

Abstract
Our objective was to present clinicopathologic evidence of anterior visual pathway involvement in patients with amyotrophic lateral sclerosis (ALS) secondary to a C9orf72 mutation. Two related patients from an extended pedigree with ALS and GGGGCC hexanucleotide repeat expansion in the C9orf72 gene (C9-ALS) underwent neuro-ophthalmologic examination. Following death and tissue donation of the younger ALS patient, histopathologic examination of the retina, optic nerve and central nervous system (CNS) was performed. Ophthalmologic examination revealed contrast sensitivity impairment in the younger C9-ALS patient. Immunohistochemistry performed on this patient’s donor tissue demonstrated p62-positive, pTDP43-negative perinuclear inclusions in the inner nuclear layer of the retina and CNS. Further colocalization with GLT-1 and recoverin suggested that the majority of retinal p62-positive inclusions are found within cone bipolar cells as well as some amacrine and horizontal cells. In conclusion, this is the first report that identifies disease-specific pathologic inclusions in the anterior visual pathway of a patient with a C9orf72 mutation. Cone bipolar cell involvement within the inner nuclear layer of the retina may explain the observed subtle visual function deficiencies in this patient. Further clinical and histopathologic studies are needed to fully characterize a larger population of C9-ALS patients and explore these findings in other forms of ALS.
Keywords: Pathology, neuropathology, ubiquitin, amyotrophic lateral sclerosis, c9orf72, retina, midget bipolar cells
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized clinically by progressive muscle weakness, muscle wasting and paralysis that is ultimately fatal, most often as the result of respiratory failure (1). ALS is primarily motor neuron degeneration; however, cognitive, extrapyramidal and neuropathic manifestations have been recognized in a minority of cases. Most of such cases have a family history of ALS with Parkinsonism and/or dementia and the causative mutations identified are in FUS, UBQLN2, TDP43 and most recently, in C9orf72 (2,3). In ALS, there may be overlapping pathology with certain forms of frontotemporal lobar degeneration (FTD), which has led to the appreciation that ALS and some forms of FTD are traits of a disease spectrum (4). The ALS/FTD patient usually has intracellular deposits positive for ubiquitin/p62, ubiquilin-2 and p-TDP43 (5,6).
Oculomotor changes that have been described in ALS patients include supranuclear gaze palsy, gaze fixation instability (7), increased saccadic latency (8–11), smooth pursuit impairment (11,12), diminished Bell’s phenomenon (13) and eyelid apraxia (14,15). These abnormalities have been attributed to involvement of supranuclear ocular motor pathways. In patients with advanced disease, clinical and pathologic involvement of oculomotor, abducens and trochlear nuclei has been described (16). Recently, binocular high and low contrast vision deficits were also demonstrated in a representative ALS cohort (15). The pathophysiological basis for these visual deficits has not been elucidated. Decreased event-related cortical potentials and decreased functional MRI activity in secondary vision areas in response to visual stimuli suggest higher order processing deficits, but do not account for decreased visual acuity (17–19). A cross-sectional study of retinal ganglion cell layer thickness in subjects with spinal-onset ALS did not show abnormalities (20). To date, there have been no reports of structural abnormalities of the retina or anterior visual pathway in association with ALS, whether in post mortem studies or in vivo.
An autosomal dominant form of ALS and FTD linked to chromosome 9p 13–22 (21) related to GGGGCC hexanucleotide repeat expansion in intron 1 of the C9orf72 gene has recently been identified as the most common pathologic mutation in familial and sporadic ALS/FTD as well as in familial ALS and familial FTD (22,23). On autopsy, multiple unique pathologic features have been identified in C9-ALS/FTD (24–27). Star and dot-shaped p62 and ubiquilin-2-positive, pTDP43-negative neuronal cytoplasmic and intranuclear inclusions in the hippocampus and neocortex are abundant in C9orf72 mutation carriers (C9-ALS/FTD) but absent in ALS/FTD without this mutation (24–26). In addition, a unique feature is dot-like inclusions in the cerebellum that are immunoreactive to p62. These characteristic inclusions also contain poly-(Gly-Ala), poly-(Gly-Pro), and poly-(Gly-Arg) dipeptide repeat proteins, likely formed from non-conventional, non-ATG initiated translation of the expanded hexanucleotide repeat (26,28). The potential contribution of dipeptide repeat protein inclusions to disease pathogenesis remains controversial. While these inclusions are considered by some to be pathological (28), a recent study by Mackenzie et al. has suggested they may be protective, as these aggregates could sequester the more toxic soluble dipeptide proteins (29).
In this study, we describe detailed clinicopathologic studies of the eye and brain in a patient diagnosed with familial ALS carrying a C9orf72 mutation, as well as functional and clinical ocular examination of the patient’s first cousin, also diagnosed with C9-ALS.
Methods
This research was approved by the institutional review boards of Northwestern University and University of Illinois at Chicago; informed consent was obtained from patients prior to enrollment in this study.
Clinical neurologic exam of III-1 and III-5: Pedigree Figure 1
Figure 1.

Pedigree of C9orf72 family. III-1 underwent complete ophthalmologic clinical exam 12 months prior to their demise, when autopsy was performed. III-5 underwent complete ophthalmologic clinical exam.
The patient (III-1) was the ninth member of a four-generation family to become affected with ALS, FTD or both. III-1 presented at age 49 years with bilateral hand weakness and atrophy, hyperreflexia in all limbs, and EMG changes consistent with probable ALS by the El Escorial criteria, and died of respiratory failure 42 months after symptom onset.
