

Abstract
Some patients with soft tissue sarcoma (STS) report a history of injury at the site of their tumor. While this phenomenon is widely reported, there are relatively few experimental systems that have directly assessed the role of injury in sarcoma formation. We recently described a mouse model of STS whereby p53 is deleted and oncogenic Kras is activated in muscle satellite cells via a Pax7CreER driver following intraperitoneal injection with tamoxifen. Here, we report that after systemic injection of tamoxifen, the vast majority of Pax7-expressing cells remain quiescent despite mutation of p53 and Kras. The fate of these muscle progenitors is dramatically altered by tissue injury, which leads to faster kinetics of sarcoma formation. In adult muscle, quiescent satellite cells will transition into an active state in response to hepatocyte growth factor (HGF). We show that modulating satellite cell quiescence via intramuscular (IM) injection of HGF increases the penetrance of sarcoma formation at the site of injection, which is dependent on its cognate receptor c-MET. Unexpectedly, the tumor promoting effect of tissue injury also requires c-Met. These results reveal a mechanism by which HGF/c-MET signaling promotes tumor formation after tissue injury in a mouse model of primary STS, and they may explain why some patients develop a STS at the site of injury.
INTRODUCTION
Soft tissue sarcomas (STS) represent a heterogeneous mix of >50 mesenchymal tumor types that together encompass approximately 1% of tumors in adults and 9% of tumors in children (1–3). To study sarcoma development, we utilized Pax7CreER/+; KrasLSL-G12D/+; Trp53flox/flox (P7KP) mice because mutations in the Ras and p53 pathways have been reported in human soft tissue sarcoma (4, 5). Following systemic administration of tamoxifen by intraperitoneal (IP) injection, P7KP mice develop sarcomas throughout the animal in 6–8 weeks (6). The most common locations for sarcoma development are the body wall, extremities, and head and neck (6). Similar to other reports, the histology of the sarcomas in P7KP mice exist along a continuum of undifferentiated pleomorphic sarcoma (UPS), myogenic UPS, and embryonal rhabdomyosarcoma (6, 7). To develop a temporally- and spatially-restricted model of STS, we injected P7KP mice with 4-hydroxytamoxifen (4OHT) directly into the gastrocnemius muscle. Remarkably, sarcomas developed at the 4OHT injection site with 100% penetrance with a median time that was approximately twice as fast as when the P7KP mice received systemic tamoxifen, which prompted us to test the hypothesis that more rapid sarcoma formation was caused by tissue injury related to 4OHT administration.
Although others have reported an association between injury and sarcoma development (8–14), P7KP mice represent a unique model system to investigate the mechanism. P7KP mice are a mammalian system in which the timing of p53 loss and Kras activation is tightly controlled in a defined population of cells (muscle satellite cells). Our experiments demonstrate that the fate of Pax7+ cells harboring oncogenic mutations is altered in the setting of muscle injury in a process dependent on HGF/c-MET signaling. HGF/c-MET signaling plays an important role in regulating the proliferation of muscle progenitors following injury (15), and we show that it is also required for rapid sarcoma formation in our model system. Therefore, we propose that the activation state of the sarcoma cell of origin serves as a barrier to tumor formation. This tumor suppressor mechanism may explain why sarcomas are relatively rare.
MATERIALS AND METHODS
Mouse Experiments
Mice were maintained on a mixed 129 S4/SvJae and C57/Bl6 background. The Pax7CreER allele (B6;129-Pax7tm2.1(cre/ERT2)Fan/J) has been described (16). R26LSL-YFP/YFP (B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J) reporter mice (17) were obtained from the Jackson Laboratory. KrasLSL-G12D (18) and Trp53flox/flox mice (19) were obtained from Tyler Jacks and Anton Berns respectively. The c-Metflox/flox mice (FVB;129P2-Mettm1Sst/J) were obtained from the Jackson Laboratory (20). All animal experiments were performed according to protocols approved by the Duke University and Carnegie Institution of Science Institutional Animal Care and Use Committee.
Sarcomas were generated in Pax7CreER/+; KrasLSL-G12D/+; Trp53flox/flox mice (P7KP) mice using intraperitoneal (IP) injections of tamoxifen (Sigma-Aldrich) dissolved in ethanol and diluted in corn oil (10 μl of 20 mg/ml tamoxifen per gram body weight). PBS, cardiotoxin (0.5 mg/mL dissolved in PBS) and HGF (2 μg/mL dissolved in PBS) were delivered intramuscularly (IM) at a volume of 25 μL. 5′-Ethynyl-2′-Deoxyuridine (EdU), a thymidine analog that serves as a marker of cell proliferation, was dissolved in PBS at a concentration of 0.5 mg/mL and administered intraperitoneally at 10 μL per g of body weight. Tumor specimens were fixed in 10% formalin/70% ethanol and paraffin embedded. 5 μm sections were stained with hematoxylin and eosin or with antibodies.
For HGF-mediated activation of satellite cells and assessment of P-MET expression in muscle, mice 2–4 months-of-age were treated with tamoxifen IP and corresponding IM injection (HGF to the tibialis anterior muscle or cardiotoxin to the gastrocnemius muscle, respectively). Mice treated with IM HGF were injected with IP EdU (10 μl 0.5 mg/mL per gram of body weight) for two subsequent days after IP tamoxifen/IM HGF. Three days after treatment, all mice were euthanized and the treated muscles were mounted on cork using Tragacanth (Sigma) and flash frozen for 30–45 seconds in 2-methylbutane (Sigma) cooled by liquid nitrogen. More specifically, in YFP/P-MET experiments, female Pax7CreER/+; R26YFP/YFP (P7Y) mice were given tamoxifen (Sigma) diluted to 20 mg/ml in corn oil, at 3 mg per 40 g body weight per intraperitoneal injection once a day consecutively for 5 days. Mice were anesthetized using 2,2,2-tribromoethanol (Sigma), which was dissolved in 2-methyl-2-butanol (Sigma). For quantification of P-MET/YFP double positive cells in injured and uninjured muscle, 25 μl of 40 μM cardiotoxin (Sigma) was injected with an insulin syringe into gastrocnemius muscles. 3 days later, mice were sacrificed and gastrocnemius muscles were harvested, fixed for 10 minutes in ice cold 4% paraformaldehyde (EMS) / 1x HALT phosphatase inhibitor (Thermo) / phosphate buffered saline (PBS; Gibco), incubated at 4°C overnight in 10% sucrose / 1x HALT phosphatase inhibitor / PBS, then overnight again at 4°C in 20% sucrose / 1x HALT phosphatase inhibitor / PBS, then flash frozen as described above.
