

Abstract
L4F, an alpha helical peptide inspired by the lipid-binding domain of the ApoA1 protein, has potential applications in the reduction of inflammation involved with cardiovascular disease as well as liver fibrosis. In addition to its biological activity, amphipathic peptides such as L4F are likely candidates to direct the molecular assembly of peptide nanostructures. Here we describe the stabilization of the amphipathic L4F peptide through fusion to a high molecular weight protein polymer. Comprised of multiple pentameric repeats, elastin-like polypeptides (ELPs) are biodegradable protein polymers inspired from the human gene for tropoelastin. Dynamic light scattering confirmed that the fusion peptide forms nanoparticles with a hydrodynamic radius of approximately 50 nm, which is unexpectedly above that observed for the free ELP (~5.1 nm). To further investigate their morphology, negative and cryogenic transmission electron microscopy were used to reveal that they are unilamellar vesicles. On average, these vesicles are 49 nm in radius with lamellae 8 nm in thickness. To evaluate their therapeutic potential, the L4F nanoparticles were incubated with hepatic stellate cells. Stellate cell activation leads to hepatic fibrosis; furthermore, their activation is suppressed by ApoA1 mimetic peptides. Consistent with this observation, L4F nanoparticles were found to suppress hepatic stellate cell activation in vitro. To evaluate the in vivo potential for these nanostructures, their plasma pharmacokinetics were evaluated in rats. Despite the assembly of nanostructures, both free L4F and L4F nanoparticles exhibited similar half-lives of approximately 1 hr in plasma. This is the first study reporting the stabilization of peptide-based vesicles using ApoA1 mimetic peptides fused to a protein polymer; furthermore, this platform for peptide-vesicle assembly may have utility in the design of biodegradable nanostructures.
Keywords: apolipoprotein, nanoparticle, protein polymer, self-assembly, elastin-like polypeptide, D4F, L4F, hydrodynamic radius, TEM, cryo-TEM, vesicle
Introduction
The prevalence of secondary diseases associated with obesity, such as non-alcoholic fatty liver disease (NAFLD), are expected to rise with the high prevalence of obesity in Western nations [1]. NAFLD, characterized by the deposition fat and the presence of inflammation, can progress to hepatic fibrosis and cirrhosis [2]. A candidate for the prevention of progression of NAFLD is the apolipoprotein A-1 mimetic peptide D4F, which prevents hepatic fibrosis in murine models [3]. D4F, a peptide comprised of D-amino acids, and its natural enantiomer L4F, made up of L-amino acids, has been extensively studied in animal models related to lipid oxidation and inflammatory diseases. Based on the 18A peptide[4], a synthetic 18 amino acid peptide designed to mimic amphipathic helices found in apolipoproteins and originally intended to displace apolipoproteins from HDL[5, 6], the 4F peptides are amphipathic and create type A α-helices due to their 4 hydrophobic phenylalanine residues. The 4F peptides bind oxidized lipids with an affinity 4–6 orders of magnitude higher than ApoA-1[7]. 4F’s anti-inflammatory properties are attributed to their affinity for pro-inflammatory oxidized lipids. L4F has been demonstrated to decrease inflammatory cytokines including IL-6, TNF-α, and IL-1β in obese mice[8, 9] as well as inhibit activation of the inflammatory transcription factor NF-κB in chronic kidney disease rat models[10]. D/L-4F’s ability to decrease systemic inflammation is the basis for its exploration in inhibiting the localized inflammation and activation of hepatic stellate cells and the progression of liver fibrosis.
While D4F is anti-fibrotic in vivo [3], we chose to develop the L-amino acid L4F peptide for three reasons. First, unlike D4F, the L4F peptide can be directly engineered onto an recombinant ELP protein polymer to modulate its self-assembly properties, which may impact its biodistribution and efficacy. Secondly, chronic use of D-amino acids results in high tissue accumulation due to impaired breakdown, which is not an obstacle using biodegradable enatiomers[4]. Lastly, when administered subcutaneously, the L form of the 4F molecule was just as effective at treating atherosclerosis as the D form[11, 12]. It is therefore likely that D4F’s anti-fibrotic mechanism is conserved in its L4F enantiomer.
To capitalize on ability of the L4F to form an amphipathic secondary structure, we here explore the addition of high molecular weight elastin-like polypeptide (ELPs) to modulate its assembly properties. ELPs are derived from the human gene for tropoelastin and are repetitive polypeptide chains of the amino acid sequence (Val-Pro-Gly-Xaa-Gly)n, where Xaa can be substituted with virtually any amino acid[13]. ELPs phase separate above a transition temperature, Tt, which can be tuned by selection of Xaa and n. ELPs are emerging as a platform to display and manipulate the assembly of protein polymer nanoparticles, partly because they are biodegradable, biocompatible, and non immunogenic[14, 15]. Since they are genetically encoded, they lend themselves to direct fusion with biologically functional proteins. In addition, the fusion of ELPs to low molecular weight peptides has been shown to both increase the half-life of the peptide as well as decrease systemic clearance[16, 17].
In this study, we report the surprising formation of L4F ELP fusion proteins into nanoparticles 50 nm in radius at 37 °C, significantly below the transition temperature of the parent ELPs. We also demonstrate L4F nanoparticles inhibit the activation of hepatic stellate cells in vitro; furthermore, we show that the pharmacokinetics of these nanoparticles are not compromised in comparison to the free peptide. We expect these data to act as a springboard for the development of a biologically active nanostructures assembled by ApoA-1 mimetic peptides.