The patient’s first cousin, III-5, presented at age 54 years with bilateral upper extremity weakness, hand atrophy, right leg weakness, hand and thigh cramping, brisk reflexes in the upper extremities, decreased right ankle jerk, and easy fatigability, consistent with probable ALS by the El Escorial criteria. A diaphragm pacer was placed 26 months after symptom onset, and now, 36 months after onset, the patient requires non-invasive ventilation and total care.
Neither patient demonstrated any clinical evidence of cognitive or behavioral impairment, although other members of the extended family (IV-1 and III-8) were cognitively impaired.
Genetic analysis of III-1 and III-5
As previously described (22,25,30), repeat-primed PCR was used to qualitatively identify C9orf72 (G4C2) repeat expansion mutations. Genomic DNA (40 ng) was used as a template and was mixed with 10% PCR buffer supplemented with 15 MM MgCl2 (Qiagen), 20% Q-solution (Qiagen), 0.2 mM dNTP3 mix, 0.2 mM 7-deaza dGTP (Affymetrix), 0.7 μM D4-flourescent labeled forward primer, 0.07 μM reverse primer consisting of three (G4C2) repeats and an anchor tail, 0.07 μM anchor primer corresponding to the anchor tail of the reverse primer, and 0.01 U/μl Taq DNA polymerase (Qiagen). The final PCR reaction volume was adjusted to 10 μl with deionized H2O. Using DNA Engine Dyad™ Peltier Thermal Cycler, a touchdown PCR cycling program was set where the annealing temperature was incrementally lowered from 70°C to 56°C with an 8-min extension time for each cycle. The resulting PCR product was then precipitated using ice-cold 100% ethanol, washed twice with 70% ethanol, and the resulting DNA pellet was air-dried, re-suspended in 40 μl foramide solution (Beckman Coulter) and mixed with 0.2 μl GenomeLabs Size Standard-600 (Beckman Coulter). Fragment length analysis was performed on a Beckman Coulter CEQ8000 Genetic Analysis System.
Primer Sequences:
| Primer | Sequence (5′ → 3″) |
|---|---|
| Forward | D4-CAACCGCAGCCTGTAGCAAGCTCTGGAAC |
| Reverse | CAGGAAACAGCTCTGACCCCCCGGCCCCGGCCCCGG |
| Anchor | CAGGAAACAGCTATGACC |
The patient’s and the cousin’s samples contained well over 30 repeats (Figure 2).
Clinical and functional ophthalmologic exam III-1 and III-5: Table I
Table I.
Visual function and ophthalmologic exam.
| III-1 OD | III-1 OS | III-1 OU | III-5 OD ETDRS | C9-III-5 OS ETDRS | |
|---|---|---|---|---|---|
| High-contrast Best-corrected vision (ETDRS Chart letters) | 20/25 (77) | 20/25 (79) | 20/20 (84) | 20/25 (81) | 20/32 (76) |
| Contrast sensitivity (Pelli Robson) | 1.50 | 1.50 | 1.50 | Bilateral: 1.95 | |
| Color vision test | 11/11 (Ishihara) | (l’anthony D15) C-index 1.3. (minor error-protanomaly) | (l’anthony D15) C-index 0.96. (Normal) | ||
| Pupils | Normal | Normal | Normal | Normal | |
| Motility | Normal | Normal | Grossly normal | Grossly normal | |
| NEI-VFQ25 | 98.18%, perfect score on neuro-ophthalmology supplement | 93% | |||
OD: right eye; OS: left eye; OU: both eyes; NEI-VFQ25: National Eye Institute visual function questionnaire with 25 sections.
III-1 underwent a complete neuro-ophthalmologic exam including best-corrected visual acuity, contrast sensitivity, color vision testing, pupil exam and ocular motility exam.
The visual function questionnaire-25 (VFQ-25) (31), a 25-item validated questionnaire that assesses vision specific quality of life was self-administered by the subject. This exam was performed approximately 30 months after onset of ALS symptoms.
III-5 underwent similar examination, except for color vision testing using L’anthony Desaturated-15 color vision test. Color confusion index (C-index) was determined for each eye by inputting participant responses into a web-based application available at http://www.univie.ac.at/Vergl-Physiologie/colortest/colortestF-en.html. A C-index value of 1.78 or higher was considered abnormal, as described by Vingrys and King-Smith (32).
Ocular tissue processing
The right globe and retrobulbar optic nerve were obtained at autopsy from III-1. An age-matched 55-years-old control donor eye was obtained from the Illinois Eye Bank. Globes were fixed in 10% neutral buffered formalin for at least 24 h. The optic nerves were sectioned 2 mm behind the globe. Eyes were sectioned along the nasal-temporal plane, with the macular region embedded separately. Blocks were embedded into paraffin wax and cross-sections obtained at a thickness of 7 μm.
Hematoxylin and eosin (H + E) was used for general morphometric studies of retinal and optic nerve cross-sections.
Immunohistochemistry
Following rehydration and antigen retrieval as described in detail in a previous study (33), sections were blocked in TBS solution containing 1% BSA and 10% donkey serum for 2 h. Retinal and nerve tissue was then incubated in various primary antibodies at appropriate dilutions overnight at 4°C followed by 1-h incubation at room temperature in various secondary antibodies or 30-s incubation in SIGMAFAST 3,3′-diaminobenzadine (DAB) solution (Sigma-Aldrich, St. Louis, MO, USA).