Immunohistochemistry and Immunofluorescence
All immunohistochemistry was performed using the Vectastain Elite ABC Kit (Mouse IgG) and 3,3′-Diaminobenzidine tetrahydrochloride (Sigma-Aldrich). Antibodies used included Pax7 (mouse monoclonal, Developmental Studies Hybridoma Bank (DSHB) 1:5 for immunofluorescence, 1:50 for immunohistochemistry), EdU (Click-iT® EdU Alexa Fluor® 647 Imaging Kit, Life Technologies, C10340), MyoD1 (Dako, clone 5.8A, 1:100), Myogenin (Dako, clone FD5, 1:100), and CD45 (BD Pharmingen clone 30-F11, 1:50). Immunofluorescence and immunohistochemistry images were captured on a Leica DM5500B microscope using Leica Application Suite software.
For P-MET/YFP double positive cell quantification experiments, primary antibodies included P-MET (rabbit anti-phospho-c-Met Y1234/1235, Cell Signaling 1:100) and GFP (chicken anti-GFP, Aves 1:500). Secondary antibodies included biotinylated goat anti-rabbit, Vector 1:200, and Alexa Fluor 633 conjugated goat anti-chicken, Invitrogen 1:500. Biotinylated antibody was detected by Alexa Fluor 568 conjugated streptavidin, Invitrogen 1:500. DAPI was used for DNA detection, and slides were mounted with Fluoromount-G (SouthernBiotech). Images were taken with a Leica SP5 confocal equipped with a 25x/.95 water objective using Leica image acquisition software. Images were processed using ImageJ software. Percentage of P-MET/YFP double positive cells was calculated for each muscle, then percentages were averaged and presented with standard deviation. n = 3 muscles for 2 mice, over 1000 cells counted for injured and uninjured muscles.
PCR
The details of the PCR protocol for detection of recombined and wild type c-Met were kindly provided by Dr. Thorgeirsson (20). Briefly, Quick-Load Taq 2X Master Mix (NEB) was used to assess c-Met recombination. The cycling parameters were 94°C for 30 sec, 56°C for 30 sec, and 68°C for 60 sec for a total of 30 cycles. Primers Met-1 and Met-2 yield a Floxed (380 bp) and a WT (300 bp) band. Primers Met-1 and Met-3 yield a PCR product corresponding to the deleted allele (650 bp).
Primers
Met-1: 5′ tta ggc aat gag gtg tcc cac 3′
Met-2: 5′ cca ggt ggc ttc aaa ttc taa gg 3′
Met-3: 5′ cag ccg tca gac aat tgg cac 3′
Statistical Analysis
Survival curves were compared with the log-rank test using GraphPad Prism software. Statistical significance of values reported on bar graphs was deterimined using the Student’s t test.
RESULTS
A temporally and spatially restricted model of STS
We previously developed a temporally and spatially-restricted mouse model of soft tissue sarcoma using intramuscular (IM) injection of an adenovirus expressing Cre recombinase (21), which has been useful for surgical and radiation therapy experiments (22, 23). However, adenovirus infects a heterogeneous population of cells. To generate a spatially-restricted tumor model where the cell of origin is better defined, we delivered 4OHT by IM injection into the gastrocnemius muscle of the left hindlimb of P7KP mice (6) to try to restrict genetic recombination and subsequent sarcoma formation to the site of injection. We treated 14 P7KP mice with an IM injection of 50 μl of 10 mg/ml 4OHT dissolved in DMSO. As expected, all mice developed a tumor at the site of injection (Figure 1A). However, we were surprised to find the kinetics of tumor onset (median = 20 days, range 13–26 days) was faster than after IP tamoxifen (median 45 days, range 33–62 days) (6) (Figure 2A). Evaluation of the tumors by histology revealed pleomorphic sarcomas characterized by spindle cells, epithelioid cells, or a mixture of both. The sarcomas were called pleomorphic rhabdomyosarcoma (RMS) if they stained positive for MyoD and myogenin (71% of tumors), and they were categorized as undifferentiated pleomorphic sarcoma (UPS) if they lacked immunoreactivity for both differentiation markers (29% of tumors) (Figure 1A,B).
Figure 1.

Soft tissue sarcomas resembling undifferentiated pleomorphic sarcoma (UPS) or pleomorphic rhabdomyosarcoma (pRMS) arise in P7KP mice following recombination with intramuscular (IM) 4-hydroxytamoxifen (4OHT). A) Representative sections are shown for sarcomas resembling UPS and pRMS. A hematoxylin and eosin (H&E) stained section is shown for each sarcoma subtype in addition to immunohistochemistry with antibodies directed against MyoD and myogenin. B) Histologic distribution of sarcomas in P7KP mice treated with IM 4OHT.
Figure 2.
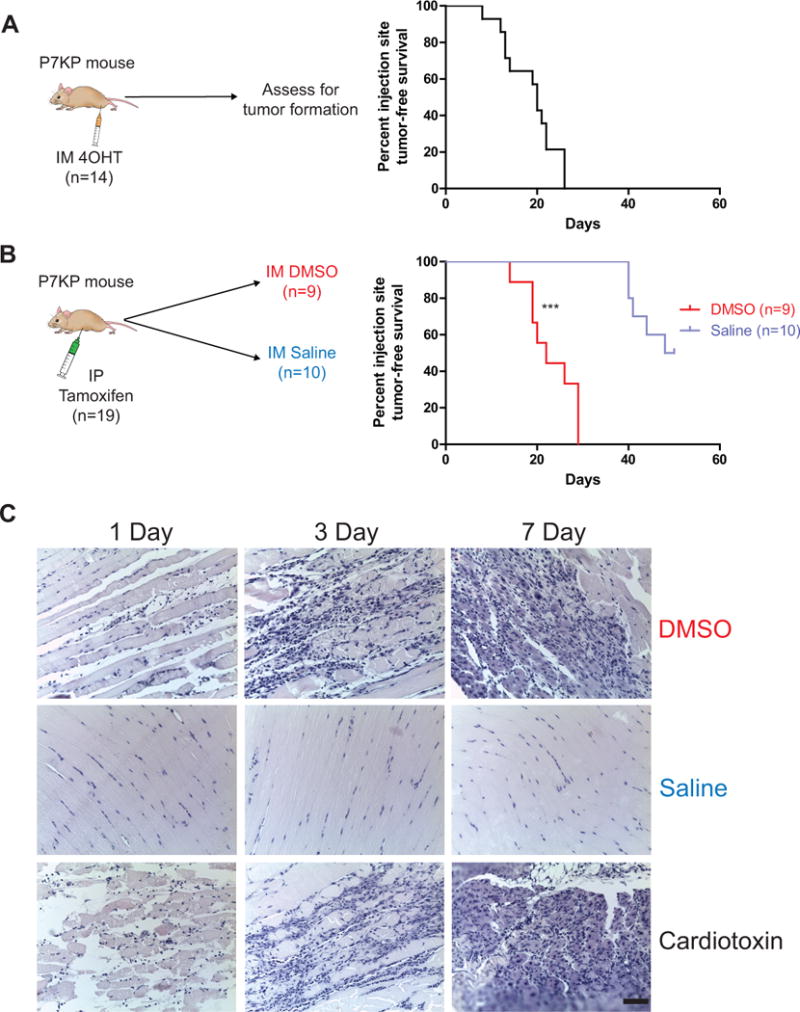
Tissue injury accelerates tumor formation in a temporally and spatially restricted mouse model of primary soft tissue sarcoma. A) Pax7CreER/+; KrasLSL-G12D/+; Trp53flox/flox (P7KP) mice were treated with an intramuscular (IM) injection of 10 mg/ml 4-hydroxytamoxifen (4OHT) in a total volume of 50 μl of DMSO in the gastrocnemius muscle of the left hindlimb. The median tumor-free survival at the injection site was 20 days. B) P7KP mice were treated with an intraperitoneal (IP) injection of 200 mg/kg tamoxifen, immediately followed by an intramuscular (IM) injection of either DMSO or saline (50 μl) in the gastrocnemius muscle of the left hindlimb. 100% of mice treated with DMSO developed a sarcoma at the injection site with a median onset of 22 days. 50% of mice treated with saline developed a sarcoma at the injection site, but with significantly delayed kinetics (median 45 days). The remaining mice developed sarcomas at other sites and had to be euthanized. C) P7KP littermates were treated with either IM DMSO, saline, or cardiotoxin and euthanized 1, 3, or 7 days following treatment. Hematoxylin and eosin stained slides show evidence of tissue injury in mice injected with DMSO or cardiotoxin (Scale bar = 150μm). *** (p < 0.001).