Materials and Methods
Construction of L4F ELP fusions
A DNA sequence encoding for the peptide L4F followed by a thrombin cleavage site and ELP insertion site (IDT Technologies, Coralville, IA) was cloned into a pET25b+vector (Clonetech, Mountain View, CA). A forward primer (TATGGATTGGTTCAAAGCGTTTTATGATAAAGT GGCGGAAAAATTCAAAGAAGCGTTCGGTCTGGTTCCGCGTGGTTCTGGTTACTGATC TCCTCG) and a reverse primer (GATCCGAGGAGATCAGTAACCAGAACCACGCGGAAC CAGACCGAACGCTTCTTTGAATTTTTCCGCCACTTTATCATAAAACGCTTTGAACCAA TCCA) were annealed, and ligated into a pET25b+ vector digested with NdeI/ BamHI to generate a 2 base pair overhang created by digestion of at an amino terminal BseRI cut site. Various ELP genes[18] were ligated downstream of the L4F encoding sequence using BseRI/BamHI cut sites in both L4F and ELP plasmids to digest and 1 μL T4 DNA ligase (Invitrogen, Carlsbad, CA) to ligate, resulting in N-L4F-thrombin-ELP-C (Table 1). The resulting fusion protein constructs were expressed in BLR E. Coli and purified using the ELP-mediated phase separation[19]. Purity was determined by running 20 μg of polymer on a 4–20% SDS-PAGE gel stained with copper chloride.
Table 1.
Recombinant protein polymers examined during this study
| label | * encoded amino acid sequence | MW [kDa] | ** Tt [°C] | Property at 37 °C | *** slope [°C/log10(μM)] | y-intercept [°C] |
|---|---|---|---|---|---|---|
| A192 | G(VPGAG)192Y | 73.7 | 61.9 | soluble | −9.2 [−9.9 to −8.6] | 74.9 [74.0 to 75.8] |
| L4F-A192 | MDWFKAFYDKVAEKFKEAFGLVPRGS G(VPGAG)192Y | 76.6 | 45.8 | nanoparticle | −2.6 [−3.6 to −1.7] | 49.6 [48.2 to 50.9] |
| V2A192 | G(VPGVG VPGAG VPGAG)64Y | 75.4 | 47.2 | soluble | −5.8 [−6.7 to −4.9] | 55.9 [54.6 to 57.1] |
| L4F-V2A192 | MDWFKAFYDKVAEKFKEAFGLVPRGS G(VPGVG VPGAG VPGAG)64Y | 79.0 | 34.3 | coacervate | −1.3 [−1.6 to −1.0] | 36.2 [35.8 to 36.6] |
Genes encoding for L4F-A192, V2A192, and L4F-V2A192 were constructed similarly to A192.
The observed transition temperature at 25 μM in PBS.
The slope, b, and Y-intercept, m, were derived from the log-linear regression analysis for transition temperature vs concentration fit to the equation Tt = m Log10[CELP] + b. mean [95% CI]
Optical characterization of the ELP phase diagram
The phase behaviors of ELPs were characterized as a function of molecular weight and concentration by measuring the solution turbidity at 350 nm of protein polymer as a function of temperature. 300 μL of protein polymers in phosphate buffered saline (PBS, 0.2 g/L KCl, 0.2 g/L KH2PO4, 8 g/L NaCl, 1.15 g/L Na2HPO4), concentrations ranging 5–100 μM were observed in a Beckman Tm microcell at a constant ramp rate of 1 °C min−1 and measurements were captured 3x min−1 by a UV visible spectrophotometer (DU800 Spectrophotometer, Beckman Coulter, CA). The maximum first derivative of the curve was defined as the transition temperature, Tt.
Dynamic Light Scattering
A Dynapro plate reader (Wyatt Technology Inc., Santa Barbara, CA) was used for all hydrodynamic radius and polydispersity measurements. Polymers were prepared at 25 μM concentrations in PBS, filtered through 0.2 μm cellulose acetate filters, and centrifuged at 4 °C, 1200 rpm to remove air bubbles. Mineral oil was added to the top of the sample to prevent evaporation. Polymers were observed over a range of temperatures from 4 to 60 °C in 1 °C increments.
Negative and cryogenic transition electron microscopy
TEM and Cryo-TEM images were captured on an FEI Tecnai 12 TWIN Transmission Electron Microscope, operating at 100 kV for TEM and 80 kV for cryo-TEM. 25 μM TEM samples were pipetted onto a carbon-coated copper grid (Electron Microscopy Sciences, Hatfield, PA). Filter paper was used to wick away excess solution. 10uL of 2 wt% aqueous uranyl acetate was used to stain samples. For cryo-TEM, 25 μM samples were pipetted onto lacey carbon coated TEM grids (LC325-Cu, Electron Microscopy Sciences) pre-treated with plasma air to make the lacey carbon film hydrophilic. Samples were vitrified by plunging them into a liquid ethane reservoir precooled with liquid nitrogen. Both TEM and Cryo-TEM images were acquired with a 16 bit 2k × 2k FEI Eagle bottom mount camera (Hillsboro, OR). Images were processed with ImageJ (NIH, Bethesda, MD). Nanoparticle size was averages from three areas of view with more than 50 particles per image for TEM and 20 particles for Cryo-TEM. Bilayer thickness was averaged across three points along the circumference of the vesicle. Numbers are presented as averages ± 95% confidence interval.