Retinal antibodies included mouse monoclonal anti-ubiquilin2 C-terminus antibody (1:1000, Novus Biologicals, Littleton, CO, USA), mouse monoclonal anti-p62 antibody (1:250, Abnova Coporation, Taipei, Taiwan), rabbit polyclonal anti- phosphorylated TDP-43 antibody (1:2000, Cosmo Bio Co, LTD, Tokyo, Japan), rabbit polyclonal anti-ubiquitin antibody (1:200, Proteintech, Chicago, IL, USA), rabbit polyclonal anti-GLT-1 n-terminal (1:10, Novus Biologicals, Littleton, CO, USA), rabbit polyclonal anti-recoverin (1:500. AB5585, Millipore, Billerica, MA, USA). Optic nerve antibodies included HSP-conjugated anti-Myelin Basic Protein (MBP), p62, and phosphorylated TDP-43. The rabbit anti-C9orf72 (poly-GA)n antibody (1:500) was prepared as follows: a polypeptide of 18 amino acids with Gly-Ala repeats (NH2-GAGAGAGAGAGAGA-GAGA-COOH) was synthesized and was used as an antigen. The antibody was raised in two rabbits. The antiserum was affinity-purified for our studies. Secondary antibodies included Alexa Fluor 488 donkey polyclonal anti-mouse IgG H&L antibody (1:500, Abcam, Cambridge, MA, USA), and Alexa Fluor 594 donkey polyclonal anti-rabbit IgG H&L antibody (1:500, Abcam, Cambridge, MA, USA). Antibodies used for brain and spinal cord included rabbit polyclonal phosphoTDP-43 (pS409/410-2) (1:5000, CosmoBio Co., Ltd. Carlsbad, CA, USA) and monoclonal p62 (anti-SQSTM1 M01, clone 2C11) (1:750, Abnova, Walnut, CA, USA).
Stained tissues were then treated for autofluorescence for 20 min in 0.1% Sudan Black solution (34) and coverslipped with ProLong-DAPI mounting medium (Life Technologies, Carlsbad, CA, USA).
Microscopy and retinal morphometric analysis
For morphometric cross-sectional comparisons of the macula, we measured total retinal thickness from the anterior side of the retinal nerve fiber layer to the posterior side of the retinal pigment epithelium, and inner nuclear layer (INL) thicknesses using the distance distance_between_ polylines.java plug-in for imageJ software (NIH, Bethesda, MD) to calculate an INL/total retina ratio. Using H&E stained macular sections, two reviewers independently measured INL thickness at three sites in the macula, about 1.5 mm temporal to the optic nerve, in order to generate the ratio of average INL to total retina thickness.
Single plane fluorescence and brightfield images were taken using a Nikon Eclipse 80 inverted microscope (Nikon Instruments Inc, Melville, NY, USA). Differential interference contrast (DIC) and confocal images were captured and analyzed using a Zeiss LSM 510 META laser scanning confocal microscope (Carl Zeiss Meditec, GmbH, Hamburg, Germany).
Results
Clinical ophthalmologic examination and imaging findings
III-1 (Table I)
At the time of the exam, the patient was 51 years of age and had an ALSFRS score of 44, with signs of motor neuron disease restricted to the limbs. The patient had normal high contrast best-corrected vision and evidence of binocular summation with more letters identified during binocular than during monocular testing (35). Pelli-Robson contrast sensitivity was impaired more than 2 SDs below published normative data (36). Examination of color vision, ocular motility and pupils was normal. Vision-specific quality of life was not impaired.
III-5 ophthalmologic exam was grossly normal (Table I)
The patient had normal high-contrast and normal binocular low-contrast best-corrected vision. Interestingly, this subject had one or less transformation of adjacent hues in the L’anthony Desaturated 15 color vision test, consistent with borderline normal change OD, and normal color vision OS.
Retinal and optic nerve morphometry and immunohistochemistry (III-1)
Immunofluorescent labeling demonstrated abundant dot and semi-lunar shaped p62-positive cytoplasmic inclusions that were almost exclusively localized to the inner nuclear layer (INL) in the macula and peripheral retina (Figure 3A–F). On DIC imaging, most p62-positive inclusions were found in a perinuclear location and were seen at all levels of the INL, with rare inclusions seen in the inner plexiform and ganglion cell layer (Figure 3J–K). No p62 + inclusions were seen in the control retina (Figure 3G–I).
Figure 3.

p62 + immunofluorescence in C9orf72 retina and absence of inclusions in control eye. Abundant p62+ (green) inclusions are seen in the inner nuclear layer of C9-macula (A–C) and peripheral retina (D–F). No p62 + inclusions are seen in control eye (G–I). Sections are counterstained with DAPI (blue). Perinuclear location and crescent morphology of p62 + inclusions in C9orf72 macula are demonstrated on DIC imaging (J, K – inset, arrow). Scale bars = 100 μm.
The (poly-GA)n dipeptide repeat colocalized with p62 to a high degree (Figure 4A–C). Additionally, these p62-positive inclusions were also immunoreactive for ubiquitin (Figure 4G–I), with a relative preponderance of p62 + compared to ubiquitin immunostaining.
Figure 4.

Colocalization of Poly-(GA)n dipeptide repeat and p62 as well as ubiquitin and p62 in inner nuclear layer of C9orf72 retina. Colocalization of (GA)n dipeptide repeat (red) and p62 (green) is seen in C9orf72 macula INL (A–C). Only background level staining in control macula (D–F). Colocalization of ubiquitin, UB (red) and p62 (green) is seen in C9orf72 macula indicated by the arrow (G–I). In contrast, no colocalization is seen in control macula, only background staining (J–L). Sections were counterstained with DAPI (blue). Scale bars = 100 μm (A–F) and 10 μm (G–L).