Intramuscular injection with DMSO injures muscle and accelerates STS formation
The kinetics of spatially restricted tumor formation in P7KP mice was significantly more rapid than that observed following systemic tamoxifen exposure, which suggested that IM injection of the DMSO vehicle accelerates sarcoma formation. To investigate this possibility, we treated P7KP mice with systemic tamoxifen along with concurrent IM injection of DMSO (vehicle for 4OHT) (Figure 2B). With this experimental approach, the recombination frequency following IP tamoxifen should be similar in Pax7-expressing cells throughout the mice. A cohort of mice was also treated with IP tamoxifen and concurrent IM saline to determine if any effect observed with DMSO was specific to the vehicle or simply related to the IM injection itself.
Remarkably, 100% of P7KP mice treated with IP tamoxifen and IM DMSO developed a palpable tumor at the IM injection site in a median of 22 days (range 14–29 days) (Figure 2B), similar to the kinetics seen with 4OHT alone (Figure 2A). In P7KP mice treated with IP tamoxifen and IM saline, 50% of mice developed an injection site sarcoma, but the time to tumor was significantly longer (median 41 days, range 40–48 days) (Figure 2B). These results reveal that that DMSO accelerates sarcoma formation in P7KP mice. Of note, sarcomas developed at sites other than the injection with similar kinetics as P7KP mice treated with IP tamoxifen alone (data not shown).
In order to determine if IM DMSO injection caused any change in the histologic appearance of muscle, littermates of the P7KP mice (P7P mice) were treated with IM DMSO and euthanized 1, 3, and 7 days following injection. IM saline was used as a negative control, and cardiotoxin, a component of cobra venom often utilized in muscle injury and regeneration studies (24), was used as a positive control. Evaluation of muscle treated with IM DMSO revealed significant cell death and neutrophil infiltration, consistent with injury to the skeletal muscle (Figure 2C). As expected, massive cell death and inflammation was seen in cardiotoxin-treated muscle, and no significant changes were seen in saline-treated muscle (Figure 2C).
Cardiotoxin-mediated injury also accelerates sarcoma formation in P7KP mice
The experiments described above indicate that IM injection of DMSO resulted in a tissue injury, leading to more rapid sarcoma formation. To further test this model, we next determined if IM injection of cardiotoxin could promote sarcoma formation in P7KP mice following systemic tamoxifen because muscle injury following administration of cardiotoxin has been extensively characterized (Figure 2C) (24). Eight P7KP mice were treated with IP tamoxifen and concurrent IM cardiotoxin (Figure 3B). Remarkably, all 8 mice developed palpable tumors at the cardiotoxin injection site with a median onset of 17 days (range 7–17 days). Thus, concurrent administration of systemic tamoxifen and injury with IM cardiotoxin in P7KP mice exerts temporospatial control over the formation of soft tissue sarcoma at the injury site.
Figure 3.
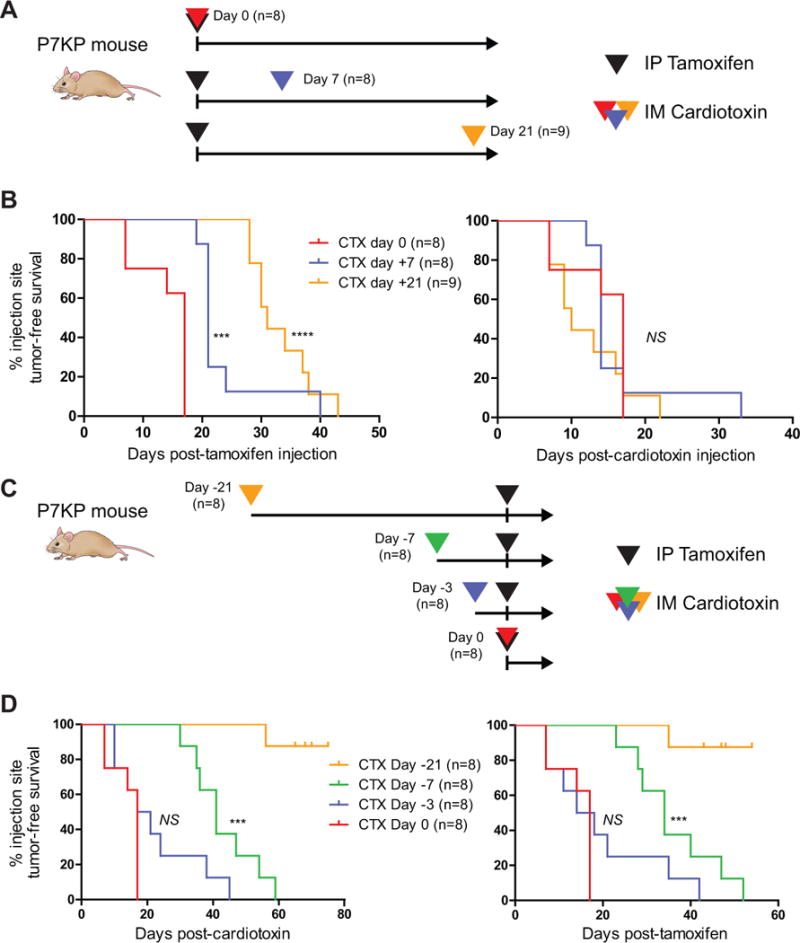
Tissue injury with cardiotoxin acts as a promoter to accelerate sarcoma formation following initiation by tamoxifen-mediated recombination. A) P7KP mice were treated with IP tamoxifen on Day 0 followed by tissue injury with cardiotoxin injected into the gastrocnemius muscle on Day 0, 7, and 21. B) Injection site tumor-free survival is shown both post-tamoxifen and post-cardiotoxin. Of note, the curves overlap on the post-cardiotoxin survival curve, consistent with injury by cardiotoxin acting as a rate limiting step for rapid sarcoma formation following recombination of Trp53flox/flox and KrasLSL-G12D in tamoxifen-treated mice. C) A similar experiment was performed where P7KP mice were treated with IM cardiotoxin 21, 7, 3, and 0 days prior to IP tamoxifen. D) Injection site tumor-free survival is shown both post-cardiotoxin and post-tamoxifen. In contrast to when cardiotoxin is administered after tamoxifen, cardiotoxin administered >3 days prior to IP tamoxifen does not accelerate sarcoma formation, even when evaluated as the time-to-tumor post-tamoxifen. Taken together, these data support a model where tamoxifen-mediated recombination of Trp53flox/flox and KrasLSL-G12D acts as a classic initiator and tissue injury by cardiotoxin acts as a promoter to accelerate sarcoma formation. NS (not significant), *** (p<0.001), **** (p<0.0001).