α-Smooth Muscle Actin Assay
Primary mouse hepatic stellate cells (HSCs) were isolated by collagenase/pronase digestion and Stractan density gradient centrifugation before being cultured on plastic for 3 days with or without L4F-A192 (Table 1) at 37 °C [3]. HSCs were fixed with 10% formaldehyde. Primary antibody staining was done using a mouse monoclonal anti-α-SMA antibody (1:100, Sigma), followed by a fluorescein isothiocyanate-conjugated goat anti-mouse secondary antibody (Sigma). Images were acquired using a Nikon PCM-2000 confocal microscope with a thickness of 0.8 μm.
Pharmacokinetics of L4F-A192 nanoparticles
All animal experiments were performed in compliance with the guidelines established by the USC Institutional Animal Care and Use Committee. Rats were administered doses of 10 nanomoles of rhodamine covalently linked to the amino terminus of recombinant L4F-A192. Rats were administered fluorescein L4F, at doses up to 50 nanomoles. L4F was chemically synthesized as follows, Met-Asp-Trp-Phe-Lys-Ala-Phe-Tyr-Asp-Lys-Val-Ala-Glu-Lys-Phe-Lys-Glu-Ala-Phe-Leu-FITC (LifeTein, South Plainfield, NJ). Rats were bled at indicated time points from the tail into plasma separator tubes with lithium heparin (Becton, Dickinson and Co, Franklin Lakes, NJ). Serum was collected by spinning down blood samples and collecting supernatant. Plasma concentrations were determined using a calibrated fluorescence microplate assay run on a BioTek Synergy H1 Hybrid Multi Mode Microplate Reader (Biotek, Winooski, VT).
Results
Characterization of L4F ELP fusions
To develop a soluble ELP carrier for the L4F peptide, two ELPs were evaluated (Table 1). Both A192 and V2A192 have the same number of 192 pentameric repeat units, resulting in similar molecular weights. L4F was ligated at the N-terminus of the ELP of interest (Fig. 1A). ELPs reversibly phase separate in response to heating (Fig. 1B); however, the goal of this project was to determine if fusion to the amphiphilic L4F peptide is sufficient to mediate the assembly of a nanoparticle of much higher hydrodynamic radius (Fig. 1C). All four ELP constructs were purified using ELP-mediated phase separation, which resulted in highly pure samples with expected molecular weights (Fig. 1D).
Figure 1. Fusion of an amphipathic peptide and an ELP designed to assemble nanostructures.

A) The amphipathic L4F peptide was appended at the amino terminus of two ELP genes called V2A192 and A192 to drive nanoparticle assembly and increase hydrodynamic radius. B) ELPs undergo temperature-mediated phase separation called coacervation, which can be used for purification. C) The amphipathic peptide L4F (triangle) mediates assembly of nanostructures that are sterically stabilized by ELPs and these structures also phase separate in response to heating. D) A library of purified protein polymers (Table 1) were evaluated for identity and purify using SDS-PAGE and stained with copper chloride. The Left lane contains molecular weight standards with the mass indicated to the left of the marker.
Next, these fusion proteins were characterized for their phase behavior using optical density measurements with respect to temperature and concentration. Below their phase transition temperature, ELPs are soluble in water and have low optical density. Above their transition temperature, there is a sharp increase in solution turbidity (Fig. 2A–D). As has been observed previously[18], the transition temperature depends on the logarithm of concentration (Fig. 2E, F). This increase signifies the phase transition, or transition temperature of the ELP. Above this temperature, the ELP become insoluble and forms microparticles called coacervate[20]. An ELP’s transition temperature is highly contingent on the hydrophobicity of the guest residue, Xaa, in the ELP sequence, Val-Pro-Gly-Xaa-Gly[21]. Guest residues that are more hydrophilic have higher transition temperatures relative to residues that are more hydrophobic. For example, an ELP with alanine guest residues will have a higher transition temperature relative to an ELP with valine guest residues[22].
Figure 2. Fusion of the L4F peptide depresses the ELP transition temperature.

Two ELPs of the same length and different transition temperatures were appended to L4F and observed using optical density as a function of temperature and concentration. A) Unmodified A192 phase separates near 60 °C. B) Addition of L4F to A192 depressed the transition temperature to about 45°C. C) Unmodified V2A192 phase separates around 47°C. D) Addition of L4F to V2A192 decreases the transition temperature to about 34°C. E–F) The concentration-temperature phase diagrams for all constructs follow a log-linear relationship. L4F suppressed both the magnitude and the slope (Table 1) of the line for both E) A192 and F) V2A192.
The ELP A192, with a high molecular weight of 74 kDa and the relatively hydrophilic guest residue alanine, phase separates between 58 and 70 °C (Fig. 2A). The fusion of the 2.2 kDa L4F to the N-terminus of the ELP has a surprisingly large effect on the transition temperature. L4F-A192, at 25 μM, phase separates above 47 °C, which is 14 °C below the parent ELP. Similarly, at 25 μM, V2A192 phase separates above 46 °C, and again the L4F peptide decreases the transition temperature to 35 °C, below body temperature (Fig. 2D). This depression of the phase transition temperature for L4F-V2A192 will prevent this formulation from remaining soluble at physiological temperatures; however, L4F-A192 is an excellent candidate for solubility during systemic administration or in vitro evaluation in cell culture.