We found no evidence of C-terminal ubiquilin-2 immunostaining in the retina, specifically none of the cells with p62 + inclusions had any staining (Figure 5A–C). Further, we did not find any p-TDP-43 immunostaining in the C9 retina (Figure 5G–I).
Figure 5.

Lack of ubiquilin-2 or pTDP-43 immunostaining in p62 + inclusions in C9orf72 macula. No inclusions seen in control macula. No specific ubiquilin-2 (green) staining was seen in C9orf72 (A–C) or control macula (D–F). There was lack of p-TDP-43 (red) colocalization with p62 + inclusions (green) in C9orf72 (G–I). There was no immunostaining in control macula (J–L). Sections were counterstained with DAPI (blue). Scale bars = 100 μm.
In order to explore the cellular identity of p62-positive cells in the INL, we used GLT-1 antibody (n-terminal), which labels flat midget and DB2 bipolar cells as well as a subpopulation of retinal amacrine and cone photoreceptor cells (37). We found that p62-positive inclusions were mostly found in GLT-1 positive cells. Based on their morphology and localization within INL, these GLT-1+, p62 + cells included bipolar cells as well as other GLT-1 positive neurons within the INL (data not shown). In order to quantify the percentage of GLT-1 cells that are P62+, we used ImageJ software cell-counter plug in to analyze the number of P62 + deposits, GLT1 + cells, and their colocalization in five retinal cross-sections (three macular and two peripheral sections). We found that an average of 40% of GLT1 + cells contained P62 + inclusions. Since these were confocal imaging slices (not z-stacks), 50% of P62 + inclusions were not in the imaging plane, and could not be verified to be within GLT1 + cells. We performed further immunostaining using recoverin, an antibody against calcium-binding protein, which is specific to photoreceptors and cone midget bipolar cells. Our goal was to confirm the colocalization of p62 deposits within these cells. Figure 6A–F shows colocalization of p62 within cell bodies of recoverin immunostained cells within the INL of the macula, further confirming the nature of these cells as cone midget bipolars.
Figure 6.

Colocalization of p62 (green) and recoverin (Red), in the inner nuclear layer of the macula in a patient with C9orf72 mutation. Figure 6A–C, scale bar = 100 micrometer. Figures 6D–F, correspond to the boxed inset in Figure C, scale bar = 20 micrometers. This figure highlights the fact that around 50% of recoverin + cells (cone midget bipolars) contain intracellular p62 deposits. This figure also illustrates that additional cell types in the INL, not immunostained with recoverin, also harbor these p62 + deposits..
H&E staining of the retina revealed normal architecture compared to control eye (Figure 7). The average macular INL to total retinal thickness ratio at this site was 0.163 in the affected retina and 0.177 in the control retina.
Figure 7.
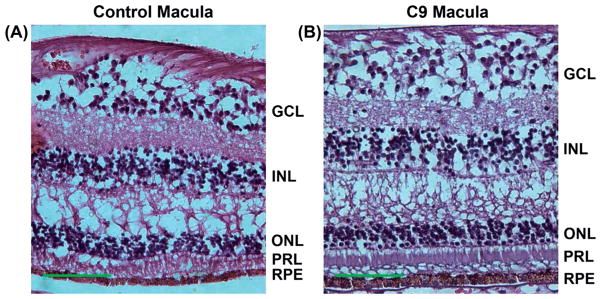
H&E staining of control (A) and C9orf72 (B) macula. No apparent morphologic differences are seen between control and C9orf72 macula. Scale bars = 100 μm.
MBP staining of the optic nerve showed normal morphology and no focal myelin loss compared to control (Figure 8). We found neither p62 + nor pTDP deposits of the optic nerve on immunostaining (data not shown).
Figure 8.

Control and C9orf72 optic nerve histology. MBP (A,B) and H&E (C,D) staining in control and C9orf72 optic nerve sections. Scale bars = 1 mm (A and B) and 100 μm (C and D).
Brain and spinal cord pathology (III-1)
The unfixed brain weighed 1380 g. Gross examination revealed no cortical, hippocampal, or caudate atrophy, mild spinal cord atrophy, moderate pallor of the substantia nigra, and no atrophy of the cerebellum. Microscopic examination of H&E sections revealed minimal to mild rarefaction of corticospinal tracts (CSTs) in motor subcortical white matter, internal capsule, cerebral peduncle, basis pontis, medullary pyramids, and ventral and lateral spinal CSTs. There was no appreciable neuronal loss in motor cortex, but in brainstem and spinal cord, neuronal loss and gliosis was moderate in the hypoglossal nucleus and severe in cervical, thoracic, and lumbar anterior horns. There was severe atrophy and degeneration of the ventral nerve roots but not the dorsal nerve roots. Bunina bodies were moderate in the hypoglossal nucleus and mild in cervical and lumbar anterior horn neurons.
TDP-43-positive skein-like inclusions were frequent in the hypoglossal nucleus, moderate in lumbar anterior horn neurons, and sparse in cervical and thoracic anterior horns. TDP-43-positive Lewy-like bodies were sparse in hypoglossal nucleus and lumbar anterior horn neurons (Figure 9). There were no TDP-43-positive inclusions in the neocortex or hippocampal dentate gyrus to suggest concomitant FTLD-TDP. Immunostaining with p62 highlighted dot-like perinuclear cytoplasmic inclusions in the cerebellar granular layer (Figure 10A), ‘star-shaped’ inclusions in the hippocampal pyramidal layer in the CA2–4 region (Figure 10B) and frontal cortex pyramidal neurons, as well as dot-like cytoplasmic inclusion in the hippocampal dentate gyrus (Figure 10C) that are specific to brains from patients with C9orf72 hexanucleotide repeat expansion. Histologic sections of optic chiasm (including pre-chiasmatic optic nerve), lateral geniculate body, superior colliculus, optic radiations, and visual cortex revealed no microscopic abnormality on H&E sections and there were no TDP-43 immunolabeled inclusions in any of these sections. p62 immunostains of visual cortex showed small cytoplasmic inclusions in granular neurons and larger inclusions, including star-shaped inclusions, in pyramidal neurons, as has been reported (29). There were no p62 immunolabeled inclusions in any other visual-related regions. In summary, the pathology was consistent with ALS, LMN predominant, without FTLD-TDP, and with p62-positive/TDP-43-negative inclusions consistent with C9orf72 hexanucleotide repeat expansion.
