

Abstract
Ras and Rho GTPases are molecular switches for various vital cellular signaling pathways. Overactivation of these GTPases often causes development of cancer. Guanine nucleotide exchange factors (GEFs) and oxidants function to upregulate these GTPases through facilitation of guanine nucleotide exchange (GNE) of these GTPases. However, the effect of oxidants on GEF functions, or vice versa, has not been known. We show that, via targeting Ras Cys51, an oxidant inhibits the catalytic action of Cdc25—the catalytic domain of RasGEFs—on Ras. However, the enhancement of Ras GNE by an oxidant continues regardless of the presence of Cdc25. Limiting RasGEF action by an oxidant may function to prevent the pathophysiological overactivation of Ras in the presence of both RasGEFs and oxidants. The continuous exposure of Ras to nitric oxide and its derivatives can form S-nitrosated Ras (Ras-SNO). This study also shows that an oxidant not only inhibits the catalytic action of Cdc25 on Ras-SNO but also fails to enhance Ras-SNO GNE. This lack of enhancement then populates the biologically inactive Ras-SNO in cells, which may function to prevent the continued redox signaling of the Ras pathophysiological response. Finally, this study also demonstrates that, unlike the case with RasGEFs, an oxidant does not inhibit the catalytic action of RhoGEF—Vav or Dbs—on Rho GTPases such as Rac1, RhoA, RhoC, and Cdc42. This result explains the results of the previous study in which, despite the presence of an oxidant, the catalytic action of Dbs in cells continued to enhance RhoC GNE.
The Ras and Rho families of small GTPases are subfamilies of the Ras superfamily.1 The Ras family of small GTPases includes Harvey Ras (HRas), Neuroblastoma Ras, and Kirsten Ras.2 Ras-dependent cellular signals control cell growth and division.3,4 Rac1 and other proteins, such as RhoA, RhoC, and Cdc42, belong to the Rho family of small GTPases.5 These Rho proteins modulate various cellular functions, including cell polarity, vesicular trafficking, and the cell cycle.5,6 Various diseases, including cancer, are linked to misregulation of the cellular signaling events associated with Ras and Rho GTPases.4,7−9
A variety of regulators govern the cycle between the biologically active GTP- and inactive GDP-bound forms of these small GTPase proteins. These regulators include guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs).10 GAPs downregulate the level of activity of small GTPases by stimulating the intrinsically slow rate of GTP hydrolysis, populating small GTPases in their inactive GDP-bound form. Conversely, GEFs upregulate the function of small GTPases by promoting the dissociation of the bound GDP from small GTPases. This dissociation allows small GTPases to bind with cellularly abundant GTP to generate the active GTP-bound state of small GTPases in vivo.
A number of Ras-specific GEF (RasGEF) proteins have been identified. These include Son of Sevenless (SOS, originally named the Drosophila gene product of Son of Sevenless),11 Ras protein-specific guanine nucleotide-releasing factor (RasGRF),12 and Ras guanyl nucleotide-releasing protein (RasGRP).13 The general architecture of these related RasGEFs is conserved sequentially and structurally within the catalytic core domain Cdc25.14 Nevertheless, both SOS and RasGRF also possess the noncatalytic regulatory domains of Dbl homology (DH) and the Pleckstrin homology (PH). However, RasGRP lacks these regulatory domains.15 The DH domains of these RasGEFs are homologuous to the catalytic domain of the Rho-specific GEF (RhoGEF) proteins that may endow these RasGEFs with Rho-specific GEF activity in addition to the RasGEF function.16 A PH domain that connects directly to a DH domain interacts with the plasma membrane.17 The current model of the mechanism for the activation of RasGEF is that, by the binding of the RasGEF to the plasma membrane, the PH/DH domain-mediated allosteric inhibition of RasGEF is released, resulting in activation of the RasGEF.18
Dbl’s big sister (Dbs) that possesses DH and PH domains is known as a RhoGEF specific to RhoA and RhoC19 as well as to Cdc42.20 Vav, another RhoGEF composed of several domains that have been implicated in protein–protein interactions in addition to the DH and PH domains, has been shown to be broadly active with several Rho GTPases, such as Rac, RhoA, and Cdc42. However, it is most active with Rac1.21
Biologically important oxidants include the superoxide anion radical (O2•–), hydrogen peroxide (H2O2), the hydroxyl radical, nitric oxide (NO), and nitrogen dioxide (•NO2).9 Among them, O2•– and •NO2 are capable of enhancing the dissociation of GDP from redox-sensitive Ras and Rho proteins.22,23 In Ras proteins, these oxidants target the site of the Cys118 (HRas numbering) in the NKCD motif.24 In Rho GTPases, the Cys18 (Rac1 numbering) in the GXXXXGK(S/T)C motif serves as their target site.23 Intriguingly, the redox-mediated enhancement of Ras GDP dissociation is often coupled with S-nitrosation at the Cys118 side chain of Ras (Ras-SNO).24,25 Despite the lack of clarity about the cellular conditions necessary to produce Ras-SNO, it is easily formed when Ras is continuously exposed to oxidants such as •NO2 in the presence of NO.26 Nonetheless, because Ras-SNO does not react with oxidants such as O2•– and •NO2, some researchers have speculated that Ras-SNO formation terminates the redox regulation of Ras GTPases.9
The mechanisms of the regulation of Ras and Rho GTPases by their GEF alone or by an oxidant alone are well-established.9 However, it is less clear how redox sensitive Ras and Rho GTPases are regulated when a GEF and oxidants are both present at the same time. Although the sensitivity of the catalytic action of Cdc25 to oxidants has been reported,27 the biological significance of the kinetic and mechanistic features of this sensitivity remains unclear. No report exists of the redox sensitivity of the catalytic action of RhoGEFs. This study examined the redox properties of the catalytic core domain Cdc25 of RasGEFs as well as those of RhoGEFs Vav2 with the DH and PH domains and of the cysteine-rich domains (Vav2 DPC) and those of the Dbs with the DH and PH domains (Dbs DH/PH). The result clarifies the regulation of Ras activity by RasGEFs associated with Ras Cys51 in the presence of an oxidant. Moreover, this study also explains some earlier enigmatic findings about the inhibitory effect of the oxidant NO in regulating the cellular activity of Ras28−30 in which NO typically upregulates the cellular activity of Ras.31−34 Finally, this study notes the redox inert features of Vav2 DPC and Dbs DH/PH.
Materials and Methods
Experimental Conditions
The presence of transition metals in an assay mixture often converts biologically relevant oxidants into other molecules via the Haber–Weiss reaction and the Fenton reaction.35,36 Removal of transition metals from the assay mixture to prevent these Haber–Weiss and Fenton reactions ensures the desired oxidant concentrations and also blocks generation of byproducts.22 Moreover, most of the biologically relevant oxidants are highly reactive with O2. The reaction of oxidants with O2 not only depletes the effective oxidant concentrations but also often generates unwanted byproducts.9 However, despite the aerobic conditions of this cellular reaction, the cellularly produced oxidant effectively targets redox-sensitive proteins such as Ras.37−39 The spatial proximity between the redox-sensitive proteins and the oxidant-producing proteins (e.g., nitric oxide synthase)40 is one of the cellular features that may be responsible for directing the oxidant to target redox-sensitive proteins while minimizing its side reaction with O2. However, such a cellular proximity feature cannot be easily mimicked in in vitro assay approaches.
Although an anaerobic experimental condition—an experimental condition that lacks O2—is apparently atypical in biologically relevant kinetic studies, it is nevertheless one recourse for the mimicking of cellular reaction conditions. This is because the anaerobic condition minimizes the reaction of an oxidant with O2 during the course of in vitro assays.22 To prevent undesirable decay or conversions of oxidants during the time periods of the experiments, all experiments were performed under transition metal-free anaerobic experimental conditions. Residual transition metals on the surface of all assay equipment, including vials and cuvettes used in experiments, were removed by soaking them in 1 N HCl for 1 day and then thoroughly rinsing them with double-distilled water. A transition metal-free assay buffer was prepared by passing a solution containing 50 mM NaCl, 0.1 mM diethylenetriaminepentaacetate, and 10 mM TrisHCl (pH 7.4) through a column packed with Bio-Rad Chelex-100 cation exchange resin. The highest grade of pure MgCl2 (5 mM) was then added to the transition metal-free solution. All purified protein samples (see below) were dialyzed with the transition metal-free assay buffer to produce transition metal-free protein samples.
O2-free rubber serum stopper-sealed assay vials containing the transition metal-free buffer and small GTPase protein samples were prepared in an N2-filled anaerobic glovebox (O2 concentrations <3 PPM). GEF protein samples (Cdc25, Vav2 DPC, and Dbs DH/PH) as well as the fluorescence-tagged Ras proteins complexed with Cdc25 stocks in O2-free rubber serum stopper-sealed assay vials were also prepared in this N2-filled anaerobic glovebox. All syringes were flushed with N2 before being used to transfer buffer, reagents, or proteins from the stock vials to the assay cuvettes.
Generation and Quantification of O2•–
KO2 was used as a source for the generation of O2•–. The KO2 stock solution (∼3.6 mM) was prepared in anhydrous dimethyl sulfoxide essentially as described in the previous study.41 Anaerobic KO2 and H2O2 stock solutions were prepared by placing the KO2 and H2O2 solutions in a sealed vial, applying a vacuum, and then using a vacuum manifold to flush them three times with N2. A fraction of the KO2 solution (e.g., ∼1–2 μL) was transferred to an anaerobically sealed assay vial that contained an assay buffer (1 mL). The O2•– concentration in the assay vial was then measured using unmodified ferricytochrome c as described in the previous study.42 In brief, a fraction of the assay solution containing KO2 was transferred to an anaerobically sealed vial that contained oxidized ferricytochrome c (10 μL). The change in wavelength of 550 nm against 557 nm that occurred because of the reduction of ferricytochrome c was then monitored with a spectrophotometer. The value of the spectra of cytochrome c (550 nm minus 557 nm) provided the concentration of O2•– derived from KO2 in the assay solution. The cytochrome c concentrations were calculated from the absorption coefficient of 21 mM–1 cm–1.43
Protein Sample Preparations
Except for the commercially available superoxide dismutase (SOD, from bovine erythrocytes, Sigma), all protein samples used were prepared using human origin constructs. The C-terminus truncated version of wild type (wt) HRas (1–166) conserves the structural features of the full-length wt Ras (i.e., Switches I and II) necessary for the binding interactions with the full-length wt HRas effectors and regulators.44−46 This C-terminus truncated wt HRas construct also is stably and abundantly expressed in Escherichia coli and thus is widely used for various in vitro studies. Within this study, unless otherwise specified, all experiments were conducted with the C-terminus truncated wt HRas. Also, in all experiments, unless otherwise specified, the term “wt Ras” describes the C-terminus truncated version of wt HRas (1–166). As with wt Ras, single cysteine mutant Ras proteins—C51S, C80S, and C118S—as well as a double cysteine mutant—C51S/C118S Ras—that were constructed using the wt HRas (1–166) were stably and abundantly expressed in E. coli. Therefore, these cysteine mutants represent versions of C-terminus truncated HRas. However, the C-terminus double cysteine mutant—C181S/C184S—was constructed using full-length wt HRas (1–189). Therefore, C181S/C184S Ras protein represents a full-length HRas mutant protein, whereas the full-length wt Ras denotes the full-length wt HRas. The expressions of the full-length wt and C181S/C184S HRas proteins in E. coli were not as abundant as in these C-terminus truncated versions; this may have been because of the instability of these C-terminus HRas residues (167–189). Nevertheless, their expressions were sufficient for the planned experiments. When necessary, the control of C181S/C184S Ras, the full-length wt Ras, is noted as it is. A Cdc25 construct of human RasGRF1 (564–1049) was used with this study. The advantage in using Cdc25, instead of the whole RasGEF (such as SOS), was to pinpoint whether the target action of the oxidant is linked directly to the catalytic action of RasGEFs. Moreover, because Cdc25 represents the catalytic core domain of these RasGEFs, the result associated with Cdc25 generally applies as well to the catalytic action of these RasGEF proteins.
