autoimmune diseases34; (3) potential protection against multiple forms of neurodegenerative stimuli such as excitotoxicity and demyelination.29
Taken together, DMF is indeed a multi-target ligand, acting primarily as an Nrf2 activator that initiates a wide variety of cellular processes. Figure 1 summarizes the multiple activities and important mechanisms of DMF action.
Figure 1.
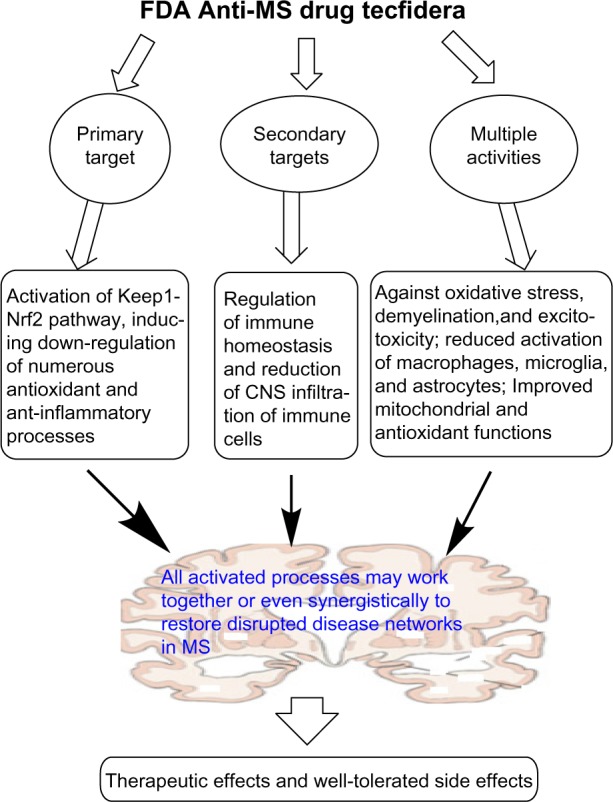
The multiple targets and diverse pharmacological activities of FDA-approved anti-MS drug Tecfidera (DMF).
The success story of the Nrf2 activator DMF for MS treatment has clearly shown that the Keap1–Nrf2 pathway is a viable target with excellent therapeutic potential. In fact, besides for treating MS, Nrf2 activators may be therapeutically advantageous in other neurodegenerative diseases including AD, Parkinson’s disease (PD), and Amyotrophic lateral sclerosis (ALS).35–37 Previous studies have revealed that the Keap1–Nrf2 pathway can regulate many processes related to AD pathogenesis, including Aβ deposition, iron homeostasis, OS, mitochondrial dysfunction, astrocyte activation, and glucose metabolism.30 HIF-1 regulators have potential to reduce Aβ-promoted astrocyte activation, prevent Aβ toxicity, and improve brain glucose metabolism. For example, Soucek et al showed that activation of HIF-1 by overexpression of a non-degradable HIF-1α prevented Aβ1–42-induced toxicity; pre-treating with the HIF-1α inducer mimosine rendered neuronal cells resistant to toxicity induced by subsequent lethal dose of Aβ through HIF-1α induction; and Aβ-induced changes in glucose metabolism were mediated by HIF-1α activation.38 In addition, Schubert et al (2009) showed that Aβ-dependent astrocyte activation led to a long-term decrease in HIF-1α expression and a reduction in the rate of glycolysis.39 The astrocyte activation and glycolytic changes were reversed by maintenance of HIF-1α levels with the conditions that prevent HIF-1α proteolysis.39 Aβ was also found to inhibit (2–6 hours) then activate (24 hours) proteasome activity, leading to a transient increase then a net decrease in HIF-1α expression.39 Some HIF-1α activators, such as DFO and curcumin, have shown promising results for AD treatment. For example, in a single clinical trial conducted in AD patients, intramuscular injection of DFO (twice daily) was found to slow cognitive decline by 50% in 2 years.40 However, DFO has poor bioavailability in the brain and potential side effects,41 which hinder its further development as an anti-AD drug.
A new generation of Nrf2 activators has been developed in order to avoid the potential side effects of most current known Nrf2 activators. DMF, like most of the current known Nrf2 activators, including endogenous metabolites and natural products, acts as an electrophile and inhibits Keap1–Nrf2 interaction through covalent S-alkylation of cysteine thiols of Keap1. This nonselective covalent modification of cysteine raises additional concerns about the potential side effects, because these Nrf2 activators potentially react with other cysteine-containing proteins and enzymes. These concerns are deepening because of the serious side effects of the electrophilic Nrf2 activator, bardoxolone methyl, which led to the termination of its clinical trials in patients with type 2 diabetes mellitus (T2DM) and stage 4 chronic kidney disease (CKD).42 To avoid the potential risk of “off-target” toxic effects because of nonspecific cysteine modification, several new Nrf2 activators have been developed. These new Nrf2 activators inhibit Keap1–Nrf2 interaction through reversibly binding to Keap1. One such example is compound 2, as shown in Figure 2. Compound 2 has been reported to be the most potent Nrf2 activator to date, which binds to Keap1 to directly disrupt the Keap1–Nrf2 interaction with an EC50 of 28.6 nM in the fluorescence polarization assay. Compound 2 was found to remarkably activate the Keap1–Nrf2 pathway and significantly increase the transcription of the Nrf2 targeting genes. However, the poor cell permeability of compound 2 may hinder its further development.43
Figure 2.