Figure 9.

Lower motor neuron skein-like inclusions and Lewy-like bodies. Phosphorylated TDP-43 positive skein-like inclusions (arrowhead) and Lewy-like body (arrow) in lumbar spinal cord anterior horn neurons. Scale bar = 50 μm.
Figure 10.
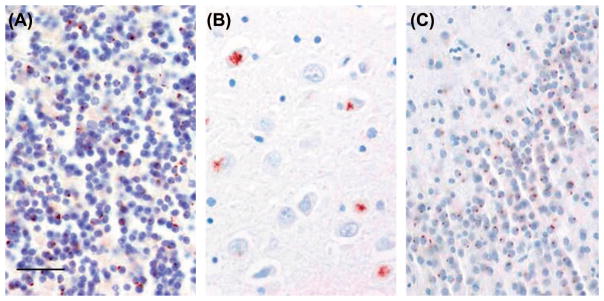
C9orf72-specific brain pathology. p62-positive small dot-like cytoplasmic inclusions in cerebellar granular neurons (A), ‘star-shaped’ cytoplasmic inclusions in hippocampal pyramidal neurons in (B), and small dot-like cytoplasmic inclusions in hippocampal dentate gyrus (C). Scale bar = 50 μm.
Discussion
This is the first report that identifies disease-specific inclusions associated with ALS/FTD in the retinal tissue of a patient with a C9orf72 mutation. Furthermore, we report the corresponding ocular functional deficits in the patient prior to her death. Together, our findings shed light on this complex disease. While previous reports identified subtle visual defects in patients with ALS, the location of the afferent visual pathway changes remained elusive. Our findings suggest that the retina represents a previously unrecognized neuro-anatomical area where this entity can manifest and lead to sensory functional defects.
In an attempt to identify the exact cellular target of the p62-positive aggregates within the inner nuclear layer, we used a specific glutamate transporter antibody, GLT-1, which labels two types of cone off-bipolar cells, the flat-midget bipolar and the diffuse bipolar type 2 (37,38), cone photoreceptors as well as some amacrine and horizontal cells (38,39). We further confirmed this finding by demonstrating colocalization of p62 deposits within cells immunostained with recoverin, a specific antibody against calcium-binding protein expressed in retinal photo-receptors and midget cone bipolar cells (Figure 6). We found that the majority of these p62 + deposits were located within the cytoplasm of GLT-1-positive cells. More specifically, we found that, on average, 40% of GLT1 + cells contain P62 + deposits. Based on their morphology, abundance in the macular region and localization at multiple levels in the INL, we believe these deposits are located within cone bipolar cells as well as some amacrine and horizontal cells. While the exact pathophysiology for this specific cellular target remains unclear, our findings help expand the spectrum of systemic involvement in C9-ALS/FTD to include retinal deposits at the level of the INL, within the cytoplasm of specific neuronal cell types in the anterior visual pathway. The findings of more abundant P62 deposits than ubiquitin or p-TDP-43 deposits in the INL is well in line with similar pathology in the dentate gyrus of this patient, a finding that is thought to be characteristic of C9-ALS.
In parallel with intracellular deposits within retinal neuronal cells, patient III-1 had visual deficits including decreased contrast sensitivity, which is well in line with the localization of the p62 + and DPR pathology in the inner nuclear layer. The inner nuclear layer harbors cell bodies of the second order neuron in the visual pathway, the bipolar cells (10–12 different types in humans). In addition, this layer also harbors cell bodies of two classes of interneuron that modulate signal transmission at the first and second visual synapse, namely the horizontal and amacrine cells (around 22 types, mostly inhibitory, GABA or glycine), respectively (40,41). An important function of the flat-midget bipolar cell is transmission of color vision to midget ganglion cells, to which they are connected through ‘private lines’, with one-one connections from a single cone in the macula (42). Given that GLT-1 flat-midget bipolars represent two-thirds of GLT-1-positive bipolar cells (43) and are hence potentially the majority of P62 + bipolar cells, as further confirmed by colocalization with recoverin, this would suggest the predominant involvement of the midget-parvocellular pathway, which is responsible for visual acuity and color perception in this patient, as opposed to the magnocellular pathway, responsible for low contrast, motion and flicker detection. While the nature of GLT1 + amacrine cells has not been adequately studied and could not be clarified from our paraffin-embedded sections, a potentially relevant amacrine cell type could be small-field diffuse calretinin-immunoreactive cells, which are found in high numbers in the foveal, rod-free area of the retina, and in lesser numbers in the peripheral retina. These cells have been suggested to participate in the reciprocal color opponency (in addition to the horizontal cells, working at the cone photoreceptor synapse) through modulating synaptic transmission between midget bipolar and midget ganglion cells (44).