L4F mediates the assembly of ELP nanoparticles
Having observed the significant depression in the transition temperature upon fusion to the amphipathic L4F peptide, we speculate that this short peptide may possess the ability to drive self-association into nanostructures prior to ELP-mediated bulk phase transition. When assaying the hydrodynamic radius using dynamic light scattering, both L4F-A192 and L4F-V2A192 form nanoparticles that are stable up to a temperature of 46 and 34 °C respectively (Fig. 3A, B). In contrast, both ELP monoblocks A192 and V2A192, which lack the L4F domain, do not exhibit any signs of nanoassembly. Monoblock ELPs are not expected to have a large hydrodynamic radius because they remain unimeric until they phase separate[18]. Since A192 and V2A192 do not assemble into nanostructures alone, the assembly of nanostructures upon fusion to the L4F peptide suggests that these amphipathic peptides mediate the assembly of these particles. In fact, when in an alpha helical secondary structure, L4F is thought to orient 4 hydrophobic phenylalanine to one face[23]. Therefore, the most straightforward explanation for this data is that the L4F-mediated hydrophobicity is responsible for the assembly of structures decorated by ELPs (Fig. 1C). Investigation of L4F nanoparticle size distribution at 37 °C further supported the difference in radius between L4F ELP fusions and plain ELPs (Fig. 3C, D). A192 and V2A192 controls have low hydrodynamic radii, both measuring only 5 nm. In contrast, both L4F nanoparticles have a much higher average radius. The particle radius for L4F-A192 is at 50 nm with a polydispersity of 9 nm. Similarly, L4F-V2A192 also has a hydrodynamic radius of 42 nm.
Figure 3. Fusion to the L4F peptide mediates nanostructure assembly.

Dynamic light scattering was used to characterize the thermal stability of the fusion proteins (25 μM, PBS). A) The hydrodynamic radius for L4F-A192 is greater than for A192 even below the ELP transition temperature. B) Similarly, the hydrodynamic radius for L4F-V2A192 is greater than V2A192 even below the transition temperature. Both C) L4F-A192 and D) L4F-V2A192 form a population of nanoparticles with a hydrodynamic radius near 50 nm at 37 °C.
To further investigate the morphology of these peptide nanoparticles, the L4F-A192 nanoparticles were subsequently imaged using transmission electron microscopy (TEM) (Fig. 4). These images support the polydispersity and median sizes measured by DLS. To verify that the imaged samples were representative of the total solution and also the filtered solution evaluated by DLS, both unfiltered and filtered samples were evaluated. For negative TEM imaging, the samples were stained using 2 % uranyl acetate. Unfiltered L4F-A192 yielded a radius of 41 ± 16 nm (Fig. 4A). Filtered through 0.2 μm pores, this radius decreases to 28 ± 11 nm. When imaged by cryo-TEM, unfiltered L4F-A192 nanoparticles averaged 36 ± 13 nm (Fig. 4C), while filtration results in a radius of 43 ± 17 nm. The differences in particle size between filtered and unfiltered samples on TEM and Cryo-TEM were found to be statistically insignificant; furthermore these results confirm that these particles can be sterile filtered without a change in their size (Supplementary Fig. S1). Additionally, cryo-TEM images reveal the wall thickness of the observed vesicular structures is consistently 8.4 ± 1.3 nm, a value that is close to twice the size of the individual fused proteins. Collectively, our results suggest that the L4F amphipathic peptide promotes the assembly of the fused proteins into unilamellar vesicles.
Figure 4. The nanostructures formed by L4F fusions are peptide vesicles.

TEM and Cryo-TEM were used to characterize purified L4F-A192 that was A–B) unfiltered and C–D) filtered through a 0.2 μm membrane. A, C) TEM images of negatively stained L4F-A192 revealed evidence of nanoparticles. Unfiltered, nanoparticles averaged 41 ± 16 nm and filtered vesicles are 28 ± 11 nm. B, D) Similarly, cryo-TEM revealed the presence of unilamellar vesicles with an average radius of 36 ± 13 nm unfiltered and 43 ± 17 nm when filtered. Consequently, the average membrane thickness of unfiltered particles is 8.4 ± 1.3 (n = 18) and 0.2 um filtered particles is 6.8 ± 0.7 nm (n = 8). The scale bar indicates 200 nm.
L4F nanoparticles prevent hepatic stellate cell activation
Having demonstrated that the L4F peptide mediates the assembly of nanoparticles stabilized by ELP, it was unclear if the peptide would remain accessible to cellular targets. To evaluate this possibility, we selected hepatic stellate cells as an in vitro model for assessing L4F activity. Quiescent hepatic stellate cells have a compact morphology with cytoplasmic lipid droplets that are lost with activation. When cultured on plastic, hepatic stellate cells spontaneously activate and secrete extracellular matrix proteins. Murine hepatic stellate cells (HSCs) were cultured on plastic for 3 days in the presence or absence of L4F-A192 and A192 alone. Control hepatic stellate cells show morphologic evidence of activation and stain positive for α-smooth muscle actin, a known marker of stellate cell activation[24] (Fig. 5A). Co-culture with A192 ELP does not appear to inhibit stellate cell activation (Fig. 5B). However, HSCs cultured in the presence of L4F-A192 retain the compact morphology and cytoplasmic lipid droplets characteristic for quiescence cells (Fig. 5C). Thus, it appears that despite the involvement of the L4F peptide in the stabilization of peptide vesicles, they retain their intrinsic ability to act on a biological target associated with their anti-inflammatory properties.
Figure 5. L4F-A192 nanoparticles inhibit activation of hepatic stellate cells (HSCs).

Murine HSCs were seeded on plastic for 3 days then stained for α-smooth muscle actin. A) Without treatment, HSC seeded on plastic lose their compact morphology and cytoplasmic lipid droplets and stain positively for α-smooth muscle actin[3]. B) 10 μM A192 added to the media resulted in minimal inhibition of HSCs activation C) 10 μM L4F-A192 treated HSCs exhibit characteristic compact morphology for quiescent stellate cells. Scale bar = 20 μM. D) The fluorescence intensity of α-smooth muscle actin was quantified and compared between HSCs treated with A192 and L4F-A192. *p<0.05; **p<0.01. Values represent the mean ± 95% CI.