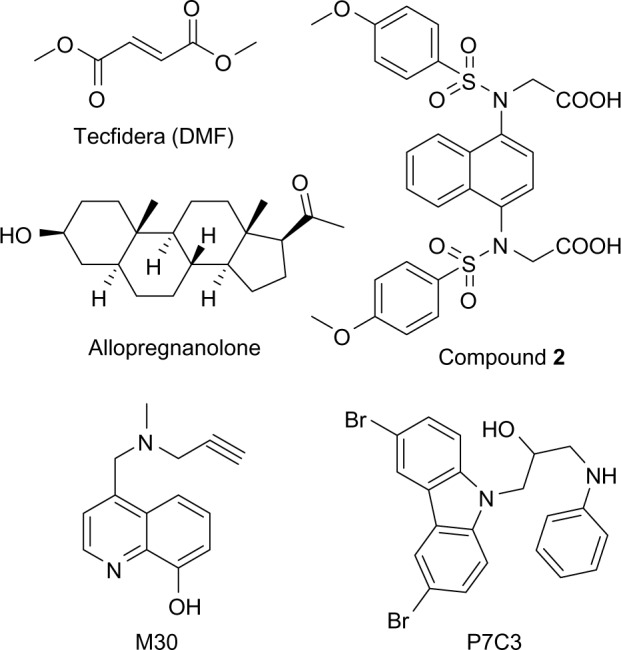
Structures of Nrf2 activators DMF and 2, neurogenic agents allopregnanolone and P7C3, and multi-target ligand M30, fumarate (13) and CDDO-Me (14).
Neurogenic Agents
AD is characterized by progressive loss of neurons in the cortical area (especially hippocampal) and cholinergic neurons with massive brain atrophy and degeneration. This neuronal loss has been correlated with cognitive deterioration and memory loss in AD patients.44 Accordingly, drugs with the ability to prevent neuron death, to salvage dying neurons, and especially to induce neuron regeneration, have great potential for AD treatment.45,46 It is known that neurogenesis, although most active during prenatal development, still occurs in adult brain, which plays an important role in various forms of learning and memory.47 Neurogenesis is impaired in AD brain, along with a concomitant decline in cognitive function.47 The impairment of neurogenesis occurred early in AD etiology, which preceded plaque and tangle formation in AD 3xTg mouse models.48 Enhancement of neurogenesis to promote neuron regeneration has shown improved cognition in AD animal models.49 However, many challenges still remain to the approaches to regenerate the lost neurons to restore cognitive function in AD patients. Neural stem cells, despite having great potential for neuron regeneration, have limited use in the clinic because of the challenge of their delivery to the brain.50,51 An alternative approach that may be more amenable to clinical application in patients may be to identify small molecules with neurogenic activity. In fact, induction of neurogenesis through the brain-permeable molecules has attracted increasing interest, and numerous molecules with neurogenic activity have been identified in vitro.52,53 However, only a few such compounds have been investigated in terms of their in vivo neurogenic activity.49,54 Among these, allopregnanolone (Fig. 2) has been investigated as a potential anti-AD drug and may serve as proof of concept for therapeutics that target endogenous regeneration.54 Allopregnanolone is a prototypic neurosteroid that exhibits an age- and AD-associated decrease in the brain. Allopregnanolone acts as a potent modulator of GABAA receptors. In vitro, allopregnanolone was found to induce neurogenesis in human and rat neural progenitors in a dose-dependent and steroid-specific manner.55 In vivo with 3xTgAD mouse models, allopregnanolone significantly increased neurogenesis within the subgranular zone of dentate gyrus and the subventricular zone. It also reversed neurogenic deficits in the hippocampus of young and aging 3xTgAD mice. In addition, allopregnanolone reversed the learning and memory deficits in the 3xTgAD mouse and restored both regenerative and cognitive function to that of the normal nontransgenic mouse.56 Chronic administration of allopregnanolone significantly increased the survival of newly generated neurons with simultaneous reduction of Aβ oligomer accumulation and microglia activation in the 3xTgAD mouse.57
Allopregnanolone is currently moving from laboratory discovery to clinical trial as a potential regenerative drug for the treatment of AD.58 However, safety concerns exist with chronic exposure to allopregnanolone because allopregnanolone possesses sedative hypnotic and/or antiseizure effects.59 Moreover, oral delivery of allopregnanolone presents a challenge because of its low solubility and metabolism in the digestive tract and liver.59 In addition, negative results of allopregnanolone effects on cognitive function in human and animal studies have been reported, in which chronic treatment with allopregnanolone was found to accelerate AD development in AβPP(Swe)PSEN1(∆E9) mice and impair episodic memory in healthy women.60,61
In summary, although allopregnanolone offers hope for AD patients, liabilities including, but not limited to, adverse side effects and poor bioavailability may limit its therapeutic utility. The novel neurogenic P7C3 class of compounds were uncovered from in vivo chemical library screen in 2011.49 Among these compounds, the lead P7C3 (Fig. 2) was found to significantly increase the number of mature neurons in the hippocampus of aged rats and concomitantly improve cognitive performance as evaluated by the Morris water maze test.49
HIF-1 Regulators
Hypoxia-inducible factor 1 (HIF-1) is a transcriptional factor, which consists of HIF-1α and HIF-1β subunits. Under normoxic conditions, prolyl hydroxylase domain (PHD) enzymes mediate HIF-1α hydroxylation resulting in an immediate ubiquitination and subsequent proteasomal degradation. Under hypoxia or in the presence of HIF-1 activators, PHDs become stable and interact with p300 coactivator, forming the active HIF complex. This complex binds to hypoxia-responsive elements in promoter regions of HIF-1-target genes, resulting in the expression of diverse genes that regulate diverse processes. The regulated processes include iron metabolism, cell cycle control, cell proliferation and death, energy metabolism, angiogenesis, and erythropoiesis. Over 100 protective genes can be activated by regulation of HIF-1 pathway, including erythropoietin (EPO), transferrin (TfR), angiogenic vascular endothelial growth factor (VEGF), glucose transporter-1 (GLUT-1), antioxidant enzymes (Mn-SOD), neurotransmitter synthesis (TH), and glycolytic enzymes aldolase and enolase–1.62 Therefore HIF-1 activators may be considered as network medicines that have great potential for treating complex diseases like AD, PD, and stroke.62 In fact, numerous HIF-1 activators have been identified, and some of them have been investigated as potential drugs for AD and PD.63,64