We next studied the temporal relationship between tamoxifen-mediated recombination of p53 and Kras and cardiotoxin-mediated tissue injury. A time course experiment was performed whereby P7KP mice were injured with IM cardiotoxin at times before, after, and coincident with IP tamoxifen delivery (Figure 3A,C). When cardiotoxin-mediated injury was performed either just prior to recombination with tamoxifen (up to 3 days before) or at any timepoint thereafter (up to 21 days after), an injection site sarcoma formed within 20 days of cardiotoxin administration with nearly 100% penetrance (Figure 3B,D). No sarcomas formed following treatment with cardiotoxin alone, as seen in other studies (14). These results are consistent with a classic initiator/promoter model of sarcomagenesis whereby recombination with IP tamoxifen provides the initiating genetic insult, and injury serves as the promoter to drive sarcoma formation.
The Hepatocyte Growth Factor/c-MET pathway promotes sarcoma formation in P7KP mice
In the time course experiment, we were intrigued by the observation that recombined Pax7-expressing cells appeared to have reduced transformation potential until they were “activated” by cardiotoxin administration. Similar to recent experiments in squamous cell carcinoma (25) and intestinal cancer (26), we hypothesized that a key function of cardiotoxin might be to alter the activation state of Pax7-expressing satellite cells. We first assessed the number of proliferating satellite cells in P7KP mice following a single dose of systemic tamoxifen. For these experiments, P7KP mice (n=3 per group) were also treated with IM saline or hepatocyte growth factor (HGF), which has been shown to promote satellite cell proliferation both in vitro and in vivo (27, 28). Of note, the IM injection volume to the tibialis anterior (TA) muscle for these experiments was reduced to 25 μl in an effort to limit trauma related to the volume of injection (Supplementary Fig. 1A,B online). The mice were given two consecutive daily doses of EdU and euthanized on day 3 after the IP tamoxifen and the IM HGF/saline injections. We collected the TA muscles and stained 10 μm frozen sections for Pax7 and EdU (Supplementary Fig. 2A,B online). We counted 400 – 600 satellite cells per mouse and determined the percentage that were EdU+ (Pax7, EdU double positive) as a measure of satellite cell activation. Interestingly, the vast majority of Pax7+ cells were not proliferating in mice treated with IM saline despite deletion of p53 and activation of Kras in Pax7-expressing cells throughout the animal (Figure 4A). Administration of IM HGF caused a >3-fold increase in EdU uptake in satellite cells (Figure 4A). Moreover, evaluation of specimens by H&E staining revealed no significant inflammatory infiltrate (Supplementary Fig. 2C online). Thus, HGF promotes proliferation of recombined satellite cells without the histological hallmarks of tissue injury.
Figure 4.

HGF signals via c-MET to activate satellite cell proliferation and increase the penetrance of injection-site tumor formation. A) P7KP mice were treated with IP tamoxifen and IM saline or 50 ng hepatocyte growth factor (HGF) in a total volume of 25 μl. The mice were treated with EdU prior to euthanasia, and the number of Pax7/EdU double positive cells was determined via immunofluorescence. A greater number of proliferating cells was seen in the P7KP mice treated with IM HGF. B) P7KP mice were treated with tamoxifen on Day 0 along with a concurrent IM injection into the gastrocnemius muscle with IM saline, cardiotoxin, or HGF. C,D) All mice treated with IM cardiotoxin (14/14) developed a sarcoma with rapid kinetics at the injection site. A minority of mice treated with IM saline (3/12) developed an injection-site sarcoma with slower kinetics. When mice were treated with IM HGF, the majority (11/13) developed an injection-site sarcoma with similar kinetics to IM saline. E) P7KP; c-Metflox/+ and P7KP; c-Metflox/flox mice were treated with IP tamoxifen and IM HGF and evaluated for sarcoma formation at the IM injection site. F,G) The increased penetrance of injection-site sarcomas observed following treatment with IP tamoxifen and IM HGF in P7KP mice is dependent on signaling through c-MET. * (p<0.05), ** (p<0.01), *** (p<0.001), **** (p<0.0001).
We next determined if activation of recombined satellite cells by HGF is sufficient to promote tumor development at the site of injection after systemic tamoxifen. A total of 39 P7KP mice were treated with IP tamoxifen along with IM injection of 25 μl of either 40 μM cardiotoxin (positive control), saline (negative control), or HGF (Figure 4B). Of note, a concentration of 40 μM cardiotoxin was used because the time-to-tumor was not statistically different than a concentration of 75 μM (Supplementary Fig. 1C–E online). All cardiotoxin-treated mice developed a sarcoma at the injection site with a median onset of 15 days, while only 3 of 12 saline-treated mice developed a sarcoma at the injection site (Figure 4C). In contrast, 11/13 mice treated with IM HGF developed a sarcoma at the injection site (Figure 4C). Of note, the sarcomas arising in HGF-treated mice occur with a median onset of 37 days, which is slower than sarcomas that arise following treatment with cardiotoxin (Figure 4D).