All Ras constructs, as well as the Cdc25 construct, were expressed as a maltose-binding protein (MBP)-fusion protein from the pMAL vector (New England Biolabs). They were purified from E. coli as described by the vendor. Ras and Cdc25 proteins were isolated by using a Q-Sepharose column after cleavage of the fusion proteins by using the protease supplied by the manufacturer. To produce a Ras-SNO, wt Ras (10 mL of ∼10 μM) was transferred for 30 min into a sealed vial (100 mL) containing a mixture of NO and O2. The mixture of NO and O2 in a sealed vial was prepared by purging NO gas for 5 min, followed by an injection of air (50 mL) into the NO-filled sealed vial while simultaneously venting the vial to release the increased air pressure that resulted. The NO/O2-treated Ras was passed through a G-25 size exclusion column (1.5 × 7 cm) to remove denatured Ras proteins. An estimation of the Ras-SNO characteristic peak at 542 nm (an extinction coefficient (ε) of 0.020 cm–1 mM–1)22 gave the fraction of Ras-SNO in the NO/O2-treated Ras sample as ∼0.7. As this fraction indicated, not all of the Ras was S-nitrosated. This was likely because a certain fraction (0.2–0.3) of the sulfur atom of the Cys118 side chain of the as-purified Ras exists in various oxidized states, such as sulfinate and sulfonate.47 These oxidized forms of Ras are redox inert; thus, they did not affect the experiment results. The Ras-SNO is stable unless otherwise illuminated by high energy UV light (i.e., >260 nm).47,48 Given that Ras-SNO was not exposed to UV light during the experiments, the decomposition of Ras-SNO was not expected.
wt Ras and its mutant proteins were tagged with rhodamine fluorescence as described in the previous study.47 The fluorescence-tagged Ras proteins complexed with Cdc25 were generated essentially as described in the previous study.49
Glutathione S-transferase (GST)-tagged wt Rho GTPases—such as Rac1 (1–177), RhoA (1–181), RhoC (1–193) in the pGEX vector—were expressed in and purified from E. coli by using the GST gene fusion system supplied by the manufacturer (GE Healthcare). Rho GTPases were isolated by using the factory-provided GSTrap followed by thrombin digestion as indicated in the vendor’s protocol. The Vav2 DPC (1–573) and The Dbs DH/PH (525–1097) construct, expression, and purification were essentially the same as described in the previous studies.19 Tritium-labeled guanosine diphosphate ([3H]GDP) was diluted with unlabeled GDP, giving ∼4000 dpm/μM [3H]GDP. When necessary, [3H]GDP was loaded on Ras and Rho GTPases as described previously.23
Kinetic Assay
A syringe flushed with N2 was used to add an anaerobically prepared oxidant or GEF protein (e.g., Cdc25) to the anaerobically sealed assay cuvettes that contained small GTPase loaded with [3H]GDP in the presence and absence of SOD in the transition metal-free assay buffer. Simultaneous treatment with GEF and an oxidant was done by introducing GEF, followed immediately by an oxidant, into the assay cuvettes containing [3H]GDP-loaded small GTPase in an assay buffer. When necessary, this sequence was reversed, first introducing an oxidant, followed immediately by GEF. Aliquots of the assay sample were then withdrawn with a syringe at specific times and spotted onto nitrocellulose membrane filters. These filters were then washed three times with an assay buffer. The radioactivity of the nitrocellulose membrane filters was measured with a scintillation counter (Beckman). When necessary to perturb any potential binding interaction between GEF and GTPase, GEF (50 nM) was pretreated with an oxidant (e.g., O2•–, 5 μM) in the presence of GTPase (1 μM) under anaerobic conditions, quenched with excess ascorbic acid (1 mM), and then treated with 100 mM (NH4)2SO4. The (NH4)2SO4-treated protein sample was concentrated with an Amicon centrifugal filter (10 kDa cut off). To isolate GTPase from GEF protein and chemicals, the concentrated protein sample was then passed through a gel filtration column (2.5 × 20 cm packed with Sephadex 75).
Depending on the assay involved, a certain fraction of [3H]GDP was dissociated from small GTPases by treatment with an oxidant and/or GEF. However, complete dissociation did not occur, even over an extended period (e.g., 1 h). One possible reason for this incomplete dissociation is that, although the GTPase protein samples used were more than 95% pure (judged by SDS-PAGE), the structure and/or redox-sensitive motif of the used GTPase samples was not entirely homogeneous. For example, a certain fraction of the redox-sensitive cysteine residues of Ras proteins that are coupled with the Ras GDP dissociation was in a redox inert oxidation state (sulfenic acid, sulfinate, and/or sulfonate states). A Ras-specific redox enzyme has not been reported; however, it is possible that in cells these redox inert states can be reversed by the action of various redox enzymes such as thioredoxin, peroxiredoxin, and sulfiredoxin. However, the in vitro assay used for this study lacked such redox enzymes. Therefore, the presence of a desensitized fraction of Ras in an assay mixture reduced the quantity of the redox-mediated dissociation of the bound [3H]GDP from Ras. It is not unprecedented for a fraction of GDP to remain unassociated from Ras when treated with an oxidant.47 Another possible explanation is that a fraction of the oxidation reaction of the redox-sensitive residue of Ras does not always couple with the mechanical perturbation of the Ras nucleotide-binding interaction. For example, a fraction of the thiyl radical generated by the reaction of a redox-sensitive cysteine of Ras with an oxidant (e.g., O2•–) reacts instead with another O2•– to produce a redox inert oxidized form of Ras. This cysteine oxidation does not couple with the dissociation of [3H]GDP from Ras.
The fluorescence-based binding assays under anaerobic conditions were performed by titration of the rhodamine-tagged Ras Cdc25 binary complex with KO2. This was described in the previous study,47 except that Cdc25 was used instead of Raf-RBD. KO2 was repeatedly injected into the anaerobically sealed assay cuvettes containing the rhodamine-tagged Ras complexed with Cdc25. The change in fluorescence emission intensity at 545 nm, followed by excitation at 490 nm because of the dissociation by KO2 of Cdc25 from the rhodamine-tagged Ras, was then monitored with a Perkin-Elmer LS 55 Fluorescence spectrometer.
Data Analyses
Three independent measurements were performed for each set of experiments (e.g., wt Ras). When necessary, the values were normalized against the value of the initial untreated sample that was set at 1.0. For purposes of graphic presentation, the mean values of each data point were then calculated, along with the corresponding standard deviations (SD) from these independent triple measurements. Depending on the experimental set, the values were plotted against either time (e.g., s) or concentration of an effector(s) (e.g., KO2). The plot was further fitted to a proper kinetic model—the one-phase exponential decay or the one-site binding (hyperbola)—to statistically analyze and then estimate various kinetic parameters. Briefly, for each data set of the triple independent measurements, Prism software was used to perform a two-tailed variance ratio test (F test) for a null hypothesis (H0) “one curve is for all data sets” and an alternative hypothesis (HA) “one curve is not for all data sets” with a P value < 0.05. This test evaluates whether the individual curves are statistically distinguishable with a 95% confidence interval. Once the result of the F test does not reject the null hypothesis, Prism software was used to find the global fit, finding one shared (among data sets) best-fit value for all triple independent measurements for each parameter. These values include the rate constant, the apparent inhibition constant (appKI), or the dissociation constant (KD). The fittings also provide the SD of each of the determined parameters as well as the regression values (r2) of the global fits.
To test the difference between the kinetic parameters obtained from each set of experiments (e.g., wt Ras versus C118S Ras), Prism software was used to perform a two-sample t test for a two-tailed H0 “one parameter is for all data sets” and an HA “one parameter is not for all data sets” with a P value < 0.05.
Kinetic parameters of reactions include the rate constant and the span. Such parameters can be obtained by fitting the reaction data to the kinetic model “one phase exponential decay” (see above). However, unlike their rate constants, spans are often omitted in the description of reactions. This is because, in the absence of any other reaction limitation, the span of the complete reaction asymptotically reaches 1 (in terms of the fraction of the total reaction substrate). The span within this study is defined as the total quantity GTPase GDP dissociation in the time period for a given reaction. Under optimal conditions, the quantity of the usage of GEF does not control the span of the GEF-mediated Ras GDP dissociation—which is expected to be near 1. This is because, up until the reaction is completed to reach span 1, the presence of any amount of whatsoever of the enzyme GEF will be continuously recycled. However, how much of an oxidant is used determines the span of the oxidant-mediated Ras GDP dissociation, which is expected to vary. This is because, unlike GEFs, the inorganic oxidant cannot be recycled but is consumed in the course of the turnover of the facilitation of the GTPase GDP dissociation. Adding more of the oxidant generates more of the GTPase GDP dissociation and thus yields a bigger span. For example, the span of the wt Ras GDP dissociation by O2•– can be further increased by multiple additions of O2•– during the assay time period or by using the xanthine oxidase system that continuously generates O2•– over the assay time period (not shown).
However, characterization of the completion of the oxidant-mediated Ras GDP dissociation is outside the scope of this study, which aims instead to clarify the effect an oxidant has on the catalytic action of GEFs on these GTPases. Nonetheless, when the quantity of oxidant to be used for assays is set, the span—an indicator of the amount of the GTPase GDP dissociation by the oxidant—can be used as a signature feature of the oxidant-mediated GTPase GDP dissociation. Accordingly, this study used a fixed concentration of an oxidant, rather than multiple additions of oxidants or of the xanthine oxidase system. A certain fraction of Ras and Rho GTPases was denatured in the presence of oxidant concentrations higher than 10 μM (see Figure 3 in Results). To minimize denaturation of these GTPases during the assay time, ∼3 μM of oxidant was uniformly used for all assays unless otherwise noted.
Figure 3.

Determination of the apparent inhibition constant of KO2 for C118S and C51S/C118S Ras in the presence and absence of Cdc25. Independent triple equilibrium titrations of [3H]GDP-loaded C118S and C51S/C118S Ras (1 μM) with various concentrations of KO2 (between 0 and 20 μM) were performed in the presence and absence of Cdc25 (50 nM). The KO2-treated samples were then incubated for 250 s, and their radioactivity values associated with the Ras-bound [3H]GDP were determined as described in Figure 2. All radioactivity values measured were normalized against the radioactivity value of the Ras sample without treatment of KO2. This radioactivity value was set at 1.0. The F tests with a linear regression model (P < 0.05), but not the hyperbola model (P < 0.05), support one curve for each set of the triple data of C118S and C51S/C118S Ras with KO2 in the absence of Cdc25. Mean values and the SD from each of the triple measurements of C118S and C51S/C118S Ras with KO2 in the absence of Cdc25 are shown in this figure. The F tests (P < 0.05) for each data set of the triple measurements of C118S and C51S/C118S Ras with KO2 in the presence of Cdc25 indicate the insignificant curve difference within each of these triple measurements. The global fits for each of these triple data with the hyperbola model (P < 0.05) determined the KO2 concentrations that give appVmin values of the catalytic action of Cdc25 on C118S and C51S/C118S Ras, respectively, to be 27 ± 7 and 6 ± 2 μM. The global fitting also gave appKI values of KO2 for C118S and C51S/C118S Ras in the presence of Cdc25, respectively, of 2.3 ± 1 and >100.7 ± 34 μM [KO2]. The r2 values associated with fit were >0.9065. Note that when the O2•– concentrations were higher than ∼10 μM, these Ras proteins were partly denatured. This Ras denaturation complicates fitting these values to a curve. Therefore, kinetic values beyond O2•– concentrations higher than ∼10 μM were ignored for ease of fit to the curve. However, as a way to present the original data, the kinetic values associated with O2•– concentrations beyond 10 μM, including 20 μM, are shown in this figure.
When the oxidant concentration is fixed, not only the rate constant but also the span of the oxidant-mediated GTPase GDP dissociation may differ from the rate constant and the span of the GEF-mediated GTPase GDP dissociation. If the spans between one reaction and another differ, visual comparison of the reaction rates (expressed as rate constants) between these two reactions is confusing. As illustrated in Figure 1, reaction 1 appears slower than reaction 3. In fact, however, these rates are the same. This illusion of difference is because of the 2-fold smaller span of reaction 1 compared with the span of reaction 3. Furthermore, reaction 2 appears slower than reaction 3. This apparent difference also is misleading. In fact, the rate of reaction 2 is 2-fold faster than the rate of reaction 3, yet the span of reaction 2 is 2-fold smaller than the span of reaction 3. Similarly, reaction 2 appears much slower than reaction 4. However, their rates are actually the same; nonetheless, the span of reaction 2 is simply 2-fold smaller than the span of reaction 4. To avoid such confusion, we provide not only the rate constant values but also the span values of the oxidant- and the GEF-mediated GTPase GDP dissociations, along with their corresponding figures.