Having determined that L4F-A192 assembles unilamellar vesicles with evidence of biological activity in a model of hepatic stellate cell activation, we next explored whether or not the assembly of these much larger structures would have a deleterious effect on the half-life of with respect to the peptide. To accomplish this, fluorescently labeled samples containing either the L4F-A192 vesicles or the free L4F peptide were administered to rats (n=3) and sampled over a period of approximately three observed half-lives (Fig. 6). Despite the assembly of a nanostructure for L4F-A192, both peptide formulations showed a surprisingly similar half-life, on the order of 1 hr (Table 2). No significant differences were observed in the half-life or clearance. The main difference observed, was that the vesicles showed approximately twice the volume of distribution observed for the free L4F peptide. Based on these findings, it appears that the assembly of these nanostructures does not lead to premature clearance from the plasma; furthermore, this study supports their potential therapeutic evaluation.
Figure 6. Pharmacokinetics of L4F-A192 nanoparticles and free L4F do not differ significantly.
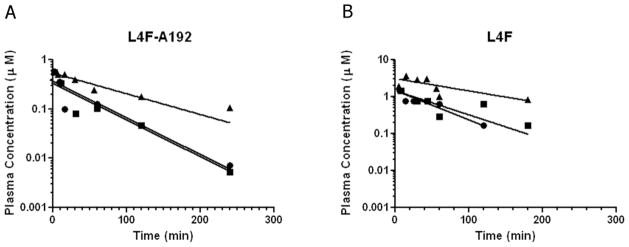
Rats were injected via the tail vein with a bolus of either fluorescently labeled L4F-A192 or the free L4F peptide. Plasma concentrations were quantified over a three-hour window and data were fit by a single exponential decay. Both treatments revealed an approximately 1 hour half life and no significant difference was detected between in clearance; however, the volume of distribution for the L4F-A192 vesicles was approximately twice that observed for the free L4F peptide (Table 2).
Table 2.
Pharmacokinetics of L4F-A192 and L4F in rats
| * Parameter | Units | L4F-A192 (n=3) | Free L4F (n=3) | ** Significance | ||
|---|---|---|---|---|---|---|
| Average | SD | Average | SD | (p-value) | ||
| Volume of distribution, V0 | mL | 26 | (4) | 12 | (2) | Yes (p=0.02) |
| Plasma half-life, thalf | min | 62 | (36) | 70 | (22) | No (p=0.77) |
| Plasma clearance, CLp | mL min−1 | 0.37 | (0.20) | 0.12 | (0.02) | No (p=0.16) |
Rat body weights were an average of 281 g.
Parameters compared by a two-tailed t test.
Discussion
Unexpectedly, the addition of a high molecular-weight protein polymer to the relatively short L4F peptide leads to the stabilization of nanoparticles. As protein polymers, ELPs were utilized due to their potential biocompatibility and biodegradability[25]. In addition, the temperature sensitivity of ELPs has recently been exploited to create micelles for the encapsulation of chemotherapeutics[26, 27]. However, these schemes rely on a temperature triggered self-assembly of a hydrophobic ELP block to create the internal portion of the micelle[18]. In contrast, the L4F ELP fusion assembles nanoparticles without the need to use heat as a trigger. The same amphipathic characteristics of L4F that make it a potent binder of oxidized lipids, also drives the hydrophobic regions of L4F to self-assemble in the absence of lipid membranes or a thermal stimulus. Most surprisingly, the addition of a large hydrophilic ELP tail to these interacting L4Fs results in unilamellar vesicles. The lamellar structures arise from a 74 kDa hydrophilic ELP linked to a 2.2 kDa hydrophobic L4F, which suggests that the ELP hydrophilic fraction exceeds 95%. When a synthetic diblock copolymer’s hydrophilic fraction exceeds 50 %, the resulting morphology is typically micellar, not vesicular[28]. In contrast, peptide diblock copolymers have been reported forming vesicles at much higher hydrophilic fractions. For example, peptide diblock copolymers composed from poly-lysine and poly-leucine have been reported that form unilamellar vesicles with a hydrophilic fraction as high as 88 % by Deming and coworkers[29]. Thus, the behavior observed for L4F-A192 is consistent with prior reports of peptide stabilized vesicles; however, the intriguing possibility remains that a region of the ELP proximal to the L4F peptide phase separates and stabilizes the lamellar phase.
DLS measurements indicate that L4F ELP fusions form nanoparticles that are stable up to the transition temperature of the fused ELP (Fig. 3). L4F-V2A192 retains a 42 nm radius up to 45 °C, above which a larger hydrodynamic radius is observed owing the imminent phase separation of the ELP V2A192. This is consistent with L4F-V2A192’s increase in optical density observed on a UV-Vis spectrophotometer (Fig. 2). Due to the greater hydrophilic nature of A192 over V2A192, the L4F-A192 fusion remains stable at temperatures slightly higher than L4F-V2A192. L4F-A192 retains a hydrodynamic radius of 50 nm up to 50 °C, again consistent with its increased in turbidity demonstrated with spectrophotometry. While the vesicles used here have been optimized for thermal stability under physiological conditions, it is certainly plausible that vesicles composed similarly to the L4F-V2A192 could be induced to deposit or release contents in response to local heating[30–32].