Next, we wished to determine if the increased penetrance of sarcoma formation seen with HGF is mediated by the c-MET receptor rather than a nonspecific effect from the IM injection. We crossed P7KP mice to c-Metflox/flox mice (20) to cause c-Met deletion by Cre in satellite cells upon tamoxifen treatment. We treated 11 P7KP; c-Metflox/flox and 13 P7KP; c-Metflox/+ littermate control mice with systemic tamoxifen and IM HGF. We observed that P7KP mice with homozygous c-Met deletion no longer had an increased penetrance of tumor formation at the IM HGF injection site as compared to P7KP mice with only one copy of c-Met deleted (Figure 4G). Of note, P7KP; c-Metflox/flox mice could not be followed beyond 2–3 months because they developed sarcomas at other sites. These data provide genetic evidence that HGF requires the c-MET receptor to increase the penetrance of sarcoma formation in P7KP mice.
c-MET has recently been shown to to play an important role in satellite cell migration in the context of cardiotoxin-mediated injury (15), so we were interested to learn if it might also play a role in injury-induced sarcomas. We injected Pax7CreER/+; R26LSL-YFPYFP (P7Y) mice with IM cardiotoxin and assessed the levels of activated c-MET (P-MET) in satellite cells contained within injured and non-injured muscle (Figure 5A). In P7Y mice injected with IP tamoxifen and IM cardiotoxin and euthanized 3 days after, the number of P-MET+ cells within the YFP+ population was 40-fold higher than in uninjured muscle (Figure 5B). This result emphasizes that c-MET is activated following acute muscle injury by cardiotoxin. Of note, the increased number of DAPI-stained nuclei seen in muscle injured by cardiotoxin (Figure 5A) has been reported by others to be related to inflammation, fibroblast infiltration, as well as an increase in the number of activated satellite cells that are proliferating within the injury site (29–32). Indeed, immunohistochemistry of muscle sections from injured P7Y mice contain numerous CD45+ cells, consistent with a significant immune infiltrate (Figure 5C). The presence of satellite cells (YFP+ cells) lacking activated c-MET suggests that alternative pathways are likely to be involved in satellite cell activation in vivo (15).
Figure 5.
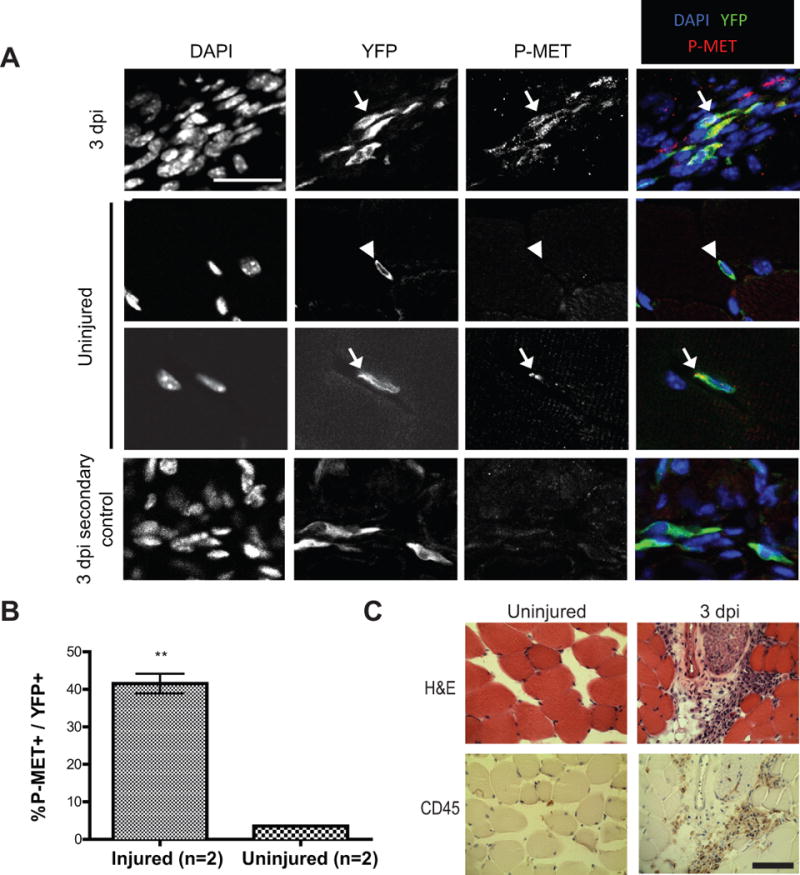
Muscle injury by cardiotoxin activates c-MET. A) c-MET is phosphorylated in satellite cells following injury. Lineage marked cells from 3 days post injury (dpi) and uninjured muscle tissue were exposed to DAPI along with antibodies to detect YFP and phosphorylated c-MET (P-MET). Arrows identify YFP+; P-MET+ cells; arrowheads identify a YFP+; P-MET- cell. Specificity of the P-MET antibody was confirmed by performing a control with the secondary antibody only (Scale bar = 25 μm). B) Bar graph summarizing the results of the aforementioned experiment shows a dramatic increase in Pax7-lineage cells that express P-MET 3 days after cardiotoxin injury (P-MET+; YFP+ cells). C) Hematoxylin and eosin (H&E) staining of muscle from 3 dpi shows an increase in hematoxylin-stained nuclei, which include CD45+ inflammatory cells. In contrast, CD45 staining is virtually absent in uninjured muscle (Scale bar = 50μm). ** (p<0.01).
We then tested whether c-MET signaling might be a critical mediator of the sarcoma-promoting effect of cardiotoxin. A total of 20 mice (10 P7KP; c-Metflox/flox and 10 P7KP; c-Metflox/+) were treated with systemic tamoxifen along with IM cardiotoxin (Figure 6A). As expected, all of the P7KP; c-Metflox/+ mice developed sarcomas at the IM injection site with rapid kinetics (Figure 6B). P7KP; c-Metflox/flox mice also developed a sarcoma at the site of IM cardiotoxin administration, but the kinetics were much slower and similar to those observed in spontaneously developing tumors after IP tamoxifen (Figure 6B, median = 42 days). This result reveals that c-MET signaling is required for cardiotoxin-mediated promotion of sarcoma formation in P7KP mice. Notably, sarcomas arising in P7KP; c-Metflox/flox mice had full recombination at both c-Met loci, confirming that sarcomas arise despite the deletion of c-Met (Figure 6D).
Figure 6.