While central visual pathway involvement has been better characterized using electrophysiologic (17) and imaging (18) approaches, our findings suggest the anterior visual pathway may also be involved. A recent cross-sectional clinical neuro-ophthalmologic evaluation of ALS patients (sporadic and familial) revealed supranuclear ocular motility findings in this population (15). Reduced high and low contrast acuity was a surprising finding of this study, which was interpreted cautiously given the small difference between patients and controls. Further, the findings did not allow a specific localization within the afferent visual pathway. While patient III-5 with confirmed C9-ALS did not show significant visual or ocular findings, it is difficult to predict the earliest functional deficit. It will be important to study the correlation between the onset of ocular and neurologic findings, since this will carry important implications for the potential to include ocular imaging and visual testing as an additional tool in ALS/FTD work-up, especially if retinal involvement is present in other ALS/FTD types.
In vivo high resolution optical retinal imaging, using optical coherence tomography, can achieve 5–10 micron axial resolution and presents an attractive tool to consider for disease detection; however, one needs to keep in mind that the INL is a 30-micron layer as seen on OCT retinal imaging. In this regard, in III-1, despite confirmed functional deficits, we were not able to detect significant thinning of the INL layer histologically (16% of total retinal thickness vs. 17% in control). Similarly, a recent OCT study of a diverse group of sporadic ALS patients found no significant thinning on various OCT parameters, including optic nerve and macular sublayer analysis (20). In contrast to our study, the authors did not obtain visual function testing on their patients, and included only sporadic ALS patients with spinal symptom onset. On this point, it is possible that ocular involvement may be specific to patients with the C9orf72 mutation, where INL thinning can be very subtle and, perhaps, a late finding.
The involvement of a single retinal layer, if confirmed in other studies, may limit the potential of OCT imaging as a potential screening tool, and predicts that functional visual tests will be more sensitive for studying this disease entity. While the effect of these deposits on neural function of the bipolar cells cannot be confirmed from our findings, it is possible that these deposits, once they reach a certain load, become functionally deleterious, impeding signal transmission within bipolar cells (as evidenced by abnormal contrast sensitivity), without cell death and structural axonal loss. It is possible that such structural changes occur at later stages of the disease. Ultimately, we believe that continued improvements in OCT technology and software analysis tools may greatly aid our understanding of ALS and other neurodegenerative diseases. To emphasize this point, Schneider et al. recently analyzed single retinal sublayer thicknesses in patient with Parkinsonian syndromes and found significant differences between patients with multiple system atrophy and progressive supranuclear palsy (45). It is therefore possible that with improved technology and larger series, including a carefully matched control population, subtle changes in the macular sublayers can be discerned.
This study highlights the importance of including a battery of functional visual testing rather than relying on screening color vision (Ishihara charts) or high contrast visual acuity tests alone (Snellen vision charts), as these only capture one aspect of visual function. The relative subtlety of visual functional defects in this specific patient suggests that future studies should use tests designed to elicit acquired color vision defects, including the Farnsworth Munsell-100 or D-15 panel, or as explored in the current study, the desaturated version of the 15 panel, L’anthony D15. The low contrast and color vision tests used in this study would be able to identify subtle functional defects affecting a select subset of retinal neural cells, such as in this patient who demonstrated p62 + deposits localized within a subset of retinal second order neurons and their interneurons in the INL. Further research is needed to explore the potential involvement of retinal midget-parvocellular pathway in patients with a C9orf72 mutation. Our findings suggest a need for larger cohort studies to further explore the prevalence, severity and course of these visual deficits in other patients with a C9orf72 mutation, as well as explore anterior visual pathway involvement in larger cohorts of ALS/FTD patients.
Figure 2.

C9orf72 (G4C2) repeat expansion detection using repeat-primed PCR. Genomic DNA was amplified using a three-primer system and touchdown PCR cycling program as detailed in the methods and previously described (22,23). In brief, the forward primer contains a 5′ fluorescently labeled tag that hybridizes upstream of the (G4C2) repeat expansion, the reverse primer consists of a non-complementary 5′ tail sequence and three (G4C2) repeat units, and the anchor primer corresponds to the anchor sequence of the reverse primer. As shown in the representative electropherogram, the initial peak corresponds to the shortest fragment containing a minimum of three (G4C2) repeats, with each successive peak corresponding to fragments expanding by one repeat unit. A characteristic saw-tooth pattern is generated as signal intensity decreases as fragment size increases. Electropherograms displaying ≥30 repeats is commonly used as the pathological cut-off value (23).
Acknowledgments
We acknowledge NIH- EY-021470 (AAF), NIH-NS050641 (TS), NIH-K12 EY 021475 (HEM) NIH-P30 EY 001792 (HEM), NIH-NIA-AG13854 (EB), Illinois Eye Bank (AAF), Eleanor Wood-Prince Grants: A Project of The Woman’s Board of NMH (AAF), Illinois Society for the Prevention of Blindness (JS, AAF) and unrestricted funds from Research to Prevent Blindness, NY (Department of Ophthalmology, Northwestern University; Department of Ophthalmology and Visual Sciences, University of Illinois at Chicago).
Teepu Siddique acknowledges the following support: Les Turner ALS Foundation, Les Turner ALS Foundation/Herbert C. Wenske Foundation Professor, Ride for Life and the Foglia Family Foundation. The authors wish to thank Drs. Steven H. DeVries and Jun Shi, of Northwestern University, who provided the recoverin antibody and critically reviewed the bipolar cell immunohistochemistry.