In our efforts to develop new multi-target anti-AD/PD drugs, a series of new compounds were designed, synthesized, and investigated. Among these compounds, M30 or 5-[N-methyl-N-propargylaminomethyl]-8-hydroxyquinoline was identified as a new promising anti-AD/PD drug for further development.65–67 M30 was found to exhibit a wide range of activities including activation of HIF-1 pathway. It acts as an HIF-1 activator to upregulate HIF-1α and significantly increases the levels of HIF-1-dependent neuroprotective genes both in vitro and in vivo. In vitro studies, M30 at 1 μM concentration significantly increased expression levels of HIF-1 protein and mRNA in rat primary cortical neurons, and subsequently induced expression of neuroprotective HIF-1- dependent genes.68 Those induced genes include VEGF, EPO, enolase-1, p21, tyrosine hydroxylase (TH), TfR, and iNOS in motor neuron-like NSC-34 cells and rat primary embryonic cortical neurons.68 In vivo studies, M30 was demonstrated to differentially induce the transcription of diverse HIF-1-related protective genes such as EPO, VEGF, GLUT-1, TfR, heme oxygenase 1, and iNOS, in various brain regions (eg cortex, striatum, and hippocampus) and spinal cord of adult mice.68,69
Previous studies reported that HIF-1 levels were decreased in AD brains compared to age-matched controls, and overexpression of HIF-1 was sufficient to protect neurons from Aβ neurotoxicity.38,70 In conformity with this, in vitro studies demonstrated that M30 induced activation of several HIF-1α-target genes (eg enolase-1, VEGF, EPO, and p21) in cultured cortical neurons and NSC-34 cells, accompanied by protecting against Aβ25–35- and mutant G93A-SOD-1-induced toxicity, respectively.71
In vivo studies found that M30 significantly extended the survival of G93A-SOD-1 ALS mice and delayed the onset of the disease.71 In addition, M30 treatment increased HIF-1 expression and reduced Aβ accumulation/plaque formation in APPswe/PSEN1 mouse model of AD.69
PHD contains a non–heme-bound Fe2+ in its catalytic center and binding to the Fe2+ leads to inhibition of PHD activity, and subsequent induction of HIF-1α target genes.62 The prototype iron chelator DFO was shown to activate HIF-1 pathway by binding to the Fe2+ in PHD catalytic center, and exert its neuroprotective effects. As M30 has been shown to be a strong iron chelator with binding affinity comparable to that of DFO,67 it is likely that this mechanism may also play an important role in the neuroprotective effects of M30. Besides acting as an HIF-1 activator, M30 also functions as an MAO A/B inhibitor. In vitro, M30 highly inhibited both MAO-A and B activity with IC50values of 0.037 ± 0.02 μM and 0.057 ± 0.01 μM, respectively, when tested in rat brain homogenates.48 In vivo, M30 selectively inhibited brain MAO-A and B activity with poor inhibition of these enzymes in the liver and small intestine.65 This unique property conferred M30 potential antidepressant activity with limited potentiation of the tyramine pressor effect.72
M30 was rationally designed by combining a metal protein attenuating and ionophore moiety and neurogenesis/neuroprotection (propargylamine) moiety from the FDA-approved anti-PD drug rasagiline.73 Rasagiline was the first drug marketed with an apparent disease-modifying effect, at a dose of 1 mg per day.74 This disease-modifying effect was shown to be caused by its key pharmacophore or propargylamine moiety that interacts with an array of neuroprotective/neurorescue pathways.75 As M30 contains propargylamine moiety in its structure, it may exhibit similar activities like rasagiline. Indeed, studies have shown that M30 possessed activities of neurogenesis, neurorescue, and neuroprotection in various cell culture and animal models.71,76 In vitro, the neurogenesis activity of M30 was examined in SH-SY5Y and PC12 cells, which showed that M30 stimulated neuronal differentiation, cell body elongation, and neurite outgrowth.77 In NSC-34 motor neuron cells, M30 induced cell elongation and stimulated neurite outgrowth.71