Figure 1.

Evaluation of the kinetic rate constant associated with the span of the reaction. Each line represents a reaction with (a) a 0.5 rate constant and a 0.5 span; (b) a 1.0 rate constant and a 0.5 span; (c) a 0.5 rate constant and a 1.0 span; and (d) a 1.0 rate constant and a 1.0 span.
Results
To assess any potential effect of oxidants on the catalytic function of the GEF proteins of Ras and Rho GTPases, two kinetic parameters—a rate constant and a span—were determined and analyzed for the GDP dissociation from Ras and Rho GTPases in the presence and absence of GEFs and/or various oxidants. When necessary, values were determined for the oxidant concentration that gives the maximal inhibition of the catalytic action of GEFs on small GTPases. This is equivalent to the minimal velocity (appVmin) of the GEF-mediated GDP dissociation from small GTPases. Values also were determined for appKI of an oxidant in its effect on the catalytic action of GEF on GTPases as well as for KD of an oxidant for small GTPases complexed with GEF. These parameters also aid evaluation of the potential effect of oxidants on the catalytic function of the GEF proteins of Ras and Rho GTPases. wt Ras and various Ras mutants as well as wt Rac1 and wt RhoC were used for this study. Cdc25 was used as a Ras GEF. Vav2 DPC and Dbs DH/PH were used for Rho GEFs.
Apparent Inhibition of the Oxidant-Mediated Catalytic Action of Cdc25 on wt Ras GTPase
wt Ras GDP dissociation was enhanced by Cdc25, which is consistent with previous reports.49 wt Ras GDP dissociation also was enhanced by KO2 alone (Figure 2). The KO2-mediated wt Ras GDP dissociation was abolished by treatment with SOD (Table 1), suggesting that O2•–—derived from KO2—is the active species to facilitate wt Ras GDP dissociation. The effect of O2•– and its nullification by SOD are consistent with the previous result.23 The values of the rate constant and the span of the Cdc25-mediated wt Ras GDP dissociation are, respectively, 5.4-fold smaller and 1.2-fold larger than the values of the rate constant and the span of the O2•–-mediated wt Ras GDP dissociation (Table 1). These rate constants indicate that, under experimental conditions, the rate of wt Ras GDP dissociation by Cdc25 is slower than the rate of wt Ras GDP dissociation by O2•–. The span values indicate that, under experimental conditions, the quantity of the wt Ras GDP dissociation by Cdc25 exceeds that by O2•–. Unlike the dissociation rate constant, the reaction span is not an intrinsic kinetic characteristic of each reaction. Nevertheless, such a span difference occurred. This occurrence is because, as detailed in the kinetic analysis section of Materials and Methods, the total treated quantity of KO2 (i.e., ∼3 μM) that produces the active radical species O2•– for the wt Ras GDP dissociation was simply insufficient to complete the wt Ras GDP dissociation. In contrast, a relatively small amount of the enzyme, Cdc25 (50 nM), was recycled during the assay time period to reach the maximal span.
Figure 2.

Effect of oxidants on the Cdc25-mediated GDP dissociation from wt Ras and C118S Ras. (A) Independent triple filter-binding assays were performed to measure the radioactivity of the [3H]GDP that remained to bind to Ras after treatment of the [3H]GDP-loaded wt Ras (1 μM) with or without a regulator(s), including Cdc25 (50 nM) and/or KO2 (∼3 μM), for the times given in Materials and Methods. (B) The identical independent triple filter-binding assays with or without the regulator(s), as described for panel A, also were performed for the [3H]GDP-loaded C118S Ras (1 μM). All of these triple measurements of radioactivity values measured at different times were normalized against the radioactivity value of the initial sample mixture (time = 0 s), which was set at 1.0. The one-phase exponential decay model (P < 0.05) was used to perform F tests on each data set of the triple measurements. The F tests indicated there was no significant curve difference within any data set of the triple measurements. Accordingly, mean values and their corresponding SD from each of the triple measurements are represented in panels A and B of this figure. The one-phase exponential decay model (P < 0.05) was used to obtain kinetic values, including kinetic rate constants and spans from the plots, for global fits for each of the triple data sets. Global fits give kinetic values and SD with r2 of fit of >0.9050. These values are summarized in Table 1.
Table 1. Kinetic Parameters for the GDP Dissociation from wt Ras, C118S Ras, and wt Ras-SNO in the Presence and Absence of Cdc25, KO2, SOD, and/or and H2O2a.
| wt Ras |
C118S Ras |
wt Ras-SNO |
||||
|---|---|---|---|---|---|---|
| treatment | GDP dissociation rate constants (10–3 s–1) | span (fraction) | GDP dissociation rate constants (10–3 s–1) | span (fraction) | GDP dissociation rate constants (10–3 s–1) | span (fraction) |
| none | 0.01 ± 0.03a | 0.05 ± 0.16 | 0.01 ± 0.02a | 0.03 ± 0.06 | 0.01 ± 0.01a | 0.03 ± 0.03 |
| Cdc25 | 0.40 ± 0.05b | 0.65 ± 0.08 | 0.37 ± 0.02b | 0.68 ± 0.04 | 0.36 ± 0.04b | 0.66 ± 0.07 |
| KO2 | 2.14 ± 0.02c | 0.55 ± 0.04 | 0.03 ± 0.03a | 0.08 ± 0.08 | 0.04 ± 0.04a | 0.07 ± 0.07 |
| KO2 + SOD | 0.04 ± 0.03a | 0.05 ± 0.09 | 0.03 ± 0.04a | 0.05 ± 0.07 | 0.03 ± 0.01a | 0.03 ± 0.07 |
| Cdc25 + KO2 | 2.31 ± 0.05d | 0.55 ± 0.01 | 0.14 ± 0.02e | 0.54 ± 0.08 | 0.15 ± 0.02e | 0.53 ± 0.02 |
| Cdc25 + KO2 + SOD | 0.42 ± 0.06b | 0.71 ± 0.01 | 0.35 ± 0.01b | 0.68 ± 0.02 | 0.36 ± 0.02b | 0.34 ± 0.03 |
| H2O2 | 0.02 ± 0.05a | 0.07 ± 0.17 | 0.03 ± 0.03a | 0.08 ± 0.08 | 0.04 ± 0.03a | 0.04 ± 0.08 |
| Cdc25 + H2O2 | 0.41 ± 0.02b | 0.69 ± 0.03 | ND | ND | ND | ND |
The values with SD of the rate constant and the span of GDP dissociation from wt Ras and C118S Ras with and without Cdc25 and/or KO2 were taken from Figure 2. The values with SD of the rate constant and the span of GDP dissociation from wt Ras and C118S Ras, with and without Cdc25 in the presence of KO2 and SOD, were obtained, with one exception, as described in Figure 2. The exception is that the Ras-containing assay vial was pretreated with SOD (5000 units), as noted in Materials and Methods. The values with SD of the rate constant and the span of GDP dissociation from wt Ras and C118S Ras, with and without Cdc25 in the presence of H2O2, were obtained as described in Figure 2, except that H2O2 (10 μM) was used instead of KO2. All listed values with SD of the rate constant and the span of GDP dissociation from wt Ras-SNO, with and without Cdc25, KO2, and/or H2O2 in the presence and absence of SOD, also were determined, with one exception, as described in Figure 2 and in this Table 2; the exception was the use of wt Ras-SNO instead of C118S Ras. To examine the significance of the potential similarity or difference among these rate constant values listed in Table 1, t tests with P < 0.05 were performed as described in Materials and Methods. Briefly, any values denoted by the letter “a” correspond to other values denoted with “a”. The same is true for the values denoted with the letters “b”, “c”, “d”, and “e”. However, any values denoted with “a” differ from the values denoted by “b”, “c”, “d”, and “e”. The same applies for “b” with “c”, “d”, and “e”; and for “c” with “d” and “e”; and for “d” with “e”. All of the t test results associated with the rate constants were exactly the same with the t test results for the corresponding span values. Therefore, for clarity of presentation, the t tests for the span value analyses are not shown. ND, not determined.
Facilitation of wt Ras GDP dissociation also was observed in the presence of both Cdc25 and O2•– together (Figure 2). If O2•– does not affect the catalytic action of the Ras GEF Cdc25 on wt Ras or vice versa, the values of the rate constant and the span of the wt Ras GDP dissociation in the presence of Cdc25 and O2•– together will be a numerical sum. This sum is the total of the rate constant and the span values of the Cdc25-mediated wt Ras GDP dissociation and the O2•–-mediated wt Ras GDP dissociation. However, the values of the rate constant and the span of the Cdc25/O2•–-mediated wt Ras GDP dissociation approximate the values of the rate constant and the span of the O2•–-mediated wt Ras GDP dissociation. Nevertheless, they are not equivalent to the sum of the values of the rate constant and the span of the Cdc25-mediated wt Ras GDP dissociation and the O2•–-mediated wt Ras GDP dissociation (Table 1).
The O2•–-dominant kinetic features can be quenched by addition to the assay of the O2•–-scavenger SOD containing both Cdc25 and O2•–. In this assay, the values of the kinetic constant and the span of the Cdc25/O2•–-mediated wt Ras GDP dissociation in the presence of SOD were similar to the values of the kinetic constant and the span of the Cdc25-mediated wt Ras GDP dissociation (Table 1). The best explanations for these results are (i) O2•– interferes with the catalytic action of Cdc25 on the wt Ras GDP dissociation, so that the kinetic characteristics of the Cdc25-mediated wt Ras GDP dissociation were fated; and (ii) O2•– continuously facilitates wt Ras GDP dissociation in the presence of Cdc25 that exhibits the signatures of the O2•–-mediated wt Ras GDP dissociation. Accordingly, we hypothesize that O2•– inhibits the Cdc25-mediated wt Ras GDP dissociation. However, Cdc25 has no effect on the O2•–-mediated wt Ras GDP dissociation.
The wt Ras GDP dissociation was not facilitated by H2O2 (Table 1), which is consistent with the previous result.23 The Cdc25-mediated wt Ras GDP dissociation also was unaffected by other oxidants, such as H2O2 (Table 1). The result suggests that both the intrinsic and the Cdc25-mediated wt Ras GDP dissociation are insensitive to H2O2. Nonetheless, the inability of H2O2 to inhibit the Cdc25-mediated wt Ras GDP dissociation clarifies that the oxidant-mediated inhibition of the catalytic action of Cdc25 on wt Ras is O2•– specific.
Deconvolution of the Oxidant-Mediated Inhibition of the Catalytic Action of Cdc25 on wt Ras GTPase
It would be challenging to validate the effect of O2•– on the Cdc25-mediated facilitation of the wt Ras GDP dissociation through monitoring of the GDP dissociation from wt Ras. This is because, although O2•– apparently inhibits the Cdc25-mediated wt Ras GDP dissociation, it directly facilitates the wt Ras GDP dissociation.
To isolate the effect of O2•– on the Cdc25-mediated wt Ras GDP dissociation from the direct effect of O2•– on the wt Ras GDP dissociation, the C118S Ras mutant and the S-nitrosated wt Ras on Cys118 side chain (wt Ras-SNO) were used instead of wt Ras. This substitution was made because O2•– is unable to facilitate the GDP dissociation from C118S Ras and wt Ras-SNO (Figure 2B and Table 1). This inability is because C118S Ras lacks the redox-sensitive Cys118, whereas the SNO moiety of wt Ras-SNO is redox inert against O2•–.47 Moreover, the values of the rate constant and span of the Cdc25-mediated GDP dissociation from C118S Ras and wt Ras-SNO were similar to those of the rate constant and span of the Cdc25-mediated wt Ras GDP dissociation (Figure 2B and Table 1). The results suggest that the mutated Ser residue in C118S Ras and the SNO moiety of the Cys118 side chain of wt Ras do not alter the catalytic function of Cdc25 on Ras. Collectively, kinetic data of C118S Ras and wt Ras-SNO can be used to deconvolute the sole potential inhibition effect of O2•– on the Cdc25-mediated wt Ras GDP dissociation from the O2•–-mediated wt Ras GDP dissociation.