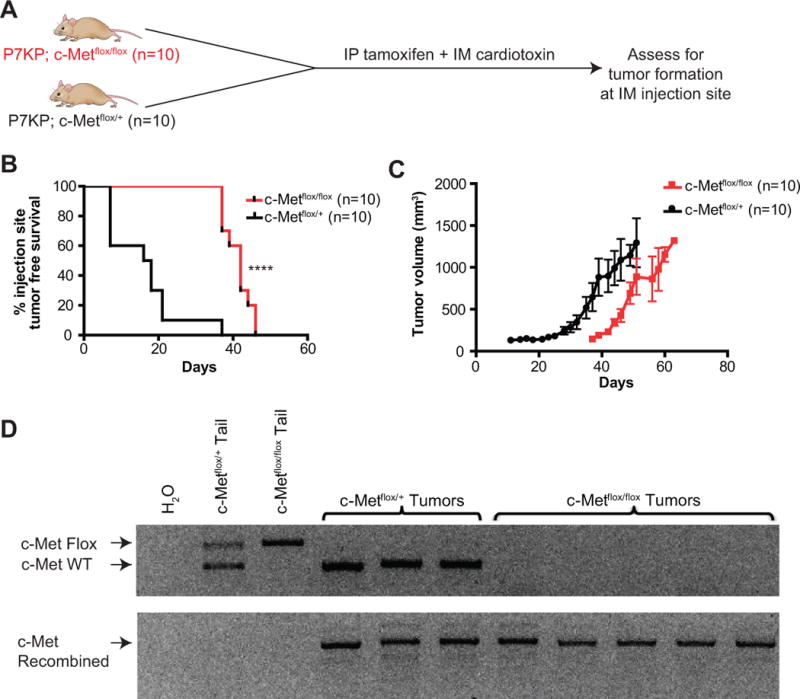
Sarcoma promotion by muscle injury requires c-Met. A) Schematic of experiment where P7KP; c-Metflox/flox and P7KP; c-Metflox/+ mice were treated with systemic IP tamoxifen and concurrent IM injection of 25 μl of 40 μM cardiotoxin. B) Sarcomas in P7KP; c-Metflox/flox mice develop with delayed kinetics, demonstrating that c-Met is required for sarcoma promotion by muscle injury. C) Tumor growth kinetics in P7KP; c-Metflox/flox vs. P7KP; c-Metflox/+ after IP tamoxifen and IM cardiotoxin. D) Cells lines were generated from primary sarcomas obtained from P7KP; c-Metflox/+ and P7KP; c-Metflox/flox mice treated with IP tamoxifen and IM cardiotoxin. After passaging the cells to deplete stroma, DNA was extracted from the cell lines and PCR was performed to assess recombination at the c-Met locus. **** (p<0.0001).
As a corollary experiment, we treated 6 P7KP; c-Metflox/flox and 10 P7KP; c-Metflox/+ controls with IM 4OHT and again observed that the median time to tumor at the site of injection was significantly faster in those mice that retained a copy of wild type c-Met (Supplementary Fig. 3 online). This further proves that the c-MET pathway is required for sarcoma promotion in the context of injury in P7KP mice.
DISCUSSION
The P7KP mouse model of STS is a unique system to study the role of injury in tumorigenesis because it provides tight control of the timing of both genetic mutation (IP injection of tamoxifen) and injury (IM injection of cardiotoxin) prior to tumor formation. Other injury-mediated sarcoma model systems have been described, but they all lack the ability to control the timing of oncogenic mutations in a genetically engineered organism. Perhaps the most noteworthy model system was described by the laboratory of Dr. Bissell (8–10). They described a series of experiments in chickens infected with Rous sarcoma virus (RSV). The chickens developed sarcomas at the site of virus injection in the wing despite systemically elevated viral titers. However, the chickens developed sarcomas in the contralateral wing after wounding with a clip or suture material. Similar findings were seen in transgenic mice constitutively overexpressing the v-jun oncogene (11–13). The transgenic mice did not have any demonstrable phenotype with the exception of sarcoma formation at the site of ear tagging and tail clipping. More recent examples include the observation that recurrent muscle injury with cardiotoxin gives rise to STS at the site of injury in p53-null mice (14). Attempts to generate urothelial tumors in a mouse model containing inducible activation of Kras and loss of p53 resulted in STS at the suture site, though this was reported as an incidental finding as the intent was to generate a genitourinary tumor (33). Lastly, neurofibromas were found to preferentially develop at the site of nerve injury in a mouse model of neurofibromatosis type I (34).
The rapid kinetics of tumor formation in P7KP mice treated with systemic tamoxifen and IM cardiotoxin demonstrates that injury acts as a classic promoter of STS in a mammalian system. Most Pax7+ cells remain quiescent despite loss of p53 and expression of oncogenic Kras (Figure 4A), but cardiotoxin breaks their quiescence and promotes tumor formation in a process dependent on c-MET signaling. HGF is also able to break quiescence of satellite cells harboring p53 and Kras mutations and promote sarcoma formation (Figure 4A–D and Supplementary Fig. 2 online), but the level of HGF used in our experiment was not sufficient to speed up the kinetics. It is also possible that the expression of other growth factors in addition to HGF that signal through c-MET are required to promote sarcoma formation. For example, it has been widely reported that the c-MET pathway has significant crosstalk with other receptor tyrosine kinase receptors such as EGFR, VEGFR and FGFR to promote cancer development as well as drug resistance (35).
Several studies in rodents have implicated the HGF/c-MET signaling pathway in satellite cell activation. For example, Allen and colleagues demonstrated that cultured rat satellite cells were activated after HGF treatment in a dose-dependent manner (36). Subsequent studies by Tatsumi and colleagues observed that rats treated with HGF had increased satellite cell proliferation, confirming that this effect was relevant in vivo. Additionally they showed that HGF is present in the extracellular matrix surrounding adult muscle fibers, while the c-MET receptor is expressed specifically in satellite cells. Upon satellite cell activation, HGF and c-MET co-localize (27). More recent experiments have shown that c-MET is required for quiescent satellite cells to respond to injury in a contralateral limb by transitioning into an alert state, which increases their propensity to cycle in vivo (37). Likewise, deletion of the c-MET receptor in Pax7+ cells results in a significant decrease in the recruitment of proliferating satellite cells to the site of cardiotoxin injury (15). In humans, the role of HGF/c-MET signaling is difficult to study in the context of acute injury because of the lack of available tissue samples. However, there was a small study of a group of human subjects that underwent a muscle biopsy of the quadriceps after completing a series of lengthening contractions (38). A trend toward increased HGF protein expression in the muscle and blood following intense exercise was noted (38).