The neurorescue activity of M30 was investigated both in vitro and in vivo. In vitro with SH-SY5Y cell serum deprivation models, serum deprivation (3 days) was found to induce an extreme apoptotic damage to SH-SY5Y cells. Treatment with M30 markedly decreased the apoptotic damage compared with control cells.78 In vivo, M30, when given post-treatment with MPTP to mice, restored the severe reduction in dopaminergic cell count, striatal dopamine content, and TH activity and expression levels, while water-treated mice did not show any spontaneous recovery.79 Further studies found that treatment with M30 in G93A-SOD1 mutant amyotrophic lateral sclerosis mice significantly increased lifespan and delayed the onset of neurological dysfunction, even when the treatment was initiated at a relatively advanced stage of the disease.71
A wide range of neuroprotection activities of M30 were demonstrated by different experiments. Some of these activities include: (1) decrease in cell death induced by serum deprivation and 6-hydroxydopamine in rat pheochromocytoma (PC12) cells66; (2) increase in the expression levels of the transcripts of brain-derived neurotrophic factor (BDNF) and growth-associated protein-43 (GAP43) in primary cortical cells; (3) protection against hydrogen peroxide or 3-morpholinosydnonimine-induced neurotoxicity in mouse NSC-34 motor neuron cells. M30 also enhanced mRNA expression levels of BDNF in the cortex and striatum and induced expression of glial cell-derived neurotrophic factor (GDNF) in the hippocampus and spinal cord of mouse brain.68
M30 shows protective actions against AD-related Aβ toxicity and improves cognitive performance in AD mouse models. For example, in vitro, M30 markedly lowered the secreted levels of Aβ in the conditioned medium and the cellular β-CTF in CHO/∆NL cells stably transfected with the APP “Swedish” mutation, and significantly increased the release of soluble APPα into the conditioned medium.80 In vivo, chronic treatment of aged mice with M30 (1 and 5 mg/kg; four times weekly for 6 months) significantly improved neuropsychiatry functions and age-related cognition impairment.80 In addition, M30 significantly reduced cerebral Aβ level and iron accumulation in the treated mice.76 Chronic administration of M30 also attenuated cerebral Aβ pathology and behavioral deficits in an APP/PS1 transgenic mouse model of AD.81
In APP and presenilin 1 (PS1) double transgenic mice, M30 significantly elevated cortical insulin and insulin receptor (InsR) transcript and protein expression, and increased the phosphorylated form of glycogen synthase kinase-3β in the frontal cortex. M30 treatment increased the hepatic protein expression levels of InsR and Glut-1 and lowered the increased blood glucose levels in the APP/PS1 mice. Since dysregulation of brain insulin/InsR and insulin signaling cascade has been related to AD pathogenesis, drugs that regulate brain glucose metabolism might be beneficial for AD therapy.69
In summary, M30 possesses diverse pharmacological properties, interacts with multiple targets, and regulates numerous neurorescue and neuroprotection pathways related to AD pathogenesis. M30 also acts as an HIF-1 activator to induce several endogenous antioxidant enzymes and transcription of numerous HIF-1- adaptive target genes. This unique profile of M30 makes it potentially valuable for AD therapy to delay further neurodegeneration. Figure 3 illustrates the multiple targets of M30 and its diverse pharmacological activities.
Figure 3.
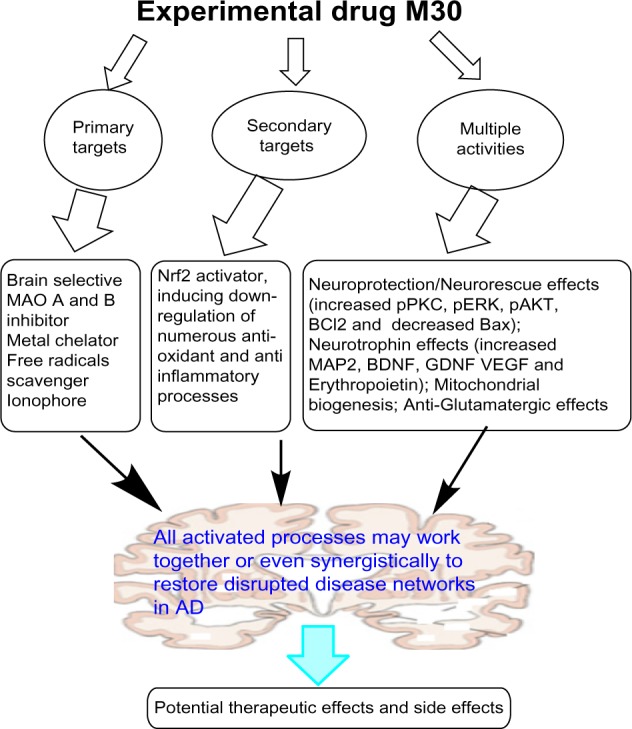
The multiple targets and diverse pharmacological activities of experimental drug M30.
Conclusions
AD is a complex brain network-disrupted disease, and the development of effective drugs for its treatment faces many challenges. The successful treatment of AD will require novel approaches not only to treat cognitive and behavior symptoms but also to rescue damaged neurons and to regenerate new neurons, and even to halt or stop neurodegeneration. To date, no current approaches simultaneously address these issues, but network medicines may offer a promising alternative. This review has briefly covered three classes of multi-target ligands (Keap1–Nrf2 regulators, endogenous neurogenic agents, and HIF-1 activators) as potential drugs for AD. These ligands act as master modulators to trigger numerous processes that may function together or even synergistically to exert their efficacy, and thus may be considered as network medicines. The fundamental principles of network medicine have been successfully applied to develop combination therapy or multi-target ligands for effectively treating complex diseases such as AIDS, cancer, and depression. Moreover, drug development further demonstrates the potential of network medicines for complex diseases, which may open up a new avenue for AD therapy.3,5,82
Footnotes
Author Contributions
Conceived and designed the concepts: HZ. Analyzed the data: HZ. Wrote the first draft of the manuscript: HZ. Agree with manuscript results and conclusions: HZ. Made critical revisions and approved final version: HZ. Made minor revisions: MF, MY. All authors reviewed and approved of the final manuscript.
ACADEMIC EDITOR: Yitzhak Tor, Editor in Chief
FUNDING: Authors disclose no funding sources.
COMPETING INTERESTS: HZ, MF and MBHY will receive royalties from Varinel Inc. (West Chester, PA, USA) if M30 described in this review reaches the market.