The values of the rate constant and the span of the Cdc25-mediated C118S Ras GDP dissociation in the presence of O2•– were 2.6- and 1.3-fold smaller than the rate constant and span of the Cdc25-mediated C118S Ras GDP dissociation in the absence of O2•– (Figure 2B and Table 1). However, treatment with SOD abolished the decreases in the rate constant and the span of the Cdc25-mediated C118S Ras GDP dissociation by O2•– (Table 1). Identical biochemical results were obtained by using wt Ras-SNO instead of C118S Ras (Table 1). These results suggest that O2•– perturbs the catalytic function of Cdc25 on C118S Ras and wt Ras-SNO. This supports a hypothesis that O2•– inhibits the catalytic action of Cdc25 on wt Ras.
The inhibition of the rate constant and span of the Cdc25-mediated C118S Ras GDP dissociation by O2•– was hardly significant (Table 1). To determine the effective inhibition concentrations of O2•– on the catalytic action of Cdc25 on C118S Ras, the mixture of C118S Ras and Cdc25 was titrated with various concentrations of O2•–. The appVmin value—equivalent to the value of the maximal O2•–-mediated inhibition—of the catalytic action of Cdc25 on C118S Ras was determined to occur at an O2•– concentration of 27 μM (Figure 3). The appKI value of O2•– for C118S Ras in the presence of Cdc25 was estimated to be 2.3 μM (Figure 3). This result explains why the default O2•– concentration (∼3 μM, see the Materials and Methods section) caused only partial inhibition of the catalytic action of Cdc25 on C118S Ras.
Intriguingly, although C118S Ras lacks Cys118 and wt Ras-SNO possesses the chemically modified redox inert Cys118 side chain, O2•– was able to perturb the Cdc25-mediated GDP dissociation from C118S Ras and wt Ras-SNO. Therefore, these results constitute another piece of critical evidence that the residue associated with the redox feature of Ras Cys118 is not involved in the O2•–-mediated inhibition of the Cdc25-mediated catalysis of the wt Ras GDP dissociation.
Role of Ras Cys51 in the Oxidant-Dependent Inhibition of the Cdc25-Mediated wt Ras GDP Dissociation
Full-length HRas has a total of five cysteines: Cys51, Cys80, Cys118, Cys181, and Cys184. As noted elsewhere, Ras Cys118 is well-known as a direct target of an oxidant such as O2•–, resulting in enhancement of the GDP dissociation from Ras.23,24 However, it is unclear if any of these Ras cysteines play a role in how O2•– inhibits the catalytic action of Cdc25.
To determine if Ras Cys51 has a role in the O2•–-mediated inhibition of the catalytic action of Cdc25 on wt Ras, we examined the kinetic properties of the Ras mutant C51S in the presence and absence of Cdc25 and/or O2•–. In contrast to the results associated with wt Ras (Figure 2A and Table 1), the rate constant and the span of the GDP dissociation from C51S Ras by Cdc25 and O2•– together approximated the sum of the individual rate constants and the spans of the GDP dissociation from C51S Ras by Cdc25 and by O2•– (Figure 4A and Table 2). Nevertheless, the values of the rate constants and the spans of the intrinsic, the Cdc25-mediated, and the O2•–-mediated C51Ras GDP dissociation (Figure 4A) were, respectively, similar to those of the values of the intrinsic, Cdc25-mediated, and O2•–-mediated wt Ras GDP dissociation. These results suggest that, although Ras Cys51 has no role in either the wt Ras GDP binding interaction or in the O2•–- and Cdc25-mediated wt Ras GDP dissociation, the Ras residue Cys51 is involved in the O2•–-mediated inhibition of the catalytic action of Cdc25 on Ras. Given that O2•– targets Ras Cys51 rather than Cdc25 to inhibit the catalytic action of Cdc25 on wt Ras, it follows that the mechanism of the O2•–-mediated inhibition of the catalytic action of Cdc25 on wt Ras is that O2•– targets Ras Cys51, which in turn renders Ras insensitive to Cdc25.
Figure 4.

Effect of KO2 on the Cdc25-mediated GDP dissociation from C51S and C51S/C118S Ras. (A) All experimental methods and data analysis procedures were identical to those described in Figure 2, except that C51S Ras was used instead of wt Ras and C118S Ras. (B) The same experiments and analyses that are described for panel A also were performed for C51S/C118S Ras instead of C51S Ras. The F tests indicate that the curve differences are insignificant within the curve of each of the triple measurements associated with C51S and C51S/C118S Ras in the presence and absence of an effector(s). Therefore, all values within this figure are shown with mean values and the SD from the independent triple measurements. Global fits for all triple data with the one-phase exponential decay model (P < 0.05) also were performed that gave kinetic values and their corresponding SD. All kinetic values determined are summarized in Table 2. The r2 values of fit were >0.9015.
Table 2. Kinetic Parameters for the GDP Dissociation from Full-Length wt Ras and Various Ras Cysteine Mutants in the Presence and Absence of Cdc25 and/or KO2a.
| C51S |
C51S/C118S |
C80S |
full-length wt Ras |
C181S/C184S |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| treatment | GDP dissociation rate constants (10–3 s–1) | span (fraction) | GDP dissociation rate constants (10–3 s–1) | span (fraction) | GDP dissociation rate constants (10–3 s–1) | span (fraction) | GDP dissociation rate constants (10–3 s–1) | span (fraction) | GDP dissociation rate constants (10–3 s–1) | span (fraction) |
| none | 0.01 ± 0.02a | 0.06 ± 0.12 | 0.01 ± 0.02a | 0.45 ± 0.90 | 0.01 ± 0.01a | 0.04 ± 0.04 | 0.01 ± 0.01a | 0.06 ± 0.06 | 0.01 ± 0.02a | 0.04 ± 0.08 |
| Cdc25 | 0.38 ± 0.03b | 0.69 ± 0.05 | 0.39 ± 0.02b | 0.72 ± 0.04 | 0.41 ± 0.03b | 0.73 ± 0.05 | 0.37 ± 0.04b | 0.70 ± 0.08 | 0.38 ± 0.03b | 0.72 ± 0.06 |
| KO2 | 2.31 ± 0.04d | 0.54 ± 0.01 | 0.02 ± 0.02a | 0.06 ± 0.06 | 2.15 ± 0.01c | 0.60 ± 0.03 | 2.09 ± 0.02c | 0.58 ± 0.06 | 1.98 ± 0.02c | 0.45 ± 0.05 |
| Cdc25 + KO2 | 2.85 ± 0.06f | 0.80 ± 0.02 | 0.36 ± 0.05b | 0.55 ± 0.08 | 2.18 ± 0.03c | 0.69 ± 0.09 | 2.11 ± 0.04c | 0.60 ± 0.01 | 2.07 ± 0.02c | 0.70 ± 0.07 |
Data for the rate constants and the spans of GDP dissociation with the SD from C51S and C51/C118S Ras mutants were taken from Figure 4. The values with SD of the rate constants and the spans of GDP dissociation from C80S and C181S/C184S Ras mutants were obtained as described in Figure 4, except that C80S and C181S/C184S Ras mutants were used instead of C51S and C51/C118S Ras mutants. As with the values in Table 1, t tests with P < 0.05 were performed to evaluate the potential similarities or differences among the kinetic values. Table 2 is a continuation of Table 1. Therefore, the letters adopted in Table 1 continue to be used in Table 2. Also, the implications of the letters in Table 2 are exactly the same as in Table 1. For example, data denoted by the letter “a” in Table 2 are the same as the values denoted with “a” in Tables 1 and 2. Conversely, data denoted with a letter “a” in both Tables 1 and 2 differ from other values denoted with other letters, such as “b”, in both Tables 1 and 2. This also applies to other letters in Tables 1 and 2. Notice that the letter “e” in Table 1 does not occur in Table 2. This is because Table 2 contains no equivalent value associated with the letter “e” of Table 1. Finally, there is no comparable value in Tables 1 and 2 for the value with the letter “f” of Table 2 within Tables 1 and 2.
A double mutant Ras C51S/C118S was introduced to further verify the potential O2•–-mediated desensitizing role that Ras Cys51 plays in the catalytic action of Cdc25 on wt Ras. The rate constant and the span of the intrinsic and the Cdc25-mediated GDP dissociation from C51S/C118S Ras (Figure 4B and Table 2) were similar to that of the intrinsic and the Cdc25-mediated GDP dissociation from wt Ras (Figure 2A and Table 1). This result suggests that the Cys51 and Cys118 residues of wt Ras are unnecessary in the Ras nucleotide binding interaction and also in the catalytic action of Cdc25. This is not unusual because the Ras nucleotide binding interaction and the catalytic action of Cdc25 were unaffected by the absence, either separately or in combination, of the Cys51 and Cys118 residue of Ras (see above). Furthermore, the rate constant and the span of the Ras C51S/C118S GDP dissociation by Cdc25 and O2•– together were similar to the rate and the span of the C51S/C118S Ras GDP dissociation by Cdc25 alone (Figure 4B and Table 2). This similarity was unchanged even in the presence of higher concentrations of O2•– (up to 20 μM) (Figure 3). These results indicate that O2•– was unable to desensitize C51S/C118S Ras to Cdc25. Given that the Ras mutant C51S/C118S lacks the Cys118 residue responsible for the direct O2•–-mediated wt Ras GDP dissociation, whatever effect O2•– has on the GDP dissociation from C51S/C118S Ras can be attributed solely to the absence of the Ras residue Cys51. Accordingly, the failure to desensitize C51S/C118S Ras to Cdc25 in the presence of O2•– appears to be caused by the absence of Cys51 in C51S/C118S Ras. These analytical results also support a hypothesis that Ras Cys51 plays a role in the O2•–-mediated desensitization of wt Ras to the catalytic action Cdc25.
Unlike Ras Cys51, Ras Cys118 was shown to be unaffected by the O2•–-mediated inhibition of the catalytic action of Cdc25 (see the section on the deconvolution of the oxidant-mediated inhibition of the catalytic action of Cdc25 on wt Ras GTPase). This result suggests that Ras Cys118 is not involved in the O2•–-mediated desensitization of wt Ras to Cdc25. However, the potential role(s) of other Ras cysteine residues—including Cys80, Cys181, and Cys184—in the O2•–-mediated desensitization of wt Ras to Cdc25 was unclear. The kinetic and redox properties associated with C80S Ras GDP dissociation were exactly the same as those with wt Ras (Table 2). The result suggests that the Cys80 of Ras has no role in the O2•–-mediated desensitization of wt Ras to Cdc25. As noted in the Materials and Methods section, the construction of C51S, C80S, and C118S Ras (1–166) excluded the Ras C-terminus. However, because Cys181 and Cys184 are located at the end of the C-terminus of the full-length wt Ras, C181S/C184S Ras was constructed with the full-length wt Ras (1–189). Therefore, full-length wt Ras serves as a control for C181S/C184S Ras. The kinetic and redox properties of C181S/C184S Ras were similar to the kinetic and redox properties of the full-length wt Ras (Table 2). It is also noteworthy that the kinetic and redox properties of the full-length wt Ras were similar to the kinetic and redox properties of the C-terminal truncated version of wt Ras (Tables 1 and 2). The results suggest that the portion of the N-terminus of the full-length wt Ras that includes the Cys181 and Cys184 residues is unnecessary for the O2•–-mediated desensitization of wt Ras to Cdc25. Taken as a whole, it appears that, as far as these five Ras cysteines are concerned, only Ras Cys51 is involved in the O2•–-mediated desensitization of the action of Cdc25 on wt Ras.
Role of Cysteines of Cdc25 in the Oxidant-Dependent Inhibition of the Cdc25-Mediated wt Ras GDP Dissociation
There are seven cysteine residues in Cdc25. None has been investigated for a potential redox role(s) of any of these Cdc25 cysteines. Given that the oxidant-dependent inhibition of the action of Cdc25 on wt Ras depends entirely on the presence of Ras Cys51, these Cdc25 cysteines are unlikely to participate in the oxidant-dependent inhibition of the Cdc25-mediated wt Ras GDP dissociation. However, the possibility that these Cdc25 cysteines have other roles, such as modulation of the activity of Cdc25, cannot be dismissed. More studies are necessary to examine this possibility.
The O2•–-Mediated Perturbation of the Binding Interaction between wt Ras and Cdc25
Typically, enzymes must bind with their substrates as a prerequisite for their catalytic action.50 In accounting for the kinetic results showing that Ras Cys51 is a central element in the O2•–-mediated desensitization of wt Ras to Cdc25, we hypothesize that the Ras Cys51-targeting action of O2•– is implicated in the perturbation of the binding interaction between Cdc25 and wt Ras that results in desensitization of wt Ras to Cdc25.