Nanoscale vesicles composed from amphiphilic copolypeptides have previously been reported[33–35]. To achieve radii below 50 nm, these authors performed multi-step extrusion to decrease vesicle diameter from a few micrometers down to the nanoscale. We present an alternate approach to the formation of nanoscale vesicles furthermore, the formation of ~50 nm radius particles from L4F ELP fusions does not require extrusion. Additionally, the majority of literature describing peptide-derived vesicles has used chemically synthesized polymers as precursors[29]. In contrast, this manuscript uses biological synthesis of protein polymers to produce vesicles that self-assemble.
The presence of L4F ELP fusion nanoparticles was confirmed using both negative TEM and Cryo-TEM. Cryo-TEM images of L4F-A192 show peptide vesicles with a 43 nm radius. This radius is consistent with the hydrodynamic radius of L4F-A192 measured using DLS. Interestingly, Cryo-TEM images also captured what appears to be a unilamellar polypeptide membrane. Unfiltered vesicles have a membrane thickness of 8.4 nm and filtered vesicles have a similar membrane thickness of 6.8 nm. Greater than the thickness of a phospholipid bilayer (4–5 nm)[36], this membrane thickness is consistent with the thickness of a bilayer composed from the L4F peptide attached to a sterically-stabilizing ELP[28].
Having discovered that the L4F peptide fusion results in the assembly of peptide vesicles, we next ascertained whether or not the peptide would be sufficiently accessible to retain its known biological activity, namely the potential to reduce inflammation resulting from oxidized lipids[3]. To demonstrate this, we employed a well-characterized assay for the activation of hepatic stellate cells, whereby HSCs are seeded on plastic and observed for changes in cellular morphology. In vivo activated HSCs deposit of collagen involved with liver fibrosis; furthermore, when plated on tissue culture polystyrene HSCs also adopt an activated morphology. Inactivated HSCs maintain a compact morphology, characteristic to quiescent stellate cells. However, in the absence of an inhibitor, HSCs seeded on plastic lose their compact morphology and cytoplasmic lipid droplets and increase in α-smooth muscle actin. Similar to past studies of D-4F[3], the addition of L4F-A192 nanoparticles to the media inhibits the activation of HSCs. This indicates that despite the addition of a 74 kDa ELP and its assembly into 50 nm radius nanoparticles, the proposed anti-fibrotic activity of L4F is retained.
For their therapeutic development, these peptide vesicles must remain in circulation for a sufficient period. Some of the best examples of long circulating nanoparticles are pegylated liposomes, which circulate for days[37]. While the pharmacokinetics of ELPs and their particles have only been explored in rodents[19], recent reports suggest that other recombinant polymeric materials have potential circulation in humans on the order of a week[38]. Despite their small size, the ApoA1 mimetic peptides (L-18A) have already been reported with a relatively long a plasma half-life of approximately 1 hour in rats[4]. In the same species, this manuscript confirms that the free L4F peptide also is cleared with a 1 hour plasma half-life (Fig. 6B). To determine if vesicle assembly promotes shorter (via increasing reticuloendothelial system clearance) or longer (via reducing renal clearance) half-life, the pharmacokinetics of L4F-A192 vesicles were similarly evaluated. Surprisingly, despite their 50 nm radius the both free L4F and L4F-A192 vesicles displayed nearly the same 1 hr plasma-have life and clearance (Table 2). The mechanisms for clearance for these L4F-A192 vesicles will require additional study if they are to be translated into therapeutic nanoparticles.
Herein we fused the amphipathic ApoA1 mimetic peptide L4F to a high molecular weight ELP. Unexpectedly, fusions to the ELP protein polymer not only increased the hydrodynamic radius, but also lead to the assembly of nanoparticles with a vesicular morphology. These vesicles maintained both in vitro biological activity and as well as a similar in vivo pharmacokinetic profile to the free peptide. To the best of our knowledge, this is the first report of a protein polymer fused to a biologically active peptide domain (L4F) that both assembles vesicles and maintains the therapeutic potential of its active domain[39].
Conclusions
Herein we present a scheme for the assembly of stable, potent nanoparticles driven by the amphipathic nature of L4F, an Apolipoprotein A1 derived peptide. We genetically engineered a L4F peptide with high molecular weight ELP protein polymers. At body temperature, L4F-A192 self assembles into nanoparticles with a hydrodynamic radius of 50 nm. This self-assembly is driven by the amphipathic nature of L4F. Using cryogenic TEM, we observed the formation of unilamellar vesicles with a membrane thickness of 6.8 nm. Despite forming nanoscale vesicles, L4F retained its anti-inflammatory properties as reported by a hepatic stellate cell activation assay. Because they assemble vesicles of about 50 nm in radius, this L4F ELP fusion strategy has the potential to serve as an assembly platform for other amphipathic peptides, and enable co-encapsulation of soluble therapeutics into the aqueous interior of the vesicle.