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. New England Journal of Medicine. 2001;344:1688–700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- 2.Giordana MT, Ferrero P, Grifoni S, Pellerino A, Naldi A, Montuschi A. Dementia and cognitive impairment in amyotrophic lateral sclerosis: a review. Neurological Sciences: official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. 2011;32:9–16. doi: 10.1007/s10072-010-0439-6. [DOI] [PubMed] [Google Scholar]
- 3.Phukan J, Pender NP, Hardiman O. Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurology. 2007;6:994–1003. doi: 10.1016/S1474-4422(07)70265-X. [DOI] [PubMed] [Google Scholar]
- 4.Mackenzie IR, Feldman HH. Ubiquitin immunohistochemistry suggests classic motor neuron disease, motor neuron disease with dementia, and frontotemporal dementia of the motor neuron disease type represent a clinicopathologic spectrum. Journal of Neuropathology and Experimental Neurology. 2005;64:730–9. doi: 10.1097/01.jnen.0000174335.27708.0a. [DOI] [PubMed] [Google Scholar]
- 5.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science (New York, NY) 2006;314:130–3. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 6.Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–11. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 7.Shaunak S, Orrell RW, O’Sullivan E, Hawken MB, Lane RJ, Henderson L, et al. Oculomotor function in amyotrophic lateral sclerosis: evidence for frontal impairment. Annals of Neurology. 1995;38:38–44. doi: 10.1002/ana.410380109. [DOI] [PubMed] [Google Scholar]
- 8.Ohki M, Kanayama R, Nakamura T, Okuyama T, Kimura Y, Koike Y. Ocular abnormalities in amyotrophic lateral sclerosis. Acta Oto-laryngologica Supplementum. 1994;511:138–42. doi: 10.3109/00016489409128318. [DOI] [PubMed] [Google Scholar]
- 9.Jacobs L, Bozian D, Heffner RR, Jr, Barron SA. An eye movement disorder in amyotrophic lateral sclerosis. Neurology. 1981;31:1282–7. doi: 10.1212/wnl.31.10.1282. [DOI] [PubMed] [Google Scholar]
- 10.Leveille A, Kiernan J, Goodwin JA, Antel J. Eye movements in amyotrophic lateral sclerosis. Archives of Neurology. 1982;39:684–6. doi: 10.1001/archneur.1982.00510230010003. [DOI] [PubMed] [Google Scholar]
- 11.Marti-Fabregas J, Roig C. Oculomotor abnormalities in motor neuron disease. Journal of Neurology. 1993;240:475–8. doi: 10.1007/BF00874116. [DOI] [PubMed] [Google Scholar]
- 12.Abel LA, Williams IM, Gibson KL, Levi L. Effects of stimulus velocity and acceleration on smooth pursuit in motor neuron disease. Journal of Neurology. 1995;242:419–24. doi: 10.1007/BF00873543. [DOI] [PubMed] [Google Scholar]
- 13.Esteban A, de Andres C, Gimenez-Roldan S. Abnormalities of Bell’s phenomenon in amyotrophic lateral sclerosis: a clinical and electrophysiological evaluation. Journal of Neurology, Neurosurgery, and Psychiatry. 1978;41:690–8. doi: 10.1136/jnnp.41.8.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Averbuch-Heller L, Helmchen C, Horn AK, Leigh RJ, Buttner-Ennerver JA. Slow vertical saccades in motor neuron disease: correlation of structure and function. Annals of Neurology. 1998;44:641–8. doi: 10.1002/ana.410440410. [DOI] [PubMed] [Google Scholar]
- 15.Moss HE, McCluskey L, Elman L, Hoskins K, Talman L, Grossman M, et al. Cross-sectional evaluation of clinical neuro-ophthalmic abnormalities in an amyotrophic lateral sclerosis population. Journal of the Neurological Sciences. 2012;314:97–101. doi: 10.1016/j.jns.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okamoto K, Hirai S, Amari M, Iizuka T, Watanabe M, Murakami N, et al. Oculomotor nuclear pathology in amyotrophic lateral sclerosis. Acta Neuropathologica. 1993;85:458–62. doi: 10.1007/BF00230482. [DOI] [PubMed] [Google Scholar]
- 17.Haverkamp S, Michalakis S, Claes E, Seeliger MW, Humphries P, Biel M, et al. Synaptic plasticity in CNGA3(−/−) mice: cone bipolar cells react on the missing cone input and form ectopic synapses with rods. The Journal of Neuroscience: the official journal of the Society for Neuroscience. 2006;26:5248–55. doi: 10.1523/JNEUROSCI.4483-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lule D, Diekmann V, Muller HP, Kassubek J, Ludolph AC, Birbaumer N. Neuroimaging of multimodal sensory stimulation in amyotrophic lateral sclerosis. Journal of Neurology, Neurosurgery, and Psychiatry. 2010;81:899–906. doi: 10.1136/jnnp.2009.192260. [DOI] [PubMed] [Google Scholar]
- 19.Munte TF, Troger MC, Nusser I, Wieringa BM, Matzke M, Johannes S, et al. Abnormalities of visual search behaviour in ALS patients detected with event-related brain potentials. Amyotroph Lateral Scler Other Motor Neuron Disord : official publication of the World Federation of Neurology, Research Group on Motor Neuron Diseases. 1999;1:21–7. doi: 10.1080/146608299300079493. [DOI] [PubMed] [Google Scholar]
- 20.Roth NM, Saidha S, Zimmermann H, Brandt AU, Oberwahrenbrock T, Maragakis NJ, et al. Optical coherence tomography does not support optic nerve involvement in amyotrophic lateral sclerosis. European Journal of Neurology: the official journal of the European Federation of Neurological Societies. 2013;20:1170–6. doi: 10.1111/ene.12146. [DOI] [PubMed] [Google Scholar]