To examine the potential O2•–-mediated perturbation of the binding interaction between Cdc25 and Ras via the targeting of Ras Cys51, a binary complex of Cdc25 with the rhodamine fluorescence-tagged wt Ras and all available cysteine mutants was titrated with O2•– in the absence of free GDP. Figure 5A shows that the rhodamine fluorescence intensity of Cdc25 complexed with wt Ras decreased hyperbolically with increases in the concentration of O2•–. This is an indicator of the dissociation of Cdc25 from wt Ras; it suggests that O2•– interferes with the binding of Cdc25 to wt Ras. The rhodamine fluorescence intensity of Cdc25 complexed with C118S Ras also decreased hyperbolically after treatment with O2•– (Figure 5A). This result suggests that Ras Cys118 is not involved in the O2•–-mediated interference with the binding interaction between Cdc25 and Ras. Identical results were obtained with other Ras cysteine mutants C80S and C181S/C184S Ras (not shown). However, the O2•–-dependent decrease in rhodamine fluorescence was not observed when either C51S or C51S/C118S Ras was used instead of wt Ras or C118S Ras (Figure 5A). This result indicates that treatment with O2•– did not enhance dissociation of Cdc25 from C51S or C51S/C118S Ras. Accordingly, this result suggests that the targeting action of O2•– on Ras Cys51, but not on Ras Cys80, Cys118, Cys181, and Cys184, is linked to the perturbation of the binding interaction between Cdc25 and wt Ras.
Figure 5.

Determination of the KO2-mediated perturbation of Cdc25 binding interaction with wt Ras, C118S Ras, and C51S/C118S Ras. (A) Cdc25 complexed with rhodamine fluorescence tagged-wt Ras, -C118S Ras, and -C51S/C118S Ras (1 μM) was treated with various concentrations of KO2 (between 0 and 20 μM), as indicated by the arrows. The corresponding changes in fluorescence intensity were monitored. When necessary, a radical quencher DMPO (1 mM) or ascorbic acid (1 mM; not shown) was added after treatment with KO2, as indicated by the arrows. (B) To ensure confidence in the results, the experiments shown in panel A were repeated two more times. The changes in fluorescence intensities of these triple measurement data sets were plotted against the KO2 concentrations. The F tests with a hyperbola model (P < 0.05) support one curve for each of triple data sets. Hence, the fluorescence values are shown in this figure with mean values and the SD from independent triplicate measurements. The global fits with a hyperbola model (P < 0.05) gave the apparent dissociation constants of KO2 for the wt Ras–Cdc25, C118S Ras–Cdc25, C51S/C118S Ras–Cdc25, and C51S Ras–Cdc25 binary complexes. These constants are, respectively, 2.3 ± 0.1, 2.2 ± 0.1, >20.6 ± 0.9, and >20.6 ± 0.8 μM [KO2] with r2 > 0.9095.
Unlike the slow pace of the O2•–-mediated wt Ras nucleotide dissociation (Figure 2A), the O2•–-mediated wt Ras Cdc25 dissociation is more likely to be immediate (Figure 5A). This is possibly because the Ras Cdc25 binding interactions are not multilayered. Also, an increase in fluorescence intensity was observed after addition of either a radical quencher 5,5-dimethyl-1-pyrroline N-oxide (DMPO) to the O2•–-treated Cdc25 complexed with the rhodamine fluorescence-tagged wt Ras or C118S Ras (Figure 5A). Treatment with ascorbic acid (1 mM), instead of DMPO, showed the same result (not shown). The thiyl radical can be quenched by DMPO and ascorbic acid.51,52 Therefore, this result supports the involvement of a thiyl radical in the O2•–-mediated perturbation of the binding interaction between Cdc25 and wt Ras or C118S Ras. Given that Ras Cys51 is the target site of the oxidant that couples with the O2•–-mediated perturbation of this binding interaction, the thiyl radical is likely formed on the Ras Cys51 side chain. This result, therefore, discloses a radical formation as an essential step involved in the O2•–-mediated perturbation of the wt Ras Cdc25 binding interaction. In addition, the fact that both DMPO and ascorbic acid reverse the effect of O2•– on the wt Ras or C118S Ras Cdc25 complex raises the possibility of the reversability of the O2•–-mediated perturbation of the binding interaction between Cdc25 and Ras.
The values of the KD of O2•– for wt Ras and C118S Ras complexed with Cdc25, respectively, were determined to be 2.3 and 2.2 μM (Figure 5B). The O2•–-dependent changes in the rhodamine fluorescence intensity of C51S and C51S/C118S Ras complexed with Cdc25 were nevertheless negligible (Figure 5B). In light of these minimal changes, the values determined for the KD of O2•– for Ras and for C118S Ras complexed with Cdc25 may represent the sensitivity of the targeting action of O2•– on Ras Cys51 that interferes with both the Ras and C118S Ras binding interaction with Cdc25. Intriguingly, these KD values of O2•– for wt Ras and for C118S Ras complexed with Cdc25 are similar to those of the appKI values of O2•– for wt Ras and for C118S Ras in the presence of Cdc25 (Figure 3). This similarity suggests a hypothesis that the Ras Cys51-targeting action of O2•– interferes with the binding interaction of Cdc25 with Ras and that this interference is linked to the O2•–-mediated desensitization of wt Ras for Cdc25.
Lack of the Oxidant-Mediated Inhibition of the RhoGEF-Mediated GDP Dissociation from Rac1 and RhoC
To determine the effect of oxidants on the catalytic function of the RhoGEFs, including Vav2 DPC and Dbs DH/PH or vice versa, we examined the rate of Rac1 and RhoC GDP dissociation in the presence and absence of these GEFs and/or H2O2 and O2•–.
wt Rac1 GDP dissociation was enhanced by its GEF Vav2 DPC or KO2 alone (Figure 6). Treatment with SOD ended the KO2-mediated wt Rac1 GDP dissociation (Figure 6). These results suggest that either Vav2 DPC or O2•– derived from KO2 enables facilitation of the wt Rac1 GDP dissociation, which is consistent with previous results.23 The values of the rate constant and the span of the Vav2 DPC-mediated wt Rac1 GDP dissociation are, respectively, 6.9-fold smaller and 1.2-fold larger than the values of the rate constant and the span of the O2•–-mediated wt Rac1 GDP dissociation (Table 3). The difference in the rate constant values indicates that, under our experimental conditions, the rate of the wt Rac1 GDP dissociation by Vav2 DPC is much slower than the rate of dissociation of wt Rac1 GDP by O2•–. The difference in the span values indicates that, under these experimental conditions, the quantity of the wt Rac1 GDP dissociation by Vav2 DPC exceeds that by O2•–. However, as noted in Materials and Methods, this is simply because the quantity of concentration treated with O2•– (∼3 μM) is insufficient to complete the wt Rac1 GDP dissociation.
Figure 6.

Effect of KO2 on the Rho GEF-mediated GDP dissociation from wt Rac1. The experimental and analytical methods were identical to those used in Figure 2, except that wt Rac1 (1 μM) and its GEF, Vav2 DPC (100 nM), were used instead of wt Ras and Cdc25. The F tests with a hyperbola model (P < 0.05) indicate that the curve differences within each of the triple data sets are insignificant. Therefore, mean values and the SD from each of the independent triple measurements are presented in this figure. The global fits for each of these triplicate data with the hyperbola model (P < 0.05) gave kinetic values and their corresponding SD. The values determined for the apparent GDP dissociation rates of Rho proteins in the presence and absence of a regulator(s) are summarized in Table 3. The r2 values of all analyses were >0.9065.
Table 3. Kinetic Parameters for the GDP Dissociation from wt Rac1 and wt RhoC in the Presence and Absence of Vav2 DPC or Dbs DH/PH and/or KO2a.
| wt Rac1 |
wt RhoC |
|||
|---|---|---|---|---|
| treatment | GDP dissociation rate constants (10–3 s–1) | span (fraction) | GDP dissociation rate constants (10–3 s–1) | span (fraction) |
| none | 0.01 ± 0.03a | 0.04 ± 0.12 | 0.01 ± 0.01a | 0.05 ± 0.05 |
| Vav2 DPC | 0.53 ± 0.01b | 0.66 ± 0.01 | ND | ND |
| Dbs DH/PH | ND | ND | 0.48 ± 0.01b | 0.66 ± 0.01 |
| KO2 | 3.64 ± 0.01c | 0.55 ± 0.02 | 3.86 ± 0.03c | 0.56 ± 0.06 |
| Vav2 DPC + KO2 | 3.71 ± 0.02c | 0.67 ± 0.04 | ND | ND |
| Dbs DH/PH + KO2 | ND | ND | 3.67 ± 0.02c | 0.68 ± 0.04 |
The kinetic values of the rate constants and the spans for GDP dissociation from wt Rac1, in the presence and absence of Vav2 DPC and KO2, were taken from Figure 6. The kinetic values of the rate constants and the spans for GDP dissociation from wt RhoC in the presence and absence of Dbs DH/PH and KO2 also were obtained as described in Figure 6, except that wt RhoC was used instead of wt Rac1. To evaluate the significance of the potential similarities and differences among the rate constant values listed within Table 3, t tests with P < 0.05 were performed as described in Materials and Methods. Any rate constant values that refer to the letter “a” are the same as other values coupled with the same letter “a”. This also applies to all other letters used within Table 3. However, any values denoted with “a” differ from the values denoted with “b” and “c”. The values denoted by “b” also differ from the values associated with the letter “c”. For presentation clarity, only the t tests for the rate constants are shown. However, all of the t test results associated with the rate constants were exactly the same as the t test results for the span values that correspond to the rate constants. ND, not determined.
wt Rac1 GDP dissociation was significantly enhanced by a combination of Vav2 DPC and O2•– (Figure 6 and Table 3). In contrast to what occurred with wt Ras with a combination of Cdc25 and O2•– (Table 1), the values of the rate constant and the span of the Vav2 DPC/O2•–-mediated wt Rac1 GDP dissociation were almost the sum of the values of the rate constant and the span of the Vav2 DPC-mediated wt Rac1 GDP dissociation plus the O2•–-mediated wt Ras GDP dissociation (Table 3). For such an approximation of the total sum to occur means neither O2•– nor Vav2 DPC, respectively, interferes with the catalytic action of Vav2 DPC or of O2•– on wt Rac1. These results, therefore, suggest that the catalytic action of Vav2 DPC and O2•– on the wt Rac1 GDP dissociation is preserved even if both Vav2 DPC and O2•– are present simultaneously.
The catalytic action of another GEF, Dbs DH/PH, on another redox-sensitive wt Rho GTPase, wt RhoC, was unperturbed by O2•– (Table 3). O2•– also failed to perturb the catalytic action of Dbs DH/PH on wt RhoA and on wt Cdc42 (not shown). Therefore, this conclusion concerning the action of Vav2 DPC on wt Rac1 GDP dissociation with the oxidant O2•– is also applicable to the catalytic action of Dbs DH/PH on the dissociation of GDP from wt RhoA, RhoC, and Cdc42 with O2•–.
The GDP dissociation from wt Rac1, wt RhoC, wt RhoA, and wt Cdc42 was not facilitated by H2O2 (not shown), which is consistent with the previous result.23 Similarly, H2O2 did not affect the Vav2 DPC or Dbs DH/PH-mediated GDP dissociation from these redox-sensitive wt Rho proteins (not shown). Taken together, these results suggest that the GDP dissociation from these redox-sensitive Rho proteins as well as the catalytic function of Vav2 DPC and Dbs DH/PH on these redox-sensitive Rho proteins are insensitive to H2O2.
Discussion
This study shows that an oxidant, O2•–, inhibits the catalytic action of Cdc25—the catalytic core domain of RasGEFs—on wt Ras. Although not shown for clarity of presentation, •NO2 can be used to mimic this inhibitory effect of O2•– on the catalytic action of Cdc25 with wt Ras and its cysteine mutants. This function of O2•– or •NO2 is an intriguing addition to the previously known role of oxidants in regulating wt Ras activity.23,24 Because the use of Ras C51S as the substrate of Cdc25 nullified the oxidant-mediated inhibition of the catalytic action of Cdc25 on wt Ras, the apparent mechanism of this inhibitory effect lies in the Ras Cys51-targeting action of the oxidant. This action appears to cause wt Ras insensitivity to the Cdc25 of RasGEFs.