Supplementary Material
Acknowledgments
This work was made possible by the University of Southern California, the National Institute of Health R21EB012281 to J.A.M., P30CA014089 to the Norris Comprehensive Cancer Center, P30DK048522 to the USC Research Center for Liver Diseases, the USC Ming Hsieh Institute for Research on Engineering-Medicine, and the USC Whittier Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rathmann W, Giani G. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:2568–2569. doi: 10.2337/diacare.27.10.2568. author reply 2569. [DOI] [PubMed] [Google Scholar]
- 2.Friedman SL. Liver fibrosis -- from bench to bedside. J Hepatol. 2003;38(Suppl 1):S38–53. doi: 10.1016/s0168-8278(02)00429-4. [DOI] [PubMed] [Google Scholar]
- 3.DeLeve LD, Wang X, Kanel GC, Atkinson RD, McCuskey RS. Prevention of hepatic fibrosis in a murine model of metabolic syndrome with nonalcoholic steatohepatitis. The American journal of pathology. 2008;173:993–1001. doi: 10.2353/ajpath.2008.070720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garber DW, Venkatachalapathi YV, Gupta KB, Ibdah J, Phillips MC, Hazelrig JB, Segrest JP, Anantharamaiah GM. Turnover of synthetic class A amphipathic peptide analogues of exchangeable apolipoproteins in rats. Correlation with physical properties. Arterioscler Thromb. 1992;12:886–894. doi: 10.1161/01.atv.12.8.886. [DOI] [PubMed] [Google Scholar]
- 5.Hristova K, Wimley WC, Mishra VK, Anantharamiah GM, Segrest JP, White SH. An amphipathic alpha-helix at a membrane interface: a structural study using a novel X-ray diffraction method. J Mol Biol. 1999;290:99–117. doi: 10.1006/jmbi.1999.2840. [DOI] [PubMed] [Google Scholar]
- 6.Mendez AJ, Anantharamaiah GM, Segrest JP, Oram JF. Synthetic amphipathic helical peptides that mimic apolipoprotein A-I in clearing cellular cholesterol. J Clin Invest. 1994;94:1698–1705. doi: 10.1172/JCI117515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Lenten BJ, Wagner AC, Jung CL, Ruchala P, Waring AJ, Lehrer RI, Watson AD, Hama S, Navab M, Anantharamaiah GM, Fogelman AM. Anti-inflammatory apoA-I-mimetic peptides bind oxidized lipids with much higher affinity than human apoA-I. J Lipid Res. 2008;49:2302–2311. doi: 10.1194/jlr.M800075-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marino JS, Peterson SJ, Li M, Vanella L, Sodhi K, Hill JW, Abraham NG. ApoA-1 mimetic restores adiponectin expression and insulin sensitivity independent of changes in body weight in female obese mice. Nutr Diabetes. 2012;2:e33. doi: 10.1038/nutd.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peterson SJ, Drummond G, Kim DH, Li M, Kruger AL, Ikehara S, Abraham NG. L-4F treatment reduces adiposity, increases adiponectin levels, and improves insulin sensitivity in obese mice. J Lipid Res. 2008;49:1658–1669. doi: 10.1194/jlr.M800046-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaziri ND, Bai Y, Yuan J, Said HL, Sigala W, Ni Z. ApoA-1 mimetic peptide reverses uremia-induced upregulation of pro-atherogenic pathways in the aorta. Am J Nephrol. 2010;32:201–211. doi: 10.1159/000316479. [DOI] [PubMed] [Google Scholar]
- 11.Watson CE, Weissbach N, Kjems L, Ayalasomayajula S, Zhang Y, Chang I, Navab M, Hama S, Hough G, Reddy ST, Soffer D, Rader DJ, Fogelman AM, Schecter A. Treatment of patients with cardiovascular disease with L-4F, an apo-A1 mimetic, did not improve select biomarkers of HDL function. J Lipid Res. 2011;52:361–373. doi: 10.1194/jlr.M011098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Lenten BJ, Wagner AC, Navab M, Anantharamaiah GM, Hama S, Reddy ST, Fogelman AM. Lipoprotein inflammatory properties and serum amyloid A levels but not cholesterol levels predict lesion area in cholesterol-fed rabbits. J Lipid Res. 2007;48:2344–2353. doi: 10.1194/jlr.M700138-JLR200. [DOI] [PubMed] [Google Scholar]
- 13.Pastuszka MK, Janib SM, Weitzhandler I, Okamoto CT, Hamm-Alvarez S, Mackay JA. A tunable and reversible platform for the intracellular formation of genetically engineered protein microdomains. Biomacromolecules. 2012;13:3439–3444. doi: 10.1021/bm301090x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mackay JA, Chilkoti A. Temperature sensitive peptides: engineering hyperthermia-directed therapeutics. Int J Hyperthermia. 2008;24:483–495. doi: 10.1080/02656730802149570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi P, Gustafson JA, MacKay JA. Genetically engineered nanocarriers for drug delivery. International journal of nanomedicine. 2014;9:1617–1626. doi: 10.2147/IJN.S53886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hassouneh W, MacEwan SR, Chilkoti A. Fusions of elastin-like polypeptides to pharmaceutical proteins. Methods Enzymol. 2012;502:215–237. doi: 10.1016/B978-0-12-416039-2.00024-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janib SM, Gustafson JA, Minea RO, Swenson SD, Liu S, Pastuszka MK, Lock LL, Cui H, Markland FS, Conti PS, Li Z, MacKay JA. Multimeric Disintegrin Protein Polymer Fusions That Target Tumor Vasculature. Biomacromolecules. 2014 doi: 10.1021/bm401622y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janib SM, Pastuszka M, Aluri S, Folchman-Wagner Z, Hsueh PY, Shi P, Yi A, Cui H, Mackay JA. A quantitative recipe for engineering protein polymer nanoparticles. Polymer chemistry. 2014;5:1614–1625. doi: 10.1039/C3PY00537B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Janib SM, Liu S, Park R, Pastuszka MK, Shi P, Moses AS, Orosco MM, Lin YA, Cui H, Conti PS, Li Z, MacKay JA. Kinetic quantification of protein polymer nanoparticles using non-invasive imaging. Integrative biology: quantitative biosciences from nano to macro. 2013;5:183–194. doi: 10.1039/c2ib20169k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Urry DW, Pattanaik A. Elastic protein-based materials in tissue reconstruction. Ann N Y Acad Sci. 1997;831:32–46. doi: 10.1111/j.1749-6632.1997.tb52182.x. [DOI] [PubMed] [Google Scholar]