Paper subject to independent expert blind peer review by minimum of two reviewers. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE). Provenance: the authors were invited to submit this paper.
REFERENCES
- 1.Alzheimer’s A. Alzheimer’s disease facts and figures. Alzheimers Dement. 2012;2012;8:131–68. doi: 10.1016/j.jalz.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Anand R, Gill KD, Mahdi AA. Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology. 2014;76(pt A):27–50. doi: 10.1016/j.neuropharm.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 3.Chandra N, Padiadpu J. Network approaches to drug discovery. Expert Opin Drug Discov. 2013;8:7–20. doi: 10.1517/17460441.2013.741119. [DOI] [PubMed] [Google Scholar]
- 4.Azmi AS. Adopting network pharmacology for cancer drug discovery. Curr Drug Discov Technol. 2013;10:95–105. doi: 10.2174/1570163811310020002. [DOI] [PubMed] [Google Scholar]
- 5.Barabasi AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011;12:56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol. 2008;4:682–90. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- 7.Kitano H. A robustness-based approach to systems-oriented drug design. Nat Rev Drug Discov. 2007;6:202–10. doi: 10.1038/nrd2195. [DOI] [PubMed] [Google Scholar]
- 8.Medina-Franco JL, Giulianotti MA, Welmaker GS, Houghten RA. Shifting from the single to the multitarget paradigm in drug discovery. Drug Discov Today. 2013;18:495–501. doi: 10.1016/j.drudis.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou J, Seeley WW. Network dysfunction in Alzheimer’s disease and frontotemporal dementia: implications for psychiatry. Biol Psychiatry. 2014;75:565–73. doi: 10.1016/j.biopsych.2014.01.020. [DOI] [PubMed] [Google Scholar]
- 10.Brier MR, Thomas JB, Snyder AZ, et al. Loss of intranetwork and internet-work resting state functional connections with Alzheimer’s disease progression. J Neurosci. 2012;32:8890–9. doi: 10.1523/JNEUROSCI.5698-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang L, Brier MR, Snyder AZ, et al. Cerebrospinal fluid Abeta42, phosphorylated Tau181, and resting-state functional connectivity. JAMA Neurol. 2013;70:1242–8. doi: 10.1001/jamaneurol.2013.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang HY, Wang SJ, Liu B, et al. Resting brain connectivity: changes during the progress of Alzheimer disease. Radiology. 2010;256:598–606. doi: 10.1148/radiol.10091701. [DOI] [PubMed] [Google Scholar]
- 13.de Haan W, van der Flier WM, Koene T, Smits LL, Scheltens P, Stam CJ. Disrupted modular brain dynamics reflect cognitive dysfunction in Alzheimer’s disease. Neuroimage. 2012;59:3085–93. doi: 10.1016/j.neuroimage.2011.11.055. [DOI] [PubMed] [Google Scholar]
- 14.Bolognesi ML. Polypharmacology in a single drug: multitarget drugs. Curr Med Chem. 2013;20:1639–45. doi: 10.2174/0929867311320130004. [DOI] [PubMed] [Google Scholar]
- 15.Zhao B, Hemann MT, Lauffenburger DA. Intratumor heterogeneity alters most effective drugs in designed combinations. Proc Natl Acad Sci U S A. 2014;111:10773–8. doi: 10.1073/pnas.1323934111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Viayna E, Sola I, Di Pietro O, Munoz-Torrero D. Human disease and drug pharmacology, complex as real life. Curr Med Chem. 2013;20:1623–34. doi: 10.2174/0929867311320130002. [DOI] [PubMed] [Google Scholar]
- 17.Atri A, Molinuevo JL, Lemming O, Wirth Y, Pulte I, Wilkinson D. Memantine in patients with Alzheimer’s disease receiving donepezil: new analyses of efficacy and safety for combination therapy. Alzheimers Res Ther. 2013;5:6. doi: 10.1186/alzrt160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kosenko EA, Solomadin IN, Tikhonova LA, Reddy VP, Aliev G, Kaminsky YG. Pathogenesis of Alzheimer disease: role of oxidative stress, amyloid-beta peptides, systemic ammonia and erythrocyte energy metabolism. CNS Neurol Disord Drug Targets. 2014;13:112–9. doi: 10.2174/18715273113126660130. [DOI] [PubMed] [Google Scholar]
- 19.Williams TI, Lynn BC, Markesbery WR, Lovell MA. Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in Mild Cognitive Impairment and early Alzheimer’s disease. Neurobiol Aging. 2006;27:1094–9. doi: 10.1016/j.neurobiolaging.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 20.Paola D, Domenicotti C, Nitti M, et al. Oxidative stress induces increase in intracellular amyloid beta-protein production and selective activation of betaI and betaII PKCs in NT2 cells. Biochem Biophys Res Commun. 2000;268:642–6. doi: 10.1006/bbrc.2000.2164. [DOI] [PubMed] [Google Scholar]
- 21.Murakami K, Irie K, Ohigashi H, et al. Formation and stabilization model of the 42-mer Abeta radical: implications for the long-lasting oxidative stress in Alzheimer’s disease. J Am Chem Soc. 2005;127:15168–74. doi: 10.1021/ja054041c. [DOI] [PubMed] [Google Scholar]