Unlike with O2•– and •NO2, H2O2 did not inhibit the catalytic action of Cdc25 on wt Ras. Taking into account that Ras Cys51 is a target site of the oxidant, this is consistent with the chemistry in which the side chain of Ras Cys51 that contains the sulfur atom does not react with H2O2 but does react with O2•– or •NO2. However, this result does not necessarily eliminate the role of H2O2 as a redox agent that modulates wt Ras activity with RasGEFs in cells. This is because H2O2 in cells can be converted into other oxidants such as a hydroxyl radical and a hydroxyl anion through the transition metal-mediated Fenton reaction.35 These free radicals from the Fenton reaction also are known to react with sulfur.9 Therefore, H2O2—through its derivation products but not in its original form—can function as an oxidant that can inhibit the catalytic action of Cdc25 on wt Ras.
Because Cdc25 is the catalytic core domain of many RasGEFs, including SOS, RasGRF, and RasGRP, the observed desensitization of wt Ras to Cdc25 by an oxidant represents the fundamental trait of the oxidant-dependent regulation of wt Ras activity associated with these RasGEFs. However, it is unclear if the oxidant has other effects on the catalytic action of Cdc25 on wt Ras through unexamined noncatalytic RasGEF domains. For example, it is uncertain whether the oxidant affects the function of the noncatalytic domain(s) of RasGEFs (e.g., SOS), such as the DH and PH domains. This study shows that, as a RhoGEF catalysis, the function of the DH domain in combination with the PH domain is unaffected by the oxidant (see the discussion below). However, this result does not necessarily indicate that an oxidant has no effect on the regulatory role of these noncatalytic domains in RasGEFs. The roles of these noncatalytic domains of RasGEFs in the current model are linked to the membrane anchorage of RasGEFs that is accompanied by RasGEF activation.18 Nevertheless, in assessing this current model, the apparent uncertainties about the oxidant-dependent regulation of the activity of RasGEFs via these noncatalytic domains of RasGEFs is not enough to overshadow the significance of the finding of the oxidant-mediated inhibition of the catalytic action of Cdc25 on wt Ras. This is because of the expected conservation of this inhibition feature regardless of any potential effects of the oxidant on these noncatalytic domains of RasGEFs. For example, if the oxidant does not perturb the functions of the noncatalytic domains of RasGEFs, the oxidant will continue to inhibit the catalytic action of Cdc25 on wt Ras. If the oxidant serves as a negative heterotropic effector by perturbing the membrane-anchorage functions of the noncatalytic domains of RasGEFs, RasGEFs cannot be recruited for the plasma membrane and activated. In this case, the oxidant inhibits the RasGEF activation before inhibition of RasGEFs’ catalytic function. Therefore, the oxidant will continue to inhibit the catalytic action of Cdc25 on wt Ras. Finally, even if the oxidant serves as a positive heterotropic effector by enhancing functions of the noncatalytic domains of RasGEFs so as to enhance RasGEF membrane binding and RasGEF activation, the oxidant ultimately counteracts the RasGEF activation by inhibiting the catalytic action of RasGEFs on wt Ras. Therefore, RasGEFs will remain in an inactive state. In summary, regardless of any instances of the action of the oxidant on the noncatalytic domains of RasGEFs, no alteration would occur in the inhibitory effect of the oxidant on the catalytic action of RasGEFs. Therefore, although the roles of the oxidant in these noncatalytic domains of RasGEFs are yet to be investigated, the observed desensitization of wt Ras to Cdc25 by an oxidant is sufficient for it to be considered a factor in regulating the wt Ras activity associated with these RasGEFs, thereby controlling the Ras-dependent cell signaling cascades.
Implication of the Oxidant-Mediated Inhibition of the RasGEF Actions on wt Ras
Although the oxidant can directly activate wt Ras via an enhancement of wt Ras GDP dissociation,23,24 the oxidant also can indirectly inhibit the RasGEF-mediated wt Ras activation through desensitization of wt Ras to the catalytic core domain Cdc25 of RasGEFs (this study). This redox regulation of wt Ras activity is more complicated than previously thought,9 but the result of this complexity in terms of wt Ras GDP dissociation is rather straightforward: The RasGEF-mediated enhancement of the GDP dissociation from wt Ras will only occur in the presence of RasGEFs when the oxidant is absent. When both the oxidant and RasGEFs are present, the oxidant desensitizes wt Ras to the catalytic core domains of RasGEFs to perturb the catalytic action of RasGEFs. At the same time, the oxidant retains its capability to enhance the dissociation of wt Ras GDP, thereby only continuing the oxidant-mediated enhancement of wt Ras GDP dissociation. These analyses infer that, even if there are two unrelated signaling events such as the nonredox and redox stimuli—for example, hormone- and oxidative stress-dependent, respectively—toward wt Ras, the Ras-dependent cellular signaling would not be overly upregulated. This would prevent the overresponse of the Ras-dependent cellular signaling cascades from multiple-signaling stimuli toward the wt Ras. Failure of the mechanism of the oxidant-mediated inhibition of the catalytic action on Ras (i.e., by Ras Cys51 mutation) triggers the two simultaneous, but unrelated, signaling events that result in overactivation of wt Ras. An overactivated wt Ras may possibly alter various Ras-dependent cellular effects, including cell survival, proliferation, and differentiation, in ways so as to produce certain diseases such as cancer.53 This analysis introduces the potential significance of the oxidant-mediated inhibition of the function of RasGEFs on wt Ras as a regulatory outlet that prevents overactivation of wt Ras as a cause of pathophysiological responses. Also, the dominant feature of the redox-dependent response of wt Ras compared with the nonredox-dependent response of wt Ras suggests that, where wt Ras-dependent cellular signaling cascades are concerned, the redox stimulus is somehow given more weight than the nonredox stimulus. The cellular meaning of this dominancy in regulating the wt Ras activity remains to be clarified.
The redox aspect of the intrinsic catalytic action of RasGEFs on the chemically modified form of wt Ras—such as wt Ras-SNO—is intriguing. The reason for this interest is because the oxidant was unable to enhance the GDP dissociation from wt Ras-SNO but nevertheless was able to desensitize wt Ras-SNO to the catalytic core domains of RasGEFs, resulting in inhibition of the RasGEF-mediated dissociation of GDP from wt Ras-SNO. Consequently, when an oxidant and RasGEFs are present simultaneously, the GDP dissociation from wt Ras-SNO cannot be accelerated. This notion implies that the oxidant can downregulate Ras-SNO regardless of the presence of RasGEFs. However, the appVmin value of the catalytic action of Cdc25 on C118S Ras occurs at a significantly high concentration of O2•– (near 30 μM). This suggests that a significantly high concentration of O2•– is required for it to complete the inhibition of the RasGEF action on wt Ras-SNO. Such a high concentration of O2•– occurs only in special cases, such as stimulated macrophages.54 Therefore, O2•– may not be completely inhibiting RasGEFs on wt Ras-SNO in the typical oxidative stress conditions of cells. However, although it could depend on cellular conditions, a complete inhibition of the RasGEF action on wt Ras is not necessary to achieve the minimal level of the cellular fraction of the GTP-bound form of wt Ras.55 Therefore, incomplete inhibition of RasGEFs by an oxidant that spurs a certain minimal level of activity by RasGEFs is sufficient to generate a basal level of cellular activity of wt Ras-SNO.
Although the cellular conditions necessary to produce wt Ras-SNO are yet to be clarified, we have found that a continuous treatment of bladder carcinoma (T24) and fibrosarcoma (HT1080) cells with the oxidant NO for at least 2 h produces wt Ras-SNO (unpublished results). Maximization of the formation of wt Ras-SNO can also be achieved by long-term treatment of NO (e.g., at least 1 day). Intriguingly, wt Ras-SNO was formed in 15 min by treatment of NIH 3T3 and PC12 cells with S-nitrosocysteine—another Ras S-nitrosation agent.56 This result in conjunction with our result suggests that, compared with the formation of wt Ras-SNO by NO, the formation of wt Ras-SNO by S-nitrosocysteine is likely effective in generating wt Ras-SNO. It is thus possible that the degree of efficiency in the formation of wt Ras-SNO depends on the type of oxidant. Nonetheless, when these findings are taken together, it can be postulated that the continuous presence of the oxidant not only results in the formation of wt Ras-SNO but also blocks GDP dissociation from the newly formed wt Ras-SNO. The outcome of this formation and blockage is production of a GDP-bound biologically inactive wt Ras-SNO. This postulation explains the enigmatic result of wt Ras inactivation by a long-term treatment of cells (>1 day) with NO.28−30 Accordingly, although a definitive finding awaits further studies, it is tempting to speculate that the extensive and continuous presence of the oxidant leads to severe oxidative stress that results in inactivation of wt Ras and shuts down wt Ras-mediated cellular signaling events. Failure to prevent prolonged oxidant-mediated wt Ras activation could result in continuation of the upregulation of the Ras-dependent cellular signaling events. Such deregulation could result in certain diseases such as cancer. Therefore, the pathophysiological significance of the formation of wt Ras-SNO and its inactivation mechanism may be to prevent continuous oxidative stress from overactivating Ras. This notion is supported by the relatively higher fraction of the GDP-bound form of wt Ras-SNO, compared with that of wt Ras, in NIH 3T3 and PC12 cells.56
Oxidant-mediated wt Ras inactivation may not occur when oxidative stress is not continuous. For example, a short burst of oxidative stress activated wt Ras instead of inhibiting it.57 In fact, a short-term treatment of cells with NO did not generate wt Ras-SNO (unpublished results). Therefore, a short and/or a one-time treatment of cells with H2O2 or other oxidants may be insufficient to generate an oxidized form of wt Ras (e.g., wt Ras-SNO) but may be sufficient to activate wt Ras via the redox-mediated wt Ras activation mechanism.9
Kinetic Mechanism of the Oxidant-Mediated Inhibition of the Catalytic Activity of RasGEFs on wt Ras
This study shows that, among other Ras cysteines, the Ras Cys51 is involved in the oxidant-mediated interference with the wt Ras binding interaction with Cdc25. This interference results in desensitization of wt Ras to Cdc25. Such desensitization apparently inhibits the catalytic action of Cdc25 on wt Ras.
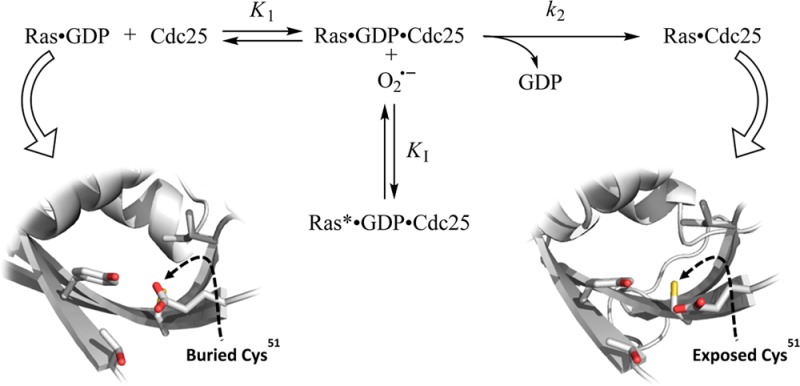
Figure 7 shows that two mechanistic steps are among the essential features of the process by which Cdc25 enhances the nucleotide exchange of wt Ras GTPase.49,58 The first of these is the binding of Cdc25 to the GDP-bound wt Ras to produce the ternary wt Ras–GDP–Cdc25 complex. The formation of the ternary complex disrupts the binding interaction between wt Ras and GDP, resulting in release of the bound GDP to produce the wt Ras Cdc25 binary complex. The second step is the binding of the cellularly abundant GTP to the wt Ras complexed with Cdc25; this binding expels the bound Cdc25 from wt Ras to produce GTP-bound wt Ras. Given that an oxidant inhibits the catalytic action of Cdc25 on wt Ras that is an enhancement of the wt Ras-bound GDP, the oxidant evidently targets the ternary complex (Figure 7). Accordingly, the pattern of the oxidant-mediated inhibition of the catalytic action of Cdc25 on wt Ras can be classified as apparent uncompetitive. This apparent uncompetitive kinetic pattern suggests that the reaction of the oxidant with the ternary complex produces the radicalized wt Ras–GDP–Cdc25 ternary complex (wt Ras*–GDP–Cdc25) through the kinetic step of KI. Formation of wt Ras*–GDP–Cdc25 dodges the k2 step—the key catalytic step of the catalytic action of Cdc25 on wt Ras (Figure 7). This kinetic scheme also suggests that the more the oxidant is proportionate to the production of the radicalized wt Ras–GDP–Cdc25 complex, but the lesser the oxidant is proportionate to the turnover of the Cdc25-mediated wt Ras GDP dissociation via the k2 step. This relation defines the disproportionate feature of the KI value over the k2 value. Notably, the appKI value (2.3 μM [KO2], Figure 3) was determined by monitoring the value of the C118S Ras GDP dissociation (the k2 value) in the presence of the oxidant. Therefore, the appKI value that was determined essentially represents the value of KI of the oxidant for wt Ras–GDP–Cdc25 to produce wt Ras*–GDP–Cdc25.