- 21.Mackay JA, Callahan DJ, Fitzgerald KN, Chilkoti A. Quantitative Model of the Phase Behavior of Recombinant pH-Responsive Elastin-Like Polypeptides. Biomacromolecules. 2010;11:2873–2879. doi: 10.1021/bm100571j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McDaniel JR, Radford DC, Chilkoti A. A unified model for de novo design of elastin-like polypeptides with tunable inverse transition temperatures. Biomacromolecules. 2013;14:2866–2872. doi: 10.1021/bm4007166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Osei-Hwedieh D. Apolipoprotein Mimetic Peptides: Mechanisms of Action as Anti-atherogenic Agents. Pharmacol Ther. 2012;130:83–91. doi: 10.1016/j.pharmthera.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamaoka K, Nouchi T, Marumo F, Sato C. Alpha-smooth-muscle actin expression in normal and fibrotic human livers. Dig Dis Sci. 1993;38:1473–1479. doi: 10.1007/BF01308606. [DOI] [PubMed] [Google Scholar]
- 25.Chilkoti A, Christensen T, MacKay JA. Stimulus responsive elastin biopolymers: Applications in medicine and biotechnology. Curr Opin Chem Biol. 2006;10:652–657. doi: 10.1016/j.cbpa.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shah M, Edman MC, Janga SR, Shi P, Dhandhukia J, Liu S, Louie SG, Rodgers K, Mackay JA, Hamm-Alvarez SF. A rapamycin-binding protein polymer nanoparticle shows potent therapeutic activity in suppressing autoimmune dacryoadenitis in a mouse model of Sjogren’s syndrome. J Control Release. 2013;171:269–279. doi: 10.1016/j.jconrel.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi P, Aluri S, Lin YA, Shah M, Edman M, Dhandhukia J, Cui H, MacKay JA. Elastin-based protein polymer nanoparticles carrying drug at both corona and core suppress tumor growth in vivo. J Control Release. 2013;171:330–338. doi: 10.1016/j.jconrel.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Discher DE, Ahmed F. Polymersomes. Annu Rev Biomed Eng. 2006;8:323–341. doi: 10.1146/annurev.bioeng.8.061505.095838. [DOI] [PubMed] [Google Scholar]
- 29.Bellomo EG, Wyrsta MD, Pakstis L, Pochan DJ, Deming TJ. Stimuli-responsive polypeptide vesicles by conformation-specific assembly. Nature materials. 2004;3:244–248. doi: 10.1038/nmat1093. [DOI] [PubMed] [Google Scholar]
- 30.McDaniel JR, MacEwan SR, Li X, Radford DC, Landon CD, Dewhirst M, Chilkoti A. Rational design of “heat seeking” drug loaded polypeptide nanoparticles that thermally target solid tumors. Nano letters. 2014;14:2890–2895. doi: 10.1021/nl5009376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park SM, Kim MS, Park SJ, Park ES, Choi KS, Kim YS, Kim HR. Novel temperature-triggered liposome with high stability: formulation, in vitro evaluation, and in vivo study combined with high-intensity focused ultrasound (HIFU) J Control Release. 2013;170:373–379. doi: 10.1016/j.jconrel.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 32.Kong G, Anyarambhatla G, Petros WP, Braun RD, Colvin OM, Needham D, Dewhirst MW. Efficacy of liposomes and hyperthermia in a human tumor xenograft model: importance of triggered drug release. Cancer research. 2000;60:6950–6957. [PubMed] [Google Scholar]
- 33.Rodriguez AR, Kramer JR, Deming TJ. Enzyme-triggered cargo release from methionine sulfoxide containing copolypeptide vesicles. Biomacromolecules. 2013;14:3610–3614. doi: 10.1021/bm400971p. [DOI] [PubMed] [Google Scholar]
- 34.Tanner P, Baumann P, Enea R, Onaca O, Palivan C, Meier W. Polymeric vesicles: from drug carriers to nanoreactors and artificial organelles. Acc Chem Res. 2011;44:1039–1049. doi: 10.1021/ar200036k. [DOI] [PubMed] [Google Scholar]
- 35.Vargo KB, Parthasarathy R, Hammer DA. Self-assembly of tunable protein suprastructures from recombinant oleosin. Proc Natl Acad Sci U S A. 2012;109:11657–11662. doi: 10.1073/pnas.1205426109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewis BA, Engelman DM. Lipid bilayer thickness varies linearly with acyl chain length in fluid phosphatidylcholine vesicles. J Mol Biol. 1983;166:211–217. doi: 10.1016/s0022-2836(83)80007-2. [DOI] [PubMed] [Google Scholar]
- 37.Woodle MC, Lasic DD. Sterically stabilized liposomes. Biochimica et biophysica acta. 1992;1113:171–199. doi: 10.1016/0304-4157(92)90038-c. [DOI] [PubMed] [Google Scholar]
- 38.Schellenberger V, Wang CW, Geething NC, Spink BJ, Campbell A, To W, Scholle MD, Yin Y, Yao Y, Bogin O, Cleland JL, Silverman J, Stemmer WPC. A recombinant polypeptide extends the in vivo half-life of peptides and proteins in a tunable manner. Nat Biotechnol. 2009;27:1186–U1155. doi: 10.1038/nbt.1588. [DOI] [PubMed] [Google Scholar]
- 39.Martin L, Castro E, Ribeiro A, Alonso M, Rodriguez-Cabello JC. Temperature-triggered self-assembly of elastin-like block co-recombinamers:the controlled formation of micelles and vesicles in an aqueous medium. Biomacromolecules. 2012;13:293–298. doi: 10.1021/bm201436y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.