- 21.Vance C, Al-Chalabi A, Ruddy D, Smith BN, Hu X, Sreedharan J, et al. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain: a journal of neurology. 2006;129:868–76. doi: 10.1093/brain/awl030. [DOI] [PubMed] [Google Scholar]
- 22.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in non-coding region of C9orf72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–56. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9orf72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–68. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, et al. p62-positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathologica. 2011;122:691–702. doi: 10.1007/s00401-011-0911-2. [DOI] [PubMed] [Google Scholar]
- 25.Brettschneider J, van Deerlin VM, Robinson JL, Kwong L, Lee EB, Ali YO, et al. Pattern of ubiquilin pathology in ALS and FTLD indicates presence of C9orf72 hexanucleotide expansion. Acta Neuropathologica. 2012;123:825–39. doi: 10.1007/s00401-012-0970-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science (New York, NY) 2013;339:1335–8. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- 27.Murray ME, DeJesus-Hernandez M, Rutherford NJ, Baker M, Duara R, Graff-Radford NR, et al. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9orf72. Acta Neuropathologica. 2011;122:673–90. doi: 10.1007/s00401-011-0907-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, et al. Unconventional translation of C9orf72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77:639–46. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mackenzie IR, Arzberger T, Kremmer E, Troost D, Lorenzl S, Mori K, et al. Dipeptide repeat protein pathology in C9orf72 mutation cases: clinicopathological correlations. Acta Neuropathologica. 2013;126:859–79. doi: 10.1007/s00401-013-1181-y. [DOI] [PubMed] [Google Scholar]
- 30.Kobayashi H, Abe K, Matsuura T, Ikeda Y, Hitomi T, Akechi Y, et al. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet. 2011;89:121–30. doi: 10.1016/j.ajhg.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mangione CM, Lee PP, Gutierrez PR, Spritzer K, Berry S, Hays RD, et al. Development of the 25-item National Eye Institute Visual Function Questionnaire. Archives of Ophthalmology. 2001;119:1050–8. doi: 10.1001/archopht.119.7.1050. [DOI] [PubMed] [Google Scholar]
- 32.Vingrys AJ, King-Smith PE. A quantitative scoring technique for panel tests of color vision. Invest Ophthalmol Vis Sci. 1988;29:50–63. [PubMed] [Google Scholar]
- 33.Deng HX, Zhai H, Bigio EH, Yan J, Fecto F, Ajroud K, et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Annals of Neurology. 2010;67:739–48. doi: 10.1002/ana.22051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schnell SA, Staines WA, Wessendorf MW. Reduction of lipofuscin-like autofluorescence in fluorescently labeled tissue. The Journal of Histochemistry and Cytochemistry : official journal of the Histochemistry Society. 1999;47:719–30. doi: 10.1177/002215549904700601. [DOI] [PubMed] [Google Scholar]
- 35.Legge GE. Binocular contrast summation – I. Detection and discrimination. Vision Research. 1984;24:373–83. doi: 10.1016/0042-6989(84)90063-4. [DOI] [PubMed] [Google Scholar]
- 36.Elliott DB, Sanderson K, Conkey A. The reliability of the Pelli-Robson contrast sensitivity chart. Ophthalmic & Physiological Optics: the journal of the British College of Ophthalmic Opticians. 1990;10:21–4. [PubMed] [Google Scholar]
- 37.Haverkamp S, Haeseleer F, Hendrickson A. A comparison of immunocytochemical markers to identify bipolar cell types in human and monkey retina. Visual Neuroscience. 2003;20:589–600. doi: 10.1017/s0952523803206015. [DOI] [PubMed] [Google Scholar]
- 38.Rauen T, Kanner BI. Localization of the glutamate transporter GLT-1 in rat and macaque monkey retinae. Neuroscience Letters. 1994;169:137–40. doi: 10.1016/0304-3940(94)90375-1. [DOI] [PubMed] [Google Scholar]
- 39.Reye P, Sullivan R, Pow DV. Distribution of two splice variants of the glutamate transporter GLT-1 in the developing rat retina. The Journal of Comparative Neurology. 2002;447:323–30. doi: 10.1002/cne.10218. [DOI] [PubMed] [Google Scholar]
- 40.Wassle H. Parallel processing in the mammalian retina. Nature Reviews Neuroscience. 2004;5:747–57. doi: 10.1038/nrn1497. [DOI] [PubMed] [Google Scholar]
- 41.MacNeil MA, Masland RH. Extreme diversity among amacrine cells: implications for function. Neuron. 1998;20:971–82. doi: 10.1016/s0896-6273(00)80478-x. [DOI] [PubMed] [Google Scholar]
- 42.Boycott B, Wassle H. Parallel processing in the mammalian retina: the Proctor Lecture. Investigative Ophthalmology & Visual Science. 1999;40:1313–27. [PubMed] [Google Scholar]
- 43.Grunert U, Martin PR, Wassle H. Immunocytochemical analysis of bipolar cells in the macaque monkey retina. The Journal of Comparative Neurology. 1994;348:607–27. doi: 10.1002/cne.903480410. [DOI] [PubMed] [Google Scholar]
- 44.Kolb H, Zhang L, Dekorver L, Cuenca N. A new look at calretinin-immunoreactive amacrine cell types in the monkey retina. The Journal of Comparative Neurology. 2002;453:168–84. doi: 10.1002/cne.10405. [DOI] [PubMed] [Google Scholar]
- 45.Schneider M, Muller HP, Lauda F, Tumani H, Ludolph AC, Kassubek J, et al. Retinal single-layer analysis in Parkinsonian syndromes: an optical coherence tomography study. Journal of Neural Transmission. 2014;121:41–7. doi: 10.1007/s00702-013-1072-3. [DOI] [PubMed] [Google Scholar]