Figure 7.
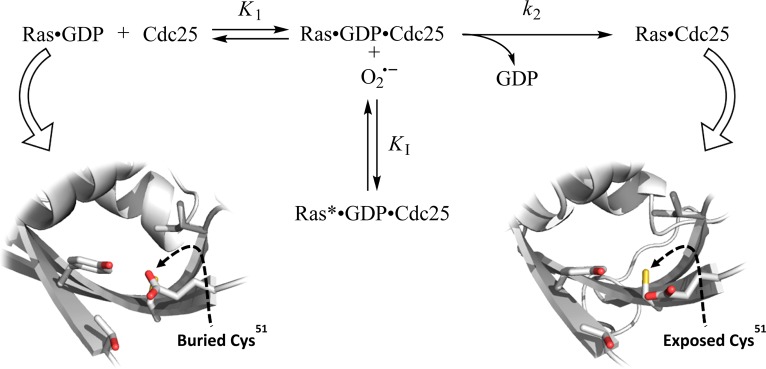
Proposed kinetic mechanism of the oxidant-mediated inhibition of the RasGEF action on Ras. An apparent uncompetitive inhibition of the oxidant for the catalytic action of Cdc25 on wt Ras is shown with several kinetic constants. The binary wt Ras–GDP complex binds Cdc25 to produce the ternary wt Ras–GDP–Cdc25 complex, and this step couples with an equilibrium association constant K1. The step represented by the rate constant k2 that produces the Ras–Cdc25 binary complex is the rate limiting step for the catalytic action of Cdc25 on wt Ras. The cellularly abundant GTP displaces the wt Ras bound Cdc25 to produce the GTP-bound wt Ras. When the inhibitor—an oxidant—exists, through the step of KI, the ternary wt Ras–GDP–Cdc25 complex reacts with the oxidant to produce the radicalized ternary wt Ras–GDP–Cdc25 complex (ternary wt Ras*–GDP–Cdc25 complex). The buried Ras Cys51 side chain in the cartoon figure of the binary wt Ras–GDP complex (PDB 1AGP) is shown with a dotted arrow (left). The solvent-exposed Ras Cys51 side chain in the cartoon figure of the binary wt Ras–Cdc25 complex (PDB 1BKD) is indicated with a dotted arrow (right). These figures were generated using The PyMOL Molecular Graphics System, Version 1.6 Schrödinger, LLC.
Potential Role of Ras Cys51 in the Oxidant-Mediated Inhibition of the Catalytic Activity of RasGEFs by the Perturbation the wt Ras–Cdc25 Binding Interaction
Although the pattern of the oxidant-mediated inhibition of the catalytic action of Cdc25 on wt Ras can be defined as apparent uncompetitive, details of the role of Cys51 in this inhibition are unclear. As a target of the oxidant, the side chain of the Ras Cys51 in the ternary wt Ras–GDP–Cdc25 complex must be accessible to solvents. This possibility cannot be inspected because the structure of the ternary complex is unknown. However, the features of the available crystal structures before and after the ternary wt Ras–GDP–Cdc25 complex (wt Ras–GDP complex) and (wt Ras–Cdc25 complex) (Figure 7), respectively, suggest that the binding interaction of wt Ras with Cdc25 exposes the side chain of the Ras Cys51 to solvents. Such binding interaction is the key to the catalytic action of Cdc25 on wt Ras. Accordingly, it can be postulated that the oxidant, via targeting Ras Cys51, interferes with the wt Ras Cdc25 binding interaction and consequently restricts the catalytic action of Cdc25 on wt Ras. Furthermore, given the similarity of the value of the KD of the oxidant for the wt Ras–Cdc25 complex and that of the appKI of the oxidant for the ternary complex, the perturbation of the wt Ras Cdc25 binding interaction is likely the main factor that determines the oxidant-mediated inhibition of the catalytic action of Cdc25 on wt Ras.
Mechanically, the oxidant-dependent protein–protein binding interaction can be perturbed by either the state- or the process-based route. The mechanism associated with the state-based route herein refers to an outcome in which a chemical modification of the redox-sensitive cysteine and/or its relevant residue results in the alteration of a protein structure. This protein structural change perturbs its protein binding interaction with its counterpart. The mechanism of the process-based route within this article denotes that the reaction process between the oxidant and the redox-sensitive cysteine associated with its relevant residue(s) perturbs the protein binding interaction with its counterpart. In this mechanism, the chemical modification of the redox-sensitive cysteine and/or its relevant residue(s) occurs at the end of the process of the perturbation of the binding interactions between the protein and its counterpart. As for the redox regulation of wt Ras, the state-based mechanism was initially postulated for the oxidant-mediated enhancement of the wt Ras GDP dissociation coupled with the Ras-SNO formation. Similarly, the process-based mechanism was postulated as an alternative of the state-based mechanism to explain the oxidant-mediated enhancement of the wt Ras and wt Rho GDP dissociation without formation of the wt Ras-SNO.59 Nonetheless, based upon the currently available data, it is difficult to predict which of these routes best describes the mechanism of the oxidant-mediated inhibition of the catalytic action of Cdc25 on wt Ras.
The chemical modification and/or oxidation state of the Ras Cys51 side chain is of interest because it could directly support the involvement of Ras Cys51 with an oxidant in the perturbation of the wt Ras–Cdc25 binding interaction. However, our mass spectrometric analyses, which used both electrospray ionization and matrix-assisted laser desorption/ionization time-of-flight approaches, did not detect any of the chemical modification/oxidation of the Ras Cys51 side chain under our experimental conditions. Nevertheless, this failure does not disprove the involvement of Ras Cys51 in the oxidant-mediated perturbation of the wt Ras–Cdc25 binding interaction. There is a precedent for this situation in which, although Ras Cys118 is typically a target of an oxidant to enhance the wt Ras GDP dissociation (which means Ras Cys118 is certainly involved in the oxidant-mediated enhancement of the wt Ras GDP dissociation), Ras Cys118 was not chemically modified/oxidized by the oxidant after the completion of the oxidant-mediated wt Ras GNE.59 It also is noteworthy that the lack of chemical modification/oxidation of the Ras Cys51 side chain does not necessarily indicate that any of these potential mechanisms fit the case of the oxidant-mediated perturbation of the wt Ras–Cdc25 binding interaction. To the contrary, the potential chemical modification/oxidation of the Ras Cys51 side chain does not support any of these mechanisms. This is because the state of the features of the chemical modification/oxidation of the Ras Cys51 side chain does not necessarily couple with the structural change in wt Ras and/or Cdc25 nor does it necessarily couple with the mechanistic perturbation of the binding interaction between wt Ras and Cdc25. This argument is again based upon the case of the oxidant-mediated enhancement of the wt Ras GDP dissociation in which the oxidant-mediated Ras chemical modification—Ras-SNO formation—couples with neither the Ras structural change nor with the mechanistic perturbation of the binding interaction between wt Ras and a nucleotide.9
Redox Regulation of wt Rho GTPases with RhoGEFs
Studies show that the GDP binding interactions of certain wt Rho GTPases, including wt Rac1 as well as wt RhoC, wt RhoA, and wt Cdc42, are redox sensitive.9,23 This study shows that, in contrast to the effect of O2•– or •NO2 on Cdc25 with wt Ras, O2•– or •NO2 is unable to inhibit the catalytic action of Vav2 DPC on wt Rac1. Vav2 DPC is the wt Rac-specific RhoGEF that possesses a catalytic domain as well as the regulatory domains of Vav. Hence, regulation of the catalytic activity of Vav associated with its regulatory domains is likely to be insensitive to O2•– and •NO2.
This study also shows that neither O2•– nor •NO2 inhibits the catalytic action of Dbs DH/PH on wt RhoC or wt RhoA. Dbs DH/PH is the catalytic domain of Dbs that is a wt RhoA- and wt RhoC-specific RhoGEF. Hence, although it is clear that the catalytic function of Dbs is unaffected by O2•– or •NO2, the potential redox role of other known or unknown regulatory domains of Dbs is of interest because the Dbs regulatory domain(s) can influence the catalytic activity of Dbs. A previous study showed that using oxidant NO in the long term treatment of SUM and wt RhoC-overexpressed primary human mammary epithelial cells did not block the loading of a nucleotide analog, 6-thioguanine nucleotide, to wt RhoC.9 Notably, these SUM cells treated with NO were exposed to O2,9 and thus, it can be safely assumed that at least some portion of the treated NO reacts with O2 to produce •NO2 and higher oxides.9 Accordingly, the generation of the 6-thioguanine nucleotide-bound wt RhoC in these cells likely was solely because the catalytic action of Dbs was uninhibited despite the presence of the oxidant •NO2. Taking into account that the Dbs expressed constitutively in these cells is a full construct that contains catalytic and regulatory domains, the results and analysis together suggest that the catalytic function of Dbs was not inhibited by an oxidant despite the presence of the Dbs regulatory domains. Hence, it can be postulated that, as with Vav, an oxidant such as O2•– or •NO2 does not influence the catalytic function of Dbs in association with its regulatory domains.