Additionally, HGF/c-MET signaling has been reported to play a role in sarcoma formation. Ink4a/Arf knockout mice overexpressing HGF under the direction of the metallothionein promoter develop embryonal rhabdomyosarcomas with a mean onset of 3.3 months. In this study, the skeletal muscle of 6–10 week-old mice had hyperplastic satellite cells, consistent with a role for HGF in tumor initiation by promoting proliferation of cells lacking Ink4a/Arf (39). HGF/c-MET signaling has also been implicated in many different types of human sarcoma. Taulli et al. showed that cell lines derived from human embryonal RMS and alveolar RMS expressed high levels of c-MET and HGF (40). Other groups have shown that the HGF/c-MET pathway is upregulated in osteosarcoma, chondrosarcoma, and leiomyosarcoma (41, 42), and elevated HGF/c-MET expression in human synovial sarcoma has been correlated with poor prognosis in patients (43).
In P7KP mice, we propose that HGF/c-MET signaling is acting as a classic promoter, activating satellite cells in the context of cardiotoxin injury. Our finding that 21 days can pass between recombination of p53 and Kras and injury by cardiotoxin suggests that satellite cells can harbor oncogenic mutations without progression to sarcoma formation. The rarity of sarcomas in humans might be partially explained by the requirement for this additional “activation” step, whereby quiescence prevents cells that harbor oncogenic mutations from forming sarcomas.
Our data also lend credence to patient reports of a localized injury causing their sarcoma (44). In his landmark 1919 textbook “Neoplastic Diseases”, James Ewing wrote that “sarcoma commonly develops after a single blow”. When patients report a history of trauma prior to the development of sarcoma at the same location, such claims are difficult to substantiate in a rigorous manner and are frequently ignored by physicians. Here we report on the development of a temporally and spatially restricted mouse model of soft tissue sarcoma (STS) that allows for the precise timing of genetic mutations and tissue injury. Our data demonstrate that injury promotes sarcoma formation in a process dependent on HGF/c-MET signaling. Although “a single blow” is not likely to cause sarcoma, our results indicate that injury might promote sarcoma development by activating quiescent progenitor cells that have acquired oncogenic mutations.
Supplementary Material
Acknowledgments
We thank members of the Kirsch laboratory for their advice and support. We thank Tyler Jacks for the KrasLSL-G12D and Anton Berns for the Trp53flox/flox mice. Research reported in this publication was supported by the Conquer Cancer Foundation of ASCO (DVM), The Hartwell Foundation (DVM), K12HD043494 (DVM), R01CA169220 (DGK), R01AR060042 (CMF), and F32AR065366 (MTW). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
The authors disclose no potential conflicts of interest.
References
- 1.Fletcher CDM, Unni KK, Mertens F, World Health Organization. International Academy of Pathology . Pathology and genetics of tumours of soft tissue and bone. Lyon Oxford: IARC Press; Oxford University Press (distributor); 2002. [Google Scholar]
- 2.Howlader N, Noone AM, Krapcho M, Neyman N, Aminou R, Waldron W, et al. SEER Cancer Statistics Review, 1975–2008. National Cancer Institute; Bethesda, MD: http://seer.cancer.gov/csr/1975_2008/, based on November 2010 SEER data submission, posted to the SEER web site, 2011–2011 [cited 2011 May 11]; [Google Scholar]
- 3.Saab R, Spunt SL, Skapek SX. Myogenesis and rhabdomyosarcoma the Jekyll and Hyde of skeletal muscle. Current topics in developmental biology. 2011;94:197–234. doi: 10.1016/B978-0-12-380916-2.00007-3. [DOI] [PubMed] [Google Scholar]
- 4.Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer discovery. 2014;4:216–31. doi: 10.1158/2159-8290.CD-13-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen X, Stewart E, Shelat AA, Qu C, Bahrami A, Hatley M, et al. Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer cell. 2013;24:710–24. doi: 10.1016/j.ccr.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blum JM, Ano L, Li Z, Van Mater D, Bennett BD, Sachdeva M, et al. Distinct and overlapping sarcoma subtypes initiated from muscle stem and progenitor cells. Cell reports. 2013;5:933–40. doi: 10.1016/j.celrep.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rubin BP, Nishijo K, Chen HI, Yi X, Schuetze DP, Pal R, et al. Evidence for an unanticipated relationship between undifferentiated pleomorphic sarcoma and embryonal rhabdomyosarcoma. Cancer Cell. 2011;19:177–91. doi: 10.1016/j.ccr.2010.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dolberg DS, Hollingsworth R, Hertle M, Bissell MJ. Wounding and its role in RSV-mediated tumor formation. Science. 1985;230:676–8. doi: 10.1126/science.2996144. [DOI] [PubMed] [Google Scholar]
- 9.Sieweke MH, Thompson NL, Sporn MB, Bissell MJ. Mediation of wound-related Rous sarcoma virus tumorigenesis by TGF-beta. Science. 1990;248:1656–60. doi: 10.1126/science.2163544. [DOI] [PubMed] [Google Scholar]
- 10.Martins-Green M, Boudreau N, Bissell MJ. Inflammation is responsible for the development of wound-induced tumors in chickens infected with Rous sarcoma virus. Cancer Res. 1994;54:4334–41. [PubMed] [Google Scholar]
- 11.Schuh AC, Keating SJ, Monteclaro FS, Vogt PK, Breitman ML. Obligatory wounding requirement for tumorigenesis in v-jun transgenic mice. Nature. 1990;346:756–60. doi: 10.1038/346756a0. [DOI] [PubMed] [Google Scholar]
- 12.Schuh AC, Keating SJ, Yeung MC, Breitman ML. Skeletal muscle arises as a late event during development of wound sarcomas in v-jun transgenic mice. Oncogene. 1992;7:667–76. [PubMed] [Google Scholar]
- 13.Shalaby F, Schuh AC, Breitman ML. Two distinct target cells for v-jun mediated wound tumorigenesis. Oncogene. 1994;9:2579–88. [PubMed] [Google Scholar]
- 14.Camboni M, Hammond S, Martin LT, Martin PT. Induction of a regenerative microenvironment in skeletal muscle is sufficient to induce embryonal rhabdomyosarcoma in p53-deficient mice. J Pathol. 2012;226:40–9. doi: 10.1002/path.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Webster MT, Fan CM. c-MET regulates myoblast motility and myocyte fusion during adult skeletal muscle regeneration. PLoS One. 2013;8:e81757. doi: 10.1371/journal.pone.0081757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lepper C, Conway SJ, Fan CM. Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nature. 2009;460:627–31. doi: 10.1038/nature08209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer cell. 2004;5:375–87. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 19.Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes & development. 2000;14:994–1004. [PMC free article] [PubMed] [Google Scholar]