- 22.Keller JN, Schmitt FA, Scheff SW, et al. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64:1152–6. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- 23.Persson T, Popescu BO, Cedazo-Minguez A. Oxidative stress in Alzheimer’s disease: why did antioxidant therapy fail? Oxid Med Cell Longev. 2014;2014:427318. doi: 10.1155/2014/427318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shinto L, Quinn J, Montine T, et al. A randomized placebo-controlled pilot trial of omega-3 fatty acids and alpha lipoic acid in Alzheimer’s disease. J Alzheimers Dis. 2014;38:111–20. doi: 10.3233/JAD-130722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng Y, Wang X. Antioxidant therapies for Alzheimer’s disease. Oxid Med Cell Longev. 2012;2012:472932. doi: 10.1155/2012/472932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haider L, Fischer MT, Frischer JM, et al. Oxidative damage in multiple sclerosis lesions. Brain. 2011;134:1914–24. doi: 10.1093/brain/awr128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mecocci P, Polidori MC. Antioxidant clinical trials in mild cognitive impairment and Alzheimer’s disease. Biochim Biophys Acta. 2012;1822:631–38. doi: 10.1016/j.bbadis.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 28.Bar-Or A, Gold R, Kappos L, et al. Clinical efficacy of BG-12 (dimethyl fumarate) in patients with relapsing-remitting multiple sclerosis: subgroup analyses of the DEFINE study. J Neurol. 2013;260:2297–305. doi: 10.1007/s00415-013-6954-7. [DOI] [PubMed] [Google Scholar]
- 29.Fox RJ, Kita M, Cohan SL, et al. BG-12 (dimethyl fumarate): a review of mechanism of action, efficacy, and safety. Curr Med Res Opin. 2014;30:251–62. doi: 10.1185/03007995.2013.849236. [DOI] [PubMed] [Google Scholar]
- 30.Suzuki T, Motohashi H, Yamamoto M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol Sci. 2013;34:340–6. doi: 10.1016/j.tips.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 31.Chen XL, Dodd G, Thomas S, et al. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am J Physiol Heart Circ Physiol. 2006;290:H1862–70. doi: 10.1152/ajpheart.00651.2005. [DOI] [PubMed] [Google Scholar]
- 32.Gerdes S, Shakery K, Mrowietz U. Dimethylfumarate inhibits nuclear binding of nuclear factor kappaB but not of nuclear factor of activated T cells and CCAAT/enhancer binding protein beta in activated human T cells. Br J Dermatol. 2007;156:838–42. doi: 10.1111/j.1365-2133.2007.07779.x. [DOI] [PubMed] [Google Scholar]
- 33.Schilling S, Goelz S, Linker R, Luehder F, Gold R. Fumaric acid esters are effective in chronic experimental autoimmune encephalomyelitis and suppress macrophage infiltration. Clin Exp Immunol. 2006;145:101–7. doi: 10.1111/j.1365-2249.2006.03094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghoreschi K, Bruck J, Kellerer C, et al. Fumarates improve psoriasis and multiple sclerosis by inducing type II dendritic cells. J Exp Med. 2011;208:2291–303. doi: 10.1084/jem.20100977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilson AJ, Kerns JK, Callahan JF, Moody CJ. Keap calm, and carry on covalently. J Med Chem. 2013;56:7463–76. doi: 10.1021/jm400224q. [DOI] [PubMed] [Google Scholar]
- 36.Petri S, Korner S, Kiaei M. Nrf2/ARE Signaling Pathway: Key Mediator in Oxidative Stress and Potential Therapeutic Target in ALS. Neurol Res Int. 2012;2012:878030. doi: 10.1155/2012/878030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Magesh S, Chen Y, Hu L. Small molecule modulators of Keap1-Nrf2-ARE pathway as potential preventive and therapeutic agents. Med Res Rev. 2012;32:687–726. doi: 10.1002/med.21257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soucek T, Cumming R, Dargusch R, Maher P, Schubert D. The regulation of glucose metabolism by HIF-1 mediates a neuroprotective response to amyloid beta peptide. Neuron. 2003;39:43–56. doi: 10.1016/s0896-6273(03)00367-2. [DOI] [PubMed] [Google Scholar]
- 39.Schubert D, Soucek T, Blouw B. The induction of HIF-1 reduces astrocyte activation by amyloid beta peptide. Eur J Neurosci. 2009;29:1323–34. doi: 10.1111/j.1460-9568.2009.06712.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crapper McLachlan DR, Dalton AJ, Kruck TP, et al. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet. 1991;337:1304–8. doi: 10.1016/0140-6736(91)92978-b. [DOI] [PubMed] [Google Scholar]
- 41.Walker JA, Sherman RA, Eisinger RP. Thrombocytopenia associated with intravenous desferrioxamine. Am J Kidney Dis. 1985;6:254–6. doi: 10.1016/s0272-6386(85)80183-9. [DOI] [PubMed] [Google Scholar]
- 42.de Zeeuw D, Akizawa T, Audhya P, et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med. 2013;369:2492–503. doi: 10.1056/NEJMoa1306033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang ZY, Lu MC, Xu LL, et al. Discovery of potent Keap1-Nrf2 protein-protein interaction inhibitor based on molecular binding determinants analysis. J Med Chem. 2014;57:2736–45. doi: 10.1021/jm5000529. [DOI] [PubMed] [Google Scholar]