Glossary
Abbreviations Used
- DMPO
5,5-dimethyl-1-pyrroline N-oxide
- DH
Dbl homology
- Dbs
Dbl’s big sister
- Dbs DH/PH
Dbs with DH and PH domains
- GAPs
GTPase activating proteins
- GEFs
guanine nucleotide exchange factors
- HRas
Harvey Ras
- PH
Pleckstrin homology
- RasGEFs
Ras-specific GEFs
- RhoGEFs
Rho-specific GEFs
- Ras-SNO
S-nitrosated Ras
- SOD
superoxide dismutase
- r2
the regression values
- [3H]GDP
tritium-labeled guanosine diphosphate and
- Vav2 DPC
Vav2 with DH, PH, and cysteine-rich domains
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Wennerberg K.; Rossman K. L.; Der C. J. (2005) The Ras superfamily at a glance. J. Cell Sci. 118, 843–846. [DOI] [PubMed] [Google Scholar]
- Colicelli J. (2004) Human RAS superfamily proteins and related GTPases. Sci. STKE 2004, RE13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahearn I. M.; Haigis K.; Bar-Sagi D.; Philips M. R. (2011) Regulating the regulator: post-translational modification of RAS. Nat. Rev. Mol. Cell Biol. 13, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y.; Grabocka E.; Bar-Sagi D. (2011) RAS oncogenes: weaving a tumorigenic web. Nat. Rev. Cancer 11, 761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heasman S. J.; Ridley A. J. (2008) Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 9, 690–701. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S.; Hall A. (2002) Rho GTPases in cell biology. Nature 420, 629–635. [DOI] [PubMed] [Google Scholar]
- Mueller B. K.; Mack H.; Teusch N. (2005) Rho kinase, a promising drug target for neurological disorders. Nat. Rev. Drug Discovery 4, 387–398. [DOI] [PubMed] [Google Scholar]
- Karnoub A. E.; Weinberg R. A. (2008) Ras oncogenes: split personalities. Nat. Rev. Mol. Cell Biol. 9, 517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo J. (2011) Redox control of GTPases: from molecular mechanisms to functional significance in health and disease. Antioxid. Redox Signal 14, 689–724. [DOI] [PubMed] [Google Scholar]
- Geyer M.; Wittinghofer A. (1997) GEFs, GAPs, GDIs and effectors: taking a closer (3D) look at the regulation of Ras-related GTP-binding proteins. Curr. Opin. Struct. Biol. 7, 786–792. [DOI] [PubMed] [Google Scholar]
- Bonfini L.; Karlovich C. A.; Dasgupta C.; Banerjee U. (1992) The Son of sevenless gene product: a putative activator of Ras. Science 255, 603–606. [DOI] [PubMed] [Google Scholar]
- Shou C.; Farnsworth C. L.; Neel B. G.; Feig L. A. (1992) Molecular cloning of cDNAs encoding a guanine-nucleotide-releasing factor for Ras p21. Nature 358, 351–354. [DOI] [PubMed] [Google Scholar]
- Ebinu J. O.; Bottorff D. A.; Chan E. Y.; Stang S. L.; Dunn R. J.; Stone J. C. (1998) RasGRP, a Ras guanyl nucleotide- releasing protein with calcium- and diacylglycerol-binding motifs. Science 280, 1082–1086. [DOI] [PubMed] [Google Scholar]
- Boriack-Sjodin P. A.; Margarit S. M.; Bar-Sagi D.; Kuriyan J. (1998) The structural basis of the activation of Ras by Sos. Nature 394, 337–343. [DOI] [PubMed] [Google Scholar]
- Stone J. C. (2011) Regulation and Function of the RasGRP Family of Ras Activators in Blood Cells. Genes Cancer 2, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimnual A. S.; Yatsula B. A.; Bar-Sagi D. (1998) Coupling of Ras and Rac guanosine triphosphatases through the Ras exchanger Sos. Science 279, 560–563. [DOI] [PubMed] [Google Scholar]
- Lemmon M. A. (2008) Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 9, 99–111. [DOI] [PubMed] [Google Scholar]
- Groves J. T.; Kuriyan J. (2010) Molecular mechanisms in signal transduction at the membrane. Nat. Struct. Mol. Biol. 17, 659–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumeister M. A.; Rossman K. L.; Sondek J.; Lemmon M. A. (2006) The Dbs PH domain contributes independently to membrane targeting and regulation of guanine nucleotide-exchange activity. Biochem. J. 400, 563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y.; Olson M. F.; Hall A.; Cerione R. A.; Toksoz D. (1995) Direct involvement of the small GTP-binding protein Rho in lbc oncogene function. J. Biol. Chem. 270, 9031–9034. [DOI] [PubMed] [Google Scholar]
- Abe K.; Rossman K. L.; Liu B.; Ritola K. D.; Chiang D.; Campbell S. L.; Burridge K.; Der C. J. (2000) Vav2 Is an Activator of Cdc42, Rac1, and RhoA. J. Biol. Chem. 275, 10141–10149. [DOI] [PubMed] [Google Scholar]
- Heo J.; Campbell S. L. (2004) Mechanism of p21Ras S-nitrosylation and Kinetics of Nitric Oxide-mediated Guanine Nucleotide Exchange. Biochemistry 43, 2314–2322. [DOI] [PubMed] [Google Scholar]
- Heo J.; Campbell S. L. (2005) Superoxide Anion Radical Modulates the Activity of Ras and Ras-related GTPases by a Radical-based Mechanism Similar to that of Nitric Oxide. J. Biol. Chem. 280, 12438–12445. [DOI] [PubMed] [Google Scholar]
- Lander H. M.; Milbank A. J.; Tauras J. M.; Hajjar D. P.; Hempstead B. L.; Schwartz G. D.; Kraemer R. T.; Mirza U. A.; Chait B. T.; Burk S. C.; Quilliam L. A. (1996) Redox regulation of cell signalling. Nature 381, 380–381. [DOI] [PubMed] [Google Scholar]
- Ibiza S.; Perez-Rodriguez A.; Ortega A.; Martinez-Ruiz A.; Barreiro O.; Garcia-Dominguez C. A.; Victor V. M.; Esplugues J. V.; Rojas J. M.; Sanchez-Madrid F.; Serrador J. M. (2008) Endothelial nitric oxide synthase regulates N-Ras activation on the Golgi complex of antigen-stimulated T cells. Proc. Natl. Acad. Sci. U. S. A. 105, 10507–10512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo J.; Hong I. (2010) Ras-targeting action of thiopurines in the presence of reactive nitrogen species. Biochemistry 49, 3965–3976. [DOI] [PubMed] [Google Scholar]
- Accorsi K.; Giglione C.; Vanoni M.; Parmeggiani A. (2001) The Ras GDP/GTP cycle is regulated by oxidizing agents at the level of Ras regulators and effectors. FEBS Lett. 492, 139–145. [DOI] [PubMed] [Google Scholar]
- Raines K. W.; Cao G. L.; Porsuphatana S.; Tsai P.; Rosen G. M.; Shapiro P. (2004) Nitric oxide inhibition of ERK1/2 activity in cells expressing neuronal nitric-oxide synthase. J. Biol. Chem. 279, 3933–3940. [DOI] [PubMed] [Google Scholar]
- Mizuno S.; Kadowaki M.; Demura Y.; Ameshima S.; Miyamori I.; Ishizaki T. (2004) p42/44 Mitogen-activated protein kinase regulated by p53 and nitric oxide in human pulmonary arterial smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 31, 184–192. [DOI] [PubMed] [Google Scholar]
- Heo J.; Raines K. W.; Mocanu V.; Campbell S. L. (2006) Redox regulation of RhoA. Biochemistry 45, 14481–14489. [DOI] [PubMed] [Google Scholar]
- Lander H. M. (1997) An essential role for free radicals and derived species in signal transduction. FASEB J. 11, 118–124. [PubMed] [Google Scholar]
- Sawyer D. B.; Siwik D. A.; Xiao L.; Pimentel D. R.; Singh K.; Colucci W. S. (2002) Role of oxidative stress in myocardial hypertrophy and failure. J. Mol. Cell Cardiol. 34, 379–388. [DOI] [PubMed] [Google Scholar]
- Oliveira C. J.; Schindler F.; Ventura A. M.; Morais M. S.; Arai R. J.; Debbas V.; Stern A.; Monteiro H. P. (2003) Nitric oxide and cGMP activate the Ras-MAP kinase pathway-stimulating protein tyrosine phosphorylation in rabbit aortic endothelial cells. Free Radic. Biol. Med. 35, 381–396. [DOI] [PubMed] [Google Scholar]
- Oliveira C. J.; Curcio M. F.; Moraes M. S.; Tsujita M.; Travassos L. R.; Stern A.; Monteiro H. P. (2008) The low molecular weight S-nitrosothiol, S-nitroso-N-acetylpenicillamine, promotes cell cycle progression in rabbit aortic endothelial cells. Nitric Oxide 18, 241–255. [DOI] [PubMed] [Google Scholar]
- Wardman P.; Candeias L. P. (1996) Fenton chemistry: an introduction. Radiat. Res. 145, 523–531. [PubMed] [Google Scholar]
- Koppenol W. H. (2001) The Haber-Weiss cycle--70 years later. Redox Rep. 6, 229–234. [DOI] [PubMed] [Google Scholar]
- Lim K. H.; Ancrile B. B.; Kashatus D. F.; Counter C. M. (2008) Tumour maintenance is mediated by eNOS. Nature 452, 646–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K.; Mitsui K.; Yamanaka S. (2003) Role of ERas in promoting tumour-like properties in mouse embryonic stem cells. Nature 423, 541–545. [DOI] [PubMed] [Google Scholar]
- Irani K.; Goldschmidt-Clermont P. (1998) Ras, Superoxide and Signal Transduction. Biochem. Pharmacol. 55, 1339–1346. [DOI] [PubMed] [Google Scholar]
- Luth H. J.; Holzer M.; Gertz H. J.; Arendt T. (2000) Aberrant expression of nNOS in pyramidal neurons in Alzheimer’s disease is highly co-localized with p21ras and p16INK4a. Brain Res. 852, 45–55. [DOI] [PubMed] [Google Scholar]
- Reiter C. D.; Teng R. J.; Beckman J. S. (2000) Superoxide reacts with nitric oxide to nitrate tyrosine at physiological pH via peroxynitrite. J. Biol. Chem. 275, 32460–32466. [DOI] [PubMed] [Google Scholar]
- Kuthan H.; Haussmann H. J.; Werringloer J. (1986) A spectrophotometric assay for superoxide dismutase activities in crude tissue fractions. Biochem. J. 237, 175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gelder B.; Slater E. C. (1962) The extinction coefficient of cytochrome c. Biochim. Biophys. Acta 58, 593–595. [DOI] [PubMed] [Google Scholar]
- Emerson S. D.; Madison V. S.; Palermo R. E.; Waugh D. S.; Scheffler J. E.; Tsao K. L.; Kiefer S. E.; Liu S. P.; Fry D. C. (1995) Solution structure of the Ras-binding domain of c-Raf-1 and identification of its Ras interaction surface. Biochemistry 34, 6911–6918. [DOI] [PubMed] [Google Scholar]
- Scheffzek K.; Ahmadian M. R.; Kabsch W.; Wiesmuller L.; Lautwein A.; Schmitz F.; Wittinghofer A. (1997) The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 277, 333–338. [DOI] [PubMed] [Google Scholar]
- Gureasko J.; Galush W. J.; Boykevisch S.; Sondermann H.; Bar-Sagi D.; Groves J. T.; Kuriyan J. (2008) Membrane-dependent signal integration by the Ras activator Son of sevenless. Nat. Struct. Mol. Biol. 15, 452–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo J.; Campbell S. L. (2006) Ras Regulation by Reactive Oxygen and Nitrogen Species. Biochemistry 45, 2200–2210. [DOI] [PubMed] [Google Scholar]
- Grossi L.; Montevecchi P. C. (2002) A kinetic study of S-nitrosothiol decomposition. Chemistry 8, 380–387. [DOI] [PubMed] [Google Scholar]
- Lenzen C.; Cool R. H.; Prinz H.; Kuhlmann J.; Wittinghofer A. (1998) Kinetic analysis by fluorescence of the interaction between Ras and the catalytic domain of the guanine nucleotide exchange factor Cdc25Mm. Biochemistry 37, 7420–7430. [DOI] [PubMed] [Google Scholar]
- Segel I. H. (1993) Enzyme Kinetics, Wiley-Interscience, New York. [Google Scholar]
- Gebicki J. M.; Nauser T.; Domazou A.; Steinmann D.; Bounds P. L.; Koppenol W. H. (2010) Reduction of protein radicals by GSH and ascorbate: potential biological significance. Amino Acids 39, 1131–1137. [DOI] [PubMed] [Google Scholar]
- Kang P. T.; Zhang L.; Chen C. L.; Chen J.; Green K. B.; Chen Y. R. (2012) Protein thiyl radical mediates S-glutathionylation of complex I. Free Radic. Biol. Med. 53, 962–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downward J. (2003) Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 3, 11–22. [DOI] [PubMed] [Google Scholar]
- James P. E.; Grinberg O. Y.; Swartz H. M. (1998) Superoxide production by phagocytosing macrophages in relation to the intracellular distribution of oxygen. J. Leukoc. Biol. 64, 78–84. [DOI] [PubMed] [Google Scholar]
- Wey M.; Lee J.; Jeong S. S.; Kim J.; Heo J. (2013) Kinetic mechanisms of mutation-dependent harvey ras activation and their relevance for the development of costello syndrome. Biochemistry 52, 8465–8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker T.; Booden M.; Buss J. (2000) S-Nitrosocysteine increases palmitate turnover on Ha-Ras in NIH 3T3 cells. J. Biol. Chem. 275, 22037–22047. [DOI] [PubMed] [Google Scholar]
- Aikawa R.; Komuro I.; Yamazaki T.; Zou Y.; Kudoh S.; Tanaka M.; Shiojima I.; Hiroi Y.; Yazaki Y. (1997) Oxidative stress activates extracellular signal-regulated kinases through Src and Ras in cultured cardiac myocytes of neonatal rats. J. Clin. Invest. 100, 1813–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzen C.; Cool R. H.; Wittinghofer A. (1995) Analysis of intrinsic and CDC25-stimulated guanine nucleotide exchange of p21ras-nucleotide complexes by fluorescence measurements. Methods Enzymol. 255, 95–109. [DOI] [PubMed] [Google Scholar]
- Heo J.; Prutzman K. C.; Mocanu V.; Campbell S. L. (2005) Mechanism of free radical nitric oxide-mediated Ras guanine nucleotide dissociation. J. Mol. Biol. 346, 1423–1440. [DOI] [PubMed] [Google Scholar]