- 20.Huh CG, Factor VM, Sanchez A, Uchida K, Conner EA, Thorgeirsson SS. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad Sci U S A. 2004;101:4477–82. doi: 10.1073/pnas.0306068101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kirsch DG, Dinulescu DM, Miller JB, Grimm J, Santiago PM, Young NP, et al. A spatially and temporally restricted mouse model of soft tissue sarcoma. Nature medicine. 2007;13:992–7. doi: 10.1038/nm1602. [DOI] [PubMed] [Google Scholar]
- 22.Moding EJ, Lee CL, Castle KD, Oh P, Mao L, Zha S, et al. Atm deletion with dual recombinase technology preferentially radiosensitizes tumor endothelium. The Journal of clinical investigation. 2014;124:3325–38. doi: 10.1172/JCI73932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mito JK, Min HD, Ma Y, Carter JE, Brigman BE, Dodd L, et al. Oncogene-dependent control of miRNA biogenesis and metastatic progression in a model of undifferentiated pleomorphic sarcoma. The Journal of pathology. 2013;229:132–40. doi: 10.1002/path.4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goetsch SC, Hawke TJ, Gallardo TD, Richardson JA, Garry DJ. Transcriptional profiling and regulation of the extracellular matrix during muscle regeneration. Physiological genomics. 2003;14:261–71. doi: 10.1152/physiolgenomics.00056.2003. [DOI] [PubMed] [Google Scholar]
- 25.White AC, Khuu JK, Dang CY, Hu J, Tran KV, Liu A, et al. Stem cell quiescence acts as a tumour suppressor in squamous tumours. Nature cell biology. 2014;16:99–107. doi: 10.1038/ncb2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Westphalen CB, Asfaha S, Hayakawa Y, Takemoto Y, Lukin DJ, Nuber AH, et al. Long-lived intestinal tuft cells serve as colon cancer-initiating cells. J Clin Invest. 2014 doi: 10.1172/JCI73434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tatsumi R, Anderson JE, Nevoret CJ, Halevy O, Allen RE. HGF/SF is present in normal adult skeletal muscle and is capable of activating satellite cells. Dev Biol. 1998;194:114–28. doi: 10.1006/dbio.1997.8803. [DOI] [PubMed] [Google Scholar]
- 28.Miller KJ, Thaloor D, Matteson S, Pavlath GK. Hepatocyte growth factor affects satellite cell activation and differentiation in regenerating skeletal muscle. Am J Physiol Cell Physiol. 2000;278:C174–81. doi: 10.1152/ajpcell.2000.278.1.C174. [DOI] [PubMed] [Google Scholar]
- 29.Tidball JG. Inflammatory cell response to acute muscle injury. Medicine and science in sports and exercise. 1995;27:1022–32. doi: 10.1249/00005768-199507000-00011. [DOI] [PubMed] [Google Scholar]
- 30.Heredia JE, Mukundan L, Chen FM, Mueller AA, Deo RC, Locksley RM, et al. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell. 2013;153:376–88. doi: 10.1016/j.cell.2013.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A special population of regulatory T cells potentiates muscle repair. Cell. 2013;155:1282–95. doi: 10.1016/j.cell.2013.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murphy MM, Lawson JA, Mathew SJ, Hutcheson DA, Kardon G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development. 2011;138:3625–37. doi: 10.1242/dev.064162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang X, La Rosa FG, Genova EE, Huber K, Schaack J, Degregori J, et al. Simultaneous activation of Kras and inactivation of p53 induces soft tissue sarcoma and bladder urothelial hyperplasia. PLoS One. 2013;8:e74809. doi: 10.1371/journal.pone.0074809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ribeiro S, Napoli I, White IJ, Parrinello S, Flanagan AM, Suter U, et al. Injury signals cooperate with Nf1 loss to relieve the tumor-suppressive environment of adult peripheral nerve. Cell reports. 2013;5:126–36. doi: 10.1016/j.celrep.2013.08.033. [DOI] [PubMed] [Google Scholar]
- 35.Maroun CR, Rowlands T. The Met receptor tyrosine kinase: a key player in oncogenesis and drug resistance. Pharmacology & therapeutics. 2014;142:316–38. doi: 10.1016/j.pharmthera.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 36.Allen RE, Sheehan SM, Taylor RG, Kendall TL, Rice GM. Hepatocyte growth factor activates quiescent skeletal muscle satellite cells in vitro. J Cell Physiol. 1995;165:307–12. doi: 10.1002/jcp.1041650211. [DOI] [PubMed] [Google Scholar]
- 37.Rodgers JT, King KY, Brett JO, Cromie MJ, Charville GW, Maguire KK, et al. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert) Nature. 2014;510:393–6. doi: 10.1038/nature13255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Reilly C, McKay B, Phillips S, Tarnopolsky M, Parise G. Hepatocyte growth factor (HGF) and the satellite cell response following muscle lengthening contractions in humans. Muscle Nerve. 2008;38:1434–42. doi: 10.1002/mus.21146. [DOI] [PubMed] [Google Scholar]
- 39.Sharp R, Recio JA, Jhappan C, Otsuka T, Liu S, Yu Y, et al. Synergism between INK4a/ARF inactivation and aberrant HGF/SF signaling in rhabdomyosarcomagenesis. Nat Med. 2002;8:1276–80. doi: 10.1038/nm787. [DOI] [PubMed] [Google Scholar]
- 40.Taulli R, Scuoppo C, Bersani F, Accornero P, Forni PE, Miretti S, et al. Validation of met as a therapeutic target in alveolar and embryonal rhabdomyosarcoma. Cancer Res. 2006;66:4742–9. doi: 10.1158/0008-5472.CAN-05-4292. [DOI] [PubMed] [Google Scholar]
- 41.Rong S, Jeffers M, Resau JH, Tsarfaty I, Oskarsson M, Vande Woude GF. Met expression and sarcoma tumorigenicity. Cancer Res. 1993;53:5355–60. [PubMed] [Google Scholar]
- 42.Scotlandi K, Baldini N, Oliviero M, Di Renzo MF, Martano M, Serra M, et al. Expression of Met/hepatocyte growth factor receptor gene and malignant behavior of musculoskeletal tumors. Am J Pathol. 1996;149:1209–19. [PMC free article] [PubMed] [Google Scholar]
- 43.Oda Y, Sakamoto A, Saito T, Kinukawa N, Iwamoto Y, Tsuneyoshi M. Expression of hepatocyte growth factor (HGF)/scatter factor and its receptor c-MET correlates with poor prognosis in synovial sarcoma. Human pathology. 2000;31:185–92. doi: 10.1016/s0046-8177(00)80218-x. [DOI] [PubMed] [Google Scholar]
- 44.Coley WB. II. The Influence of Injury upon the Development of Sarcoma. Ann Surg. 1898;27:259–84. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.