- 44.Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–80. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 45.Felsenstein KM, Candelario KM, Steindler DA, Borchelt DR. Regenerative medicine in Alzheimer’s disease. Transl Res. 2014;163:432–8. doi: 10.1016/j.trsl.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abdipranoto A, Wu S, Stayte S, Vissel B. The role of neurogenesis in neurodegenerative diseases and its implications for therapeutic development. CNS Neurol Disord Drug Targets. 2008;7:187–210. doi: 10.2174/187152708784083858. [DOI] [PubMed] [Google Scholar]
- 47.Mu Y, Gage FH. Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol Neurodegener. 2011;6:85. doi: 10.1186/1750-1326-6-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hamilton LK, Aumont A, Julien C, Vadnais A, Calon F, Fernandes KJ. Widespread deficits in adult neurogenesis precede plaque and tangle formation in the 3xTg mouse model of Alzheimer’s disease. Eur J Neurosci. 2010;32:905–20. doi: 10.1111/j.1460-9568.2010.07379.x. [DOI] [PubMed] [Google Scholar]
- 49.MacMillan KS, Naidoo J, Liang J, et al. Development of proneurogenic, neuroprotective small molecules. J Am Chem Soc. 2011;133:1428–37. doi: 10.1021/ja108211m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fan X, Sun D, Tang X, Cai Y, Yin ZQ, Xu H. Stem-cell challenges in the treatment of Alzheimer’s disease: a long way from bench to bedside. Med Res Rev. 2014;34:957–78. doi: 10.1002/med.21309. [DOI] [PubMed] [Google Scholar]
- 51.Taupin P. Adult neurogenesis, neural stem cells and Alzheimer’s disease: developments, limitations, problems and promises. Curr Alzheimer Res. 2009;6:461–70. doi: 10.2174/156720509790147151. [DOI] [PubMed] [Google Scholar]
- 52.Taupin P. Neurogenic drugs and compounds to treat CNS diseases and disorders. Cent Nerv Syst Agents Med Chem. 2011;11:35–7. doi: 10.2174/187152411794961103. [DOI] [PubMed] [Google Scholar]
- 53.Rishton GM. Small molecules that promote neurogenesis in vitro. Recent Pat CNS Drug Discov. 2008;3:200–8. doi: 10.2174/157488908786242425. [DOI] [PubMed] [Google Scholar]
- 54.Brinton RD. Neurosteroids as regenerative agents in the brain: therapeutic implications. Nat Rev Endocrinol. 2013;9:241–50. doi: 10.1038/nrendo.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang JM, Johnston PB, Ball BG, Brinton RD. The neurosteroid allopregnanolone promotes proliferation of rodent and human neural progenitor cells and regulates cell-cycle gene and protein expression. J Neurosci. 2005;25:4706–18. doi: 10.1523/JNEUROSCI.4520-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang JM, Singh C, Liu L, et al. Allopregnanolone reverses neurogenic and cognitive deficits in mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2010;107:6498–503. doi: 10.1073/pnas.1001422107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen S, Wang JM, Irwin RW, Yao J, Liu L, Brinton RD. Allopregnanolone promotes regeneration and reduces beta-amyloid burden in a preclinical model of Alzheimer’s disease. PLoS One. 2011;6:e24293. doi: 10.1371/journal.pone.0024293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Irwin RW, Brinton RD. Allopregnanolone as regenerative therapeutic for Alzheimer’s disease: translational development and clinical promise. Prog Neurobiol. 2014;113:40–55. doi: 10.1016/j.pneurobio.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Irwin RW, Wang JM, Chen S, Brinton RD. Neuroregenerative mechanisms of allopregnanolone in Alzheimer’s disease. Front Endocrinol (Lausanne) 2011;2:117. doi: 10.3389/fendo.2011.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bengtsson SK, Johansson M, Backstrom T, Wang M. Chronic allopregnanolone treatment accelerates Alzheimer’s disease development in AbetaPP(Swe) PSEN1(DeltaE9) mice. J Alzheimers Dis. 2012;31:71–84. doi: 10.3233/JAD-2012-120268. [DOI] [PubMed] [Google Scholar]
- 61.Kask K, Backstrom T, Nilsson LG, Sundstrom-Poromaa I. Allopregnanolone impairs episodic memory in healthy women. Psychopharmacology (Berl) 2008;199:161–168. doi: 10.1007/s00213-008-1150-7. [DOI] [PubMed] [Google Scholar]
- 62.Correia SC, Moreira PI. Hypoxia-inducible factor 1: a new hope to counteract neurodegeneration? J Neurochem. 2010;112:1–12. doi: 10.1111/j.1471-4159.2009.06443.x. [DOI] [PubMed] [Google Scholar]
- 63.Zhang Z, Yan J, Chang Y, ShiDu Yan S, Shi H. Hypoxia inducible factor-1 as a target for neurodegenerative diseases. Curr Med Chem. 2011;18:4335–43. doi: 10.2174/092986711797200426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weinreb O, Amit T, Mandel S, Kupershmidt L, Youdim MB. Neuroprotective multifunctional iron chelators: from redox-sensitive process to novel therapeutic opportunities. Antioxid Redox Signal. 2010;13:919–49. doi: 10.1089/ars.2009.2929. [DOI] [PubMed] [Google Scholar]
- 65.Gal S, Zheng H, Fridkin M, Youdim MB. Novel multifunctional neuroprotective iron chelator-monoamine oxidase inhibitor drugs for neurodegenerative diseases. In vivo selective brain monoamine oxidase inhibition and prevention of MPTP-induced striatal dopamine depletion. J Neurochem. 2005;95:79–88. doi: 10.1111/j.1471-4159.2005.03341.x. [DOI] [PubMed] [Google Scholar]
- 66.Zheng H, Gal S, Weiner LM, et al. Novel multifunctional neuroprotective iron chelator-monoamine oxidase inhibitor drugs for neurodegenerative diseases: in vitro studies on antioxidant activity, prevention of lipid peroxide formation and monoamine oxidase inhibition. J Neurochem. 2005;95:68–78. doi: 10.1111/j.1471-4159.2005.03340.x. [DOI] [PubMed] [Google Scholar]
- 67.Zheng H, Weiner LM, Bar-Am O, et al. Design, synthesis, and evaluation of novel bifunctional iron-chelators as potential agents for neuroprotection in Alzheimer’s, Parkinson’s, and other neurodegenerative diseases. Bioorg Med Chem. 2005;13:773–83. doi: 10.1016/j.bmc.2004.10.037. [DOI] [PubMed] [Google Scholar]
- 68.Kupershmidt L, Weinreb O, Amit T, Mandel S, Bar-Am O, Youdim MB. Novel molecular targets of the neuroprotective/neurorescue multimodal iron chelating drug M30 in the mouse brain. Neuroscience. 2011;189:345–58. doi: 10.1016/j.neuroscience.2011.03.040. [DOI] [PubMed] [Google Scholar]
- 69.Mechlovich D, Amit T, Bar-Am O, Mandel S, Youdim MB, Weinreb O. The novel multi-target iron chelator, M30 modulates HIF-1alpha-related glycolytic genes and insulin signaling pathway in the frontal cortex of APP/PS1 Alzheimer’s disease mice. Curr Alzheimer Res. 2014;11:119–27. doi: 10.2174/1567205010666131212112529. [DOI] [PubMed] [Google Scholar]
- 70.Ogunshola OO, Antoniou X. Contribution of hypoxia to Alzheimer’s disease: is HIF-1alpha a mediator of neurodegeneration? Cell Mol Life Sci. 2009;66:3555–63. doi: 10.1007/s00018-009-0141-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kupershmidt L, Weinreb O, Amit T, Mandel S, Carri MT, Youdim MB. Neuroprotective and neuritogenic activities of novel multimodal iron-chelating drugs in motor-neuron-like NSC-34 cells and transgenic mouse model of amyotrophic lateral sclerosis. FASEB J. 2009;23:3766–779. doi: 10.1096/fj.09-130047. [DOI] [PubMed] [Google Scholar]
- 72.Gal S, Abassi ZA, Youdim MB. Limited potentiation of blood pressure in response to oral tyramine by the anti-Parkinson brain selective multifunctional monoamine oxidase-AB inhibitor, M30. Neurotox Res. 2010;18:143–50. doi: 10.1007/s12640-009-9128-8. [DOI] [PubMed] [Google Scholar]
- 73.Zheng H, Fridkin M, Youdim M. From single target to multitarget/network therapeutics in Alzheimer’s therapy. Pharmaceuticals (Basel) 2014;7:113–35. doi: 10.3390/ph7020113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.lanow CW, Rascol O, Hauser R, et al. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med. 2009;361:1268–78. doi: 10.1056/NEJMoa0809335. [DOI] [PubMed] [Google Scholar]
- 75.Weinreb O, Amit T, Bar-Am O, Youdim MB. Rasagiline: a novel anti- Parkinsonian monoamine oxidase-B inhibitor with neuroprotective activity. Prog Neurobiol. 2010;92:330–4. doi: 10.1016/j.pneurobio.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 76.Kupershmidt L, Amit T, Bar-Am O, Youdim MB, Weinreb O. Neuroprotection by the multitarget iron chelator M30 on age-related alterations in mice. Mech Ageing Dev. 2012;133:267–74. doi: 10.1016/j.mad.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 77.Avramovich-Tirosh Y, Reznichenko L, Mit T, et al. Neurorescue activity, APP regulation and amyloid-beta peptide reduction by novel multi-functional brain permeable iron- chelating- antioxidants, M-30 and green tea polyphenol, EGCG. Curr Alzheimer Res. 2007;4:403–11. doi: 10.2174/156720507781788927. [DOI] [PubMed] [Google Scholar]
- 78.Avramovich-Tirosh Y, Amit T, Bar-Am O, Zheng H, Fridkin M, Youdim MB. Therapeutic targets and potential of the novel brain- permeable multifunctional iron chelator-monoamine oxidase inhibitor drug, M-30, for the treatment of Alzheimer’s disease. J Neurochem. 2007;100:490–502. doi: 10.1111/j.1471-4159.2006.04258.x. [DOI] [PubMed] [Google Scholar]
- 79.Gal S, Zheng H, Fridkin M, Youdim MB. Restoration of nigrostriatal dopamine neurons in post-MPTP treatment by the novel multifunctional brain-permeable iron chelator-monoamine oxidase inhibitor drug, M30. Neurotox Res. 2010;17:15–27. doi: 10.1007/s12640-009-9070-9. [DOI] [PubMed] [Google Scholar]
- 80.Kupershmidt L, Amit T, Bar-Am O, Weinreb O, Youdim MB. Multi-target, neuroprotective and neurorestorative M30 improves cognitive impairment and reduces Alzheimer’s-like neuropathology and age-related alterations in mice. Mol Neurobiol. 2012;46:217–20. doi: 10.1007/s12035-012-8304-7. [DOI] [PubMed] [Google Scholar]
- 81.Kupershmidt L, Amit T, Bar-Am O, Youdim MB, Weinreb O. The novel multi-target iron chelating-radical scavenging compound M30 possesses beneficial effects on major hallmarks of Alzheimer’s disease. Antioxid Redox Signal. 2012;17:860–77. doi: 10.1089/ars.2011.4279. [DOI] [PubMed] [Google Scholar]
- 82.Erler JT, Linding R. Network medicine strikes a blow against breast cancer. Cell. 2012;149:731–3. doi: 10.1016/j.cell.2012.04.014. [DOI] [PubMed] [Google Scholar]